 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spiers Memorial Lecture: Understanding reaction mechanisms in heterogeneously catalysed reactions
Graham J.
Hutchings

Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: hutch@cf.ac.uk
First published on 16th March 2021
Abstract
Heterogeneous catalysis lies at the heart of the chemical and fuel manufacturing industries and hence is a cornerstone of many economies. Many of the commercially operated heterogeneous catalysts have remained basically unchanged for decades, undergoing small but important optimisation of their formulations. Yet we all acknowledge that there is a continuous drive towards improved catalysts or designing new ones. At the heart of these studies has been the need to gain an improved understanding of the reaction mechanism for these important reactions since this can unlock new ways to improve catalyst design and, of course, the ultimate aim is to design catalysts based on the detailed understanding of the reaction mechanism. These advanced studies have been aided in the last decade by two key factors, namely: (a) access to advanced characterisation techniques based on synchrotron methods and aberration-corrected microscopy that can probe the nature of the active site, and (b) the application of high-level computational methods to understand how the reactants and products interact at the active site. In this paper this theme will be explored using two examples to bring out the complexity in gaining an understanding of a reaction mechanism. Using the zeolite H-ZSM-5 as an example of a single site catalyst, the mechanism of the conversion of methanol to the first hydrocarbon carbon–carbon bond will be discussed. In this section the use of model reactants and reaction probes will be used to try to differentiate between different mechanistic proposals. The second example explores the use of gold catalysts for CO oxidation and acetylene hydrochlorination. In both these examples the importance of advanced characterisation and theory will be highlighted.
Introduction
Understanding reaction mechanisms in catalysed reactions is an immensely important field that continues to attract immense research interest. However, do we really need to know the mechanism of a catalysed reaction? It is an important question and has only two answers, either yes or no. I first asked this question in 1985 at a small industrial meeting to discuss fundamental aspects of a catalysed reaction when I was in the early days of my academic career. There were about twenty people present and I was the only academic. Immediately, the most senior industrial representative present answered no. I then revealed my next slide which for the answer of no stated go to coffee now. It was a bit of fun and the meeting soon settled down to an in-depth discussion. I tried the same opening at an International Catalysis Congress meeting in Vienna in 1992. Now there were several hundred present and the topic I had been invited to speak on was the mechanism of carbon–carbon bond formation in the methanol to gasoline reaction. Again, the industrial contingent said no, quite forcibly, and so I revealed the next slide stating go to coffee now. This time I think several did leave, so this is probably not a good ploy if you want to retain an audience. However, it is not a simple question and at one level it is quite correct to state no; after all, many commercial catalysed processes have been successfully commercialised without any detailed understanding of the reaction mechanism. For example, ammonia synthesis, the Fischer–Tropsch reaction and methanol synthesis have all been commercialised without knowledge of the reaction mechanism at the time of commercialisation and as such incurred no significant problems. All these processes operate at a very large scale globally today and there are still interesting debates on the nature of the mechanisms. At another level, the real question is perhaps not that we would like to know the mechanism of a catalysed reaction but rather why don’t we know the reaction mechanisms for so many heterogeneously catalysed reactions that are operated today? What new thinking, techniques and methodologies are required to address this problem? For if we can understand a reaction mechanism at the molecular level, we could possibly design an improved catalyst for the reaction and that would represent a huge advance of both scientific and social benefit.At the outset of trying to determine a reaction mechanism it is essential to have some knowledge of the structure of the active site. For some catalysts this is relatively easy, for example the Brønsted acid site of a zeolite1 or well dispersed supported metal atoms or cations.2 The latter have recently been described as single atom catalysts,3 but as the well dispersed single atom is supported on a matrix of atoms, I personally prefer the term single site catalysis,4 although the term single atom catalysis is now very prevalent. For such single site catalysts, the structure of the active site is readily determined and so understanding reaction kinetics is greatly facilitated as turnover frequencies and total turnover numbers are easily determined.
However, for many heterogeneous catalysts the determination of the structure of the active site is more difficult. For example, Co based Fischer–Tropsch synthesis catalysts comprise 20–30% Co by mass and yet the active species is proposed to be Co nanoparticles 6 nm in diameter.5 Why is so much Co required to be present when the active structures are nanoparticles? Cu/ZnO catalysts, which are the active materials for the commercial methanol synthesis and low temperature water–gas shift reactions, comprise 30–50% Cu by mass but in this case the catalyst activity correlates with the Cu surface area of the reduced catalyst6 and again it is proposed that supported Cu nanoparticles are the active species. These are prepared in situ by reduction of a copper zinc hydroxycarbonate prepared by coprecipitation and malachite is the preferred precursor for the current industrial catalyst. Recently, an amorphous copper zinc hydroxycarbonate prepared using supercritical CO2 as an antisolvent7 produced much smaller Cu nanoparticles on reduction which were more disordered, and these have greatly enhanced activity for the low temperature water–gas shift reaction, and this was determined using environmental transmission electron microscopy7 (Fig. 1).
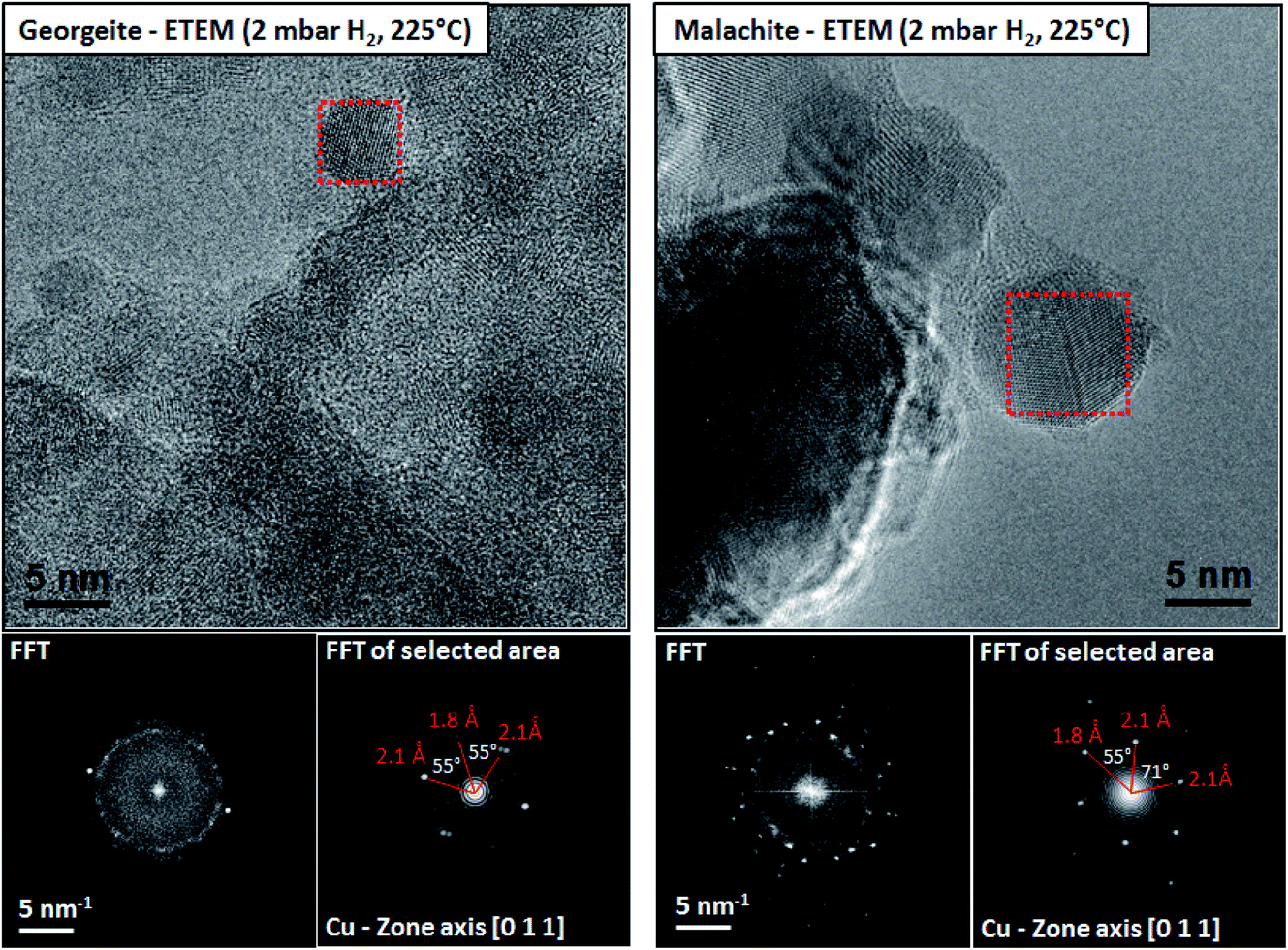 | ||
| Fig. 1 The microstructures of the reduced georgeite and malachite precursors characterized by ETEM in 2 mbar H2 at 225 °C. Both samples reveal distinct Cu nanoparticles distributed on ZnO as confirmed by FFT analysis. The reduced malachite sample reveals in general larger Cu nanoparticles compared to the georgeite sample. Reproduced with permission from ref. 7. | ||
In this paper three methods to aid the determination of reaction mechanisms will be described, namely (i) the use of model reagents to probe possible reaction mechanisms, (ii) the use of advanced in situ/operando microscopy and spectroscopy, and (iii) the use of computational methods to probe reaction pathways. Similar approaches can be used for heterogeneous, homogeneous and bio-catalysts, but the discussion in this paper will be restricted to examples of heterogeneous catalysts. The approaches will be illustrated by (i) methanol conversion to hydrocarbons, and (ii) gold catalysis.
Methanol conversion to hydrocarbons
When the conversion of methanol using the zeolite H-ZSM-5 as a catalyst was first described by Chang and Silvestri in 1977 (ref. 8), the reaction was not new. It had been known for over a century at that time and some early catalysts had been based on zinc halides9 and aluminium sulfate,10 and phosphorus pentoxide had also been used.11 However, it was the publication by Chang and Silvestri8 that started major research efforts in both industry and academia to investigate this fascinating reaction. Initially termed the methanol to gasoline (MTG) reaction, its discovery came at a time when oil prices were high and rising and so the quest for alternative synthetic fuels was a real research target. The process was commercialised in New Zealand,12 but production was not maintained as oil prices decreased and the production of methanol was more economic. However, a variant of this catalysis is very relevant today, as the methanol to olefins (MTO) process, which uses smaller pore nanoporous materials such as SAPO-34, is currently commercially operated in China.13 One of the key aspects of this chemistry is how is the initial carbon–carbon bond formed? This is a topic that has fascinated catalysis scientists since the discovery of the reaction. To date over twenty mechanistic proposals have been made, and many of these have been made without any experimental evidence to support them. In this section four of these mechanisms will be discussed and the evidence in their favour and against will be considered. Initially, after describing the basic experimental evidence for this reaction, this topic will be described using model reactants to probe the mechanism and then the more recent computational and spectroscopic studies will be introduced.Methanol conversion over zeolite H-ZSM-5: the basic experimental data
At the outset of any investigation of a reaction mechanism it is important to determine the nature of the product distribution and the nature of the primary products. For methanol conversion with H-ZSM-5 as catalyst this is shown in Fig. 2. As expected from the stoichiometry, water is the major product. The initial product formed is dimethyl ether, and subsequently hydrocarbons are formed.8 The reaction is found to be autocatalytic in nature; at low temperatures there is a distinct induction period for methanol conversion. This effect can make it very difficult to study the primary reactions occurring, as once the product containing the initial carbon–carbon bond is formed, this then rapidly reacts with methanol or dimethyl ether to form higher hydrocarbon products. By studying the reaction at a low temperature where the impact of the secondary reactions can be minimised and varying the reactant flow rate it was determined that ethene is the primary product.1413C labelling studies for the reaction of dimethyl ether showed that the two carbon atoms of dimethyl ether do not react to form ethene and therefore an intermolecular reaction, as opposed to an intramolecular reaction, is occurring.15 In addition, D/H labelling studies showed that ethene and dimethyl ether share a common intermediate.16 So the initial reaction scheme can be represented by Scheme 1.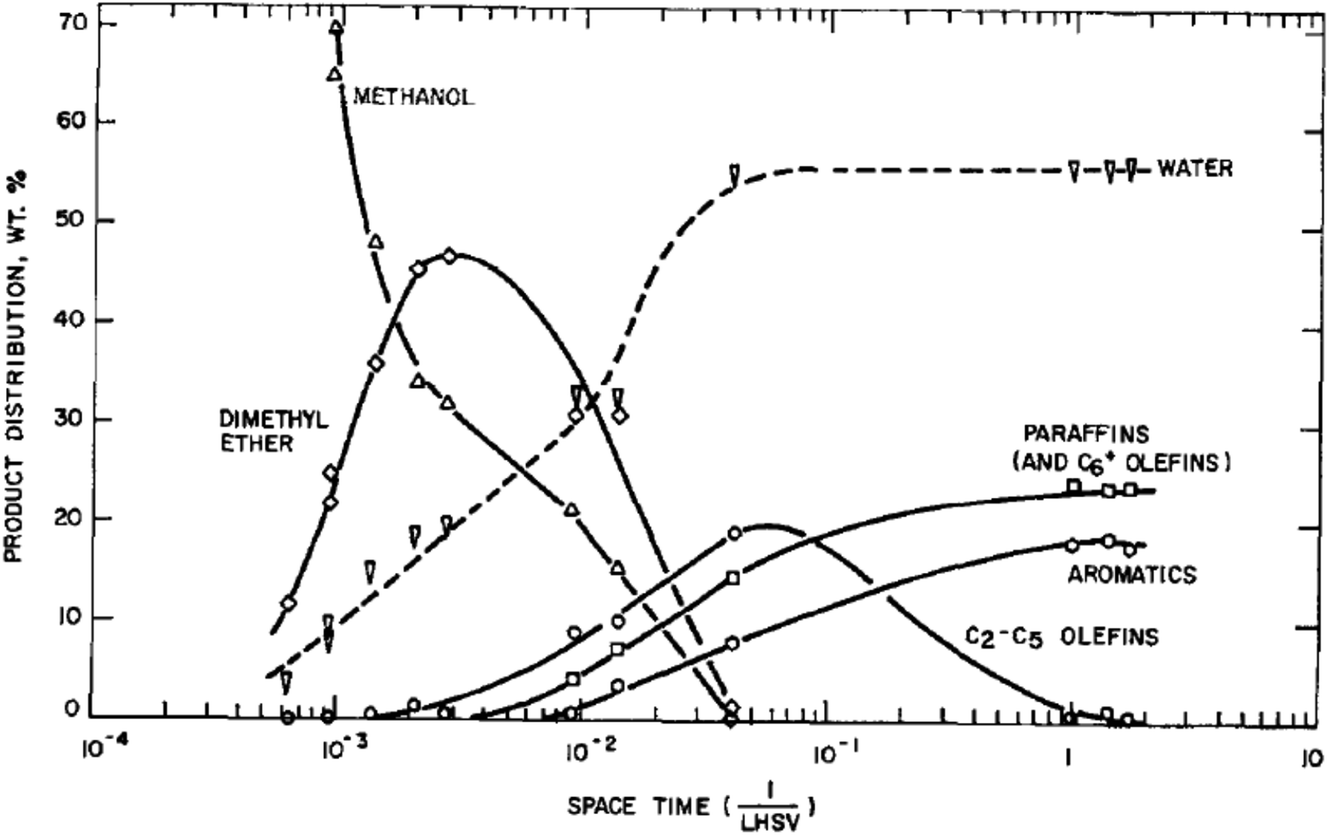 | ||
| Fig. 2 Reaction pathway for methanol conversion to hydrocarbons with H-ZSM-5 at 371 °C. Reproduced with permission from ref. 8. | ||
 | ||
| Scheme 1 | ||
The question then remains as to the mechanism by which ethene is formed from methanol/dimethyl ether. In the next sections four of the proposed mechanisms will be discussed focussing on the use of model reagents to probe whether the mechanistic proposal has merit. An important consideration is that if a model mechanism proposes a particular intermediate, then just because these species are adsorbed on the surface of a heterogeneous catalyst it does not mean that basic reactivity patterns can be changed. For example, if a species is nucleophilic it is unlikely to react with itself; i.e. the normal rules for chemical reactions still apply. One proposal for the mechanism of carbon–carbon bond formation in the Fischer–Tropsch reaction was proposed to involve the surface dimerization of two hydroxycarbenes,17 a very unlikely reaction. A more likely pathway would be for the hydroxycarbene to react with an electrophilic species such as a carbene.18 The work presented for the four mechanisms deals with research carried out in the early phase of interest in this reaction mechanism. Interest has grown again in recent years, mainly due to the commercialisation of the MTO process in China,13 and the approach to understanding the mechanism focuses on advanced in situ spectroscopy and computational methods. However, the mechanistic work carried out in the initial phase has laid the foundation on which these subsequent advanced experiments have been based and these will be discussed in a subsequent section.
The carbene mechanism
This mechanism was proposed by Chang and Silvestri in their initial paper.8 They proposed a concerted electron transfer mechanism (Fig. 3) in which the Brønsted conjugate base site in ZSM-5 deprotonates the C–H bond of methanol making a Brønsted acid site. At the same time the Brønsted acid site interacts to make water and remake the conjugate base site. This results in a carbene species that then inserts into a C–H bond of methanol or dimethyl ether to make a carbon–carbon bond.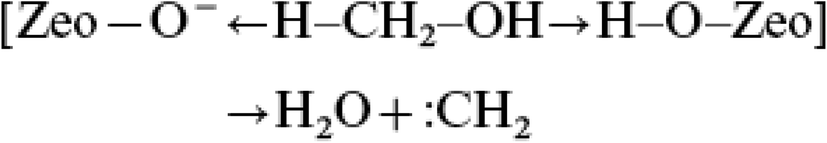 | ||
| Fig. 3 Proposed mechanism for the formation of a carbene species from methanol with H-ZSM-5. Redrawn from ref. 8. | ||
The evidence in favour of this proposal is that CH2N2, a potential source of a carbene, reacts over ZSM-5 to give ethene.19 However, there is a problem with this as CH2N2 can dimerise in the gas phase to give ethene without the intervention of a surface. A further problem is that, as pointed out by Olah,20 in acidic conditions CH2N2 is an electrophilic methylating agent and so with the Brønsted acid form of ZSM-5, CH2N2 would methylate the surface to form a surface methoxyl. Of course, a further carbene could insert into the C–H bond of the methoxyl, leading to carbon–carbon bond formation.
To overcome the problems associated with CH2N2 it is possible to use a substituted carbene CHXN2. We used ethyldiazoacetate and reacted this with both the acidic form H-ZSM-5 and its conjugate base Na-ZSM-5.21 At high temperatures (190–210 °C) and high conversions (20–45%), ethene was observed as the major product. But as these conditions did not mirror those associated with the early stages of the reaction, at lower temperatures (80 °C) and at 0.1% conversion, both ethene and propene were observed. With Na-ZSM-5 at 125 °C and 2% conversion, over 80% propene was formed as the main product. These results were rationalised in a surface catalysed reaction (Fig. 4) in which ethyldiazoacetate adsorbs as surface CHCO2C2H5 which can exist as keto and enol isomers which go on to react to form ethene and propene. The results of this study suggest a surface reaction is involved. The reaction of diazoacetate models C–C bond formation, albeit C1 → C3 and that a C1 intermediate is involved.
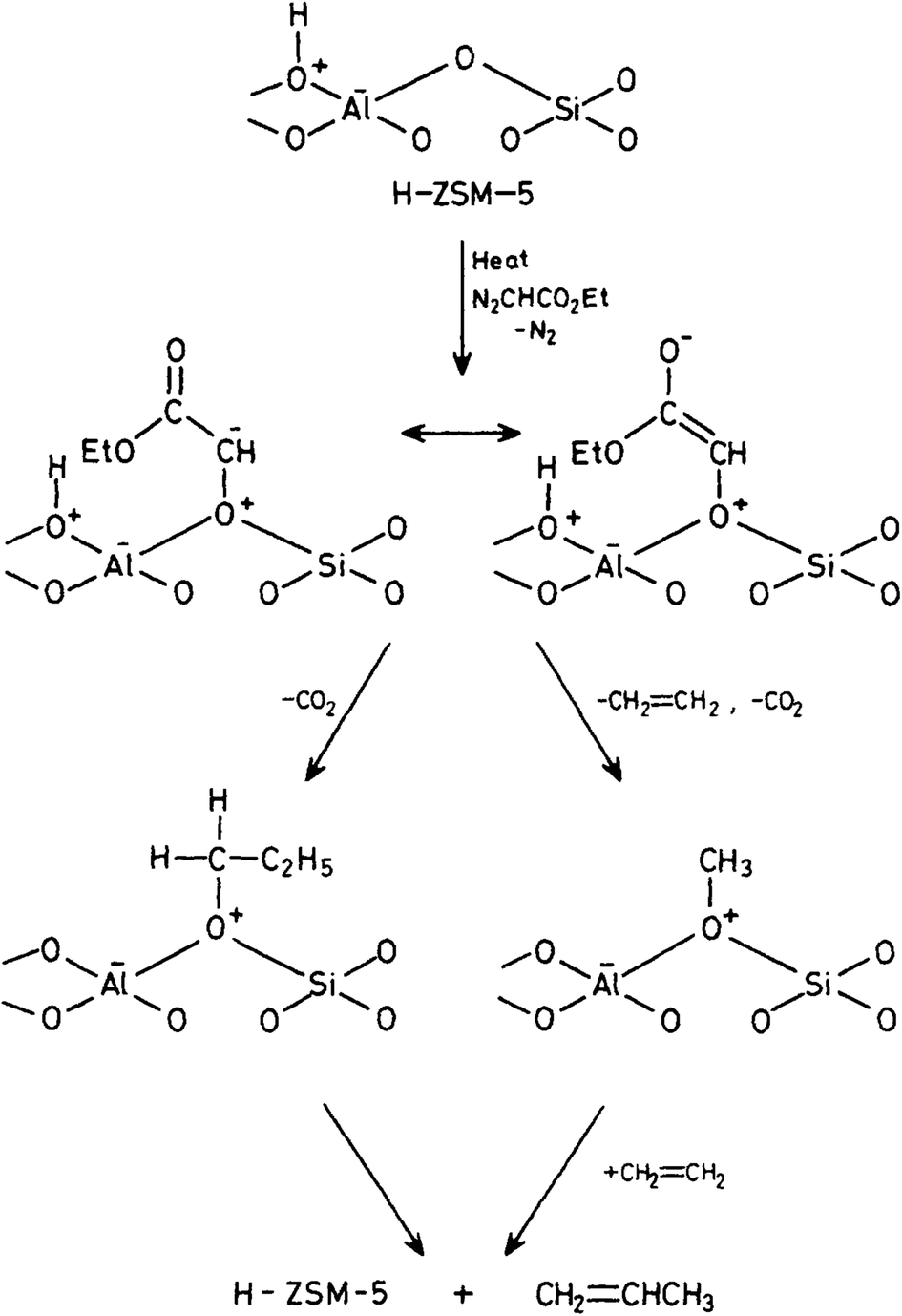 | ||
| Fig. 4 Proposed reaction mechanism for the reaction of ethyldiazoacetate with H-ZSM-5. Reproduced with permission from ref. 21. | ||
There are two additional problems associated with this mechanism; first, reaction of methanol and H2 over H-ZSM-5 does not lead to methane formation, but if a gas phase carbene species was present, this would lead to methane.22 Secondly, the main problem concerns whether the conjugate base of a strong acid, such as H-ZSM-5, is a sufficiently strong base to deprotonate the methyl group of methanol.
The trimethyloxonium-ylide mechanism
With ZSM-5 as catalyst, this was initially proposed by Engelen et al.23 In this reaction scheme, methanol is sequentially methylated to form a trimethyloxonium species. This is then deprotonated by the conjugate base site of ZSM-5, in an analogous manner to the deprotonation proposed in the carbene mechanism, to form a methylide (CH3)2OCH2−. The methylide can then react by two pathways. In a known reaction of such ylides, it can undergo a Stevens rearrangement forming methyl ethyl ether which can eliminate ethene, or it can react with methanol to form a dimethyl ethyl oxonium species that can also subsequently form ethene (Fig. 5). Support for this mechanism came from Rimmelin et al.24 who showed that methyl ethyl ether is formed by treating trimethyloxonium hexachloroantimonate with the strongly hindered base 2,2,6,6-tetramethylpiperidyl-lithium at 70 °C. They attributed this reaction to the formation of an oxygen ylide followed by a Stevens-type rearrangement or intermolecular methylation. While demonstrating that a strongly hindered base facilitated the reaction, the authors raised a question concerning the nature of the basic site in ZSM-5. | ||
| Fig. 5 Reaction scheme for the trimethyloxonium methylide mechanism. | ||
The central question as raised by Rimmelin et al.24 is whether the conjugate base of ZSM-5 is sufficiently strong to enable deprotonation. This is the same question as that raised for the carbene mechanism. However, there is a second piece of evidence that raises concerns for this mechanism. S analogues of trimethyloxonium salts form ylides much more readily than oxonium salts, but CH3SH is a poor substrate for ZSM-5 (ref. 25) and furthermore dimethyl sulfate, a substrate that cannot form a trimethyloxonium intermediate, readily reacts over H-ZSM-5 to form ethene and higher hydrocarbons.25
The hydrocarbon pool mechanism
A key feature of the methanol conversion reaction catalysed by H-ZSM-5 is the induction period due to the autocatalytic nature of the reaction.8 In this initial period the primary product is formed and then rapidly reacts in the secondary reactions to form higher alkenes and aromatics. This mechanism was first proposed by Kolboe26 and later fully described by Haw et al.27 However, there were earlier studies by Mole and co-workers28 and Langner29 that could form the basis of this approach. Mole28 co-reacted methanol and toluene and observed an enhancement in the rate of methanol conversion (Fig. 6a). Langer29 co-reacted higher alcohols with methanol and also observed an enhancement in the rate of methanol conversion. Hence the addition of molecules that already contained carbon–carbon bonds led to a reduction in the induction period (Fig. 6b). Kolboe26 proposed that in the initial phase of the reaction a hydrocarbon pool was established, denoted by (CH2)n (Fig. 6c). This pool then reacted with methanol by methylation followed by cracking at the acidic sites of the zeolite to produce the light alkenes that are desorbed from the zeolite pores. As noted by Haw,27 there was a much earlier study by Sullivan and co-workers30 which demonstrated that ring contraction of hexamethylbenzene could produce propene under acidic conditions which again supports the concept of a hydrocarbon pool of highly alkylated aromatic structures that produce alkenes that can react to form the higher hydrocarbon products that are observed. The nature of the hydrocarbon pool was fully described in very elegant in situ NMR spectroscopy studies by Haw et al.27 as well as pulse flow studies to study the induction period of the reaction, and the hydrocarbon pool comprises a very complex array of alkylated aromatic structures adsorbed within the pores of the zeolite.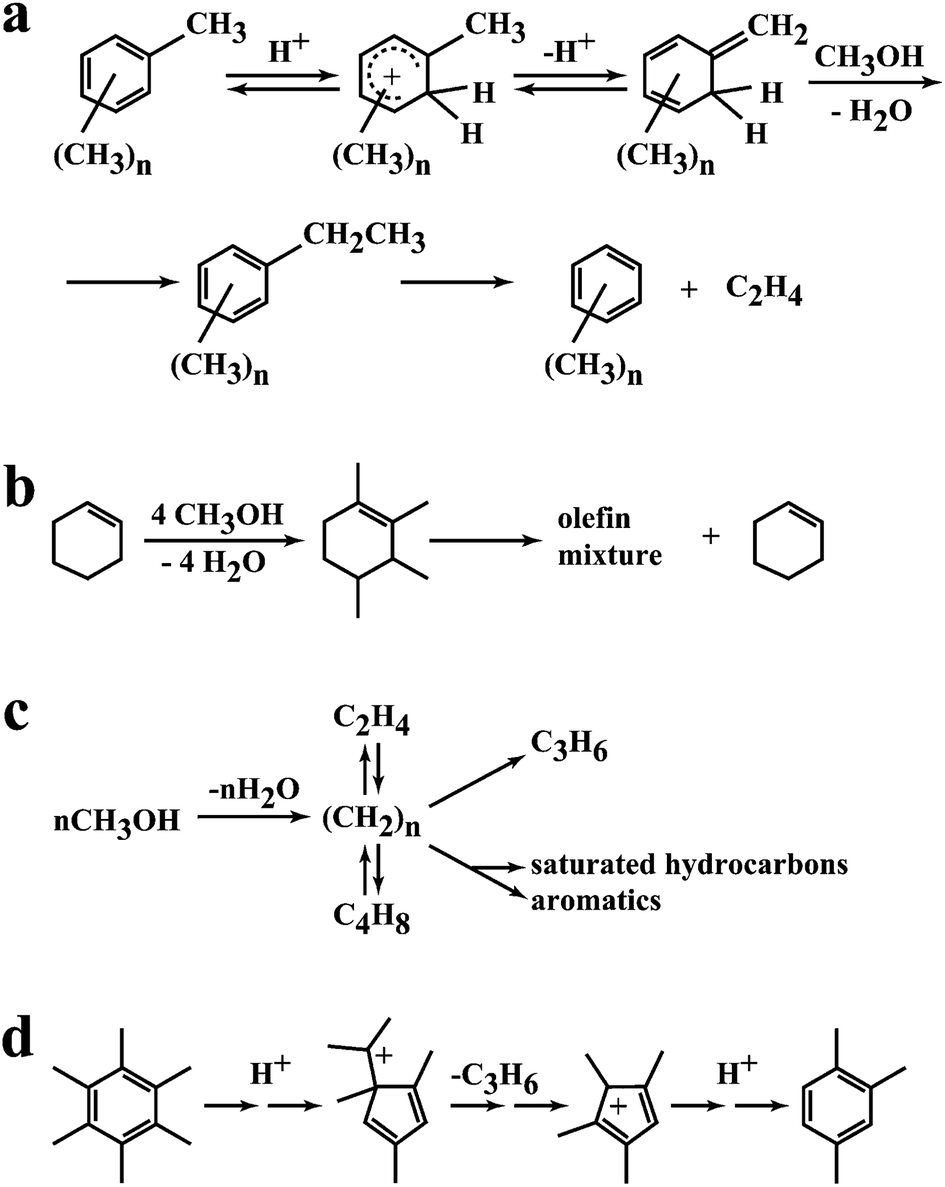 | ||
| Fig. 6 Representations of the hydrocarbon pool. (a) Proposal by Mole,28 (b) effect of co-fed alcohols by Langner,29 (c) initial proposal of the hydrocarbon pool by Kolboe,26 (d) pairing mechanism of Sullivan.30 Reproduced with permission from ref. 27. Copyright (2003) American Chemical Society. | ||
Therefore, the essence of the hydrocarbon pool mechanism is that ethene, the primary product observed with H-ZSM-5 as catalyst, originates from the secondary reactions of this hydrocarbon pool (Scheme 2).
 | ||
| Scheme 2 | ||
However, this mechanism does not explain the mechanism by which the initial carbon–carbon bonds are formed in the hydrocarbon pool. If we want to understand the mechanism of the methanol to hydrocarbon reaction fully, we need to determine how the initial carbon–carbon bonds are formed. Of course, given the studies of Mole28 and Langer29 these could be introduced in the methanol feed as impurities, but this is not a particularly elegant and satisfying solution to this age-old problem.
The surface methoxy-methylide mechanism
This mechanistic proposal is based on the observation that methanol is an effective methylating agent in the presence of a Brønsted acid, as is CH2N2.20 In the methoxy-methylide mechanism, protonated methanol acts as a methylating agent and forms a surface bound methoxyl and water (Scheme 3). The surface methoxyl is then deprotonated to form a surface bound methylide which is isoelectronic with a gas phase carbene.25 Indeed, using the principle of Occam’s razor, the surface bound methylide is a much simpler proposal than the oxonium methylide. The methylide then inserts into a C–H bond of methanol to form the initial carbon–carbon bond. The concept was to integrate parts of the carbene and oxonium methylide mechanisms as both had positive points, although they shared a common problem, namely is the conjugate base site of H-ZSM-5 sufficiently basic to enable the deprotonation?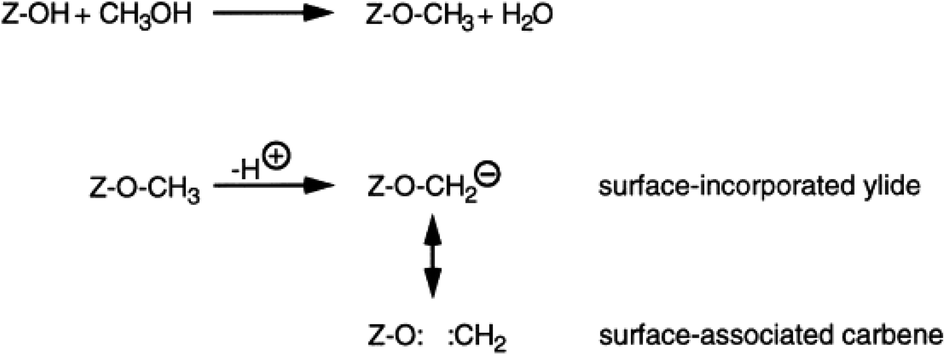 | ||
| Scheme 3 | ||
The most important evidence in support of this mechanism is that dimethyl sulfate and methyl iodide, both of which are more potent methylating agents than methanol, produce similar product distributions to methanol when used as substrates with H-ZSM-5 (Fig. 7).25,31 Also, it should be noted that neither dimethyl sulfate nor methyl iodide can form trimethyloxonium ions. These findings support the proposal that the initial step in the methanol conversion reaction with H-ZSM-5 is the formation of a surface methoxyl and that methanol is effectively a methylating agent for H-ZSM-5.
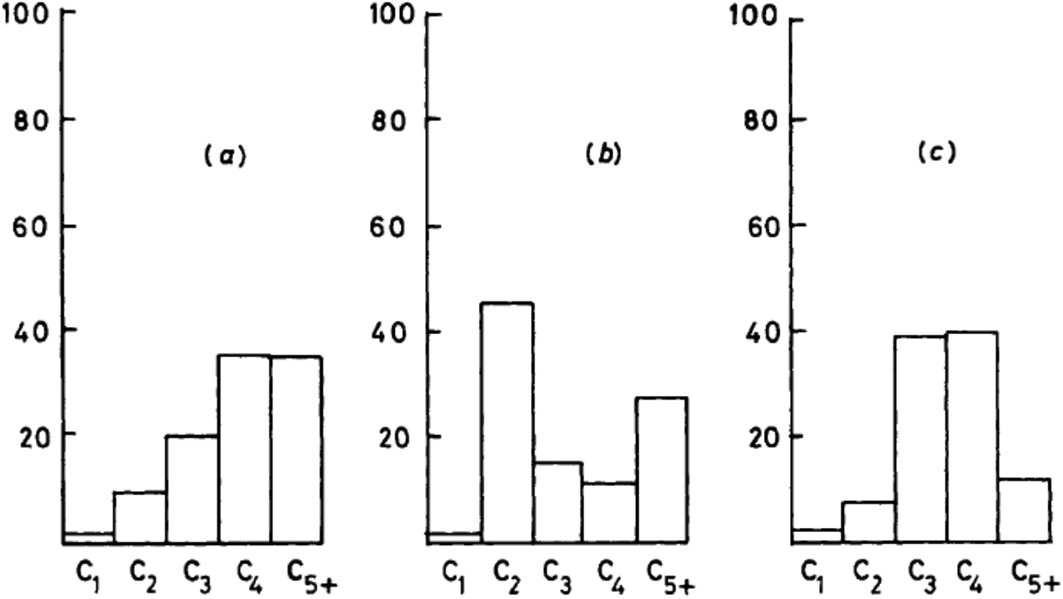 | ||
| Fig. 7 Comparison of the product selectivities for the reaction of methanol and methylating agents over H-ZSM-5 at 250 °C. (a) CH3OH (WHSV 0.005 h−1, 87% conversion), (b) (CH3)2SO4 (WHSV 0.075 h−1, conversion 21%), (c) CH3I (WHSV 0.06 h−1, conversion 0.1%). Reproduced with permission from ref. 25. | ||
To investigate the role of the conjugate base in methanol conversion, a set of experiments were carried out using a model compound for the conjugate base of the zeolite.32 Reacting LiAl(OiPr)4 with ultra-dry methylating agents led to methylation but no products from ylide formation were observed. Again, this supports methylation as the initial step, but also shows that the conjugate base cannot enable ylide formation with a non-surface bound reaction intermediate. To investigate the methylation mechanism further, methanol was reacted over zeolite β, which interestingly gave very high selectivities for C4 hydrocarbons.33 This was explained mechanistically by initial interaction of a methanol hydrogen bonded to a site adjacent to a surface methoxyl, and the reaction occurs via a concerted electron transfer such that the surface bound methylide is not formally formed (Fig. 8). This proposal may be a viable way for the initial carbon–carbon bond to be formed, as it may involve a cluster of hydrogen bonded methanol molecules within the confined space of the pore structure of H-ZSM-5, since theoretical studies have shown that the direct formation of a surface stabilised carbene species is energetically disfavoured with an energy barrier of 215–232 kJ mol−1.34
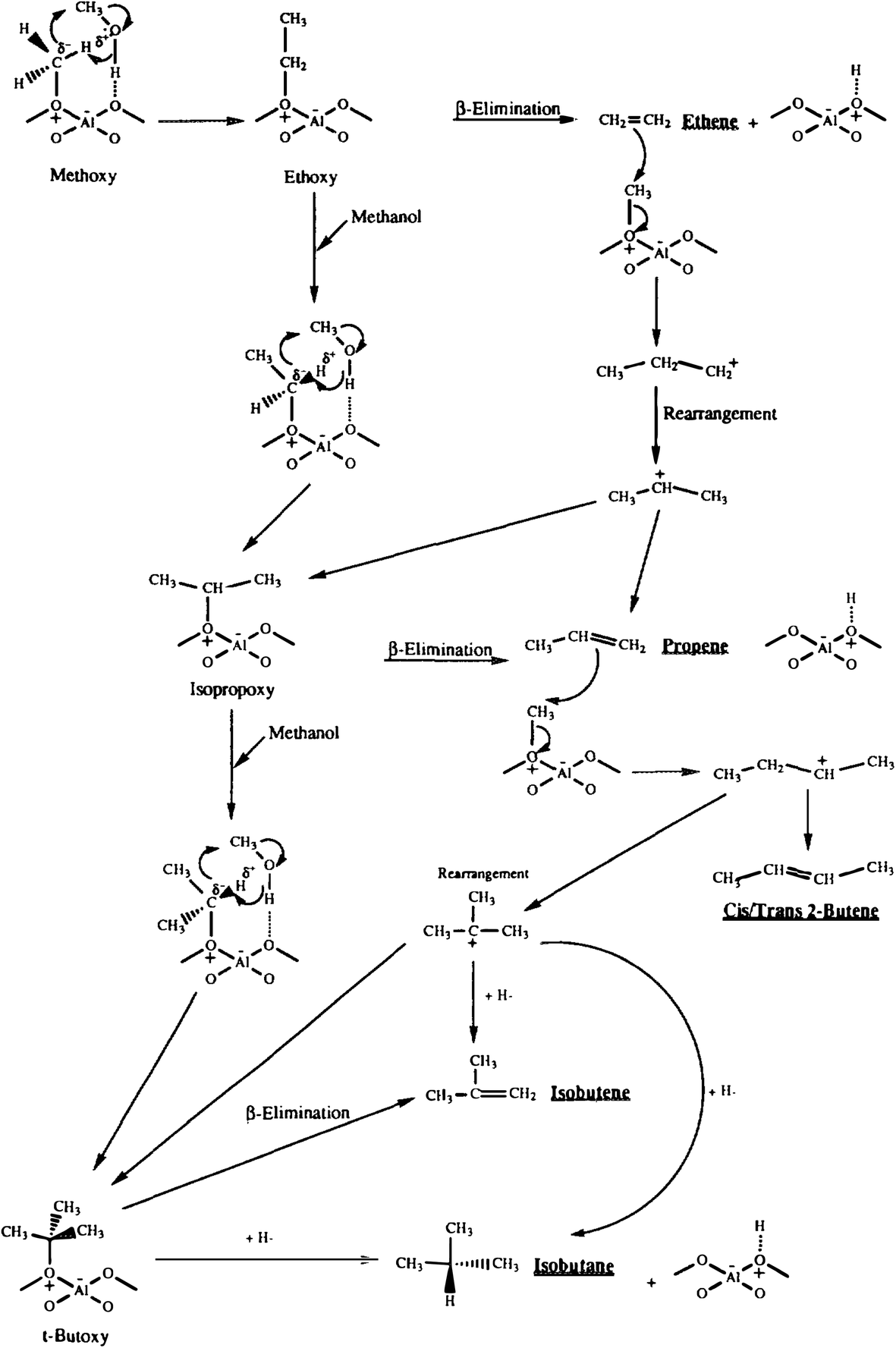 | ||
| Fig. 8 Proposed mechanism for the formation of C4 hydrocarbons for the reaction of methanol with zeolite β. Reproduced with permission from ref. 33. | ||
Computer modelling and advanced spectroscopy studies of the reaction mechanism
The use of advanced computational methods has been very important in gaining an understanding of the mechanism of carbon–carbon bond formation. Using computational modelling, Catlow, Logsdail and co-workers have shown that the initial interaction between methanol and the protonated ZSM-5 leads to a surface methoxyl.35 This confirms that with an acidic zeolite, methanol acts as a methylating agent. However, as noted above, the energetic barrier to the formation of an isolated gas phase carbene remains high.Advanced spectroscopy is now extensively used. Wang and Hunger36 reviewed earlier in situ NMR spectroscopy studies discussing the reactions of the surface methoxyl species. More recently, Weckhuysen and co-workers37 have reviewed the recent literature for the use of advanced spectroscopy to study this reaction. For example, Weckhuysen and co-workers38 and Schmidt et al.39 have shown the elegant use of atom probe tomography to map the elements in single crystals of ZSM-5 and follow coke formation. The coke formation, a key aspect of catalyst deactivation for this reaction, is noted to occur in clusters which would be consistent with the formation of the hydrocarbon pool. It was also noted that the aluminium distribution in the large zeolite crystals was not uniform. The question of the role of extra-framework aluminium is an interesting one. Although most samples of H-ZSM-5 contain very little of this species, Wang et al.40 have shown that extra-framework Al can interact with methanol to form a surface methoxy species that is bound to this extra-framework Al.† They used advanced 13C-{27Al} double-resonance solid-state NMR spectroscopy. They proposed that this species was very reactive and could lead to the formation of an ethoxy species. Hence extra-framework Al could play a role in this reaction, although most consider that it is the Brønsted acid site rather than a Lewis acid site that dominates the formation of the surface methoxyl species.
Howe and co-workers,41 following up on their earlier in situ infra-red spectroscopy studies of this reaction,42 have used operando synchrotron infrared microspectroscopy with high temporal resolution (down to 0.25 s) to identify the initial events occurring when methanol vapor is in contact with a crystal of zeolite H-ZSM-5 (Fig. 9). They were able to follow the formation of dimethyl ether and hydrocarbons in the initial period of the reaction that establishes the hydrocarbon pool. In addition, they indicated the possibility of a CH2 species being involved. Subsequently,43 they studied the effects of crystal size on methanol to hydrocarbon conversion over single crystals of ZSM-5 using operando synchrotron infrared microspectroscopy. They studied methanol conversion with coffin-shaped H-ZSM-5 crystals of different sizes: large (∼250 × 80 × 85 μm3), medium (∼160 × 60 × 60 μm3) and small (∼55 × 30 × 30 μm3). The induction period, for direct alkene formation by deprotonation of surface methoxy groups, was found to decrease with decreasing crystal size and with increasing reaction temperature. Experiments with a continuous flow of dimethyl ether showed that evolution of the hydrocarbon pool and indirect alkene formation are also strongly dependent on crystal size.
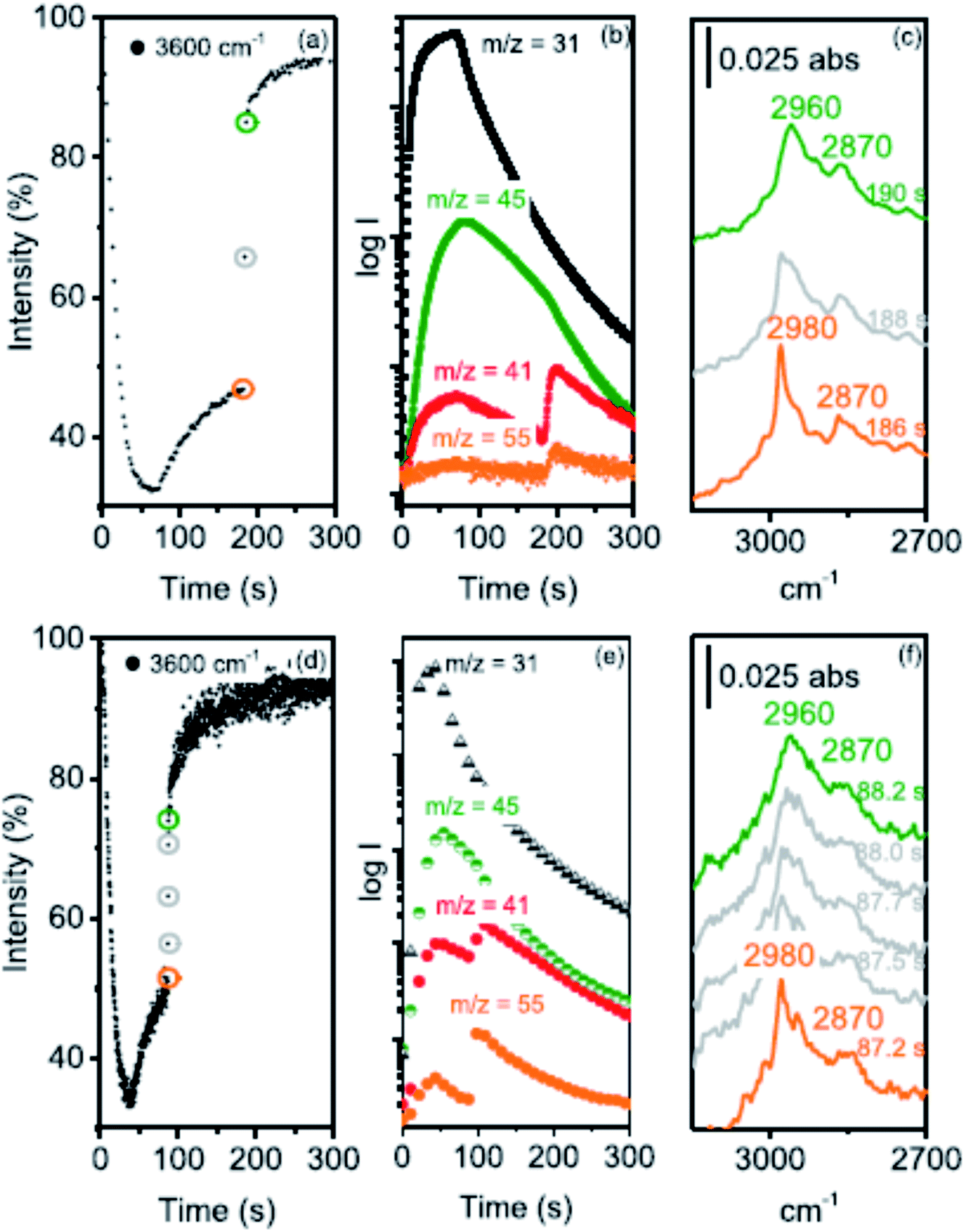 | ||
| Fig. 9 (a) Time course of the ν(OH) 3600 cm−1 band intensity relative to an activated crystal recorded at 2 s intervals during the first 8 μL methanol pulse injected into a N2 flow of 100 mL min−1 over an H-ZSM-5 crystal at 300 °C. (b) MS traces recorded during this experiment: m/z = 31 measures methanol, m/z = 45 DME, m/z = 41 propene (with a contribution from DME fragmentation), and m/z = 55 butene. (c) Evolution of the CH stretching region between 186 and 190 s. (d) The same experiment performed with 0.25 s time resolution during a 4 μL methanol pulse over a crystal from the same batch at 300 °C, (e) the corresponding MS traces and (f) evolution of the CH stretching region between 87.2 and 88.2 s after injection. Reproduced with permission from ref. 41. Copyright (2019) American Chemical Society. | ||
It is clear that advanced spectroscopies, especially those using synchrotron techniques, coupled with computational methods will in the future be able to give much more valuable insights into this fascinating reaction mechanism. At present it is clear that the initial interaction of methanol or dimethyl ether with H-ZSM-5 leads to the formation of a surface methoxyl species as initially proposed based on experimental evidence with methylating agents as model reactants.25 How this surface methoxyl is then involved in carbon–carbon bond formation continues to be a matter of debate. Certainly, the formation of an isolated carbene species is not energetically possible.34 However, perhaps the possibility that the formation of the surface associated carbene species involves a concerted electron transfer with a cluster of hydrogen bonded methanol molecules could be explored computationally.
Gold catalysis
There has been interest in gold for many years, especially colloidal gold since Faraday first demonstrated its synthesis and stability.44 The use of gold as a catalyst, however, was not particularly successful for many years. It proved difficult to prepare small nanoparticles that would be important for catalysis since gold is the most noble of metals and is readily reduced to the metal and sinters into larger particles of no or limited activity. Hence other metals such as Pt or Pd were favoured as small nanoparticles could be readily formed and stabilised. It was not until the 1980s that gold catalysis became an exciting field of catalysis when gold was found to be the best catalyst for CO oxidation and acetylene hydrochlorination.CO oxidation using gold catalysts
Haruta and co-workers45 found that gold supported on iron oxide when prepared by coprecipitation is an exceptionally active catalyst for CO oxidation. The catalysts were observed to be active at temperatures below −70 °C (Fig. 10). This exceptional activity caught the attention of the catalysis community and soon the quest was on to determine the nature of the active site and the reaction mechanism. Early high resolution transmission electron microscopy (HRTEM) revealed the presence of small gold nanoparticles in active catalysts and that there was a relationship between the nanoparticle size and the activity, with the most active catalysts comprising the smallest Au nanoparticles.46 In 2004 Chen and Goodman47 reported a model catalyst in which a molybdenum metal flat surface was used as a template for titania, and on this, either extended monolayers or bilayers of gold were deposited (Fig. 11). Using these model catalysts in a flow reactor, they showed that the bilayers were exceptionally active but the monolayers were inactive. The activity of the bilayers exhibited a turnover frequency (TOF) of 3.7 s−1 which is about two orders of magnitude higher than that of the best Au/FeOx catalyst prepared by coprecipitation. The understanding of gold catalysis was significantly enhanced by the advent of aberration-corrected scanning transmission electron microscopy (AC-STEM), as it was possible to observe atomically dispersed metal species.48 With this new technique it was now possible to observe that the gold catalysts not only comprised Au nanoparticles, but also atomic/cationic gold and nanoclusters, as well as nanoparticles of various size ranges.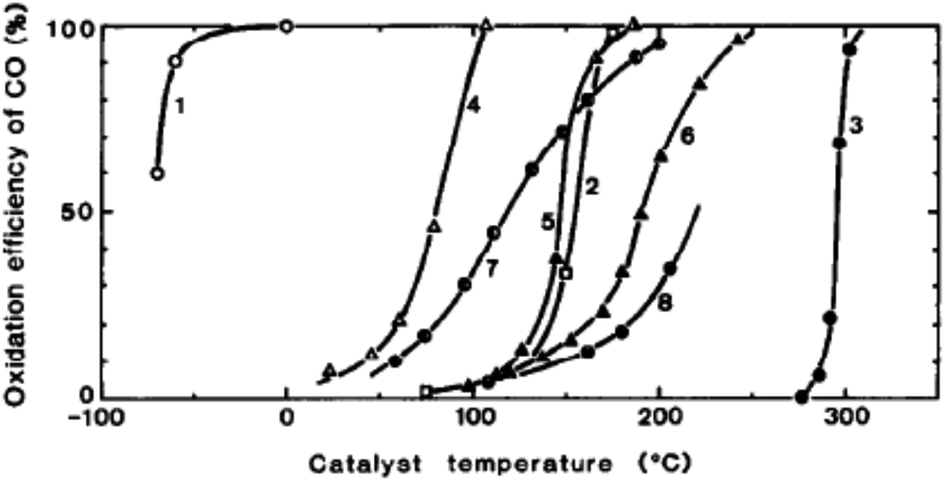 | ||
Fig. 10 Conversion of CO as a function of temperature. (1) Au/α-Fe2O3 (Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe = 1:9, coprecipitation, 400 °C), (2) 0.5 wt% Pd/γ-Al2O3 (impregnation, 300 °C), (3) Au fine powder, (4) Co3O4 (carbonate, 400 °C), (5) NiO (hydrate, 200 °C), (6) α-Fe2O3 (hydrate, 400 °C), (7) 5 wt% Au/α-Fe2O3 (impregnation, 200 °C), (8) 5 wt% Au/γ-Al2O3 (impregnation, 200 °C). Reproduced with permission from ref. 45. :Fe = 1:9, coprecipitation, 400 °C), (2) 0.5 wt% Pd/γ-Al2O3 (impregnation, 300 °C), (3) Au fine powder, (4) Co3O4 (carbonate, 400 °C), (5) NiO (hydrate, 200 °C), (6) α-Fe2O3 (hydrate, 400 °C), (7) 5 wt% Au/α-Fe2O3 (impregnation, 200 °C), (8) 5 wt% Au/γ-Al2O3 (impregnation, 200 °C). Reproduced with permission from ref. 45. | ||
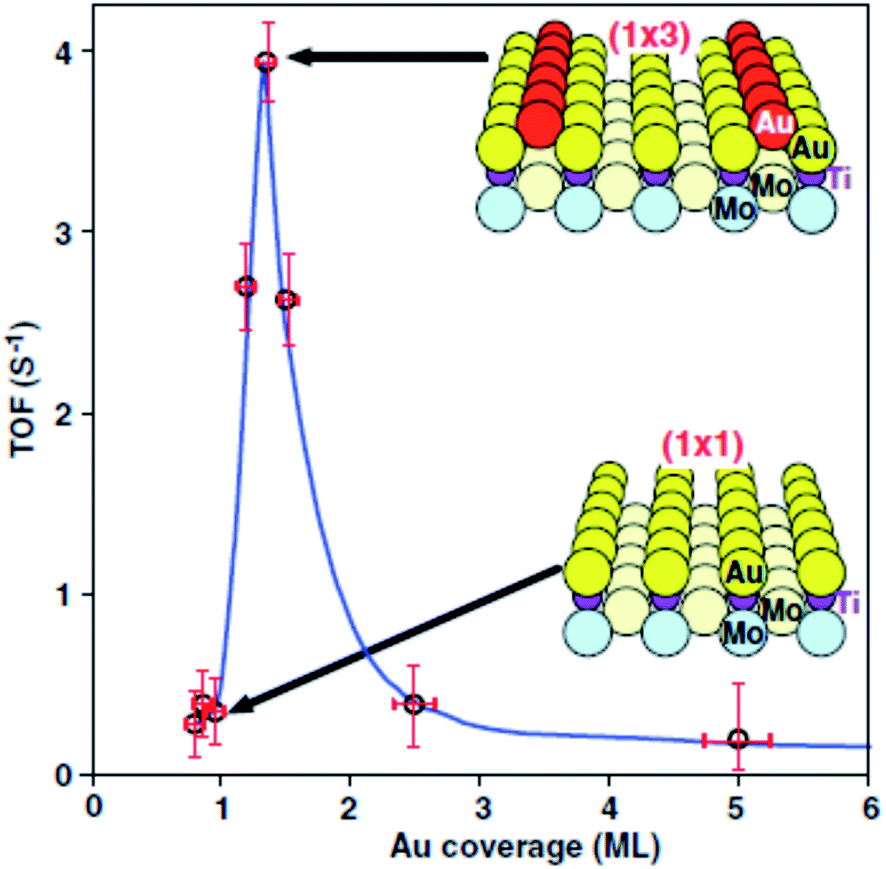 | ||
| Fig. 11 CO oxidation over model catalysts comprising monolayers and bilayers of Au. Reproduced with permission from ref. 47. | ||
An interesting observation was made by Herzing et al.,49 in which two Au/FeOx catalysts were prepared by coprecipitation and dried in different ways. Both were dried at 120 °C for 16 h, but the one dried in flowing air was exceptionally active for CO oxidation at 25 °C, whereas the material dried in static air was inactive. According to conventional HRTEM, the two materials showed near identical populations of gold nanoparticles. However, according to AC-STEM, the sample calcined in flowing air had bilayer clusters that were not present in the sample prepared by calcination in static air (Fig. 12). This prompted a study where the highly active material was further calcined at a range of higher temperatures49 to prepare a set of Au/FeOx materials that exhibited a range of decreasing activities. Examination by AC-STEM showed that the samples contained well dispersed gold atoms in addition to monolayer and bilayer clusters as well as gold nanoparticles, but the activity mapped the population density of the bilayer Au clusters (Fig. 13). If the activity of this catalyst was solely due to the bilayer clusters, then the activity of this Au/FeOx catalyst was determined to be 3.5 s−1 (ref. 50) which is very similar to the value reported by Chen and Goodman47 in their model studies on the activity of extended Au bilayers. These two studies therefore provide a basis for theoretical studies to investigate the reaction mechanism.
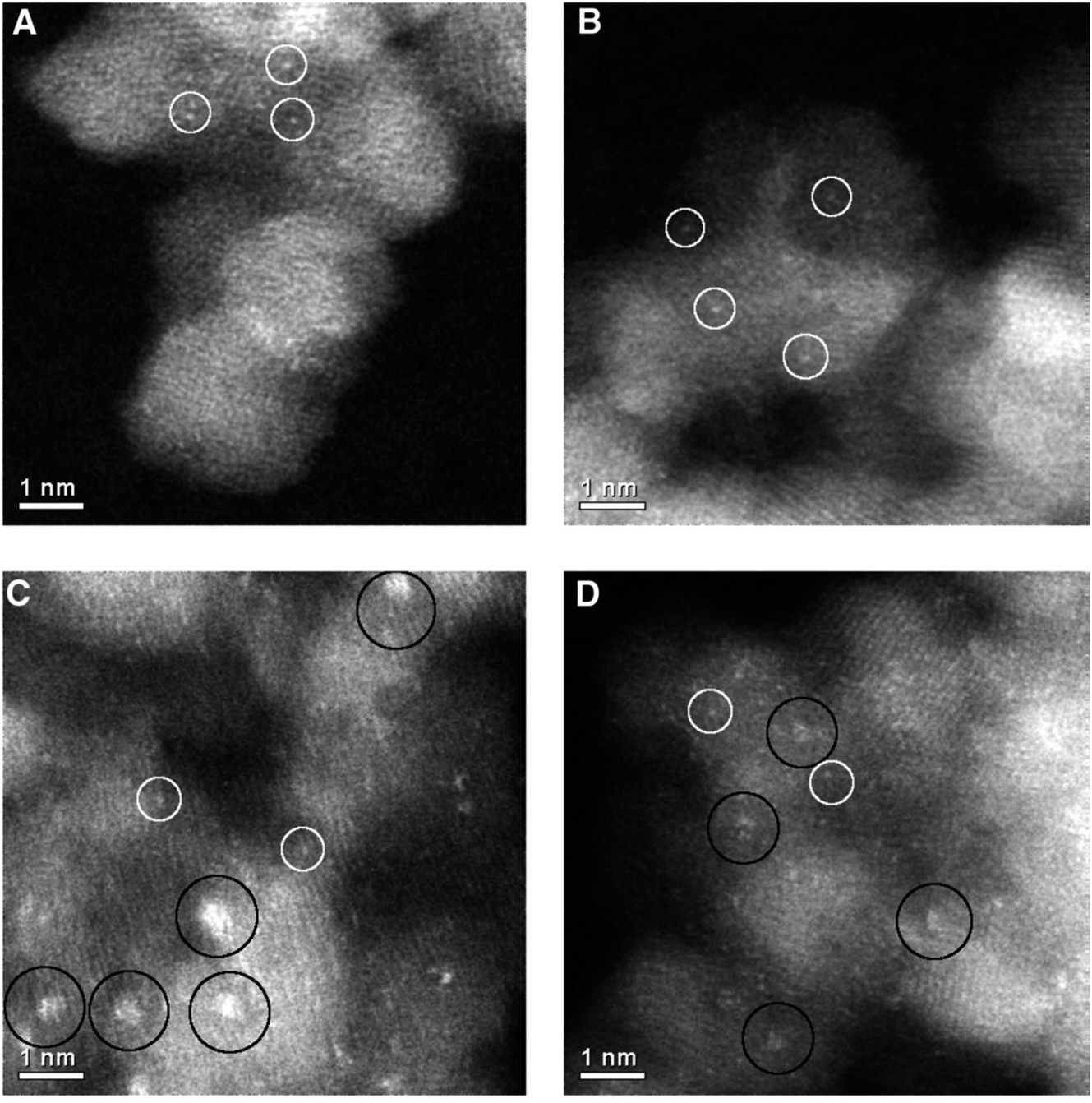 | ||
| Fig. 12 High-magnification aberration-corrected STEM-HAADF images of (A and B) the inactive and (C and D) the active Au/FeOx catalysts acquired with AC-STEM. The white circles indicate the presence of individual Au atoms, whereas the black circles indicate sub-nanometer Au clusters consisting of only a few atoms. Reproduced with permission from ref. 49. | ||
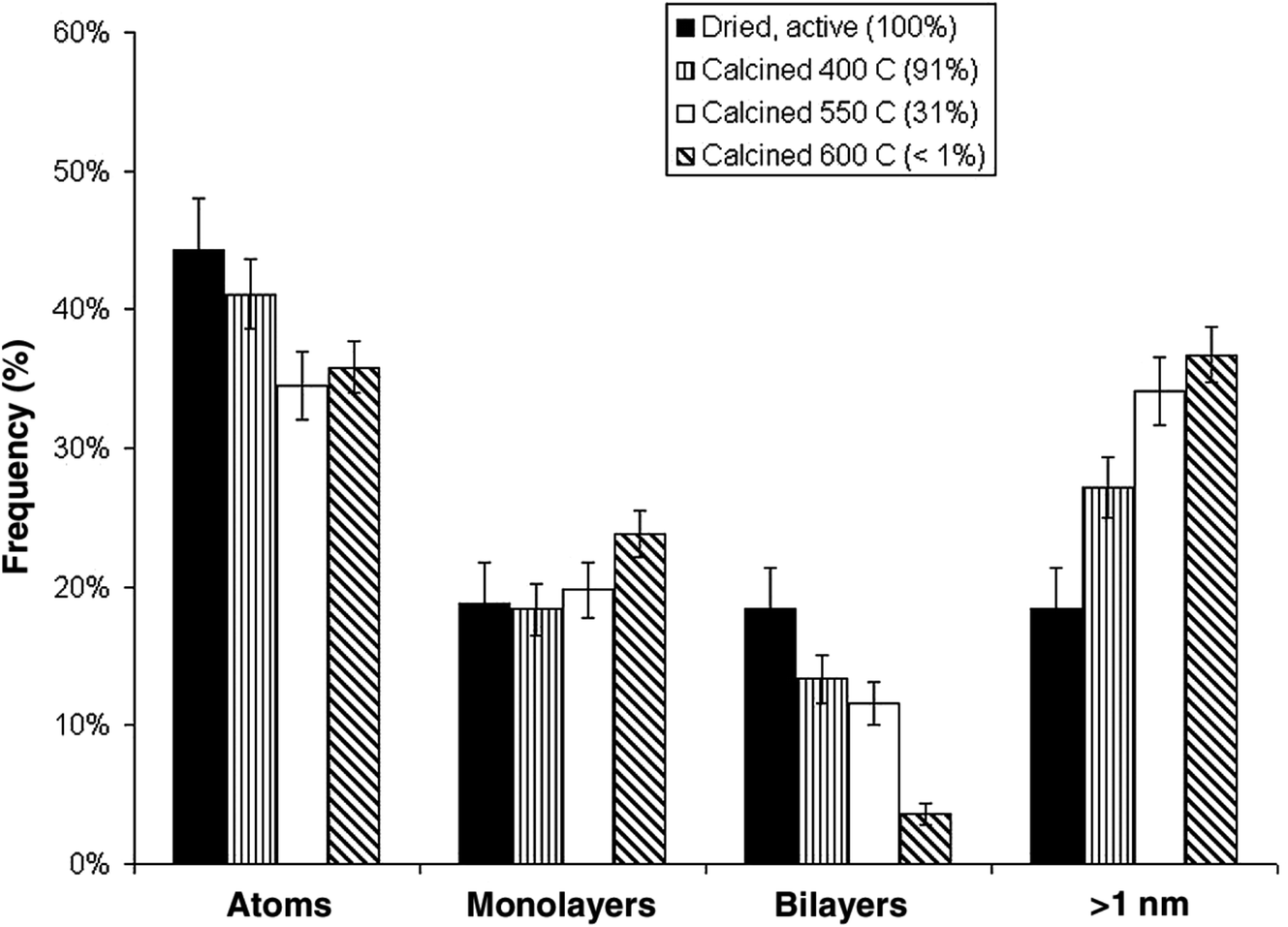 | ||
| Fig. 13 Relative populations of (i) dispersed Au atoms, (ii) 0.2 to 0.3 nm monolayer Au clusters, (iii) 0.5 nm bilayer Au clusters, and (iv) Au nanoparticles > 1 nm in diameter, as a function of the catalyst calcination temperature and measured CO conversion. The error bars correspond to two standard deviations on the size measurements. Reproduced with permission from ref. 49. | ||
The origin of the high activity of FeOx catalysts was explored in a joint study by Haruta, Hutchings and co-workers.51 It was noted that the two groups had been using very different approaches to the coprecipitation method. Haruta and co-workers used a method in which the acidic solution (Fe(NO3)3 + HAuCl4 in water) was added to the base (Na2CO3 in water) rapidly, whereas Hutchings and co-workers added the acidic solution slowly to the basic solution. Two sets of materials were prepared using these methods, with both being dried at 120 °C (denoted by CP-1 for adding the acid into the base and CP-2 for adding the base into the acid) and then calcined at 300 °C (denoted by CP-3 for adding the acid into the base and CP-4 for adding the base into the acid), giving four samples in all. The catalysts were then tested for CO oxidation, and the effect of reaction temperature on CO conversion is shown in Fig. 14. The two catalysts dried at 120 °C gave, within experimental error, identical CO conversion versus time profiles. However, the two catalysts calcined at 300 °C behaved very differently. CP-3 improved in activity, whereas CP-4 displayed a markedly poorer catalytic activity (Fig. 14). Detailed examination of the catalysts with electron microscopy using a new counting algorithm showed that the two dried catalysts had very different populations of gold species present on the support surface, yet their activity was almost identical. It was concluded that it was not possible to assign just one type of Au species as being active, while the others are inactive, in order to explain all the sets of data. Instead, it was proposed that an activity hierarchy for the different Au species was present. This readily explains the observed behavior since the co-existence of a wide range of Au nanostructures each having a different intrinsic activity needs to be considered (Fig. 15). Hence the final reported activities of these catalysts should be the weighted sums of the activities of each of the different species present, combined with their relative population densities (i.e. total activity 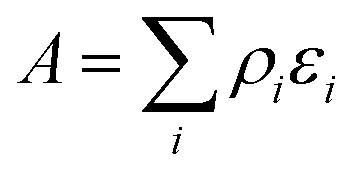 , where ρi and εi represent the population fraction and intrinsic activity, respectively, for the ith active species). This hierarchy of activities can then be used to explain the observed activities for the wide range of gold catalyst preparation methods. It is noted that bilayer clusters have a higher intrinsic activity than nanoparticles which in turn have a higher intrinsic activity than gold atoms. The reason for the enhancement in activity for CP-3 was also explained.51 As precipitation is rapid, the FeOx particles that are formed contain embedded Au species which on calcination migrate to the surface of the support particles, and these newly formed Au species are highly active. The FeOx formed by the slow precipitation method does not have such an Au reservoir and so there is no replenishment of active Au species and the Au species present on the surface are sintered into larger inactive nanoparticles.
, where ρi and εi represent the population fraction and intrinsic activity, respectively, for the ith active species). This hierarchy of activities can then be used to explain the observed activities for the wide range of gold catalyst preparation methods. It is noted that bilayer clusters have a higher intrinsic activity than nanoparticles which in turn have a higher intrinsic activity than gold atoms. The reason for the enhancement in activity for CP-3 was also explained.51 As precipitation is rapid, the FeOx particles that are formed contain embedded Au species which on calcination migrate to the surface of the support particles, and these newly formed Au species are highly active. The FeOx formed by the slow precipitation method does not have such an Au reservoir and so there is no replenishment of active Au species and the Au species present on the surface are sintered into larger inactive nanoparticles.
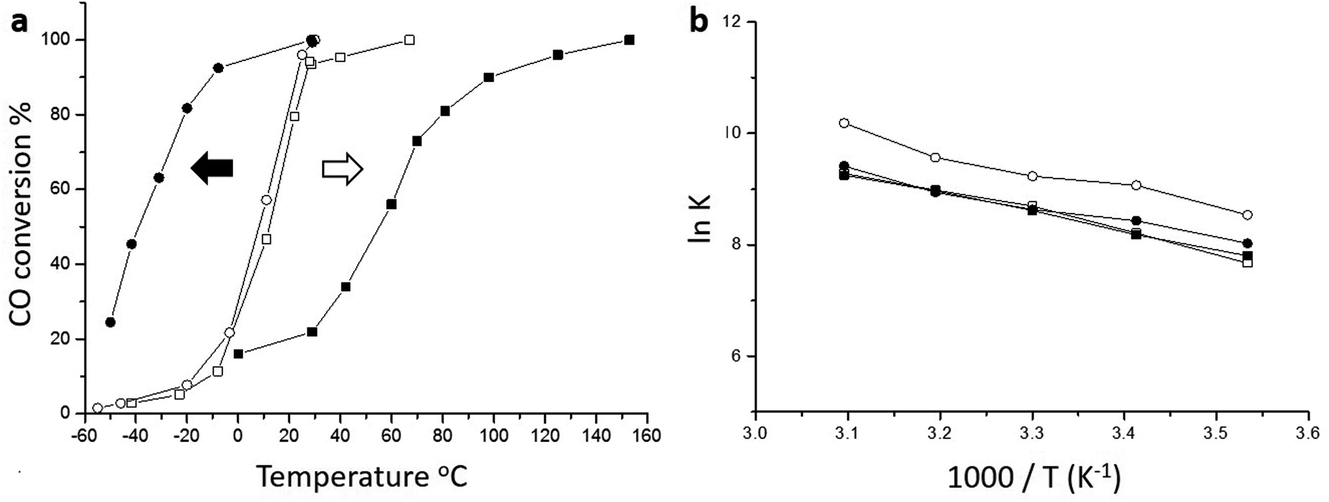 | ||
| Fig. 14 (a) CO conversion at various temperatures. Catalyst mass 150 mg, gas flow 50 mL min−1 1 vol% CO in air; (b) Arrhenius plots obtained under low conversion conditions showing that the catalysts all exhibit similar activation energies. Empty circles ○ (CP-1, dried, 6 wt% Au by ICP), filled circles ● (CP-3, calcined, 6 wt% Au by ICP), empty squares □ (CP-2, dried, 3.5 wt% Au by ICP), filled squares ■ (CP-4, calcined, 3.5 wt% Au by ICP). The arrows shown in (a) represent the thermal activation behaviour (black arrow) of the CP-3 catalyst and the thermal deactivation behaviour (white arrow) of the CP-4 catalyst. Reproduced with permission from ref. 51. | ||
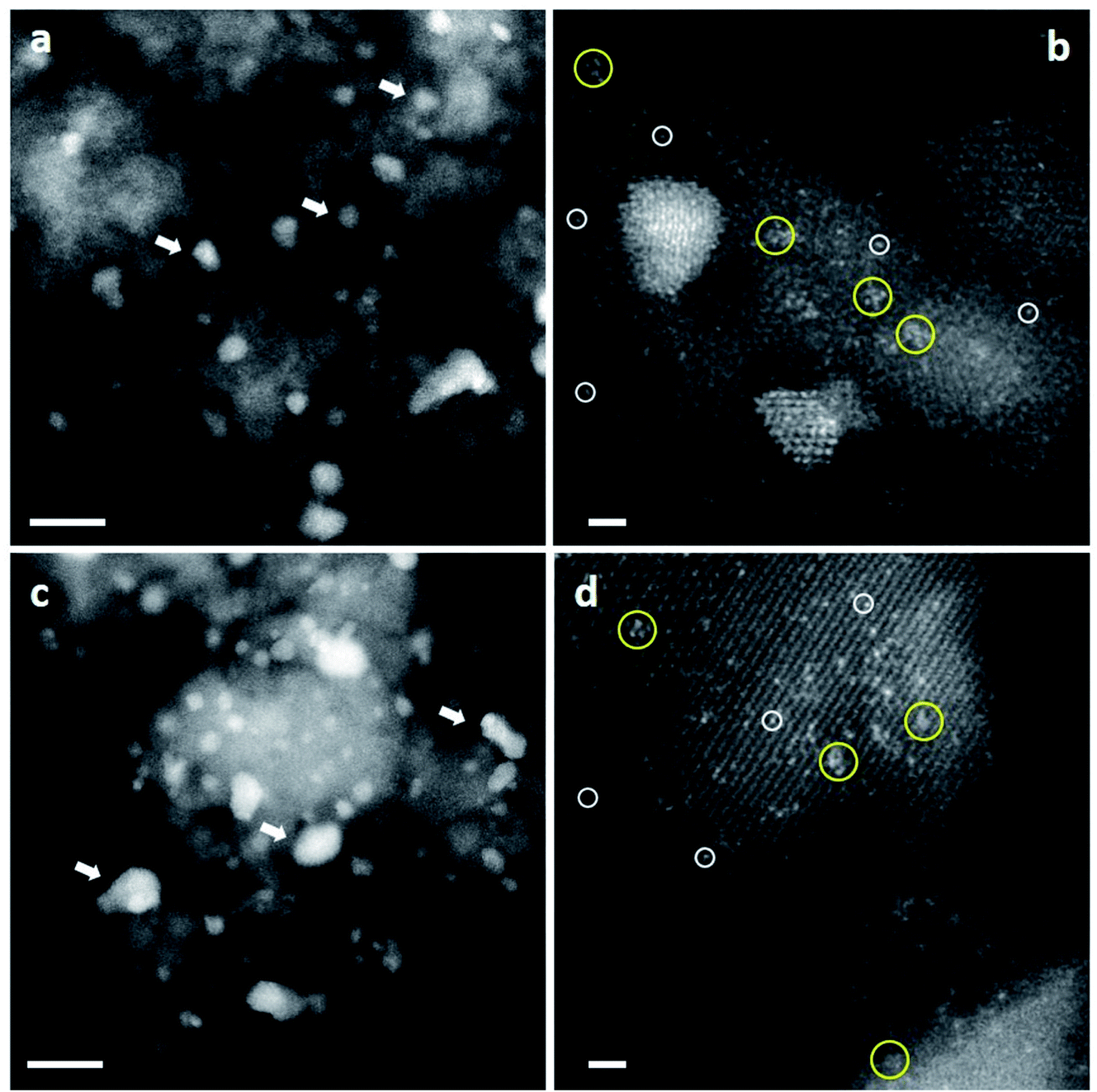 | ||
| Fig. 15 Representative HAADF-STEM images of CP-1 and CP-3 catalysts (acid-into-base). Images from ‘dried-only’ (a and b) and ‘calcined’ catalysts (c and d) showed the co-existence of nanoparticles of various sizes, sub-nm clusters and isolated atoms. Au nanoparticles – white arrows; sub-nm Au clusters – yellow circles; and isolated Au atoms – white circles. The scale bars in (a) and (c) represent 10 nm. The scale bars in (b) and (d) represent 1 nm. Reproduced with permission from ref. 51. | ||
The mechanism of low temperature CO oxidation with supported Au has been widely studied, but a proposal made by Bond and Thompson52 in 2000, based on all the information to hand at that time, can be considered to be a representative model (Fig. 16). The surface atoms of the Au nanostructure at the periphery in contact with the support carry a net positive charge. CO adsorbs on a low coordinate Au0 atom on the upper layer while a hydroxyl formed from water adsorption at a defect site adjacent to the gold nanoparticle transfers to the cationic Au peripheral site, creating an anion vacancy on the surface of the support. The CO and OH− form a carboxylate group, and an oxygen molecule occupies the anion vacancy as O2−. This then oxidizes the carboxylate group by abstracting a H atom, forming CO2, and the resulting hydroperoxide ion HO2− then oxidizes a further carboxylate species, forming another CO2 and restoring two hydroxyl ions to the support surface, completing the catalytic cycle.
 | ||
| Fig. 16 Schematic representation of the mechanism of CO oxidation on a supported gold catalyst. Reproduced with permission from ref. 52. | ||
Gold catalysts for acetylene hydrochlorination
In the early 1980s, while working in industry, I was asked to find a better catalyst than mercury for the hydrochlorination of acetylene, a reaction that makes vinyl chloride. The acetylene route to vinyl chloride is a coal-based route that is now currently operated extensively in China. Hence this early work has immense importance today. Based on available data in the literature, a correlation was made between the activity of a series of carbon-supported metal chlorides and the standard electrode potential of the cation (Fig. 17). This predicted that gold would be the best catalyst for this reaction.53 The prediction was subsequently validated.54,55 The problem with the mercury catalyst, which is 10% HgCl2/C, is that under the reaction conditions, the HgCl2 sublimes and is eventually lost from the reactor, and every year over 1000 tons of Hg is lost to the environment. Therefore, a non-mercury catalyst is essential. A number have been considered but Au is the most active and durable catalyst.56 The early catalysts54,55 were prepared using aqua regia as a solvent for the Au precursor and also used 1–2% Au. This is not a viable preparation procedure for a commercial catalyst. Subsequently, a preparation using water with gold thiosulfate was found to be superior and this required very low levels of Au. In commercial trials,56 the catalyst was found to give high sustained activity and Johnson Matthey have now commercialised the catalyst in China.56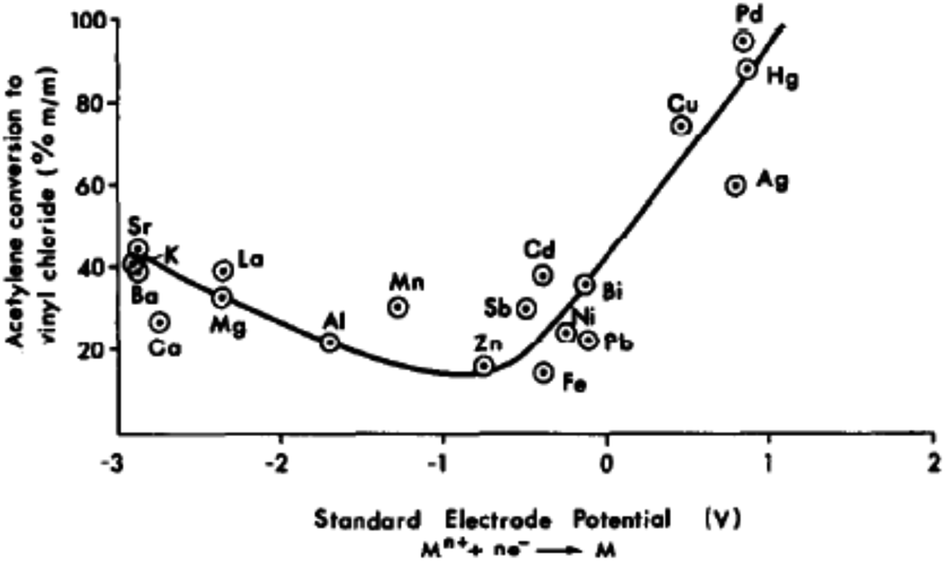 | ||
| Fig. 17 Correlation of the activity of carbon-supported metal chloride catalysts for acetylene hydrochlorination. Reproduced with permission from ref. 53. | ||
While the correlation predicts that Au cations are the active species, early microscopy and XPS ex situ investigations showed the presence of Au metal nanoparticles and so for many years the active sites were considered to be Au cations located at the periphery of the nanoparticle and the support. However, with the advent of AC-STEM and the use of synchrotron XAS and XANES it has been possible to show that in situ the Au is present as wholly dispersed cations and no nanoparticles are present (Fig. 18).2 There is a correlation between the white line height in the L3 edge XAFS and the catalyst productivity, showing that the higher the concentration of Au+ in the catalyst, the higher the activity, and the catalyst cycle involves Au+ and Au3+. These findings therefore confirm the original prediction based on the standard electrode potential.53 Based on this knowledge the mechanism of the hydrochlorination reaction was investigated.57 The role of Au(I) was studied with density functional theory (Fig. 19). The interaction of HCl with supported Au+ was studied using AuCl and the energetics of its interaction with HCl to form AuCl2H, which has more Au3+ character as determined by the Hirshfeld charge. The formation of vinyl chloride then occurs via the interaction of the surface AuCl2H with acetylene, and this represents a facile low energy pathway for the reaction.
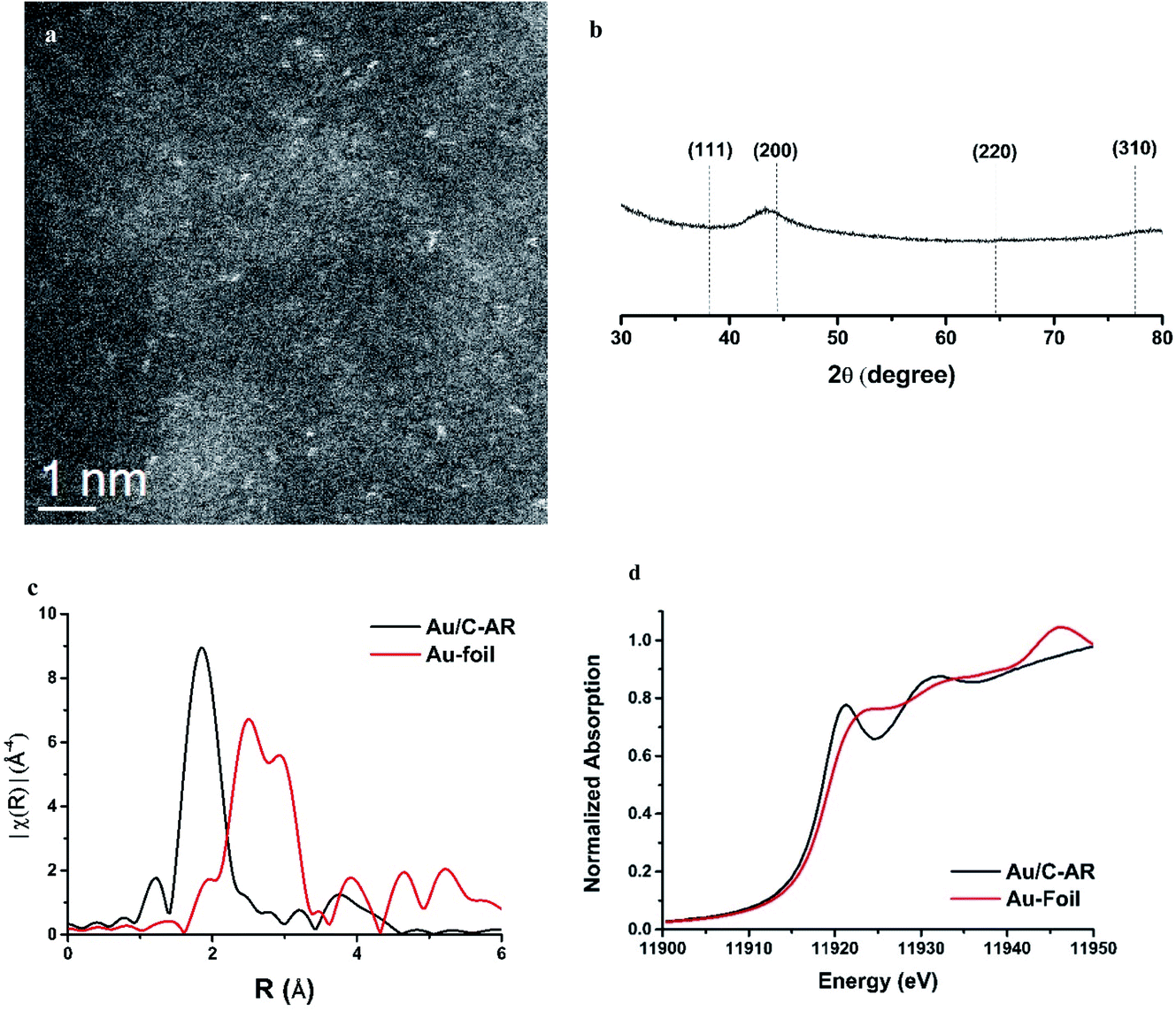 | ||
| Fig. 18 Characterization of a freshly prepared 1 wt% Au/C catalyst prepared from aqua regia solvent. (a) Representative HAADF-STEM image showing isolated Au species. (b) Powder X-ray diffraction data for this catalyst. (c) Fourier transform of k3 weighted χ EXAFS ex situ data for the sample and a gold foil reference. The variation in the magnitude of the Fourier transform is plotted against the distance R from the Au absorber. (d) Ex situ Au L3-edge normalised XANES spectra of the sample and a gold foil reference material. Reproduced with permission from ref. 2. | ||
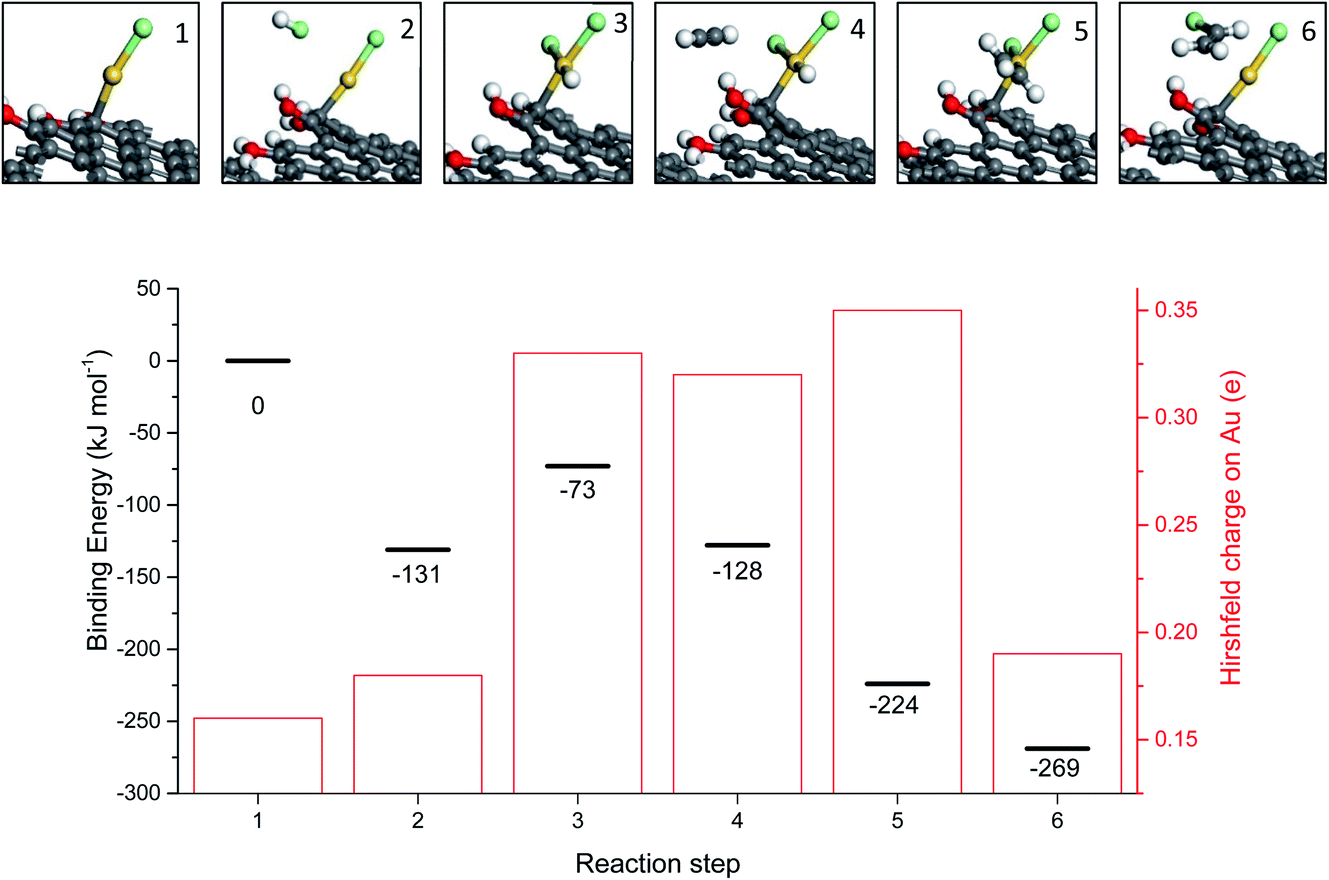 | ||
| Fig. 19 Mechanism for the transformation of AuCl to AuCl2H and the formation of vinyl chloride, reproducing AuCl. Key: Au atoms (gold), Cl atoms (green), H atoms (white), C atoms (grey) and O atoms (red). Energies are referenced to the geometry optimized configuration of AuCl on the carbon support and gas phase acetylene and HCl. Binding energies for each energy minimum and the Hirshfeld charge on each Au atom are shown for each step. Reproduced with permission from ref. 2. | ||
Conclusion
Investigating the mechanisms of catalysed reactions requires the use of many techniques and approaches. First, key experimental data on the effect of reaction conditions needs to be assembled. It is important that all the products are identified so that the reaction pathway can be established. For complex reactions there may be many by-products and their formation can provide valuable insights into the mechanism. Indeed, it is in this situation that the use of model reagents can be particularly valuable. Second, the array of in situ spectroscopies, diffraction techniques and environmental microscopies need to be fully employed. These are now becoming increasingly important, especially when accessing synchrotron facilities. Third, these experimental studies need to be coupled with computational modelling as this can provide valuable insights into the mechanism. It is crucial that all three of these methods are fully employed in the quest to understand catalysed reaction mechanisms.Looking to the future, where can we expect further advances? There is a great need for improved time resolution for the in situ techniques that are available to us. Inroads into this have been made, for example with the use of synchrotron infrared microspectroscopy where a time resolution of 0.25 s has been achieved,41,43 but we can expect further advances in this area. For example, it can be expected for oxidation reactions that EPR spectroscopy will play an enhanced role in the future since radical species are often involved in oxidations. It would be ideal to be able to combine techniques in such a way that the bulk and surface structure of a catalyst can be investigated in real time as the products are being analysed. It is clear that many are trying to make these advances. Indeed, the use of in situ techniques coupled with high level computational modelling is a major theme of the Faraday Discussion on reaction mechanisms in catalysis.
Conflicts of interest
The author has no conflicts of interest.Acknowledgements
Aspects of the research described in this paper have been supported by the EPSRC and UK Catalysis Hub (grants EP/K014714/1, EP/K014668/1, EP/K014706/1, EP/H000925/1 and EP/I019693/1).References
- G. J. Hutchings and R. Hunter, Hydrocarbon formation from methanol and dimethyl ether: a review of the experimental observations concerning the mechanism of formation of the primary products, Catal. Today, 1990, 6, 279–306 CrossRef CAS
.
- G. Malta, S. A. Kondrat, S. J. Freakley, C. J. Davies, L. Lu, S. Dawson, A. Thetford, E. K. Gibson, D. J. Morgan, W. Jones, P. P. Wells, P. Johnston, C. R. A. Catlow, C. J. Kiely and G. J. Hutchings, Identification of single-site gold catalysts in acetylene hydrochlorination, Science, 2017, 355, 1399–1402 CrossRef CAS PubMed
- A. Wang, J. Li and T. Zhang, Heterogeneous single-atom catalysis, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS
- J. M. Thomas, R. Raja and D. W. Lewis, Single-site heterogeneous catalysis, Angew. Chem., Int. Ed., 2005, 44, 6456–6582 CrossRef CAS PubMed
- W. van Deelen, J. J. Nijhuis, N. A. Krans, J. Zečević and K. P. de Jong, Preparation of cobalt nanocrystals supported on metal oxides to study particle growth in Fischer–Tropsch catalysts, ACS Catal., 2018, 8, 10581–10589 CrossRef
- G. C. Chinchen, K. C. Waugh and D. A. Whan, The activity and state of the copper surface in methanol synthesis catalysts, Appl. Catal., 1986, 25, 101–107 CrossRef CAS
- S. A. Kondrat, P. J. Smith, P. P. Wells, P. A. Chater, J. H. Carter, D. J. Morgan, E. M. Fiordaliso, J. B. Wagner, T. E. Davies, L. Lu, J. K. Bartley, S. H. Taylor, M. S. Spencer, C. J. Kiely, G. J. Kelly, C. W. Park, M. J. Rosseinsky and G. J. Hutchings, Stable amorphous georgeite as a precursor to a high activity catalyst, Nature, 2016, 531, 83–88 CrossRef CAS PubMed
- C. D. Chang and A. J. Silvestri, The conversion of methanol and other O-compounds to hydrocarbons over zeolite catalysts, J. Catal., 1977, 47, 249–259 CrossRef CAS
-
A. V. Grosse and J. C. Snyder, US Pat., 2492984, 1950 Search PubMed
- L. Kim, M. M. Wald and S. Brandenberger, One-step catalytic synthesis of 2,2,3-trimethylbutane from methanol, J. Org. Chem., 1978, 43, 3432 CrossRef CAS
- D. E. Pearson, Conversion of methanol into hydrocarbons, J. Chem. Soc., Chem. Commun., 1974, 397 RSC
-
C. J. Maiden, The New Zealand Gas-to-Gasoline Project, Methane Conversion, ed. D. M. Bibby, C. D. Chang, R. F. Howe and S. Yurchak, Elsevier, Amsterdam, 1988, pp. 1–16 Search PubMed
- P. Tian, Y. Wei, M. Ya and Z. Liu, Methanol to olefins (MTO) from fundamentals to commercialisation, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS
- W. O. Haag, R. M. Lago and P. G. Rodewald, Aromatics, light olefins and gasoline from methanol: mechanistic pathways with ZSM-5 zeolite catalyst, J. Mol. Catal., 1982, 17, 161–169 CrossRef CAS
- G. Perot, F. X. Cormerais and M. Guisnet, The role of ethers in the methanol–hydrocarbon conversion of silica-alumina and ZSM-5 zeolite, J. Mol. Catal., 1982, 17, 255–260 CrossRef CAS
- T. Behsring, T. Mole, P. Smart and R. J. Western, Homologation of olefins by methanol over ZSM-5 zeolite, J. Catal., 1986, 102, 151–159 CrossRef
-
G. Henrici-Olivé and S. Olivé, The Chemistry of the Catalyzed Hydrogenation of Carbon Monoxide, Springer-Verlag, Berlin, 1984 Search PubMed
- G. J. Hutchings, M. van der Riet and R. Hunter, CO hydrogenation using cobalt/manganese oxide catalysts: comments on the reaction mechanism of carbon–carbon bond formation, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2875–2890 RSC
- C. S. Lee and M. M. Wu, Reaction of carbenoid species diazomethane decompositionover
zeolite ZSM-5, J. Chem. Soc., Chem. Commun., 1985, 250–252 RSC
- G. A. Olah, G. K. S. Prakash, R. W. Ellis and J. A. Olah, Remarks on the mechanism of ethylene formation from methyl alcohol, J. Chem. Soc., Chem. Commun., 1986, 9–10 RSC
- R. Hunter and G. J. Hutchings, Reaction of ethyl diazoacetate with the zeolite catalyst H-ZSM-5: a model study of carbon–carbon bond formation, J. Chem. Soc., Chem. Commun., 1986, 1006–1008 RSC
- R. Hunter and G. J. Hutchings, Hydrocarbon formation from methanol using WO3/Al2O3 and zeolite H-ZSM-5 catalysts: further evidence on the reaction mechanism, J. Chem. Soc., Chem. Commun., 1987, 337–379 Search PubMed
- C. Engelen, J. Wolthuizen and J. van Hoof, On the mechanism of C–C bond formation from methanol over H-ZSM-5. New evidence for trimethyloxonium ion intermediacy, J. Chem. Soc., Chem. Commun., 1985, 300–301 RSC
- P. Rimmelin, H. Taghavi and J. Sommer, J. Chem. Soc., Chem. Commun., 1984, 1210–1211 RSC
- G. J. Hutchings, L. Jansen van Rensburg, W. Pickl and R. Hunter, Hydrocarbon formation from methanol and dimethyl ether using WO3/Al2O3 and H-ZSM-5 catalysts: a mechanistic investigation using model reagents, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 1311–1328 RSC
- S. Kolboe, Methanol reactions on ZSM-5 and other zeolite catalysts, Acta Chem. Scand., Ser. A, 1986, 40a, 711–713 CrossRef
- J. F. Haw, W. Song, D. M. Marcus and J. B. Nicholas, The mechanism of methanol to hydrocarbon catalysts, Acc. Chem. Res., 2003, 36, 317–326 CrossRef CAS PubMed
- T. Mole, G. Bett and D. Seddon, Conversion of methanol to hydrocarbons over ZSM-5 zeolite – an examination of the role of aromatic hydrocarbons using C-14-labeled and deuterium labeled feeds, J. Catal., 1983, 84, 435–445 CrossRef CAS
- B. E. Langner, Reactions of methanol on zeolites with different pore structures, Appl. Catal., 1982, 2, 289–302 CrossRef CAS
- R. F. Sullivan, G. E. Langolis and R. P. Sieg, A new reaction that occurs in the hydrocracking of certain aromatic hydrocarbons, J. Am. Chem. Soc., 1961, 83, 1156–1160 CrossRef CAS
- R. Hunter and G. J. Hutchings, Hydrocarbon formation from methylating agents over the zeolite catalyst H-ZSM-5 and its conjugate base: evidence against the trimethyloxonium ion-ylide mechanism, J. Chem. Soc., Chem. Commun., 1985, 1643–1645 RSC
- R. Hunter and G. J. Hutchings, LiAl(OiPr)4 as a model compound for the conjugate base of the zeolite catalyst H-ZSM-5 and its reaction with various methylating agents, J. Chem. Soc., Chem. Commun., 1985, 886–887 RSC
- G. J. Hutchings, P. Johnston, D. F. Lee, A. Warwick, C. D. Williams and M. Wilkinson, Conversion of methanol and other O-compounds to hydrocarbons over zeolite β, J. Catal., 1994, 147, 177–185 CrossRef CAS
- P. E. Sinclair and C. R. A. Catlow, Generation of carbenes during the methanol conversion over Brønsted acid aluminosilicates, J. Phys. Chem. B, 1997, 101, 295–298 CrossRef CAS
- S. K. Matam, S. A. F. Nastatse, A. Logsdail and C. R. A. Catlow, Methanol loading dependent methoxylation in zeolite H-ZSM-5, Chem. Sci., 2020, 11, 6805 RSC
- W. Wang and M. Hunger, Reactivity of surface alkoxy species on acidic zeolite catalysts, Acc. Chem. Res., 2008, 41, 895–904 CrossRef CAS PubMed
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Recent trends and fundamental insights in the methanol-to-hydrocarbons process, Nat. Catal., 2018, 1, 398–411 CrossRef CAS
- D. E. Perea, I. Arslan, J. Liu, Z. Ristanović, L. Kovarik, B. W. Arey, J. A. Lercher, S. Bere and B. M. Weckhuysen, Determining the location and nearest neighbours of aluminium in zeolites with atom probe tomography, Nat. Commun., 2015, 6, 7589 CrossRef PubMed
- J. E. Schmidt, J. D. Poplawsky, B. Mazumder, Ö. Attila, D. Fu, D. A. M. de Winter, F. Meirer, S. Bere and B. M. Weckhuysen, Coke formation in a zeolite crystal during the methanol-to-hydrocarbons reaction as studied with atom probe tomography, Angew. Chem., Int. Ed., 2016, 55, 11173–11177 CrossRef CAS PubMed
- C. Wang, Y. Chu, J. Xu, Q. Wang, G. Qi, P. Gao, X. Zhou and F. Deng, Extra-framework aluminum-assisted initial C–C bond formation in methanol-to-olefins conversion on zeolite H-ZSM-5, Angew. Chem., 2018, 130, 10354–10358 CrossRef
- I. B. Minova, S. K. Matam, A. Greenaway, C. R. A. Catlow, M. D. Frogley, G. Cinque, P. A. Wright and R. F. Howe, Elementary steps in the formation of hydrocarbons from surface methoxy groups in HZSM-5 seen by synchrotron infrared microspectroscopy, ACS Catal., 2019, 9, 6564–6570 CrossRef CAS
- T. R. Forrester and R. F. Howe, In situ FTIR studies of methanol and dimethyl ether in ZSM-5, J. Am. Chem. Soc., 1987, 109, 5076–5082 CrossRef
- I. B. Minova, S. K. Matam, A. Greenaway, C. R. A. Catlow, M. D. Frogley, G. Cinque, P. A. Wright and R. F. Howe, Effects of crystal size on methanol to hydrocarbon conversion over single crystals of ZSM-5 studied by synchrotron infrared microspectroscopy, Phys. Chem. Chem. Phys., 2020, 22, 18849–18859 RSC
- M. Faraday, Bakerian lecture, experimental relations of gold and other metals to light, Philos. Trans. R. Soc. London, 1857, 147, 145–181 CrossRef
- M. Haruta, N. Yamada, T. Kobayashi and S. Idima, Gold catalysts prepared by co-precipitation for low-temperature oxidation of hydrogen and of carbon monoxide, J. Catal., 1989, 115, 301–309 CrossRef CAS
- N. Lopez, T. V. W. Janssens, B. S. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. K. Norskov, On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation, J. Catal., 2004, 223, 232–235 CrossRef CAS
- M. S. Chen and D. W. Goodman, The structure of catalytically active gold on titania, Science, 2004, 306, 252–255 CrossRef CAS PubMed
- S. N. Rashkeev, A. R. Lupini, S. H. Overbury, S. J. Pennycook and S. T. Pantelides, Role of the nanoscale in catalytic CO oxidation by supported Au and Pt nanostructures, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 35438 CrossRef
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Identification of active gold nanoclusters on iron oxide supports for CO oxidation, Science, 2008, 321, 1331–1335 CrossRef CAS PubMed
- G. J. Hutchings, Nanocrystalline gold and gold palladium alloy oxidation catalysts: a personal reflection on the nature of the active sites, Dalton Trans., 2008, 5523–5536 RSC
- Q. He, S. J. Freakley, J. K. Edwards, A. F. Carley, A. Y. Borisevich, Y. Mineo, M. Haruta, G. J. Hutchings and C. J. Kiely, Population and hierarchy of active species in gold iron oxide catalysts for carbon monoxide oxidation, Nat. Commun., 2016, 7, 12905 CrossRef CAS PubMed
- G. C. Bond and D. T. Thompson, Gold-catalysed oxidation of carbon monoxide, Gold Bull., 2000, 33, 41–50 CrossRef CAS
- G. J. Hutchings, Vapour phase hydrochlorination of acetylene: correlation of catalytic activity of supported metal chloride catalysts, J. Catal., 1985, 96, 292–295 CrossRef CAS
- B. Nkosi, N. J. Coville and G. J. Hutchings, Reactivation of a supported gold catalyst for acetylene hydrochlorination, J. Chem. Soc., Chem. Commun., 1988, 71–72 RSC
- B. Nkosi, N. J. Coville and G. J. Hutchings, Vapour phase hydrochlorination of acetylene with Group VIII and 1B metal chloride catalysts, Appl. Catal., 1988, 43, 33–39 CrossRef CAS
- P. Johnston, N. Carthey and G. J. Hutchings, Discovery, Development and Commercialisation of Gold Catalysts for Acetylene Hydrochlorination, J. Am. Chem. Soc., 2015, 137, 14548–14557 CrossRef CAS PubMed
Footnote |
| † When Dr D. J. Willock asked a question relating to the role of extra-framework Al, this made me consider this matter and ref. 39 has now been included in this paper, but it was not part of the opening lecture or the subsequent discussion. |
| This journal is © The Royal Society of Chemistry 2021 |