 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterogeneity of cell membrane structure studied by single molecule tracking†
Gregory I.
Mashanov
 *a,
Tatiana A.
Nenasheva
b,
Alla
Mashanova
c,
Remigijus
Lape‡
d,
Nigel J. M.
Birdsall
a,
Lucia
Sivilotti
d and
Justin E.
Molloy
*a
*a,
Tatiana A.
Nenasheva
b,
Alla
Mashanova
c,
Remigijus
Lape‡
d,
Nigel J. M.
Birdsall
a,
Lucia
Sivilotti
d and
Justin E.
Molloy
*a
aThe Francis Crick Institute, 1 Midland Road, London, NW1 1AT, UK. E-mail: Gregory.mashanov@crick.ac.uk; Justin.molloy@crick.ac.uk
bKoltzov Institute of Developmental Biology, 26 Vavilova Str., Moscow, 119334, Russia
cSchool of Life and Medical Sciences, University of Hertfordshire, College Lane, Hatfield, Hertfordshire AL10 9AB, UK
dDepartment of Neuroscience, Physiology and Pharmacology, Division of Biosciences, University College London, Gower St., London, UK
First published on 14th October 2021
Abstract
Heterogeneity in cell membrane structure, typified by microdomains with different biophysical and biochemical properties, is thought to impact on a variety of cell functions. Integral membrane proteins act as nanometre-sized probes of the lipid environment and their thermally-driven movements can be used to report local variations in membrane properties. In the current study, we have used total internal reflection fluorescence microscopy (TIRFM) combined with super-resolution tracking of multiple individual molecules, in order to create high-resolution maps of local membrane viscosity. We used a quadrat sampling method and show how statistical tests for membrane heterogeneity can be conducted by analysing the paths of many molecules that pass through the same unit area of membrane. We describe experiments performed on cultured primary cells, stable cell lines and ex vivo tissue slices using a variety of membrane proteins, under different imaging conditions. In some cell types, we find no evidence for heterogeneity in mobility across the plasma membrane, but in others we find statistically significant differences with some regions of membrane showing significantly higher viscosity than others.
Introduction
It is now thought that structural heterogeneity of the plasma membrane plays a critical role in a variety of cell functions.1–2 This contrasts with our classical view of the plasma membrane as a fluid mosaic bilayer of phospholipids and other amphipathic molecules within which proteins can diffuse in an unhindered manner.3 The path taken by a diffusing transmembrane protein should follow a random Brownian walk and a plot of its mean-squared displacement against time interval (MSD vs. dT) should be linear,4 with a gradient determined by Dlat, the lateral diffusion coefficient. Dlat should be a linear function of membrane viscosity and thickness but should have only a weak dependence (i.e. log-function) on protein radius and viscosity of the bounding aqueous media (i.e. cytosol and extra cellular fluid).5,6 Recently a revised picture of the plasma membrane has emerged in which it is thought to have a more heterogenous structure compared to the classical fluid mosaic model, and there is biochemical and biophysical evidence for submicron, phase-separated, domains7 that are enriched with particular lipid species8 (“lipid rafts”) and regions of protein crowding or clustering. In addition, interaction of membrane proteins with the cortical cytoskeleton9 or extracellular matrix can lead to obstructions to free-diffusion10 (“picket-fences”). Together these features contribute to anomalous diffusive behaviour11,12 whereby MSD vs. dT plots are non-linear and show distinct downward curvature. The features that contribute to membrane heterogeneity are thought to be transient in nature and to extend over a length scale <200 nm, making them notoriously difficult to study in live cells. However, the advent of super-resolution, single molecule imaging methods2,13 allows “rafts”, “picket fences”, protein clustering and other heterogeneous properties to be studied in live cells and tissue slices in real-time.14,15 An advantage of single molecule studies is that they eliminate problems of bulk averaging that can mask short-lived, sub-resolution structural heterogeneities. Individual transmembrane proteins have been used as probes of membrane structure because they are of uniform size, can be labelled with high-specificity and known stoichiometry, they target to known membrane compartments and genetic manipulation allows structural alterations. However, tracking of single fluorophores in live cells is technically challenging because the spatial resolution and temporal precision of tracking is limited by photon emission rate and photobleaching of the fluorescent moiety. A useful figure of merit for a given fluorophore is the average number of photons emitted before photobleaching, giving rise to the idea of a “photon budget”.16 For a given experiment, best use of the photon budget requires a compromise to be made between tracking precision and measurement bandwidth. To measure small-scale, rapid movements, high laser power is required so that single fluorophores are bright and can be tracked with high-precision at fast video frame rate, but the cost is they soon photobleach. At low laser power, fluorophores can be tracked for longer but tracking resolution is reduced because fewer photons are emitted per second. It is also important to consider fluorophore density across the sample field of view (see ESI of ref. 14†) because at high density, fluorophore tracking becomes difficult as paths overlap, whereas at low density fewer molecules pass through a given region of interest making it difficult to accumulate sufficient data for statistical analysis. In general, fluorescent fusion proteins (like eGFP) photobleach faster than synthetic organic fluorophores (like rhodamine or Cy-dyes) and offer a lower photon budget.The aim of the current study has been to test whether live mammalian cell membranes exhibit heterogeneity in viscosity on the micron length-scale. We have explored whether heterogeneity in protein mobility is evident in immortalised cell lines, primary cell culture and live tissue samples using different protein probes. Total internal reflection fluorescence microscopy (TIRFM)17 was used to image membrane proteins labelled with single fluorophores and studied their diffusive behaviour to infer local membrane properties. We used a quadrat sampling method that enables statistical tests to be performed on data obtained from different regions of the cell. Trajectories were segmented and diffusion coefficients computed from ≥10 data points (∼0.5 second) sections of each trajectory. The image of the cell was then sub-divided into a checkerboard pattern of sample quadrats (∼1–4 μm2 area each), and local statistics were calculated for trajectory segments that resided within each quadrat so statistical tests could be performed across different regions of the cell membrane. For most of the specimens tested, protein mobility was homogenous across the cell surface but in some cells we found evidence for spatial heterogeneity. We conclude that lipid rafts (if they exist) are evenly distributed across the plasma membrane of most cells, but in some cell types, they might show heterogeneity in density forming a “raft of rafts”.
Experimental
All chemicals were obtained from Sigma-Aldrich, UK, unless stated otherwise.Cultured cell transfection and tissue labelling
All of the cells and tissue materials were imaged using a custom-made imaging chamber described previously.18 Human umbilical vein endothelial cells (HUVECs) (PromoCell GmbH, Heidelberg, Germany) derived from a pooled, primary cell culture were transfected with M2-eGFP muscarinic receptor fusion protein (kind gift of Prof. J. W. Wells, University of Toronto, Canada) using nucleofection according to the manufacturer’s instructions (Amaxa GMBH). CHO-K1 cells were transfected with cDNAs encoding the mouse α1, β1, and δ nicotinic receptor subunits subcloned within vector pRBG4 (kind gift of S. M. Sine, Mayo Clinic, MN, USA). The mouse γ-subunit was tagged with eGFP inserted into the cytoplasmic loop between transmembrane helices M3 and M4 and subcloned in pRK5 plasmid (kind gift of V. Witzemann, Max-Plank-Institute, Heidelberg, Germany). Primary cardiomyocytes were prepared as described previously14 and allowed to settle on to the imaging coverslip before labelling with Cy3B-telenzepine (10 nM for 1 h at 23 °C). The HL1 cardiomyocyte cell-line was cultured for 24 h before labelling with Cy3B-telenzepine (1 nM for 3.5 h at 23 °C). Zebrafish heart was removed and washed in room temperature PBS (pH 7.2) (Thermo Fisher Scientific, UK) solution supplemented with 10 U ml−1 heparin and 100 U ml−1 penicillin–streptomycin for 5 min. After washing, the heart was moved into an ice-cold “relaxing solution” consisting of PBS supplemented with; 1 mM EDTA, 2.5 mM KOH, and 3 mM MgCl2 for 5 min. A cutting block, with a 2 mm diameter cavity, and a single cutting-slot was used to bisect the heart along the ventricle axis.15 Mouse embryonic heart slices were prepared using procedures described previously.14 The prepared tissue slices were placed in relaxing PBS solution containing 10 nM of Cy3B-telenzepine for 1 h at 4 °C. This procedure labelled >95% of M2 muscarinic receptors with the tight-binding fluorescent ligand. The tissue slice was fixed against the coverslip with a fine nylon mesh grid (∼0.5 × 0.5 mm2) stretched in a stainless-steel tambour. The chamber was filled with Hank’s Balanced-Salt solution supplemented with 20 mM HEPES (pH 7.4). All imaging was performed at 23 °C.TIRF imaging
A custom-built TIRF microscope was used, as described previously.19 Briefly, the beam from a 100 mW, 488 nm laser or 150 mW, 561 nm laser (Light HUB-6, Omicron, Germany) was expanded using a Galilean beam expander and focused at the back focal-plane of a high numerical aperture, oil-immersion, objective lens (PlanApo, 100×, NA 1.45, Olympus, Japan) using a small, aluminium-coated mirror (3 mm diameter, Comar Optics, UK) placed at the edge of the back-aperture of the objective lens. The average laser power at the specimen plane was adjusted to ∼0.5 μW μm−2 and the incident laser beam angle was adjusted to ∼63° to create the evanescent field at the glass–aqueous medium interface. A second small mirror was placed at the opposite edge of the objective lens back-aperture to remove the returning (internally-reflected) laser beam from the microscope and a narrow band-pass emission filter FF01-525/50 or FF01-593/40 (Semrock, Rochester, NY) was used to block the scattered 488/561 nm laser light and other unwanted light. An EMCCD camera (iXon897BV, Andor, UK) captured video sequences at a rate of 20–50 fps, and the data were stored on a computer hard drive for analysis.Video data analysis
Video image sequences were analysed using GMimPro software (http://www.mashanov.uk), which employs an automatic single particle tracking algorithm (described previously20) to detect and track individual fluorophores. The position of each fluorescent spot was localised with sub-pixel resolution using a Gaussian fitting method and systematic drift was corrected via cross-correlation. Individual particle trajectories were output as a table of x,y coordinates measured at each video frame (i.e. each time point) for every fluorophore detected in the sample. From the tabulated data, the mean-squared displacement (MSD) of each fluorophore was computed over all possible time intervals (dT) (e.g. 1,2,3…n frames). MSD vs. dT plots were then generated to examine the particle motion; a linear-relationship is expected for simple Brownian motion, but curvature indicates anomalous diffusion (discussed later). To extract statistically meaningful mobility data from the trajectory analysis, we analysed trajectories where single fluorescent spots could be tracked for at least 10 consecutive frames with a permitted spot displacement of ≤0.7 μm between frames.20Single molecule mobility maps
We created a map to depict the lateral diffusion coefficient of our labelled protein probes by taking sequential 10-frame time windows (∼400 ms) along each single molecule trajectory and estimating the local Dlat value from the gradient of the MSD vs. dT plot. For display purposes, tracks were dilated to be 5 pixels wide (0.5 μm) and coded to produce a pseudo-colour heat map of membrane viscosity. At positions where trajectories overlapped, the pixel intensity was averaged. This approach is similar, but not identical, to single particle velocimetry.Measuring the local heterogeneity of cell membrane
We divided the image of each cell into a checkerboard pattern of sample quadrats and selected segments of single molecule trajectories that resided within each quadrat region. The local trajectory segments (at least 5 trajectories, ≥10 data points each per segment) were used to build an average MSD vs. dT plot. The initial slope of the plot was used to estimate the local Dlat (i.e. slope/4, for 2-dimensional diffusion). Local Dlat estimates were binned and histogrammed to build a global mobility map across the cell surface. The distribution of Dlat values of individual trajectories is described by a Gamma distribution due to the limited number of data points in each trajectory, but, according to the central limit theorem, the distribution of mean Dlat values measured at each quadrat sample should be normally distributed (i.e. Gaussian). We tested for heterogeneity across the checkerboard pattern of quadrat samples against an expectation based on the global distribution. The normality of the distributions was checked using the Kolmogorov–Smirnov (KS) test. The pairwise comparisons were done using Student’s t-test when both distributions were normal, and Mann–Whitney U (MW) test when at least one distribution was not normal.21 Calculations were performed using open-source R software.22Results
Comparison of membrane viscosity in HL1 cell-line vs. primary cardiomyocytes
We first imaged HL1 cells, a cardiac muscle cell-line, that natively-express M2 muscarinic acetylcholine receptors (Fig. 1A and Movie 1†). Receptors were labelled using the tight-binding fluorescent ligand Cy3B-telenzepine14 under conditions that gave an optimal density of labelled receptors (∼0.8 receptors per μm2) for single molecule imaging and tracking purposes. Under the conditions used here, the Cy3B photobleaching rate was 0.19 s−1, (Fig. S1A†) which allowed us to track individual molecules for around 5 s. Many trajectories were prematurely truncated because fluorophores overlapped during a single frame. This caused one track to terminate and a new track to be generated when the fluorophores separated. A small number of, non-specifically, surface-bound Cy3B-telenzepine molecules were completely immobile throughout the video recording and these objects were removed from the dataset. The projected image of all single molecule trajectories shows the degree of coverage across the cell surface (Fig. 1A(ii) and S2†). The cell periphery has a higher track density because fresh (unbleached) fluorophores diffuse into this region from the apical cell surface which is beyond the evanescent field.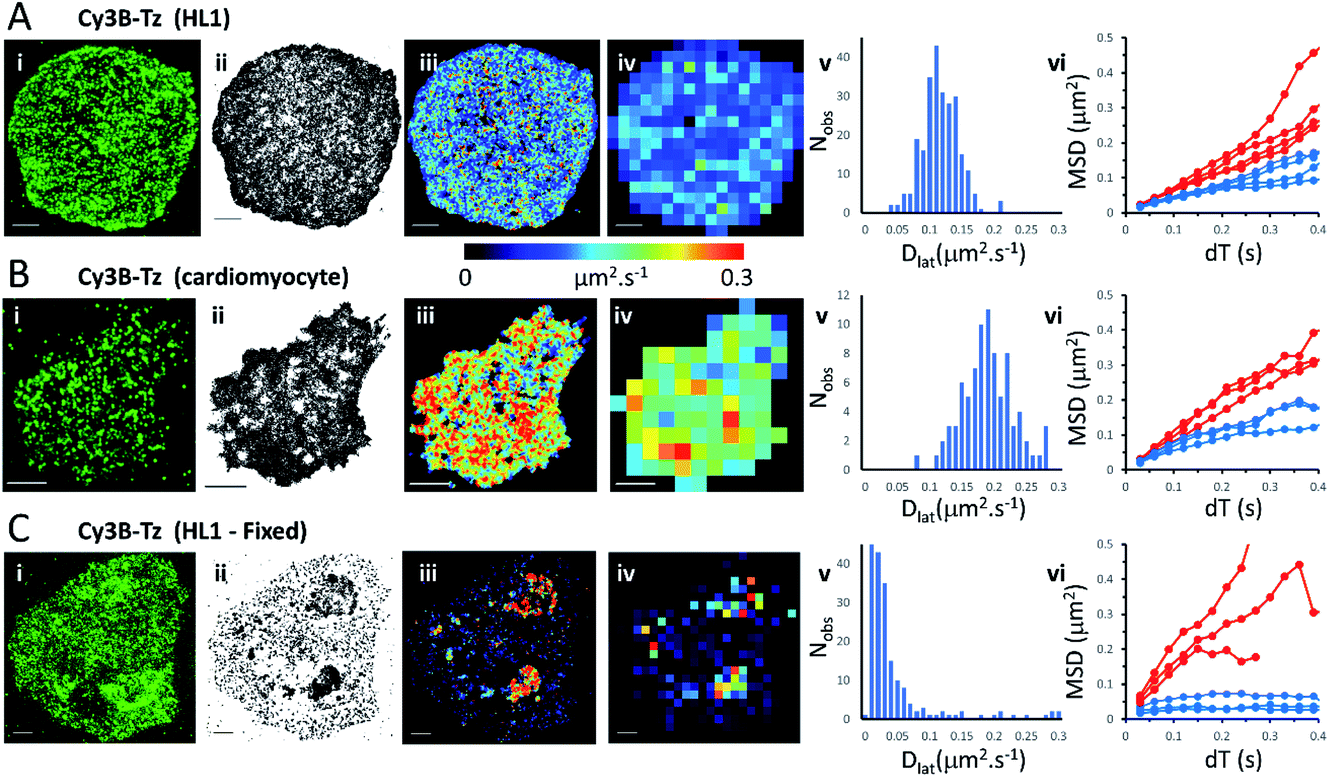 | ||
| Fig. 1 M2 muscarinic acetylcholine receptor diffusion on the plasma membrane of HL1 cells and primary cardiomyocytes. (A(i)) First video frame showing an HL1 cell-line with Cy3B-telenzepine labelled M2 receptors. (ii) A projection of all single molecule trajectory paths. (iii) Heat-map showing locally averaged Dlat values; mean Dlat = 0.12 μm2 s−1, n = 7526 trajectories. (iv) Heat map of the quadrat-sampled (2 × 2 μm2) Dlat values. (v) Distribution of Dlat measured in each quadrat region across the cell: 250 compartments, overall mean (of the local mean values) Dlat = 0.112 ± 0.028 μm2 s−1 (±SD). (vi) MSD vs. dT plots for typical fast (where Dlat > mean +1.5 × SD; red) and slow (where Dlat < mean −1.5 × SD; blue) quadrat regions. Video was recorded at 33 fps. (B) Panels as for A except: (i) primary cardiomyocyte. (iii) Mean Dlat = 0.203 μm2 s−1, n = 3133 trajectories. (v) Quadrats: 86 (2 × 2 μm2); mean Dlat = 0.188 ± 0.043 μm2 s−1. (vi) See panel A. (C) Panels as for A except: (i) HL1 cell chemically fixed with 1% paraformaldehyde. (ii) Trajectory paths show that most molecules have very restricted motion following fixation. Some regions show receptor diffusion (dark areas where molecular trajectories greatly overlap). (iii) Most M2 receptors are fixed, but some isolated regions show rapid receptor diffusion; average Dlat = 0.132 μm2 s−1, n = 6673 trajectories. (iv) Quadrats: 327 (2 × 2 μm2). (v) Distribution of Dlat values with overall mean Dlat = 0.029 ± 0.05 μm2 s−1. Fixed regions indicate the tracking noise floor (see main text and Fig. S1 C and D†). (vi) See panel A. | ||
Analysis of individual trajectories (see methods) leads to estimates of Dlat for each molecule tracked and these values can be plotted as a pseudo-colour map (Fig. 1A(iii)) indicating M2 receptor mobility at different regions across the cell surface. To test for homogeneity, the mobility map is divided into a checker-board pattern of sample quadrats (Fig. 1A(iv)). Each quadrat is then colour-coded to represent the average Dlat value within that region. According to the central limit theorem the distribution of mean Dlat values should be Gaussian (Fig. 1A(v)), and in the following text “mean Dlat” refers to the “mean of the quadrat means”. To investigate whether the diffusion of molecules in quadrats with extreme low or high Dlat values was anomalous we plotted MSD vs. dT diagrams from those, selected, quadrats (Fig. 1A(vi), S3A, B† and Table 1).
| Figure | Sample | Mean (μm2 s−1) | SD | n | K–S D-statistic | p-Value | Conclusion |
|---|---|---|---|---|---|---|---|
| a Significance testing against normal distribution: *p-value < 0.05 (e.g. >95% confidence); **p-value < 0.01 ***p-value < 0.001. | |||||||
| Fig. 1 | M2-Cy3B HL1 | 0.112 | 0.028 | 250 | 0.045 | 0.704 | Normal |
| M2-Cy3B primary myocyte | 0.188 | 0.043 | 86 | 0.073 | 0.747 | Normal | |
| M 2 -Cy3B HL1 fixed | 0.029 | 0.050 | 327 | 0.283 | <0.0001 | Not-normal*** | |
| M2-Cy3B inter-cell HL1 | 0.129 | 0.015 | 29 | 0.121 | 0.793 | Normal | |
| Fig. 2 | M 2 -eGFP-HUVEC | 0.204 | 0.086 | 150 | 0.178 | 0.0002 | Not-normal*** |
| Fig. 3 | M2-Cy3B zebrafish cell 1 and cell 2 | 0.224 | 0.116 | 49 | 0.185 | 0.069 | Normal |
| Zebrafish cell 1 | 0.291 | 0.144 | 20 | 0.198 | 0.367 | Normal | |
| Zebrafish cell 2 | 0.179 | 0.056 | 29 | 0.131 | 0.699 | Normal | |
| M 2 -Cy3B mouse slice (all cells) | 0.427 | 0.457 | 79 | 0.294 | <0.0001 | Not-normal*** | |
| Mouse slice cell 1 | 0.533 | 0.518 | 27 | 0.253 | 0.052 | Normal | |
| Mouse slice cell 2 | 0.534 | 0.590 | 18 | 0.370 | 0.010 | Not-normal** | |
| Mouse slice cell 3 | 0.285 | 0.228 | 34 | 0.260 | 0.020 | Not-normal* | |
| Fig. 4 | Nicotinic-eGFP-CHO | 0.114 | 0.078 | 181 | 0.191 | <0.0001 | Not-normal*** |
| Fig. 5A | Model | 0.188 | 0.034 | 227 | 0.046 | 0.721 | Normal |
| Fig. 5B | Model (both regions) | 0.111 | 0.080 | 226 | 0.192 | <0.0001 | Not-normal*** |
| Model (non-raft) | 0.188 | 0.030 | 120 | 0.056 | 0.847 | Normal | |
| Model (raft region) | 0.031 | 0.021 | 116 | 0.160 | 0.005 | Not-normal** | |
We next imaged M2 receptors in primary cultured mouse cardiomyocytes that had been labelled the same way as the HL1 cells (Fig. 1B and Movie 2†). We found receptor diffusion was significantly faster (Dlat = 0.2 μm2 s−1) compared to the HL1 cell-line (t-test with unequal variances (p < 0.0001), t(111) = −15.4, p < 2.2 × 10−16), implying membrane viscosity was lower in the primary cultured cells. The molecular tracks were analysed as before and binned to give a checkerboard pattern of quadrat samples across the cell (Fig. 1B(i–vi) and Table 1).
In order to measure localisation and tracking errors (i.e. the “noise floor” of our measurements), we imaged HL1 cells that had been chemically fixed using 1% paraformaldehyde. Interestingly, after several minutes treatment with fixative, many cells still had isolated regions, or “islands”, of membrane that appeared sealed off from the fixative and where the Cy3B-telenzepine-labelled receptors continued to diffuse (Fig. 1C and Movie 3†). This fixation artefact gave the opportunity to generate diffusion maps in cells that showed extreme heterogeneity in receptor mobility due to incomplete fixation. Cell regions where chemical fixation was effective allowed individual molecules to be selected and tracked in order to give an estimate of the localisation and tracking error measured under our imaging conditions. The sum of all noise sources gave 33 nm root mean squared deviation (rms) (i.e. MSD = 1 × 10−3 μm2) (Fig. S1C and D).† However, the quadrat sampling method gave a three-fold higher estimate of the noise floor: rms ∼100 nm (MSD = 1 × 10−2 μm2) (Fig. 1C(v)). This difference arose because mobile molecules were often present within a given quadrat region. The quadrat mapping of Dlat values showed a heterogeneous distribution of values which is readily explained by the incomplete fixation, whereby some islands of plasma membrane seemed to be isolated and protected from the paraformaldehyde treatment. The mean Dlat values determined for each of the quadrat sample regions were histogrammed as before. The Dlat values showed an exponential distribution (Fig. 1C(iv)). Typical slow-moving quadrat regions (blue lines in Fig. 1C(vi)) exhibited MSD vs. dT plots that were highly anomalous, consistent with the molecules being either totally immobile or trapped in a confined area. The faster moving, outlier, Dlat values were from unfixed regions of membrane and MSD vs. dT plots (red lines in Fig. 1C(vi)) showed a variety of behaviours, with some quadrat regions showing a relatively linear slope of MSD vs. dT plots and others showing a distinct downward curvature consistent with anomalous diffusive behaviour (Table 1).
Mapping membrane viscosity using eGFP-tagged M2 receptors
We next examined the mobility of a C-terminally-tagged M2-receptor eGFP-fusion protein where the fluorophore is positioned on the intracellular-side of the molecule in contrast to the extrinsic, synthetic labelling with Cy3B-telenzepine used in the previous experiments. HUVECs were transiently transfected with a recombinant M2-eGFP fusion construct in a mammalian cell expression vector and cells were then selected for imaging based on their receptor expression level (target level ∼0.8 receptors per μm2). The eGFP-tagged receptors moved rapidly and in a seemingly unhindered fashion across the plasma membrane (Fig. 2 and Movie 4†). The photobleaching rate of the eGFP fluorophore was 0.4 s−1 (Fig. S1B†) which is twice as fast as the Cy3B fluorophore. In addition to that, fluorophores could only be tracked for ∼0.5 s, because the eGFP signal-to-noise intensity ratio is lower than for Cy3B and this leads to premature track termination (i.e. tracking can fail before the fluorophore bleaches). Individual static molecules were selected and tracked in order to give an estimate of the localisation and tracking error measured under our imaging conditions. The sum of all noise sources gave 26 nm root mean squared deviation (rms) (i.e. MSD = 1 × 10−3 μm2) (Fig. S1C and D†), similar precision to Cy3B. The average rate of receptor diffusion (Dlat = 0.2 μm2 s−1) was similar to the primary cardiomyocytes (MW, U = 6265, p = 0.715) but the trajectories appear more compact (Fig. 2(ii)vs.Fig. 1A and B panel (ii)) simply because they are of shorter duration. The short tracks mean that there is less coverage (i.e. fewer unique tracks per unit area) when membrane viscosity maps are generated using a 4 × 4 μm2 quadrat size (Table 1). | ||
| Fig. 2 eGFP-tagged M2 muscarinic acetylcholine receptor diffusing at the plasma membrane of HUVECs. (i) Single image from the beginning of the record shows individual eGFP-tagged M2 receptors at the plasma membrane. (ii) Trajectory map; 5031 trajectories. (iii) Heat-map of Dlat; mean Dlat = 0.197 μm2 s−1. (iv) Dlat quadrat map; 129; 4 × 4 μm2 quadrats (black indicates <5 trajectories, removed from later analysis). (v) Distribution of Dlat values; Dlat = 0.204 ± 0.086 μm2 s−1 (mean ± SD). (vi) MSD vs. dT plots for typical fast (where Dlat > mean +1.5 × SD; red), intermediate (where Dlat ≈ mean; green) and slow (where Dlat < mean −1.5 × SD; blue) quadrat regions. Video was recorded at 33 fps. | ||
Comparison of Cy3B-telenzepine labelled M2 receptors in zebrafish tissue slice vs. mouse tissue slice
We investigated the M2 muscarinic receptor mobility in live tissue slices using two model cardiac systems (zebrafish and mouse), using Cy3B-telenzepine to fluorescently label the proteins. An initial observation was that the spread-area of the cells was noticeably smaller than for isolated primary cardiomyocytes and the HL1 cell-line; presumably because cells are held within the tissue and are less able to spread across the coverslip surface. Receptor density was similar in the tissue slices compared to the isolated cultured cells (∼1 receptor per μm2) (Fig. 3A, B and Movie 5† for comparison), but, the rate of diffusion was significantly faster in mouse tissue slices (mean Dlat = 0.43 μm2 s−1) compared to primary myocytes (MW, U = 1683, p = 2.3 × 10−8) and HL1 cell-line (MW, U = 2135, p < 2.2 × 10−16) and was similarly fast in the zebrafish tissue (mean Dlat = 0.33 μm2 s−1), but the median values were significantly different (MW, U = 1261, p = 0.001). Rapid receptor movement in both specimens produced a dense network of single molecule tracks so a small quadrat size (1 × 1 μm2) could be used and this gave a higher-resolution map of membrane viscosity (Fig. 3(iv)). A histogram of mean Dlat values derived from each quadrat sample was normally distributed (Fig. 3(v)). MSD vs. dT plots for three of the “slower” quadrat samples compared to 3 of the “faster” regions showed that MSD increased linearly with dT for both fast and slow samples and did not show substantial curvature (that would indicate anomalous diffusion) (Fig. 3(vi)). | ||
| Fig. 3 Cy3B-telenzepine labelled M2 muscarinic receptors in zebrafish and mouse cardiac tissue slices. (A(i)) First video frame showing individual Cy3B-telenzepine labelled M2 receptors on the plasma membrane of zebrafish cardiomyocytes in a tissue slice. (ii) Map showing the trajectory paths of individual molecules (see Movie 3).† (iii) Pseudo-colour heat-map showing locally averaged M2 receptor Dlat values. The overall mean Dlat = 0.33 μm2 s−1, 2518 trajectories. White rectangle shows profile plotted on the insert in the right-top corner. (iv) Heat map of the quadrat-sampled Dlat values; 49 quadrats (1 × 1 μm2) (black indicates <5 trajectories, removed from later analysis). (v) Distribution of Dlat measured in each quadrat region (red = “cell 1”, blue = “cell 2”): overall mean Dlat = 0.224 ± 0.116 μm2 s−1 (±SD): “cell 1” Dlat = 0.291 ± 0.144 μm2 s−1, “cell 2” Dlat = 0.179 ± 0.056 μm2 s−1. (vi) MSD vs. dT plots for typical “cell 1” (red), “cell 2” (blue) quadrat regions. (B) As for Panel A except: (i) mouse cardiomyocytes in a tissue slice. (iii) Mean Dlat = 0.428 μm2 s−1, 2336 trajectories. (iv) Dlat map: 83 quadrats (1 × 1 μm2). (v) Distribution of Dlat (“cell 1”(red); “cell 2” (green); “cell 3” (blue): mean Dlat = 0.427 ± 0.457 μm2 s−1). (vi) MSD vs. dT plots for typical quadrat regions (where Dlat ≈ global mean Dlat); within “cell 1” (red); “cell 2” (green); “cell 3” (blue). | ||
Although the global average Dlat was significantly faster in tissue slices (Dlat = 0.33 μm2 s−1) than isolated cultured cells, the difference was smaller in the Dlat maps because faster trajectories were underrepresented and the mean Dlat value fell to 0.22 μm2 s−1 for the zebrafish tissue slices. Consistent with this, when quadrat size was increased to 2 × 2 μm2, the mean Dlat value increased to 0.27 μm2 s−1. Interestingly, we found significant differences in mobility between neighbouring cells (Fig. 3(iii)), implying that there are significant differences in membrane composition between cardiomyocytes within a given tissue sample. In the example shown here, one cell gave Dlat = 0.29 μm2 s−1 the other, Dlat = 0.18 μm2 s−1 (t-test with unequal variances, t(23) = 3.224, p = 0.004).
Mapping membrane viscosity using nicotinic acetylcholine ion channels
Our expectation was that the nicotinic acetylcholine receptor, which is a hetero-pentameric ion channel, should exhibit unrestricted diffusive motion at the plasma membrane. However, we found it did not localise strongly to the plasma membrane and most molecules were retained in the endoplasmic reticulum (ER) following transfection. This might arise because of its hetero-pentameric nature, comprising four different polypeptides; β1, δ, γ and two α1 subunits, that must assemble correctly following co-transfection of the α1, β1, and δ subunits, together with the eGFP-γ-subunit. It is likely that a fraction of eGFP-γ subunits failed to assemble correctly into the heteromeric complex and are retained in the ER. So, we see a mixture of fluorophores at the ER network and also at the plasma membrane. Receptors that had localised correctly to the plasma membrane were most evident beneath the cell nucleus where the ER was excluded. We tracked all fluorophores as before and generated mobility maps to investigate mobility across the cell (Fig. 4). Our data show, perhaps unsurprisingly, that the histogram of Dlat values derived from the quadrat maps is not normally distributed, and while MSD vs. dT plots produced from “fast-moving” quadrat regions (plasma membrane) are consistent with an unconstrained Brownian walk, “slow-moving” regions show anomalous diffusive behaviour (MSD vs. dT plots show a distinct downward curvature) because γ subunits are confined to the ER membrane network. | ||
| Fig. 4 Nicotinic acetylcholine receptors were present at the plasma membrane and the ER of CHO-K1 cells: diffusion of nicotinic receptors was highly anomalous because the eGFP tagged γ-subunit only partially targeted the plasma membrane and the majority was found in the ER. (A(i)) Average projection of the video stack. (ii) Standard-deviation projection shows the reticulated network and also darker regions (presumably below the nucleus) where the ER was excluded and receptors could be seen moving at the plasma membrane. (iii) The overall MSD vs. dT plot showed evidence of anomalous diffusion with initial gradient (determined by least-squares’ linear regression) indicating Dlat = 0.16 μm2 s−1. (B(i)) First video frame showing single fluorophores. (ii) Projection of all the single fluorophore trajectories. (iii) Dlat map from trajectory segments. (iv) Dlat quadrat map (black indicates <5 trajectories, removed from later analysis). (v) Distribution of quadrat Dlat means (note that the histogram is not normally distributed). (vi) MSD vs. dT for fast (where Dlat > mean +1.5 × SD; red) and slow (where Dlat < mean −1.5 × SD; blue) regions. The fast-moving regions were located under the cell nucleus where receptors diffused freely at the plasma membrane (MSD vs. dT plots were linear with initial gradient (determined as in A) giving Dlat = 0.15 μm2 s−1). The slow-moving regions were from the network (ER) regions where receptors showed anomalous diffusion (note distinct downward curvature). | ||
Monte Carlo simulation of membrane protein random walks
We used an object-based, Monte Carlo stochastic model23 to generate sequences of simulated TIRFM videos (Fig. 5 and Movie 6†). The random movement of single molecules were realistically reproduced with known intensity levels, stochastic shot-noise, background noise and photobleaching behaviour consistent with our experimental TIRFM imaging modality. The simulated video data sets were analysed with the same image processing software used for our real data sets. We first checked that our localisation and tracking software gave consistent results over a wide range of simulated Dlat values (see ESI Fig. S5†). The example simulation in Fig. 5A shows trajectories detected in a sequence of images simulating molecules moving at Dlat = 0.2 μm2 s−1. The Dlat map (Fig. 5A(iii)) appears similar to our cell imaging data (Fig. 1A, B, 2 and 3). The local Dlat map calculated for a 2 × 2 μm2 quadrat size showed a normal distribution of diffusion coefficients (Fig. 5A(v)). Note here, that any superficial appearance of heterogeneous behaviour is simply due to statistical variation across sample quadrats, and demonstrates the necessity for statistical tests of heterogeneity. In fact, there is no deviation from normality (Table 1).The MSD vs. dT plots (Fig. 5A(vi)) were linear for the “fast”, “intermediate” and “slow” quadrat samples (note: we define “fast” as Dlat > mean +1.5 × SD, “slow” Dlat < mean −1.5 × SD and “intermediate” as Dlat ≈ mean). We also simulated the presence of lipid raft regions 300 × 300 nm2, dispersed randomly across the membrane (Fig. 5B, on the left-hand side of the modelled membrane) in which molecules were confined (Dlat = 0.02 μm2 s−1) and these regions adjoined others where molecules diffused freely with Dlat = 0.2 μm2 s−1 (on the right-hand side of the membrane). Now, the distribution of quadrat mean Dlat values were no longer normally distributed (Fig. 5B(v)). “Slow” regions showed anomalous diffusion with downward curvature, because molecules were confined within the modelled “lipid rafts”, whereas “fast” regions had linear MSD vs. dT plots.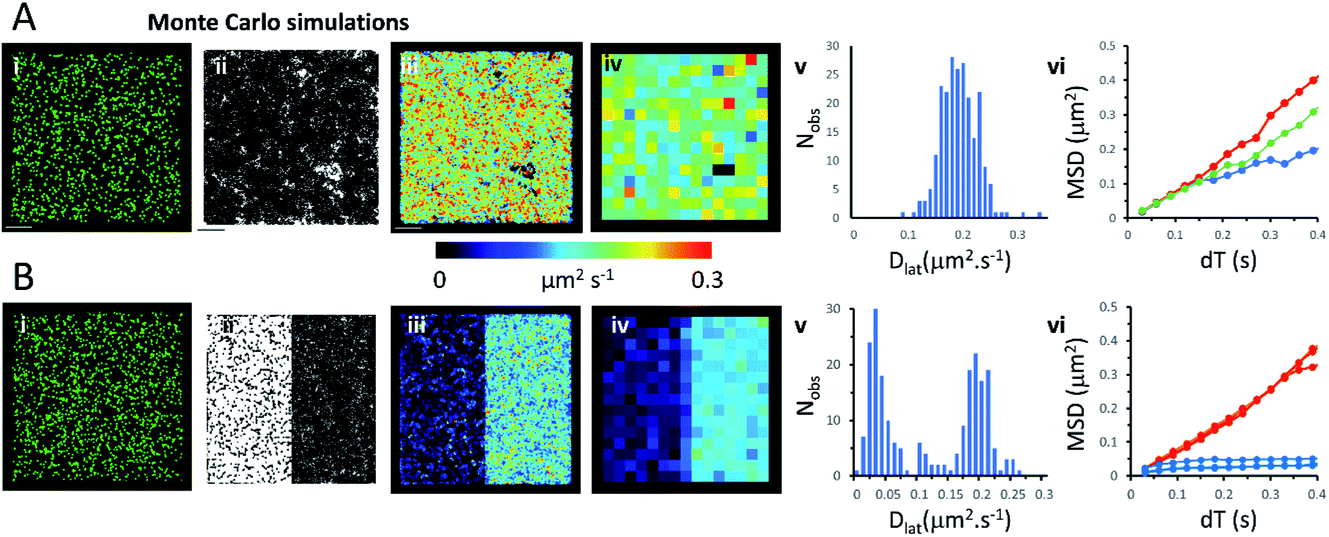 | ||
| Fig. 5 Monte Carlo simulation of single, fluorescently-tagged molecules moving at the plasma. (A(i)) Single image from the beginning of the simulated video recording shows individual fluorescent molecules (initial density 0.8 molecules per μm2). The simulated molecules are free to move in an unrestricted manner. (ii) Trajectory map consisting of 4434 trajectories. Fluorophore intensity, noise levels and photobleaching rate (0.2 s−1) were chosen to closely mimic our real data sets. (iii) Dlat map. (iv) Quadrat sampled Dlat map, 227 quadrats; 2 × 2 μm2, black squares contain <5 trajectories rejected from further analysis. (v) Histogram of quadrat Dlat values, is normally distributed and consistent with a single population. (vi) MSD vs. dT plots for typical fast (where Dlat > mean +1.5 × SD; red), intermediate (where Dlat ≈ mean; green) and slow (where Dlat < mean −1.5 × SD; blue) quadrat regions. (B) As for Panel A except: (i) the left-hand side of the simulation molecules are confined to lipid rafts; right-hand side molecules are free to move in an unrestricted manner, (ii) trajectory map, (iii) Dlat map, comprises two distinct regions. (iv) Quadrat sampled Dlat map. (v) Histogram of quadrat Dlat values, is now clearly bi-modal and not consistent with a single population. (vi) MSD vs. dT plots for typical fast (where Dlat > mean +1.5 × SD; red) and slow (where Dlat < mean −1.5 × SD; blue) quadrat regions. The “slow” (blue curves) regions show anomalous diffusive behaviour because molecules are trapped within modelled rafts of 300 × 300 nm2 area. | ||
Discussion
In the spirit of Faraday meetings our intention is to provoke discussion on the structure of biological membranes: the classical view of the plasma membrane is that it comprises a fluid-mosaic of lipids, proteins and other amphipathic molecules.3 This view has been challenged and it is now proposed that there is structural heterogeneity arising from lipid de-mixing and phase-separation which results in the formation of sub-micron sized microdomains or “lipid rafts”, enriched with saturated lipids, sphingolipids and cholesterol.24 Our view of the plasma membrane has therefore changed from a fully-mixed fluid-mosaic sheet to a cholesterol- and protein-stabilised, oil-in-oil, emulsion that resembles a two-dimensional “mayonnaise”. The small-size and short-lived, nature of lipid microdomains has led to controversy25–27 and as technologies have improved, microdomain size has fallen from 1 micrometre to as small as 10 nm diameter. Although phase separation has been demonstrated in model lipid systems7,28 there are few, direct observations of heterogeneity in live mammalian cells at physiological temperature.29,30 In a previous study, we found the mobility of M2 receptors in HL1, CHO-K1 and primary cardiomyocytes showed no evidence of a phase transition over a −5 °C to +45 °C temperature range.14 Stability of lipid microdomains may be impacted by in-plane, protein–protein and protein–lipid interactions and there may be additional interactions that extend out of the membrane plane onto the intracellular cytoskeleton and/or extracellular matrix. These interactions may act to stabilise raft structures and corral or otherwise interfere with the free diffusion of membrane proteins10 which may either partition into the raft or the surrounding isotropic lipid, or may “hop” between the two regions.31Notwithstanding the controversy surrounding the existence of lipid rafts and membrane microdomains, their proposed presence is thought to affect both the distribution and mobility of transmembrane proteins, with important consequences in neurobiology, virology, immunology and membrane–peptide interactions. One manifestation of membrane heterogeneity is that diffusive motion of transmembrane proteins becomes anomalous and MSD vs. dT plots are non-linear and show distinct downward inflection, indicating that molecules diffuse rapidly over short time and length scales, but more slowly over longer time intervals and distances.12,28,32 A significant problem with use of MSD vs. dT plots is that raw data derived from single molecule imaging experiments rarely enables meaningful analysis to be made on a single molecular trajectory.11,33 In earlier work, we found that muscarinic acetylcholine receptors (a class of membrane-spanning, 7-helix, G-protein coupled receptor) undergoes unrestricted diffusion at the plasma membrane.34 So, we chose this protein as a prototype probe of membrane viscosity and structural heterogeneity. Here, we have investigated whether molecules diffuse at different speeds in different regions of the plasma membrane and whether in regions where molecules appear to move more slowly they also exhibit anomalous diffusive behaviour. We divided the cell membrane into 1 × 1 μm2 or 2 × 2 μm2 quadrats and analysed (≥5) independent molecular trajectories in each quadrat region so we could test for significant variation between cell membrane regions. We also tested if the mean Dlat values estimated at each quadrat were homogeneous and normally distributed, as expected by the central-limit theorem. We then specifically compared MSD vs. dT plots from “slow” and “fast” moving regions to see if the plots were linear or showed evidence of anomalous diffusive behaviour.
Measurement precision to some extent depends on imaging conditions, including the type of cell, choice of membrane protein and the fluorescent tag that has been employed. The density of single molecule trajectories (or “tracks”) and the spatial resolution of local Dlat maps, depends on the lateral diffusion (Dlat) and track duration; which are limited mainly by the fluorophore photobleaching rate. To generate a local Dlat estimate we constructed MSD vs. dT plots from single particle tracks that extended for ≥10 consecutive video frames.35 Excitation laser power and imaging rate were optimised to allow molecules to be unambiguously tracked with high spatial and temporal resolution over a sufficient number of video frames. The density of unique single molecule trajectories that were accumulated over the entire imaging period (∼1 min) varied between samples even though the starting fluorophore density was similar (∼1 molecule per μm2). For most specimens a quadrat size of 2 × 2 μm2 was used, but some (with high track density) allowed the use of a smaller quadrat size (1 × 1 μm2). For randomly moving molecules (free diffusion), a histogram of Dlat values obtained by fitting all of the individual single molecule MSD vs. dT plots over the entire cell surface is best described by a gamma distribution.11,36 However, we show here that a histogram of mean Dlat values, obtained by averaging individual tracks within each quadrat sample area, obeys the central limit theorem and shows a normal distribution. Departure from normality implies heterogeneity in the data set at the level of the quadrat sample size; this can be assessed by Kolmogorov–Smirnov analysis (summarised in Table 1).
We found that the diffusive motion of M2 receptors at the plasma membrane of HL1 cells, primary cardiomyocytes and zebrafish cardiac tissue slices all gave pooled MSD vs. dT plots (i.e. taking all molecular trajectories) that were linear (Fig. S4†), with no obvious evidence of anomalous diffusion. When trajectories were segmented and subdivided into checkerboards of quadrat samples, the histograms of mean Dlat values were found to be normally distributed. Thus, there was also no evidence for heterogeneity in receptor mobility across the plasma membrane of these specimens. When we further analysed the data by examining MSD vs. dT plots on quadrat sample trajectories drawn from seemingly “fast” and “slow” moving regions we found the plots have no obvious downward curvature. Together these findings are consistent with variation in individual Dlat values, being the result of random sampling of a homogeneous population. So, in these cases, lipid rafts must not cause anomalous diffusive motion and/or must be monodisperse and evenly distributed across the plasma membrane (see Table 1 and Fig. 1–3).
We found three specimens that convincingly exhibited Dlat histograms that were not normally distributed; implying that the quadrat samples were not drawn from a simple homogeneous population. We were interested to compare our findings for nicotinic acetylcholine receptors with another recent study37 in which ion channel motion was tracked in frog embryonic muscle fibres using quantum dot labels and where MSD vs. dT plots were essentially linear once immobile objects were removed from the data sets and MSD vs. dT plots were essentially linear. However, an increasing deviation from unity gradient for log(MSD) vs. log(dT) plots at shorter times and smaller distances, indicated the receptors were diffusing in an anomalous manner that was best fit by an exponential distribution of diffusion coefficients. The authors point out that a distribution of diffusion coefficients is inconsistent both with simple lipid raft or cytoskeletal picket fence model. The same type of effect was seen in another high-resolution, quantum-dot tracking study.38
In the current study, we found downward curvature of the MSD vs. dT plots when we analysed data collected across the whole CHO-K1 cells transfected with the eGFP-tagged γ-subunit and all other component polypeptides of the nicotinic receptor. This indicates a highly anomalous diffusive behaviour of the ion channel. However, the explanation for our finding is rather simple because a standard-deviation projection of the video data revealed that many of the eGFP-tagged subunits were associated with the ER, and when regions that were rich in ER were examined separately (using our quadrat sampling method), diffusion was slow and highly anomalous. However, regions of the cell where the ER was excluded (e.g. beneath the nucleus) the ion channels moved rapidly and exhibited linear MSD vs. dT plots. The rate of diffusion in the fast-moving regions was 10× greater than values reported in the earlier study using frog muscle cells.37
We show that data averaging between the cells should be applied with caution, or avoided altogether, because variation in viscosity between the cells can be higher than variations within a single cell (see the viscosity map profile values on Fig. 3A(iii)). The Dlat quadrat distributions found between two neighbouring myocytes have very different K–S test scores when considered either separately or together (Fig. 3A(iv) and Table 1). Also, membrane heterogeneity within an individual endothelial cell expressing eGFP-tagged M2 receptors was evidenced by the “non-normal” distribution of local viscosity values (Fig. 2(v) and Table 1). Visual inspection of the quadrat map (Fig. 2(iv)) revealed that mobility in the lower-left region of the cell was significantly lower than for the rest of the cell.
Conclusion
We have presented a method to analyse single fluorophore tracking data, which employs a quadrat sampling technique to partition data into localized maps of transmembrane protein mobility. We have shown that, in many cases, the plasma membrane has a uniform viscosity and homogeneous structure, and quadrat mean values are normally distributed as expected by the central limit theorem. However, and perhaps as expected, proteins that localised to two different membrane systems (nicotinic receptors found at the ER and plasma membrane or CHO-K1 cells) or following incomplete chemical fixation (M2 receptors in HL1 cell) showed distinct heterogeneity. In one specimen, M2 receptors in an endothelial cell, we found spatial variation in membrane viscosity across an individual cell, and while the bulk MSD vs. dT plot was perfectly linear (Fig. S3B†) the quadrat mean Dlat values were not normally distributed (Table 1). Closer inspection of the MSD vs. dT plots for “fast” and “slow” diffusing regions showed the difference in mobility was not due to anomalous diffusion (i.e. microscopic variation in structure) but more likely to a bulk variation in membrane viscosity (a “raft of rafts”) that impacts the macroscopic Dlat value but has minimal effect on microscopic, anomalous diffusive motion of individual M2 receptors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr Alan Ling (Mechanical Engineering Workshop, Francis Crick Institute) for manufacturing some of the equipment used in this study, and Dr Qiling Xu (Neural Development Laboratory, Francis Crick Institute) for providing advice on zebrafish preparation. This work was supported by the Francis Crick Institute which receives its core funding from Cancer Research UK (FC001119), the UK Medical Research Council (FC001119), and the Wellcome Trust (FC001119). For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission. RL and LC were supported by Leverhulme Trust, project grant RPG-2016-407.References
- K. Jacobson, O. G. Mouritsen and R. G. W. Anderson, Nat. Cell Biol., 2007, 9, 7–14 CrossRef CAS PubMed.
- E. Sezgin, I. Levental, S. Mayor and C. Eggeling, Nat. Rev. Mol. Cell Biol., 2017, 18, 361–374 CrossRef CAS PubMed.
- S. J. Singer and G. L. Nicolson, Science, 1972, 175, 720–731 CrossRef CAS PubMed.
- H. C. Berg, Random walks in biology, Princeton University Press, Chichester, 1993 Search PubMed.
- P. G. Saffman and M. Delbruck, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 3111–3113 CrossRef CAS PubMed.
- Y. Gambin, R. Lopez-Esparza, M. Reffay, E. Sierecki, N. S. Gov, M. Genest, R. S. Hodges and W. Urbach, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2098–2102 CrossRef CAS PubMed.
- T. Baumgart, A. T. Hammond, P. Sengupta, S. T. Hess, D. A. Holowka, B. A. Baird and W. W. Webb, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3165–3170 CrossRef CAS PubMed.
- K. Simons and E. Ikonen, Nature, 1997, 387, 569–572 CrossRef CAS PubMed.
- M. Edinin, S. C. Kuo and M. P. Sheetz, Science, 1991, 254, 1379–1382 CrossRef PubMed.
- A. Kusumi, C. Nakada, K. Ritchie, K. Murase, K. Suzuki, H. Murakoshi, R. S. Kasai, J. Kondo and T. Fujiwara, Annu. Rev. Biophys. Biomol. Struct., 2005, 34, 351–378 CrossRef CAS PubMed.
- H. Qian, M. P. Sheetz and E. L. Elson, Biophys. J., 1991, 60, 910–921 CrossRef CAS PubMed.
- M. J. Saxton and K. Jacobson, Annu. Rev. Biophys. Biomol. Struct., 1997, 26, 373–399 CrossRef CAS PubMed.
- D. M. Owen, A. Magenau, D. Williamson and K. Gaus, BioEssays, 2012, 34, 739–747 CrossRef CAS PubMed.
- T. A. Nenasheva, M. Neary, G. I. Mashanov, N. J. M. Birdsall, R. A. Breckenridge and J. E. Molloy, J. Mol. Cell. Cardiol., 2013, 57, 129–136 CrossRef CAS PubMed.
- G. I. Mashanov, T. A. Nenasheva, T. Mashanova, C. Maclachlan, N. J. M. Birdsall and J. E. Molloy, J. Gen. Physiol., 2020, 153, e202012657 CrossRef PubMed.
- S. Shashkova and M. C. Leake, Biosci. Rep., 2017, 37(4), BSR20170031 CrossRef CAS PubMed.
- D. Axelrod, Methods Cell Biol., 1989, 30, 245–270 CrossRef CAS.
- T. A. Nenasheva, T. Carter and G. I. Mashanov, J. Microsc., 2012, 246, 83–88 CrossRef CAS PubMed.
- G. I. Mashanov, D. Tacon, A. E. Knight, M. Peckham and J. E. Molloy, Methods, 2003, 29, 142–152 CrossRef CAS PubMed.
- G. I. Mashanov and J. E. Molloy, Biophys. J., 2007, 92, 2199–2211 CrossRef CAS PubMed.
- M. J. Crawley, Statistics: An Introduction Using R, John Wiley & Sons, Chichester, 2nd edn, 2015 Search PubMed.
- R_Core_Team, R: A language and environment for statistical computing, http://www.R-project.org/ Search PubMed.
- G. I. Mashanov, J. R. Soc. Interface, 2014, 11, 20140442 CrossRef PubMed.
- D. Lingwood and K. Simons, Science, 2010, 327, 46–50 CrossRef CAS PubMed.
- S. Munro, Cell, 2003, 115, 377–388 CrossRef CAS PubMed.
- J. F. Hancock, Nat. Rev. Mol. Cell Biol., 2006, 7, 456–462 CrossRef CAS PubMed.
- B. Nichols, Nature, 2005, 436, 638–639 CrossRef CAS PubMed.
- R. Metzler, J. H. Jeon and A. G. Cherstvy, Biochim. Biophys. Acta, 2016, 1858, 2451–2467 CrossRef CAS PubMed.
- C. Eggeling, C. Ringemann, R. Medda, G. Schwarzmann, K. Sandhoff, S. Polyakova, V. N. Belov, B. Hein, C. von Middendorff, A. Schonle and S. W. Hell, Nature, 2009, 457, 1159–1162 CrossRef CAS PubMed.
- D. M. Owen, D. J. Williamson, A. Magenau and K. Gaus, Nat. Commun., 2012, 3, 1256 CrossRef.
- K. Suzuki, K. Ritchie, E. Kajikawa, T. Fujiwara and A. Kusumi, Biophys. J., 2005, 88, 3659–3680 CrossRef CAS.
- K. Ritchie, X. Y. Shan, J. Kondo, K. Iwasawa, T. Fujiwara and A. Kusumi, Biophys. J., 2005, 88, 2266–2277 CrossRef CAS.
- M. J. Saxton, Biophys. J., 1993, 64, 1766–1780 CrossRef CAS.
- J. A. Hern, A. H. Baig, G. I. Mashanov, B. Birdsall, J. E. T. Corrie, S. Lazareno, J. E. Molloy and N. J. M. Birdsall, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2693–2698 CrossRef CAS.
- X. Michalet, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 82, 041914 CrossRef.
- M. J. Saxton, Biophys. J., 1997, 72, 1744–1753 CrossRef CAS.
- W. He, H. Song, Y. Su, L. Geng, B. J. Ackerson, H. B. Peng and P. Tong, Nat. Commun., 2016, 7, 11701 CrossRef CAS.
- A. V. Weigel, B. Simon, M. M. Tamkun and D. Krapf, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6438–6443 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Movie 1: M2 muscarinic receptors labelled with Cy3B-telenzepine moving on the plasma membrane of cultured HL1 cardiomyocyte and imaged at 33 fps (23 °C). Real time movie, Movie 2: M2 muscarinic receptors labelled with Cy3B-telenzepine moving on the plasma membrane of primary, cultured, cardiomyocyte extracted from embryonic mouse heart and imaged at 33 fps (23 °C). Movie 3: M2 muscarinic receptors labelled with Cy3B-telenzepine on the plasma membrane of cultured HL1 cardiomyocyte treated with 1% of paraformaldehyde and imaged at 20 fps (23 °C). Some patches of plasma membrane adjacent to the substrate were sealed off by PFA and retained low viscosity allowing individual molecules to move freely inside these patches. Movie 4: M2 muscarinic receptors tagged with eGFP transfected into a HUVEC and imaged at 33 fps (23 °C). Real time movie. Movie 5: tracking individual muscarinic receptors on the surface of a heart slice from the zebrafish reveals the fine viscosity map of the plasma membranes when imaged at 50 fps (23 °C). The trajectory map is inserted at the end of Movie 1 to show all the trajectories detected in this record (×0.5 real time). Movie 6: model simulating the random movements (Dlat = 0.2 μm2 s−1) of fluorescent molecules on the surface of a rectangular cell (30 × 30 × 5 μm3. TIRFM illumination mode). A trajectory map is inserted at the end of Movie 1 to show all the trajectories detected in this record (×0.5 real time). See DOI: 10.1039/d1fd00035g |
| ‡ Current address: MRC LMB, Francis Crick Avenue, Cambridge, CB2 0QH, UK. |
| This journal is © The Royal Society of Chemistry 2021 |