 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Concluding remarks: Reaction mechanisms in catalysis: perspectives and prospects
C. Richard A.
Catlow
 abc
abc
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Park Place, Cardiff, CF10 1AT, UK. E-mail: CatlowR@Cardiff.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon St., London, WC1H 0AJ, UK
cUK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Laboratory, Harwell, Didcot, Oxon OX11 0FA, UK
First published on 7th April 2021
Abstract
We consider the current status of our understanding of reaction mechanisms in catalysis in the light of the papers presented in this Discussion. We identify some of the challenges in both theoretical and experimental studies, which we illustrate by considering three key reactions.
Introduction
This Faraday Discussion has clearly demonstrated both the importance of developing an understanding of the mechanisms of catalytic reaction and the rapid growth in our ability to develop detailed mechanistic models. The Discussion has also highlighted the challenges in the field. This article cannot attempt to present a comprehensive survey, but will focus on four themes which have been particularly prominent in the Discussion. The first concerns the role of theory and modelling; the second, the relationship between structure and mechanism; thirdly we consider developments in in situ/operando techniques; and finally, we focus on three key reactions: methanol to gasoline (MTG); ammonia synthesis; and CO2 hydrogenation. We conclude by considering the future prospects for the field.The role of theory and modelling
Theory and modelling are now essential tools in catalytic science; and methods based on Density Functional Theory (DFT) are now very widely used in developing mechanistic models. There are several excellent examples in this Discussion, including the work of Santos-Carballal et al. (DOI: 10.1039/C9FD00141G) on CO2 activation by iron sulphides and of Millán and Boronat (DOI: 10.1039/C9FD00126C) on hydrogenation of nitroaromatics. Both these studies reveal the power of DFT techniques in identifying key intermediates and in producing mechanistic energy profiles. However, the rapid growth in the utilisation of DFT raises a number of issues that we will now discuss in more detail.Accuracy of DFT methods
As argued above, DFT has unquestionably a major role to play in computational catalysis, but it is nevertheless important to note that it is an approximate method and that there are inaccuracies in energies and more especially activation energies calculated using standard functionals. The paper of Yang and Yang in this Discussion (DOI: 10.1039/C9FD00122K) makes an important contribution by allowing us to quantify the impact of these uncertainties on proposed mechanisms; and in many applications the inaccuracies may be unimportant as only qualitative information and trends are needed. But if more quantitative information is required, the limitations of standard DFT approaches may become problematic. One approach is to use more sophisticated functionals, especially hybrid functionals which mix in a proportion of exact exchange into the exchange correlation functional, thereby providing improved accuracy at the cost of greater computational expense. An alternative approach is to use quantum mechanical/molecular mechanical (QM/MM) methods, which are discussed in greater detail below.QM/MM methods
The application of atomistic modelling techniques in the study of heterogeneous catalysis has been dominated by the use of periodic methods; and in DFT studies the use of periodic, plane wave methods has been very extensive, with the use of the VASP1 code being widespread. QM/MM methods adopt an alternative approach, as illustrated in Fig. 1. The concept is simple. The region of interest, which in the case of a catalytic process will be the active site, reacting molecule and surrounding atoms, are described quantum mechanically (QM) and embedded in a more approximate interatomic potential or molecular mechanical (MM) description, which provided the chemistry is localised within the QM regions will be adequate. The method has some distinct advantages over periodic approaches: first, it avoids the artificial interactions between periodically repeated sorbed molecules; secondly, there is a significant technical advantage in calculations of e.g. ionisation potentials and proton affinities, in that the vacuum level is clearly defined, which is less apparent with periodic methods; but in the present context, perhaps the most significant advantage is that it is possible to use very high level quantum mechanical methods in the QM region, which would be impractical with periodic techniques. An excellent example is the landmark publication of Piccini et al.,2 in which by the use of QM/MM techniques with a high level (MP2) QM core, achieved chemical accuracy in modelling the reaction of methanol with alkenes in zeolites. Such accuracy is needed to calculate accurate reaction rates, although for many applications identifying trends and qualitative aspects, lower accuracy may, as noted, be quite acceptable.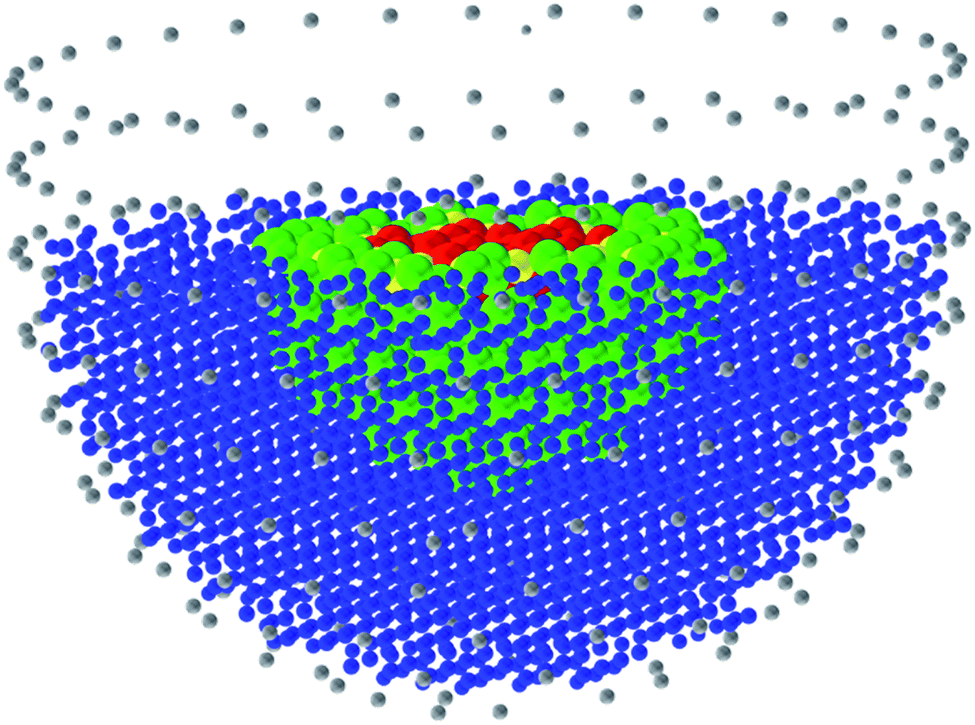 | ||
| Fig. 1 The QM/MM methodology in applications to solid state and surface modelling. The inner region (red) is described by a QM technique, while the outer region (blue) is modelled using an MM description. An appropriate description of the interface region (green) between the QM and MM regions is important. In modelling ionic crystals, an outer layer of point charges (grey) is added to ensure that the electrostatic potential in the QM region is accurately described. | ||
The use of QM/MM methods in catalytic science will continue to grow, while periodic methods will, of course, continue to make wide ranging and important contributions.
Microkinetic modelling
This increasingly used technique explores the mechanistic consequences of calculated reaction energy profiles by linking the calculated activation energies to a Monte-Carlo routine. It allows the relative contributions of different mechanistic pathways to be explored as a function of temperature and pressure and for patterns of reactant and product coverages on reacting surfaces to be predicted. Good illustrations are provided by the papers of Sinha et al. (DOI: 10.1039/C9FD00140A) and of Réocreux et al. (DOI: 10.1039/C9FD00134D) in this Discussion and a recent review is available from Ratnasamy and Wagner.3 A further example is provided by the recent work of Chutia et al.,4 who modelled the widely studied water gas shift (WGS) reaction on a palladium metal catalyst. The first stage was to perform DFT calculations both of the key elementary reactions summarised and illustrated in Fig. 2, as well as the relevant adsorption and diffusion processes. These were then input into the kinetic Monte-Carlo (KMC) algorithm available in the Zacros code.5 The analysis shows that under reaction conditions, the surface is dominated by adsorbed CO and H. Moreover, the work gives valuable mechanistic insights. It shows that the most common events are H2O and OH decomposition which are reversible; it finds that OH radicals react with CO to form a carboxyl species, which can subsequently decompose to generate CO2; it also shows that O formed by the dissociation of OH can react directly with CO to form CO2. It appears therefore the WGS reaction on Pd involves both direct oxidation and carboxyl pathways. Many other examples will be found in the recent literature illustrating the power of this approach.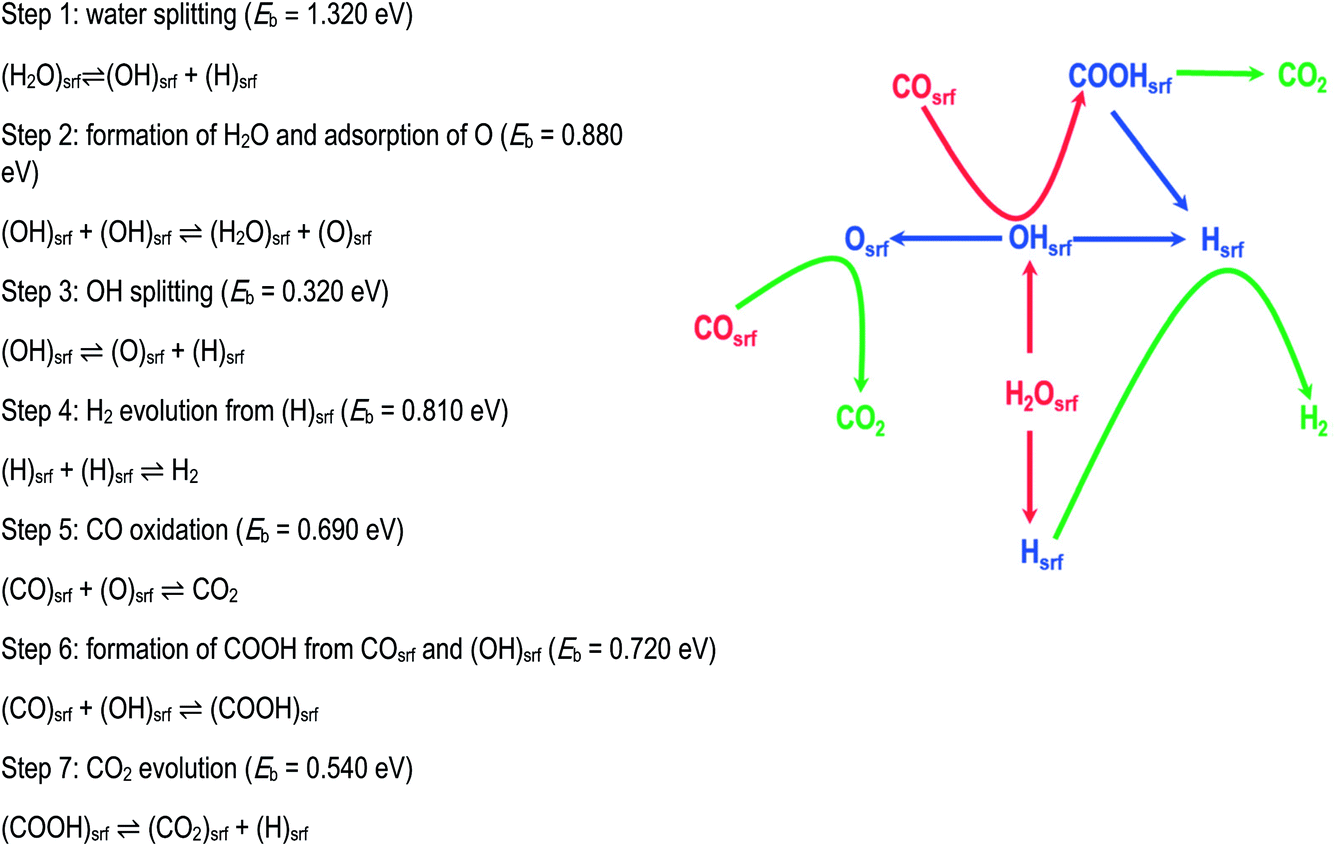 | ||
| Fig. 2 WGS mechanism comprising seven core elementary steps involving both redox and carboxyl mechanisms. For simplicity the adsorption and desorption steps are not shown. Adapted from ref. 4 with permission from the PCCP Owner Societies. | ||
Metadynamical simulations
Metadynamics is a powerful tool, which allows the investigation of activated processes which would be inaccessible to conventional molecular dynamics, enabling free energy barriers to be calculated. A review of its application to catalytic processes is available from Van Speybroeck et al.6 A recent example is provided by the work of Nastase et al.,7 who applied metadynamical simulation techniques to the study of the reactivity of methanol in ZSM-5. Ab initio dynamics first established that methanol clusters can deprotonate framework acid sites. Metadynamical simulations were then able to model the energetics of framework methoxylation. Methanol loading is found strongly to influence the activation energy of this key reaction, especially in the presence of a second acid site; the second site polarises the hydrogen bonded network and can reduce the activation barrier by ∼40 kJ mol−1.Modelling of solvation
Inclusion of solvent effects is a challenge to computational chemistry and surface science in general and to catalytic science in particular as shown in this Discussion by the papers of Raman et al. (DOI: 10.1039/C9FD00146H) and Sinha et al. (DOI: 10.1039/C9FD00140A). The commonest approach is to represent the solvent as a continuum; but with the continuing growth of computer power, explicit solvent modelling will become increasingly viable. QM/MM techniques will also have an important role to play in meeting this challenge.Structure and mechanism
Our comments here will be brief. We wish simply to emphasise that accurate structural models are essential for the development of detailed mechanistic models, which poses a particular challenge in heterogeneous catalysis, where there are often uncertainties in the structure and composition of surfaces or of nano-particle structures. A good illustration of the latter is provided by the work of Malta et al.8 on acetylene hydrochlorination catalysis by a supported Au catalyst; the work showed that, in contrast to the previous widely held view that the active sites were gold nano-particles, single gold cations were present in the operating catalyst.The complexity of oxide surface structures is illustrated by both experimental9 and computational10 studies of the polar surfaces of zinc oxide, which despite the simplicity of the bulk structure are highly complex with triangular vacancy clusters as illustrated in Fig. 3.
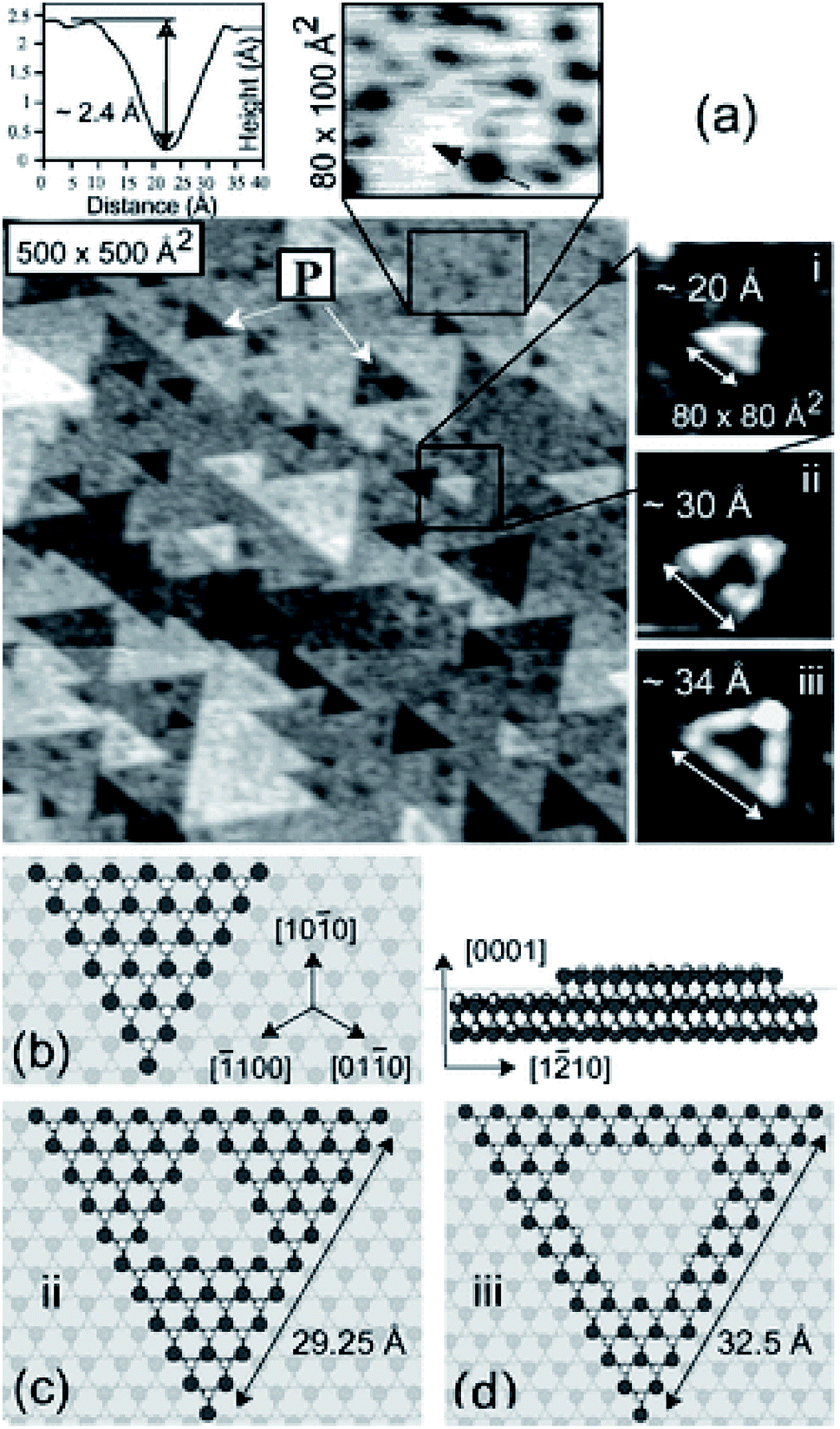 | ||
| Fig. 3 Surface microscopy image obtained by Dulub et al.9 of the Zn terminated polar surface of ZnO, revealing triangular vacancy clusters. Reproduced from ref. 9 by kind permission of the American Physical Society. | ||
The need for detailed structural models emphasises the role of advanced characterisation techniques, particularly those highlighted in the next section.
In situ/operando techniques
Several papers in this Discussion, including those of Pokochueva et al. (DOI: 10.1039/C9FD00138G), Matras et al. (DOI: 10.1039/C9FD00142E), Bugaev et al. (DOI: 10.1039/C9FD00139E) and Ziemba et al. (DOI: 10.1039/C9FD00133F), have shown very clearly the vital role of in situ/operando techniques in developing structural and mechanistic models of real catalytic systems; and the techniques are particularly powerful when deployed in conjunction with synchrotron and neutron scattering based techniques.An important recent development is the ability to undertake “in reactor” and spatially resolved experiments. The former is nicely illustrated by the paper in this Discussion of Nieuwelink et al. (DOI: 10.1039/D0FD00006J) which was able to probe single particles in a microreactor during multiphase hydrogenation reactions. To illustrate the latter, we refer to the recent work of Decarolis et al.11 who performed isothermal spatially resolved measurements on Pd/Al2O3 during NH3 selective catalytic oxidation (SCO). Their approach is illustrated in Fig. 4; and the results show clearly that in situ and operando experiments that rely on single point analysis can miss vitally important information. The study finds that at the inlet of the reactor, the Pd speciation is predominantly PdNx; that as the reaction exotherm progresses towards the outlet, the bulk interstitial N become mobile and the presence of a surface nitride structure is identified; that whilst the nitride is present there is still appreciable selectivity toward N2; and as the particles become more oxidised, there is a change in selectivity toward NOx products. Finally, at 400 °C, the data suggest that not only the NH3-SCO but also an NH3 selective catalytic reduction like behaviour is happening on the catalyst, converting the NO produced by the oxidation to N2O. The need for full spatial analysis is increasingly apparent and will assume additional importance as our ability to perform this type of experiment develops.
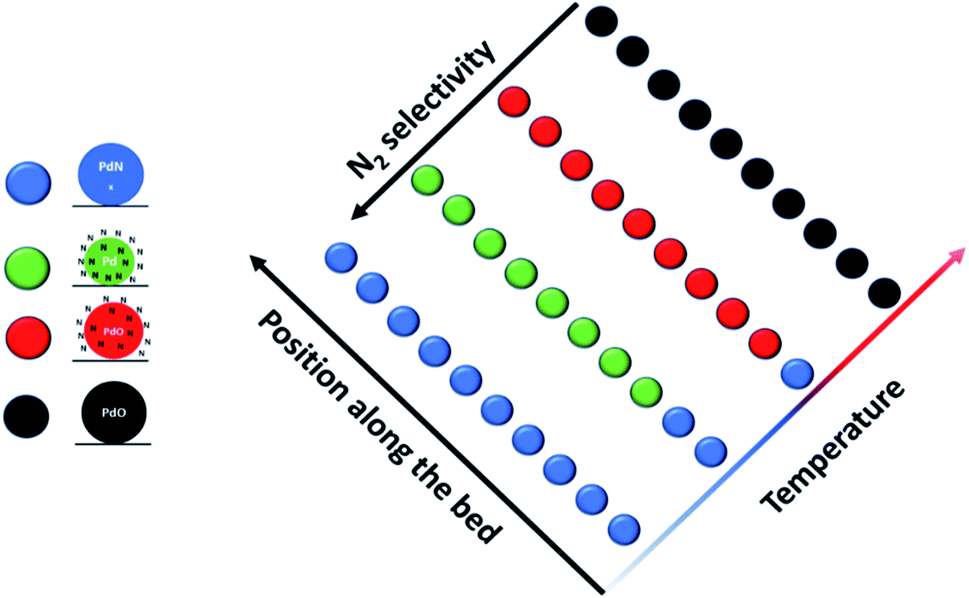 | ||
| Fig. 4 Results from spatially resolved experiment11 on Pd/Al2O3 during NH3 oxidation. During the experiment four Pd phases were identified, marked with different colours: PdNx (blue); metallic Pd with mobile surface nitrogen species (green); PdO with mobile surface nitrogen species (red); and bulk PdO (black). The predominant structure as a function of temperature and position along the bed is illustrated. The data show that N2 selectivity decreases as the sample moves more towards an oxidic phase. Figure taken from ref. 11 (https://pubs.acs.org/doi/abs/10.1021/acscatal.0c05356). Readers wishing permission related to the material excerpted should contact the ACS. | ||
Key systems: mechanistic challenges
The three key catalytic systems and processes discussed here will illustrate some of the progress and challenges in understanding reaction mechanisms in catalysis.Methanol to gasoline (MTG)
This widely studied reaction catalysed by zeolite ZSM-5 is discussed in detail in the introductory article of Hutchings (DOI: 10.1039/D1FD00023C). Here we focus on one aspect: the role of carbenes and related species in the formation of the initial C–C bond, the understanding of which is possibly the greatest challenge in unravelling the complex mechanism of MTG catalysis. As argued by Hutchings, several pieces of evidence have pointed to a significant role of carbene like species with further support being provided by recent synchrotron based IR studies of Minova et al.12 However, calculations of Sinclair and Catlow13 found high formation energies of 215–232 kJ mol−1 for the deprotonation of methoxy groups to form carbenes. The problem has recently been re-examined by Nastase et al.14,15 using QM/MM techniques. The carbene structures examined are illustrated in Fig. 5. The calculations find even higher energies than those obtained previously. They show, moreover, that both methoxy and carbene species are immobile. The results suggest that, if, as indicated by experiment, carbenes play a significant role, the mechanism cannot be stepwise; rather a concerted process involving the simultaneous formation and reaction of carbenes may operate. Clearly there is need for more computational and experimental work if we are to understand the crucially important mechanism of the first C–C bond formation.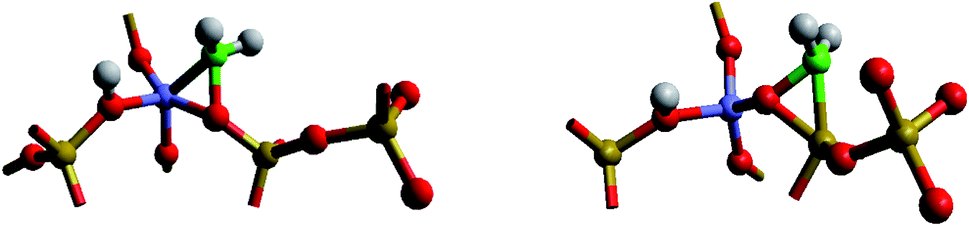 | ||
| Fig. 5 Carbene configurations adjacent to the acid site in ZSM-5; blue – Al; yellow – Si; red – O; green – C; white – H. The configuration on the left is calculated as lower in energy than that on the right by 34 kJ mol−1. | ||
Nitrogen activation
There can be no more striking example of the global importance of catalytic science and technology than ammonia synthesis, with the nutritional requirements of half the world’s population being dependent on nitrogen fixed by the Haber Bosch process, the energy intensive nature of which has, however, provided a stimulus for the development of catalysts that can operate under milder conditions. Transition metal nitrides have been extensively studied, including recent work on Li doped Mn nitrides by Laassiri et al. in this Discussion (DOI: 10.1039/C9FD00131J), in which ammonia is synthesised by chemical looping, while earlier work of Hargreaves and co-workers, summarised in a perspective article,16 showed the efficacy of Co3Mn3N in ammonia synthesis catalysis.The key problem in nitrogen activation is the strength of the nitrogen–nitrogen triple bond – the strongest chemical bond. Haber Bosch catalysis works by a “brute force” mechanism in which this bond must first dissociate before the dissociated nitrogen atoms are hydrogenated. Is a more subtle and gentle mechanism possible? Work of Zeinalipour-Yazdi et al.17 suggests that this may be the case. In computational work on the Co3Mo3N catalyst, the mechanism summarized in Fig. 6 was proposed. It involves the initial formation of surface vacancies, which can trap and activate nitrogen molecules, which are then hydrogenated without dissociation. The N–N bond is only broken when it has been degraded from a triple to a single bond. The development of this kind of associative mechanism may, we consider, be the answer to achieving nitrogen activation under relatively mild conditions.
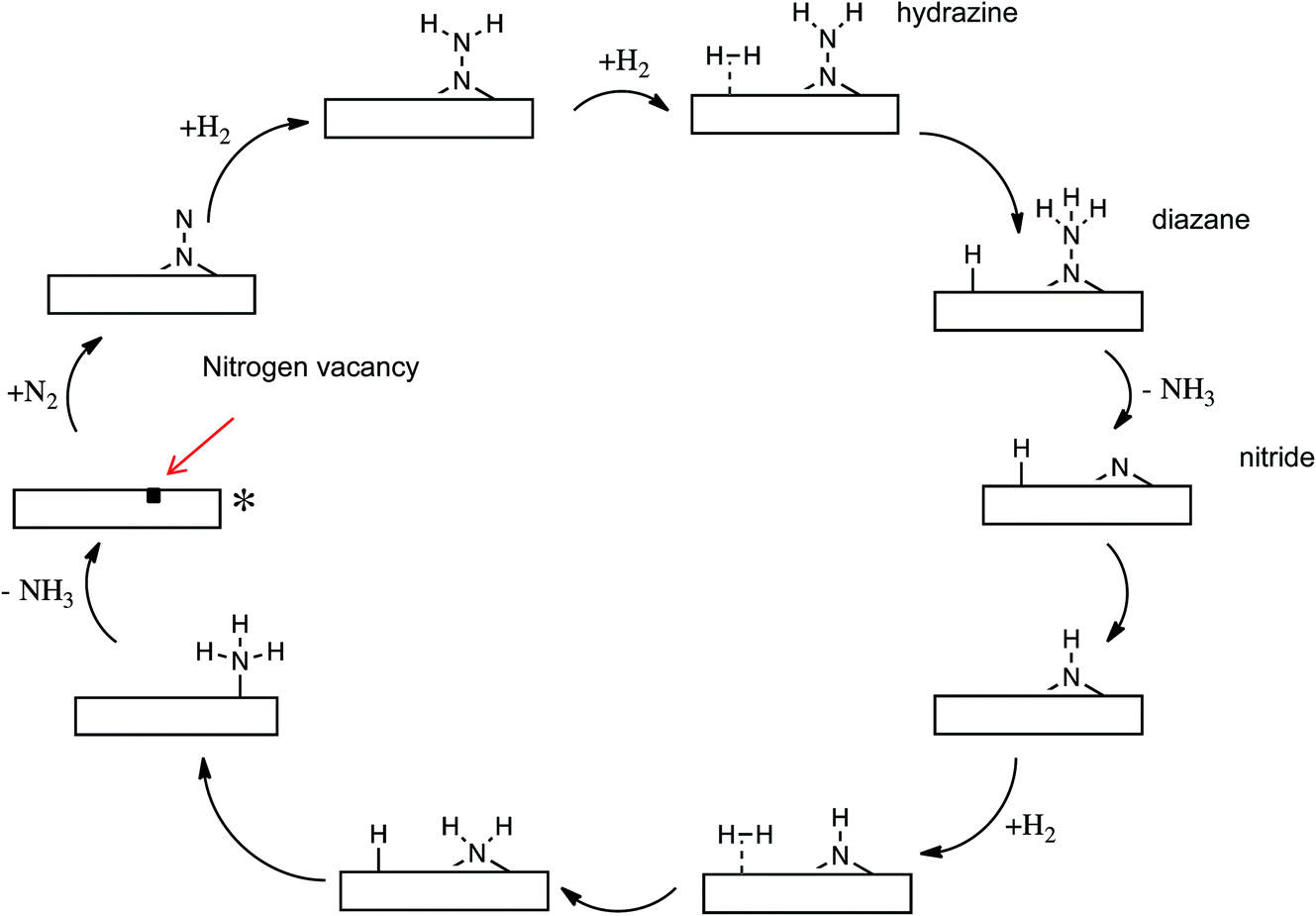 | ||
| Fig. 6 Proposed mechanism for ammonia synthesis on Co3Mo3N. The nitrogen molecule is trapped end on by a surface vacancy and then hydrogenated in a non-dissociative mechanism. Adapted from ref. 16 with permission from the PCCP Owner Societies. | ||
CO2 hydrogenation
As is well known, effective catalytic processes for carbon dioxide hydrogenation are essential for the development of sustainable chemicals and fuels. Mechanisms of hydrogenation are, however, widely debated; see, for example, the recent review of Nitopi et al.18 The Cu/ZnO catalyst used in methanol synthesis from both CO2 and syngas has been intensively studied, without the emergence of clear consensus as to mechanistic aspects. Recently, as a precursor to a mechanistic investigation of the Cu/ZnO system, a detailed computational (DFT) study was reported of the mechanism of CO2 hydrogenation over metallic copper surfaces.19 The mechanistic possibilities are summarized in Fig. 7. There are two distinct routes: one via CO2 dissociation and subsequent hydrogenation of the resulting CO; the other is non-dissociative. The two routes converge at later stages.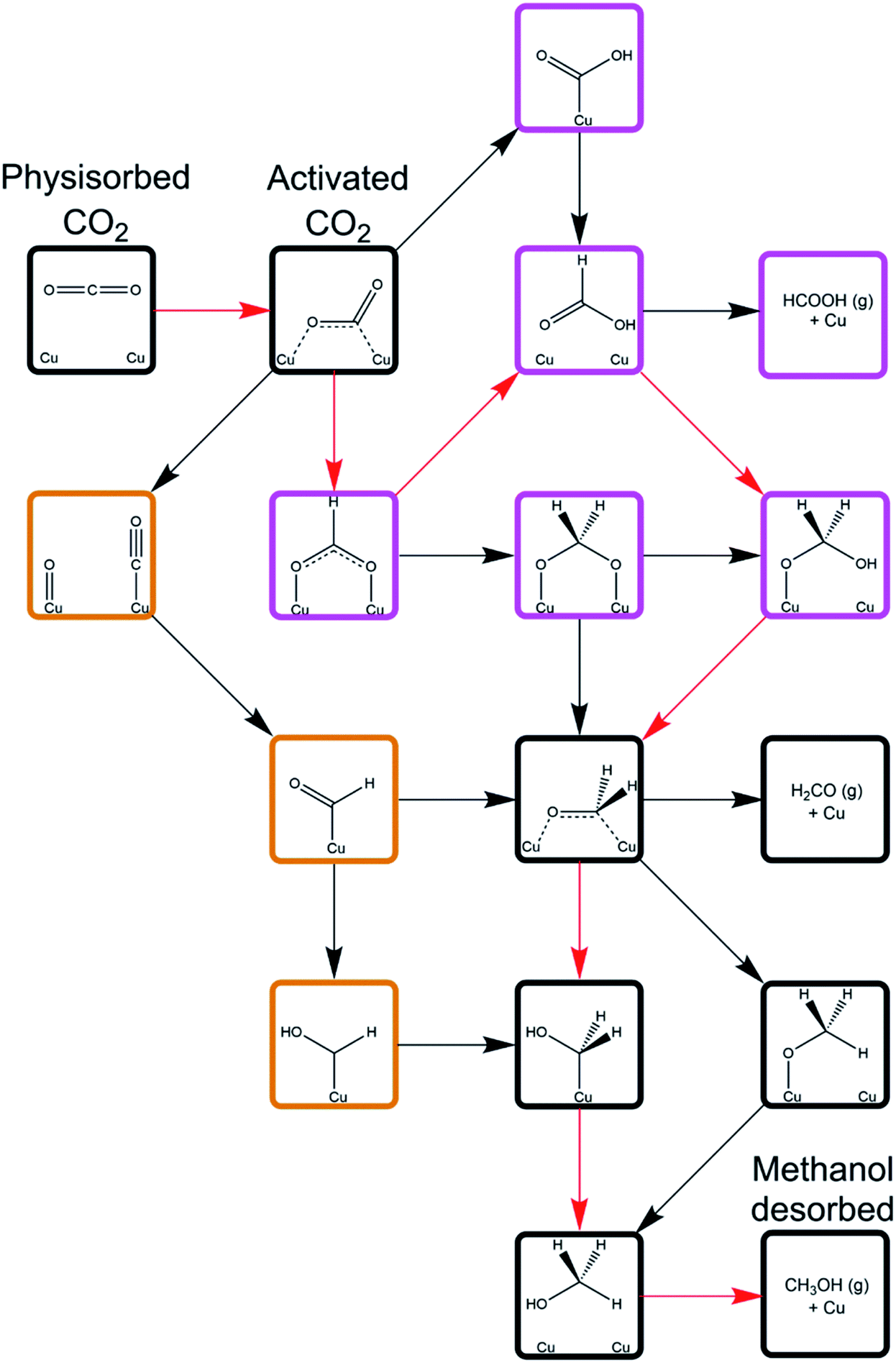 | ||
| Fig. 7 Schematic depicting all of the mechanistic pathways for CO2 hydrogenation to methanol over Cu(110) and Cu(100) surfaces investigated by Higham et al.19 Intermediates framed with orange borders correspond to those unique to the CO hydrogenation processes subsequent to CO2 dissociation, whilst the intermediates framed with purple borders are unique to direct CO2 hydrogenation processes. The species and intermediates framed with black borders are common to all of the mechanistic pathways investigated. The mechanism defined by the red arrows corresponds to the least activated pathway as determined by the calculations, with the reaction proceeding through CO2 hydrogenation to formate, formic acid, and oxy hydroxymethylene, which dissociated to yield formaldehyde, which can subsequently be hydrogenated to methanol via either methoxy or hydroxymethylene, with the former being marginally less activated overall. Reproduced from ref. 19 with permission from the Royal Society of Chemistry. | ||
The first and crucial step is CO2 adsorption and activation. Interestingly both physisorbed and chemisorbed states are possible. The latter is metastable with a bent configuration, and with electron transfer from the metal activating the adsorbed molecule. Hydrogen dissociation is mildly exothermic with a low activation barrier. The calculations suggest that the non-dissociative route is more favourable. Reaction and activation energies for individual steps for this mechanism on the Cu(110) surface are given in Table 1. The results show that HCOO* formation is less activated than COOH* formation; and that HCOOH* formation from both HCOO* and COOH* is the least activated subsequent process. Hence OCH2OH* formation can be achieved via either the HCOO* or COOH* route through HCOOH* hydrogenation and subsequent dissociation to formaldehyde; the next steps are common with the CO pathway. The results also show significant differences between the reactivity on different surfaces. Current work is extending the mechanistic analysis using models of Cu nano-clusters adsorbed on polar ZnO surfaces recently developed by Higham et al.20 The work is assisting us to unravel the mechanistic complexities of this widely studied reaction.
| Elementary process | ΔE/eV | E a/eV | v/cm−1 |
|---|---|---|---|
| Cu(100) | |||
| CO2(g) → CO2* (phys.) | −0.391 | — | — |
| CO2* (phys.) → CO2* (act.) | 0.446 | 0.535 | 181.67 |
| CO2* (act.) + H* → HCOO* | −1.174 | 0.403 | 565.90 |
| HCOO* + H* → HCOOH* | +0.437 | 0.925 | 634.64 |
| HCOOH* + H* → OCH2OH* | −0.029 | 0.813 | 729.63 |
| OCH2OH* → H2CO* + OH* | +0.337 | 0.826 | 160.06 |
| H2CO* + H* → CH3O* | −0.905 | 0.046 | 311.80 |
| CH3O* + H* → CH3OH* | −0.148 | 0.698 | 1029.91 |
| CH3OH* → CH3OH(g) | +0.613 | — | — |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Cu(110) | |||
| CO2(g) → CO2* (phys.) | −0.148 | — | — |
| CO2* (phys.) → CO2* (act.) | 0.254 | 0.419 | 190.97 |
| CO2* (act.) + H* → HCOO* | −0.978 | 0.246 | 702.30 |
| HCOO* + H* → HCOOH* | +0.349 | 0.938 | 1061.70 |
| HCOOH* + H* → OCH2OH* | −0.194 | 0.844 | 606.19 |
| OCH2OH* → H2CO* + OH* | +0.037 | 0.899 | 136.67 |
| H2CO* + H* → CH3O* | −0.622 | 0.388 | 328.28 |
| CH3O* + H* → CH3OH* | −0.004 | 0.960 | 926.04 |
| CH3OH* → CH3OH(g) | +0.771 | — | — |
Conclusions and future prospects
This Discussion volume has clearly shown that the field is developing rapidly and that we have an increasingly powerful set of computational and experimental techniques for probing reaction mechanisms in catalysis. Computational studies will become increasingly predictive with our growing ability to model real complex systems and with improvements in the accuracy of the calculations. Experimental work will have a growing reliance on advanced characterization techniques used in operando; and experiment and theory will be used increasingly in a concerted manner. An improved ability in understanding mechanisms will be vital in optimising and designing catalytic processes.Conflicts of interest
I have no conflicts of interest to declare.Acknowledgements
I am grateful for discussions and advice on the preparation of this article to Justin Hargreaves, Graham Hutchings, Michael Higham, Donato Decarolis, Peter Wells, Stefan Nastase, Andrew Logsdail, Arun Chutia, Constantinos Zeinalipour-Yazdi, Alberto Roldan, David Santos-Carballal and Nora de Leeuw. Much of the work discussed here has been supported by the UK Catalysis Hub funded by EPSRC grants, EP/R026645/1, EP/R026939/1, EP/R026815/1, EP/R027129/1. We are grateful to the GCRF START project (ST/R002754/1) which funded the work on CO2 hydrogenation on copper.References
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. M. Piccini, M. Alessio and J. Sauer, Angew. Chem., Int. Ed., 2016, 55, 5235–5237 CrossRef CAS PubMed.
- C. Ratnasamy and J. P. Wagner, Catal. Rev., 2009, 51, 325–440 CrossRef CAS.
- A. Chutia, A. Thetford, M. Stamatakis and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2020, 22, 3620–3632 RSC.
- J. Nielsen, M. D’Avezac, J. Hetherington and M. Stamatakis, J. Chem. Phys., 2013, 139, 224706 CrossRef PubMed.
- V. Van Speybroeck, K. de Wispelaere, J. van der Mynnsbrugge, M. Vandichel, K. Hemelsoet and M. Varoquier, Chem. Soc. Rev., 2014, 43, 7326–7357 RSC.
- S. A. Nastase, P. Cnudde, L. Vanduyfhuys, K. De Wispelaere, V. Van Speybroeck, C. R. A. Catlow and A. J. Logsdail, ACS Catal., 2020, 10, 8904–8915 CrossRef CAS PubMed.
- G. Malta, S. A. Kondrat, S. J. Freakley, C. J. Davies, L. Lu, S. Dawson, A. Thetford, E. K. Gibson, D. J. Morgan, W. Jones, P. P. Wells, P. Johnston, C. R. A. Catlow, C. J. Kiely and G. J. Hutchings, Science, 2017, 355, 1399–1403 CrossRef CAS PubMed.
- O. Dulub, U. Diebold and G. Kresse, Phys. Rev. Lett., 2003, 90, 016102 CrossRef PubMed.
- D. Mora-Fonz, T. Lazauskas, M. R. Farrow, C. R. A. Catlow, S. M. Woodley and A. A. Sokol, Chem. Mater., 2017, 29, 5306–5320 CrossRef CAS.
- D. Decarolis, A. H. Clark, T. Pellegrinelli, M. Nachtegaal, E. W. Lynch, C. R. A. Catlow, E. K. Gibson, A. Goguet and P. P. Wells, ACS Catal., 2021, 11, 2141–2149 CrossRef CAS PubMed.
- I. B. Minova, S. K. Matam, A. Greenaway, C. R. A. Catlow, M. D. Frogley, G. Cinque, P. A. Wright and R. F. Howe, ACS Catal., 2019, 9, 6564–6570 CrossRef CAS.
- P. E. Sinclair and C. R. A. Catlow, J. Phys. Chem. B, 1997, 101(3), 295–298 CrossRef CAS.
- S. F. Nastase, PhD thesis, Cardiff University, 2020.
- S. F. Nastase, C. R. A. Catlow and A. J. Logsdail, to be published.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves, S. Laassiri and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2018, 20, 21803–21808 RSC.
- C. D. Zeinalipour-Yazdi, J. S. Hargreaves and C. R. A. Catlow, J. Phys. Chem. C, 2018, 122, 6078–6082 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- M. D. Higham, M. G. Quesne and C. R. A. Catlow, Dalton Trans., 2020, 49, 8478–8497 RSC.
- M. D. Higham, D. Mora-Fonz, A. A. Sokol, S. M. Woodley and C. R. A. Catlow, J. Mater. Chem. A, 2020, 8, 22840–22857 RSC.
| This journal is © The Royal Society of Chemistry 2021 |