 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Virus-inspired designs of antimicrobial nanocapsules
Carlos H. B.
Cruz†
,
Irene
Marzuoli‡
 and
Franca
Fraternali
*
and
Franca
Fraternali
*
Randall Centre for Cell and Molecular Biophysics, King’s College London, London, UK. E-mail: franca.fraternali@kcl.ac.uk
First published on 13th August 2021
Abstract
Antimicrobial resistance is becoming a serious burden for drug design. The challenges are in finding novel approaches for effectively targeting a number of different bacterial strains, and in delivering these to the site of action. We propose here a novel approach that exploits the assembly of antimicrobial peptidic units in nanocapsules that can penetrate and rupture the bacterial membrane. Additionally, the chemical versatility of the designed units can be tailored to specific targets and to the delivery of genetic material in the cell. The proposed design exploits a β-annulus (sequence ITHVGGVGGSIMAPVAVSRQLVGS) triskelion unit from the Tomato Bushy Stunt Virus, able to self assemble in solution, and functionalised with antimicrobial sequences to form dodecahedral antimicrobial nanocapsules. The stability and the activity of the antimicrobial β-annulus capsule is measured by molecular dynamics simulations in water and in the presence of model membranes.
Introduction
For most of the last century, the development of new drugs has focused on the paradigm that a drug is a small inorganic compound (of mass up to 900 Da), which intervenes with a specific target (usually a protein) of a mammal or bacterial cell.Challenges have included identifying a target unambiguously, especially when the drug binds to proteins assembled in complexes or to a number of closely related gene products.1 An aspect of concern in the development pipeline has been the efficient delivery of the drug to the site of action: this has been puzzling pharmacologists and chemists for a while, and the aim to overcome these hurdles has inspired the work of synthetic biologists and material scientists. To complicate this further, the last few years have seen a dramatic increase in the development of antimicrobial resistance (AMR). The severity of the AMR threat is such that it has been raised to the status of national emergency in several countries, including the UK.2 Indeed, strict regulations in the health, agricultural and food industry sector must be enforced to prevent the misuse of antibiotics, as we are leaving the century in which these miracle drugs were discovered, to enter a phase in which we count the number losing their efficacy.3 AMR has become one of the major stimuli to overcome traditional approaches to drug-development.
One of the best nature-designed gene delivery systems are viruses, as they have evolved a number of efficient mechanisms to penetrate cell membranes and implement effective processes to transfer their genomic material to target cells. Recently, we have shown that engineered virus-like nanocapsules derived from synthetic multi branched peptides are able to promote bacterial membrane poration and are, at the same time, suitable for gene delivery.4,5
Altogether, the recent progress in understanding the mechanisms of antimicrobial resistance has helped in directing the development of new drugs. In particular, it has promoted the modification of existing compounds to overcome the resistance developed by bacteria.6
One crucial aspect in the efficacy of antimicrobials is their interaction and recognition of the cellular membrane and their ability to selectively perturb microbial versus mammalian membrane integrity.5 Therefore, modelling such interactions is very important to select the right lipid composition mixture and to fine-tune and characterise the specific peptide–lipid interactions.7
We focused our attention towards membrane active peptides as host-defence peptides belonging to the class of antimicrobial peptides (AMPs). These are naturally produced by Eukarya, either as stand-alone sequences or embedded in larger proteins, as a first, weak, and broad-spectrum defence against bacteria.8
To exploit their potential and engineer AMP-like molecules, a careful characterisation and classification of such peptides must be done. This task has been carried out over the last few decades, but because of its complexity, at present there are still many peptides with ascertained antimicrobial activity for which the mode of action is not fully understood.10 However, some general characteristics of these sequences and some of the mechanisms they employ have been ascertained.
Unsurprisingly, AMPs are heterogeneous in sequence, structure, targets and mode of action, to tackle the different challenges bacteria pose. Their size can vary between 6 and 59 amino acids:11 despite being small with respect to the average size of a protein in the human body, these macromolecules are hundreds of times larger than small molecule drugs and, as such, they have a different mode of action on bacteria.
As already mentioned, the target of AMPs is the bacterial membrane. Many of them cause disruption of the microbial membrane, while others translocate into the cytoplasm to act on intra-cellular targets, and the combination of the two is not uncommon either.12 In general, it is widely accepted that membrane interaction and the resulting membrane disruption are key factors for the antimicrobial activity of AMPs.9
The main determinant driving the interaction between AMPs and bacterial membranes is the positive charge that many AMPs present, as opposed to the negative charge of the membrane.9,13
Most AMPs have a positive charge which facilitates binding to the membrane via charge–charge recognition; accordingly, arginine and lysine residues are usually abundant in AMP sequences. However, the disruptive action takes place through the interaction of AMPs with the hydrophobic core of the membrane, therefore their sequences also contain hydrophobic aromatic residues, especially tryptophan, which favours anchoring to the lipid core.14
In terms of folds adopted by AMPs, these are rich in α-helix or β-sheet structures. Amphiphatic α-helices present a charged side which is tailored to face towards the phospholipid head groups, and a hydrophobic side which is favourably buried into the acyl chain core.
Several models have been proposed to describe the exact mechanism of AMPs penetration after they bind to the cytoplasmic membrane, and how their combined action leads to membrane permeabilisation.9,11
The picture becomes complex for oligomer-mediated insertion, i.e. when the action is triggered by the combined action of many copies of the peptide. At low peptide to lipid ratio, the favourable configuration is represented by peptides lying parallel to the membrane plane as described previously.15 An increase in peptide concentration triggers the transition to an inserted state: the organisation of AMPs inside the membrane core can assume different configurations, as previously described.9,11,13,16
We propose an alternative to this: engineered nanocapsules that contain multiple copies of AM peptides in their structure and that display an overall positive surface. As a constituent, we focus on an AM peptide derived from bovine lactoferricin that has known antimicrobial properties and acts synergistically with other antimicrobial agents by affecting the transmembrane potential and proton-motive force, resulting in inhibition of ATP-dependent multi-drug efflux pumps.17 A step further in engineering peptidic structures is the design of self-assembling functional blocks from first principles. Indeed, self-assembling peptides can form nanostructures ranging from nanoparticles to nanotubes, nanofibers, nanorods and hydrogels (Fig. 1).18
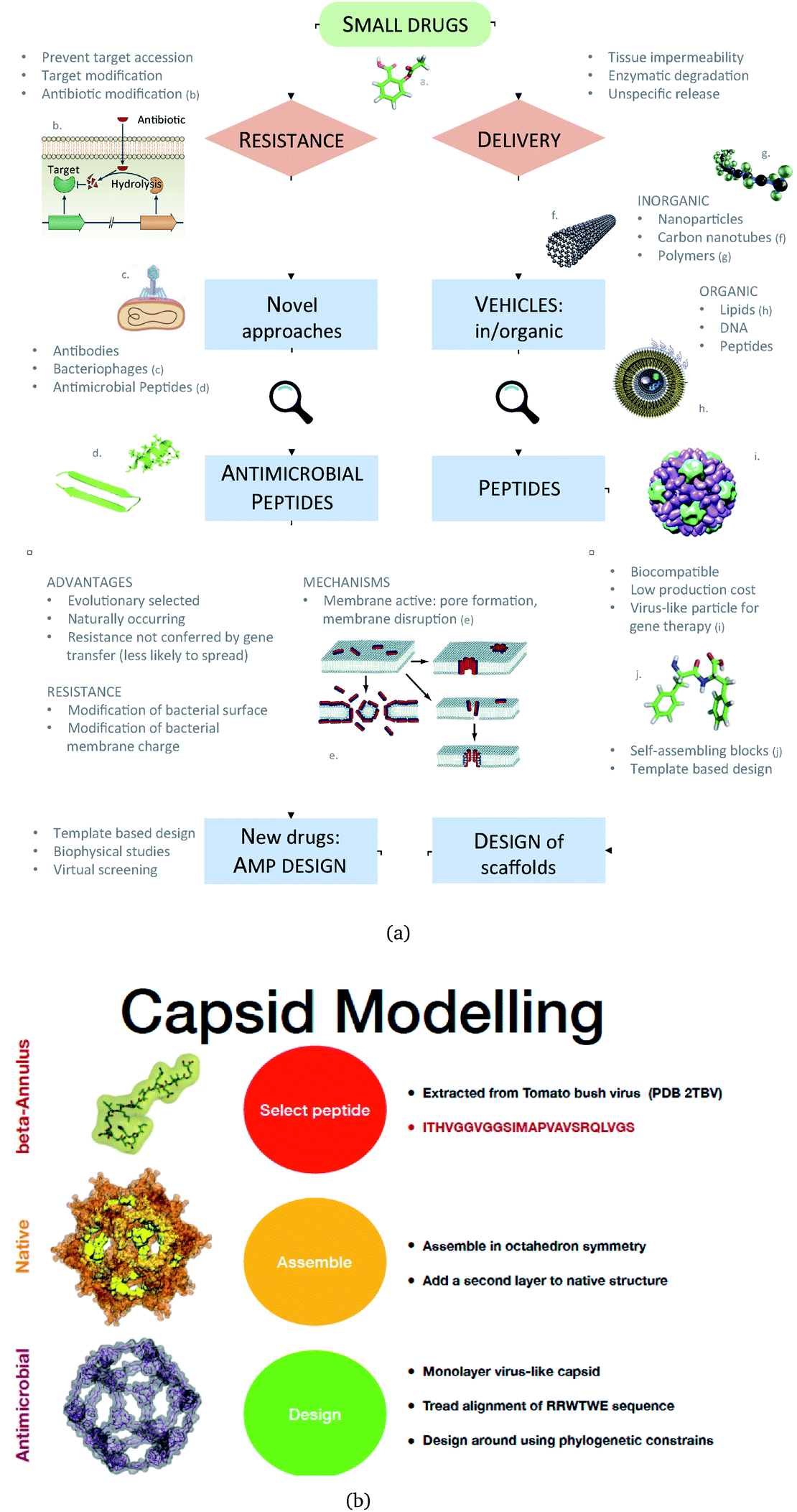 | ||
| Fig. 1 (a) A Schematic of the approaches used for AM peptide design and delivery strategies. (b) Strategy adopted to build the AM-β-annulus capsule. In red is the representation of the β-annulus triskelion units within the 2TBV structure. These were extracted and modelled to form a pentagonal geometry unit. This was the constituting block for the dodecahedron supra-molecular geometry of the monolayer and bilayer capsules. These were composed of 20 units for the internal later, and 60 units in the external layer. Figures adapted from ref. 8a–g. | ||
The strength of such molecular designs is in singling out the interactions that are crucial for a mechanism, clarifying whether they can be transferred to a different system or environment, and optimising their combined use in performing the desired function. Such principles can be used in the design, at the atomistic level, of novel and more effective AMPs.
We have previously described some of these engineered AM nanocapsules,4,5,19 here we want to describe a protocol to design a self-assembling nanocapsule inspired to the Tomato Bushy Structure by extracting some essential building blocks that infer the fullerene geometry and functionalise them with peptides that carry the AM activity.
The atomistic details of the nanocapsule assembly, necessary for the antimicrobial and gene delivery activities, are not accessible by experimental techniques. Therefore, the nanocapsule stability in water and its interaction with a model membrane was studied through molecular dynamics simulations, comparing the results with the available experimental data.4,19
Methods
Following up from previous work,4 we performed coarse-grained simulations of β-annulus (native-like and AM) capsules in solution and on model membranes, using the coarse-grained description provided by SIRAH.20,21 All simulations were performed with the GROMACS software, version 5.5 and 2016.22–24SIRAH simulations
The assembled capsules were solvated in an aqueous solution of sodium chloride at physiological concentration (0.15 mM) to achieve neutrality in the system. After energy minimisation, the systems were heated from 0 to 300 K over 10 ns at constant volume (10 fs time step), with protein coordinates constrained to their initial position.Distances between the paired arms of AM peptides within the capsule were restrained with a harmonic potential of 1000 kJ (mol nm2)−1, and progressively released over 80 ns (20 fs time step), then the systems were simulated for about 1 μs. The pressure and temperature were kept constant at 1 bar and 300 K, controlled respectively using a V-rescale thermostat and a Berendsen barostat with 2 ns collision-frequency and 8 ps pressure-coupling time. Short-range interactions were treated with a 1.2 nm cut-off radius, whereas the long-range interactions used the Particle Mesh Ewald (PME) method.
We used the recent lipid parametrization in the SIRAH force field21 united with its characteristic medium grained resolution between the atomistic and the MARTINI coarse-grained one,20 for the simulations of antimicrobial capsules on membranes (as reported in our previous work).5 For the simulation of the capsules on membranes, POPC and POPS (in ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were selected among the SIRAH available lipids, as POPC is zwitterionic and POPS is negatively charged, reflecting the characteristics of the DLPC and DLPG lipid used in other force-fields for representing the microbial membrane.
:1) were selected among the SIRAH available lipids, as POPC is zwitterionic and POPS is negatively charged, reflecting the characteristics of the DLPC and DLPG lipid used in other force-fields for representing the microbial membrane.
The SIRAH parameters and coarse-grained coordinates were assigned to the protein molecules through the cgconv script, distributed with the force field. For the membrane equilibration procedure please refer to Marzuoli et al.5
Virus inspired assembly: β-annulus design
We extracted the β-annulus (sequence ITHVGGVGGSIMAPVAVSRQLVGS) triskelion structure from the Tomato Bushy Stunt Virus (TBSV, PDB code: 2TBV25) (Fig. 2).
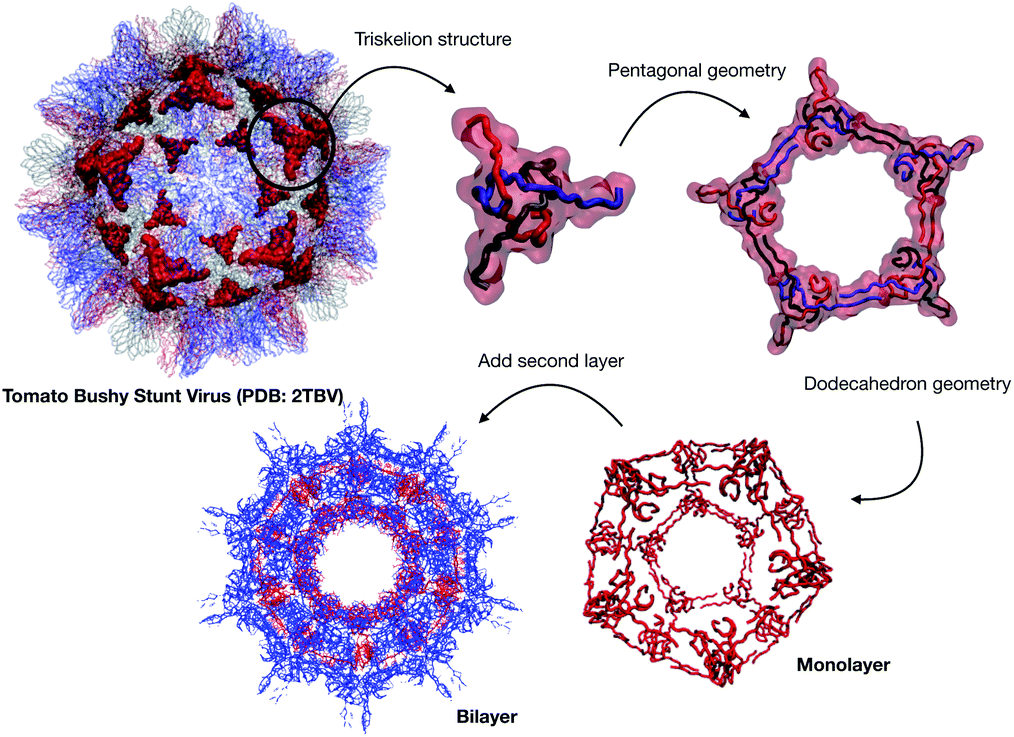 | ||
| Fig. 2 Structural components of the designed capsule. | ||
This unit has been demonstrated to assemble in a dodecahedron structure when decorated with a number of diverse nanoparticles, preserving the arrangement of the TBSV capsid structure.26 We therefore modelled the β-annulus only assembly (native like) in a dodecahedral geometry composed of twenty triskelion units. As one can notice from the figure representing the virus structure, the β-annulus triskelion co-assembles with other viral proteins, therefore extracting it from its original environment results in many hydrophobic residues exposed on the surface of the assembled dodecahedral nanocapsule (native-like monolayer). To stabilise this assembly we therefore generated a second layer of triskelion units.
Antimicrobial β-annulus design
We aimed to design a soluble single layer antimicrobial capsule (Fig. 3). This was challenging as we saw that the native single layer β-annulus capsule was not stable per se. To generate an AM capsule, we inserted the antimicrobial sequence RRWTWE at the end of the ‘arms’ of the β-annulus sequence. This generated a hybrid sequence containing the β-annulus and the antimicrobial stretch at the branching points. This was threaded onto the single layer capsule structure via the suite AlignedThread from Rosetta.27 The AM sequence RRWTWE structurally aligned with the RQLVS stretch at the C-terminal of the β-annulus sequence (Fig. 4). We joined the C-terminal fragments through anti-parallel β-sheets using the Chimera program28 to form a stable scaffold reminiscent of the capzip capsule.4 | ||
| Fig. 3 Pipeline of the design procedure adopted for both beta-annulus capsules (native and AM). | ||
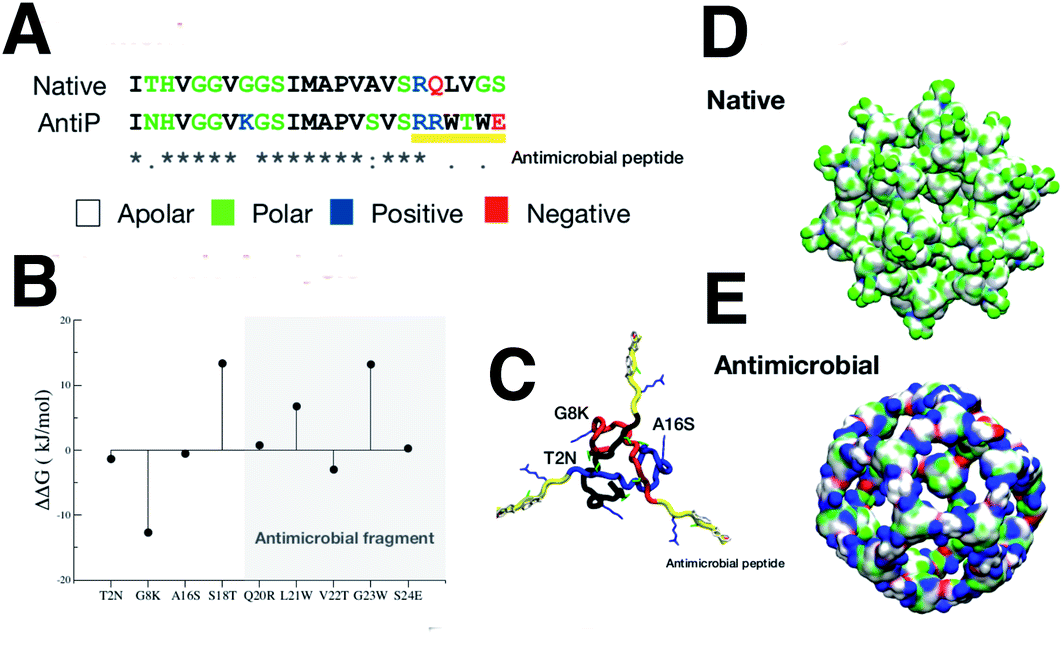 | ||
| Fig. 4 (A) Sequence alignment of the native and antimicrobial peptides. The colours of the amino acids reflect their properties, with apolar, polar, positively and negatively charged amino acids coloured, respectively, white, green, blue and red. (B) single mutational energy. The mutational energy was determined by the energy difference between the native and mutant. Mutations with positive energy not belonging to the antimicrobial fragment, were accepted. (C) Triskelion structure is shown with the three composing peptides backbone coloured in black, blue and red. The antimicrobial fragment was marked in yellow with the accepted mutations indicated in the picture. (D and E) the native internal layer and the monolayer antimicrobial capsule, are shown with surface representations reflecting the residue properties (colour scheme as in A). | ||
Monte Carlo cycles coupled with PSSM (Position Specific Mutation Score)-based mutagenesis rounds followed by energy minimization were performed, evaluating the energy within the full capsule assembly with ROSETTA scripts.27,29,30 Of these sampled mutations, only the ones with negative ΔΔG energy were accepted (Fig. 4).
The resulting structure (AM-β-annulus capsule) was simulated with the SIRAH force field in water for 1 μs (simulation conditions as before). The interaction with a microbial model membrane was tested in simulations with a POPC/POPS 3:1 bilayer.
The equilibrated AM-β-annulus capsule was initially placed at 2 nm of distance from the membrane surface, in a 150 mM NaCl solution (simulation conditions as above, except with semi-isotropic pressure coupling). The system was equilibrated for 0.6 μs, with the protein coordinates constraint in the initial positions. The constraints (1000 kJ (mol nm2)−1) were then released. Binding to the membrane occurred after about 1 μs, and the simulation was continued for an additional 1.5 μs to observe stability. In order to stimulate poration, a small electric field was applied across the z-axis (4 mV nm−1) to accelerate the capsule penetration in the lipid bilayer (see discussion points), and the simulation was continued for 3 μs.
Results
Design of an antimicrobial capsule
The native sequence and the modified AM with the RRWTWE peptide from lactoferrin are shown aligned in Fig.4A.In the figure the 3D structure of the native monolayer capsule exposing the hydrophobic (apolar) residues on the surface is shown. This was used as a template for the threading procedure (Fig. 4D).
The results of the mutagenesis are displayed in the ΔΔG plot in the figure. The negative ΔΔG values point to residues that better fit in the β-annulus triskelion structure. In total, 3 mutations are necessary to better stabilise the AM-β-annulus within the capsule (T2N, G8K and A16S).
The prediction of antimicrobial activity of the designed β-annulus sequences was performed via the AntiBP server,31 the results of which are reported in Table 1. The position column refers to the starting position in the 15 aa stretch used for the prediction of antibacterial activity. The score is the sum of the antimicrobial predicted probability over each residue within the sequence fragment from the evaluated peptide sequence. The first two fragments present predicted antibacterial activity: NHVGGVKGSIMAPVS and GSIMAPVSVSRRWTW. The second one has 67% of the amino acids making up the whole external surface of the designed capsid AM-β-annulus. Therefore, we speculate that the designed nanosphere will be able to interact with a bacterial membrane and perturb its structure. There is a candidate sequence with a better score (first row in the table), but this is very different from the AM lactoferrin sequence we wanted to test, and for which we have collected experimental results.4,19 The resulting AM capsule has dodecahedron geometry with the right balance of positive residues on the surface (Fig. 4E).
| Peptide sequence | Start position | Score | Antibacterial activity |
|---|---|---|---|
| NHVGGVKGSIMAPVS | 2 | 0.940 | Yes |
| GSIMAPVSVSRRWTW | 9 | 0.690 | Yes |
| GVKGSIMAPVSVSRR | 6 | 0.340 | No |
| HVGGVKGSIMAPVSV | 3 | 0.270 | No |
| VKGSIMAPVSVSRRW | 7 | 0.120 | No |
| INHVGGVKGSIMAPV | 1 | 0.000 | No |
| VGGVKGSIMAPVSVS | 4 | 0.000 | No |
| GGVKGSIMAPVSVSR | 5 | 0.000 | No |
| KGSIMAPVSVSRRWT | 8 | 0.000 | No |
Capsule stability in water
In Fig. 5 we observe that both the native (blue continuous line) and the antimicrobial (blue dotted line) capsules’ RMSD stabilises at around 1000 ns. The corresponding radii of gyration are subject to some shrinking during the first 500 ns of simulation, with the AM nanocapsule radii (red dotted line) adopting smaller values. The average size of the inner pore of the native capsule measured during the trajectory is around 0.80 nm, while for the AM capsid it is around 0.6 nm. The pores and the overall geometry of the capsule appear to be stable throughout the simulated time. | ||
| Fig. 5 Native and antimicrobial capsule stability. | ||
Having tested the stability of the designed capsules in solution, we measured the time-averaged atom density projected onto a 2D plane for both systems via the GROMACS densmap tool. Colours are more intense and localised for the double layer native capsule, while the AM-β-annulus single layer capsule shows a more diffuse pattern, indicative of a relatively higher flexibility (Fig. 6).
 | ||
| Fig. 6 Atom density projected onto the 2D plane of the designed capsules calculated along the trajectories. (A and D) Double-layer native capsid. (B and C) Single-layer designed antibacterial capsid. | ||
Analysis of the secondary structure content maintained during the simulation (Fig. 7) shows that the values for the AM-β-annulus are in between the values measured for the outer and inner layer of the native β-annulus capsule. We therefore strike a balance between flexibility and overall structural stability for the designed AM capsule.
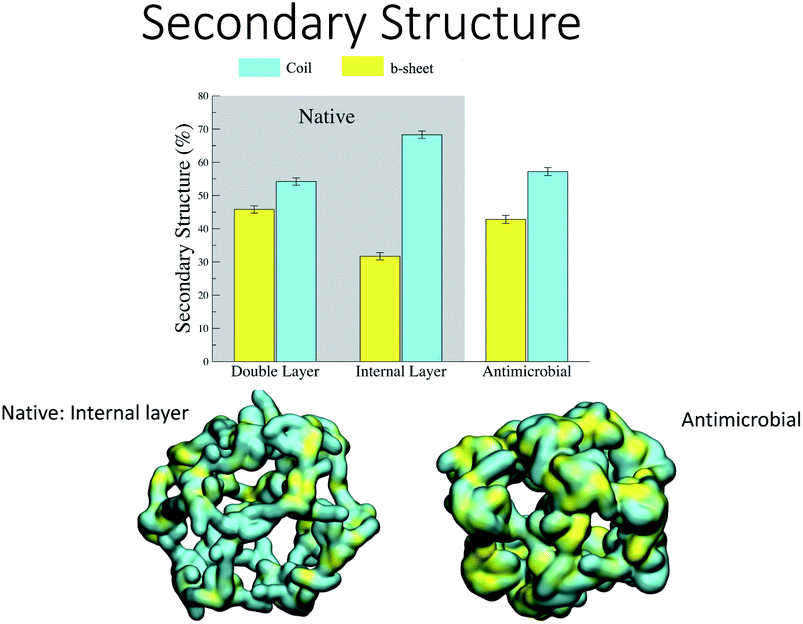 | ||
| Fig. 7 Secondary structure stability of the designed capsules. | ||
Interaction with model microbial membrane
These properties were tested by placing the AM-β-annulus near a model bacterial membrane constituting POPC and POPS lipids (in ratio 3:1).5
During the course of the unrestrained simulation, the capsule moves towards the membrane and starts to interact with it at around 1 μs, as shown by the number of protein–membrane contacts as a function of time (Fig. 8). This number increases quickly to 30 after 1 μs, and remains constant till 2.5 μs, at which point the electric field is switched on. We observe the formation of a pore within 1.5 μs from the field switch, and some atoms of the capsule reach the inner core of the membrane. The number of protein–membrane contacts increases consistently during the electric field driven simulation, reaching about 150 in total, 5 times larger than that observed in the simulation in the absence of an electric field. A similar trend was observed in our previous simulations with the capzip capsule5 in the interaction with a model membrane (DPLC/DPLG ratio 3:1), using a coarse-grained Martini parametrization. Clearly, the number of specific interactions is important not only for the effective attachment of the capsule on the surface but also in driving efficient penetration into the membrane (Fig. 9).
 | ||
| Fig. 8 Protein–lipid contact numbers across the entire simulated production time, before (black line) and after the application of the electric field (red-line). | ||
 | ||
| Fig. 9 (A) Poration of the model membrane. Purple spheres represent the phosphate lipid heads populating the pore. The sodium ions are represented by cyan spheres. Water molecules are represented in orange by small spheres. The capsule positioning onto the membrane at pore formation is displayed in the inset. (B) Frequency of contacts with lipid heads (red) and tails (blue) per aa (Sirah’s codes) without (top) and with electric field (bottom). (C) Radius of the pore measured along the z-axis of the membrane. Purple spheres are a pictorial representation of the lipid heads. | ||
In a previous study,5 we observed strong membrane invagination for the microbial-mimicking membrane when applying the electric field but no penetration. On the other hand, we did not observe attachment to the modelled mammal membrane, this was reassuring evidence that the capsule could discriminate between membranes increasing its effective AM activity on the microbial membrane. To evaluate the energetics of membrane penetration and the different process of penetration between these two membranes, we did resort to pulling the capsule through the two model membranes and computed the PMF profiles, highlighting a sensibly higher energy barrier for the mammalian model.
For the β-annulus simulations with and without electric field, reported here, in particular we observe lysine residues interacting favourably with the head groups of the POPS lipids, and these interactions increase in number once the capsule is in contact with the transmembrane lipidic portion.
The area per lipid (ApL), thickness of the membrane and lateral diffusion of lipid heads were calculated when the protein does not interact with the membrane (0.0–1.0 μs), after the interaction (1.5–2.0 μs) and under the effect of the electric field (4.5–5.5 μs), as shown in Table 2. The membrane shows similar ApL, thickness and lateral diffusion in the simulation without electric field before and after the capsule attachment, suggesting these interactions do not perturb the membrane. On the contrary, the electric field deforms the membrane and leads to a decrease in the lipid lateral diffusion in the presence of the capsule (Table 2). It is therefore speculated that the membrane–capsule interactions reduce the lateral mobility and elicit pore formation in the presence of an electric field. However, once the pore is stabilised, the order of the lipid tails shows only a slight decrease with respect to the value before the capsule attachment (Fig. 10).
 | ||
| Fig. 10 Order parameter of the POPC and POPS acyl chains 1 (circles) and 2 ( triangles) for the simulations without electric field and before capsule interaction (open symbols), and with electric field and a stable capsule interaction, in the last μs of simulation (filled symbols). | ||
| System | ApL, nm2 | Thickness, nm | LD, 10−6 cm2 s−1 |
|---|---|---|---|
| a BCI: before capsule interaction; ACI: after capsule interaction; ACI–EF: after capsule interaction with electric field. | |||
| BCI | 0.633 ± 0.0005 | 3.90 ± 0.07 | 0.251 ± 0.016 |
| ACI | 0.633 ± 0.0003 | 3.88 ± 0.05 | 0.210 ± 0.001 |
| ACI–EF | 0.631 ± 0.0011 | 3.73 ± 0.16 | 0.162 ± 0.012 |
Similar simulations (SIRAH force field, 4 mV nm−1 electric field, POPC/POPS 3:1 membrane) for the capsule tested in ref. 4, 5 and 19 (capzip) showed attachment to the membrane but no disruption. This is particularly relevant as capzip has experimentally proven antimicrobial activity. In this context, the simulations would suggest higher AM potency for the β-annulus capsule. This will have to be tested experimentally in future work. The capzip capsule relies on the same antimicrobial sequence, a similar triskelion structure, but a shorter linker (namely Lys–Lys–βAla). This results in a higher charge-to-hydrophobic residues ratio, which promotes attachment to the negatively charged membranes but allows for less plasticity. Additionally the capzip capsule is double layered and therefore intrinsically less bendy and malleable. The β-annulus capsule instead, displays more flexibility, and the simulation shows plastic rearrangement of its structure, contributing to pore initiation. For the capzip capsule the interaction with the membrane is especially mediated by the positively charged residues (arginine), analogously to what is observed in the present study. In both cases, the positive residues interact with the negative heads of the POPS lipids.5
Furthermore, it is interesting to observe how different force fields require different intensities of the electric field to achieve poration. In our previous work, where capzip molecules were simulated on a model microbial membrane, a field of 130 mV nm−1 was necessary to porate the membrane in an atomistic description with a value of 40 mV nm−1 for a MARTINI description (simulation length of few hundreds of nanoseconds). These values were not sufficient for poration in absence of the capsule. The simulations performed with the β-annulus capsule can instead be compared with the work of Zonta and coworkers,32 who tested different values of an electric field on a SIRAH membrane with a hemichannel structure inserted. In this work, a field of 100 mV nm−1 was sufficient for poration, while values in the range of 30 mV nm−1 were not able to perturb the simulated system. Therefore, the value used in the present work can accelerate poration in conjunction with the antimicrobial capsule, however it is unlikely to be sufficient to disrupt the membrane per se. The role of coarse-grained parametrization in responding to external perturbations like the electric force field explored here, should be investigated in more detail in the future.
For the data presented here, further work is needed to prove the activity of the β-annulus capsule: in line with previous work,4,19 time-resolved imaging of the supported lipid bilayer transfected with the capsule would verify if the capsule breaks simple membranes. Such experiments are the ones most closely related to the simulations. Additionally, the viability of bacterial cells exposed to the capsule can be measured. If the compound effectively acts as an antimicrobial, high-resolution imaging would give information on how the membrane is porated.4
Conclusions
We have proposed a framework for the in silico design of an antibacterial nanocapsule that is predicted to self-assemble in solution and is able to porate bacterial membranes. The originality of the design is in the combination of already recognised self-assembling triskelion β-annulus units with antibacterial peptidic sequences that have been demonstrated to form supramolecular arrangements rich in β-sheet structures. The AM-β-annulus dodecahedral nanocapsule is stable as a monolayer assembly: this allows for increased solubility and flexibility when compared to previous designs. The designed nanocapsule is able to interact with a bacterial model membrane and to cause poration through a stable pore induced by the application of a small electric field. The pore remains stable for the last 1 μs of simulation, as measured from the protein and lipid membrane atomic contacts occurring in the simulation. Future in silico studies will explore the delivery of small drugs within the model membrane and, in parallel, the synthesis of the designed units and their assembly in vitro.Author contributions
CHBC and IM performed the molecular design, computational mutagenesis and molecular simulations. FF conceptualized the project, supervised the methodology and data interpretation and wrote the paper with IM and CHBC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
IM and FF acknowledge funding by the Engineering and Physical Sciences Research Council (EPSRC) through the Centre for Doctoral Training ∼ Cross Disciplinary Approaches to Non-Equilibrium Systems ∼ (CANES, Grant No. EP/L015854/1). CHBC acknowledges funding from CAPES – Proc. no. 88881.197985/2018-01. This project made use of the following resources: computer time on ARCHER granted via the UK High-End Computing Consortium for Biomolecular Simulation, HECBioSim (http://www.hecbiosim.ac.uk), supported by EPSRC (grant no. EP/R029407/1); resources provided by the Cambridge Service for Data Driven Discovery (CSD3) operated by the University of Cambridge Research Computing Service (http://www.csd3.cam.ac.uk/), provided by Dell EMC and Intel using Tier-2 funding from the Engineering and Physical Sciences Research Council (capital grant EP/P020259/1), and DiRAC funding from the Science and Technology Facilities Council (https://www.dirac.ac.uk).Notes and references
- R. Santos, O. Ursu, A. Gaulton, A. P. Bento, R. S. Donadi, C. G. Bologa, A. Karlsson, B. Al-Lazikani, A. Hersey, T. I. Oprea and J. P. Overington, Nat. Rev. Drug Discovery, 2017, 16, 19–34 CrossRef CAS PubMed.
- J. H. Kwon and W. G. Powderly, Science, 2021, 373, 471 CrossRef CAS PubMed.
- J. O’Neill, Tackling Drug-Resistant Infections Globally: Final Report and Recommendations the Review on Antimicrobial Resistance, https://amr-review.org/sites/default/files/160525{\_g}Finalpaper{\_g}withcover.pdf, 2016 Search PubMed.
- I. E. Kepiro, I. Marzuoli, K. Hammond, X. Ba, H. Lewis, M. Shaw, S. B. Gunnoo, E. De Santis, U. Lapinska, S. Pagliara, M. A. Holmes, C. D. Lorenz, B. W. Hoogenboom, F. Fraternali and M. G. Ryadnov, Engineering chirally blind protein pseudo-capsids into nanoprecise antibacterial persisters, ACS Nano, 2020, 14(2), 1609–1622 CrossRef CAS PubMed.
- I. Marzuoli, C. H. B. Cruz, C. D. Lorenz and F. Fraternali, Nanoscale, 2021, 13, 10342–10355 RSC.
- G. Annunziato, Int. J. Mol. Sci., 2019, 20, 5844 CrossRef CAS PubMed.
- S. A. Kirsch and R. A. Böckmann, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 2266–2277 CrossRef CAS PubMed.
- (a) J. M. A. Blair, M. A. Webber and A. J. Baylay, et al. , Nat. Rev. Microbiol., 2015, 13(1), 42–51 CrossRef CAS PubMed; (b) J. Kim, Phage as a therapeutic agent, 2017, https://medium.com/@thryve/phage-as-a-therapeutic-agent-ed4c466302e5 Search PubMed; (c) M. D. Torres, S. Sothiselvam and T. K. Lu, et al. , J. Mol. Biol., 2019, 431(18), 3547–3567 CrossRef CAS PubMed; (d) The Innovation Society, 2019, http://innovationsgesellschaft.ch/en/carbon-nanotubes-can-be-toxic-to-aquatic-animals/; (e) Mettler Toledo, 2018, https://www.mt.com/au/en/home/applications/L1_AutoChem_Applications/L2_ReactionAnalysis/L2_Polymerization.html#publications; (f) Wikipedia, 2015, https://en.wikipedia.org/wiki/Liposome; (g) L. Schoonen and J. C. M. van Hest, Nanoscale, 2014, 6(13), 7124–7141 RSC.
- L. T. Nguyen, E. F. Haney and H. J. Vogel, Trends Biotechnol., 2011, 29, 464–472 CrossRef CAS PubMed.
- A. Ebbensgaard, H. Mordhorst, M. T. Overgaard, C. G. Nielsen, F. M. Aarestrup and E. B. Hansen, PLoS One, 2015, 10, e0144611 CrossRef PubMed.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed.
- R. E. W. Hancock and H.-G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS PubMed.
- M. Mahlapuu, J. Håkansson, L. Ringstad and C. Björn, Front. Cell. Infect. Microbiol., 2016, 6, 194 Search PubMed.
- D. I. Chan, E. J. Prenner and H. J. Vogel, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1184–1202 CrossRef CAS PubMed.
- L. Yang, T. A. Harroun, T. M. Weiss, L. Ding and H. W. Huang, Biophys. J., 2001, 81, 1475–1485 CrossRef CAS PubMed.
- T. Ebenhan, O. Gheysens, H. G. Kruger, J. R. Zeevaart and M. M. Sathekge, BioMed Res. Int., 2014, 2014, 867381 Search PubMed.
- J. L. Gifford, H. N. Hunter and H. J. Vogel, Cell. Mol. Life Sci., 2005, 62, 2588–2598 CrossRef CAS PubMed.
- T. Fan, X. Yu, B. Shen and L. Sun, J. Nanomater., 2017, 2017, 1–16 Search PubMed.
- V. Castelletto, E. de Santis, H. Alkassem, B. Lamarre, J. E. Noble, S. Ray, A. Bella, J. R. Burns, B. W. Hoogenboom, M. G. Ryadnov, J. Vandesompele and C. T. Wittwer, Chem. Sci., 2016, 7, 1707–1711 RSC.
- M. R. Machado, E. E. B. Guisasola, F. Klein, M. Sóñora, S. Silva and S. Pantano, J. Chem. Theory Comput., 2019, 15, 2719–2733 CrossRef CAS PubMed.
- E. E. Barrera, M. R. Machado and S. Pantano, J. Chem. Theory Comput., 2019, 15, 5674–5688 CrossRef CAS PubMed.
- H. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- M. J. Abraham, D. van der Spoel, E. Lindahl, B. Hess and the GROMACS development team, GROMACS User Manual, 2021 Search PubMed.
- P. Hopper, S. Harrison and R. Sauer, J. Mol. Biol., 1984, 177, 701–713 CrossRef CAS PubMed.
- S. Fujita and K. Matsuura, Nanomaterials, 2014, 4, 778–791 CrossRef PubMed.
- S. Ovchinnikov, L. Kinch, H. Park, Y. Liao, J. Pei, D. E. Kim, H. Kamisetty, N. V. Grishin and D. Baker, eLife, 2015, 4, e09248 CrossRef PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- S. J. Fleishman, A. Leaver-Fay, J. E. Corn, E.-M. Strauch, S. D. Khare, N. Koga, J. Ashworth, P. Murphy, F. Richter, G. Lemmon, J. Meiler and D. Baker, PLoS One, 2011, 6, e20161 CrossRef CAS PubMed.
- A. Goldenzweig, M. Goldsmith, S. E. Hill, O. Gertman, P. Laurino, Y. Ashani, O. Dym, T. Unger, S. Albeck, J. Prilusky, R. L. Lieberman, A. Aharoni, I. Silman, J. L. Sussman, D. S. Tawfik and S. J. Fleishman, Mol. Cell, 2016, 63, 337–346 CrossRef CAS PubMed.
- S. Lata, B. K. Sharma and G. P. Raghava, BMC Bioinf., 2007, 8, 1–10 CrossRef PubMed.
- F. Zonta, D. Buratto, G. Crispino, A. Carrer, F. Bruno, G. Yang, F. Mammano and S. Pantano, Front. Mol. Neurosci., 2018, 11, 170 CrossRef PubMed.
Footnotes |
| † Present address: Instituto de Tecnologia Química e Biológica António Xavier, Universidade Nova de Lisboa, Oeiras, Portugal. |
| ‡ Present address: Process Chemistry and Catalysis, Biocatalysis, F. Hoffmann-La Roche Ltd, Basel, Switzerland. |
| This journal is © The Royal Society of Chemistry 2021 |