On the completeness of three-dimensional electron diffraction data for structural analysis of metal–organic frameworks†
Received
19th February 2021
, Accepted 4th May 2021
First published on 4th May 2021
Abstract
Three-dimensional electron diffraction (3DED) has been proven as an effective and accurate method for structure determination of nano-sized crystals. In the past decade, the crystal structures of various new complex metal–organic frameworks (MOFs) have been revealed by 3DED, which has been the key to understand their properties. However, due to the design of transmission electron microscopes (TEMs), one drawback of 3DED experiments is the limited tilt range of goniometers, which often leads to incomplete 3DED data, particularly when the crystal symmetry is low. This drawback can be overcome by high throughput data collection using continuous rotation electron diffraction (cRED), where data from a large number of crystals can be collected and merged. Here, we investigate the effects of improving completeness on structural analysis of MOFs. We use ZIF-EC1, a zeolitic imidazolate framework (ZIF), as an example. ZIF-EC1 crystallizes in a monoclinic system with a plate-like morphology. cRED data of ZIF-EC1 with different completeness and resolution were analyzed. The data completeness increased to 92.0% by merging ten datasets. Although the structures could be solved from individual datasets with a completeness as low as 44.5% and refined to a high precision (better than 0.04 Å), we demonstrate that a high data completeness could improve the structural model, especially on the electrostatic potential map. We further discuss the strategy adopted during data merging. We also show that ZIF-EC1 doped with cobalt can act as an efficient electrocatalyst for oxygen reduction reactions.
Introduction
During the past two decades, three-dimensional electron diffraction (3DED) has been steadily developed as a complementary technique to single-crystal X-ray diffraction (SCXRD) for structural elucidation of nanocrystals.1–4 The early development of 3DED techniques such as ADT/PEDT,5,6 RED,7,8 and EDT9 utilize stepwise rotation, where a crystal is tilted along an arbitrary axis in a transmission electron microscope (TEM), and electron diffraction (ED) patterns are collected at each angle. Recently, new protocols of 3DED, such as continuous rotation electron diffraction (cRED),10 fast ADT/EDT,11,12 microED,13 and rotation electron diffraction,14 have been developed. They are based on continuous data acquisition where the goniometer continuously rotates at a constant speed and the detector records a movie of ED patterns. The rapid development of 3DED has made large impacts on structural characterization of new materials, such as zeolites,15–20 metal oxides21–24 and quasicrystal approximants.25
Metal–organic frameworks (MOFs) or porous coordination polymers (PCPs) are a class of hybrid materials linking inorganic metal building units and organic ligands.26,27 The almost unlimited combination of inorganic and organic components has led to an ever-expanding family of MOFs with versatile structures and properties.28 However, due to the reversible coordination bonds, MOFs are sensitive to radiation damage by electron beam, which hampers their structural analysis using 3DED. This challenge has been tackled by the development of continuous rotation data collection, using one means a 3DED dataset can be acquired in less than a few minutes, with a dose rate lower than 0.1 e s−1 Å−2. The fast data collection minimizes loss of crystallinity due to beam damage, and consequently the quality of 3DED data, such as resolution, has been improved significantly.
Benefitting from continuous rotation electron diffraction, a growing number of MOF structures have been determined, with their unique properties revealed.29–38 However, the geometric constraints in a TEM impose a physical limitation on the tilt range on the goniometer. Even using a specialized tomography sample holder, 3DED data can be acquired only from −70° to +70°. The angular coverage of a maximum 140° represents ca. 78% sampling volume of reciprocal space, where the remaining 22% of reciprocal space is not accessible. In practice, the tilt range is lower than the maximum value due to the movement of the target crystal, and also overlaps the target crystal with other crystals or the TEM grid. The limited sampling therefore leads to incomplete 3DED data, known as the missing cone problem. The incomplete data could hinder an accurate structure determination, which is more severe for MOF crystals with low symmetry.
To solve this problem, Gruene et al. developed a specialized 3D TEM grid for 3DED data collection.39 As the data processing and structure determination using 3DED data are similar to those using SCXRD data, we present a strategy to improve data quality, particularly data completeness, by merging data from different individual crystals. We use ZIF-EC1 as an example to study the missing data problem. ZIF-EC1 was first discovered as a minor phase in a MOF mixture by continuous rotation electron diffraction (cRED). It crystallizes in a monoclinic space group P21/c with a = 13.462(2) Å, b = 14.659(3) Å, c = 14.449(2) Å, β = 118.12(1)°. The structure of ZIF-EC1 was first solved and refined using a single cRED data point obtained from a phase mixture, and then using merged data from nine crystals of its pure phase.40 Here we present a systematic study of the influence of data completeness on the structural analysis of ZIF-EC1. We show that data completeness can be improved after data merging. As each crystal has a different orientation and particle size, we further discuss the use of correlations of cRED intensity among different datasets, to choose the best combination to improve data quality. Moreover, merging of 3DED data is performed using different algorithms adapted from X-ray crystallography. We show that the structures could be solved and refined with high precision from individual datasets with a completeness as low as 44.5%. With increased data completeness, the structural model of ZIF-EC1 could be further improved, and peaks corresponding to atoms in the electrostatic potential map have well-defined isotropic shapes. By knowing the atomic structure and the underlying property, ZIF-EC1 was doped with cobalt and utilized as an electrocatalyst for the oxygen reduction reaction (ORR).
Experimental section
Synthesis
In a typical synthesis of ZIF-EC1, 0.125 mL of a 3.84 M aqueous solution of 2-methylimidazole (HmIM, TCI Chemicals) was mixed with 1.875 mL of deionized water. 1 mL of 0.24 M aqueous solution of Zn(OAc)2·2(H2O) (EMSURE, Merck) was added into the above solution under vigorous stirring. The stirring condition was then maintained for at least 4 h at RT. The products were washed in deionized water at least six times. After centrifugation, the products were re-suspended in deionized water. ZIF-EC1(Zn,Co) was synthesized in a modified condition. Instead of using 0.24 M aqueous solution of Zn(OAc)2·2(H2O), a mixture of 0.16 M Zn(OAc)2·2(H2O) and 0.08 M Co(NO3)2·6(H2O) was used, with a Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co molar ratio of 2:1.
:Co molar ratio of 2:1.
cRED data collection
The samples for cRED data collection were crushed in a mortar and dispersed in deionized water. A droplet was then taken from the suspension, put on a copper grid covered with lacey carbon, and dried in air at room temperature. cRED data were collected on a JEOL JEM 2100 microscope operated at 200 kV (Cs 1.0 mm, point resolution 0.23 nm). cRED data collection was controlled using the data acquisition software Instamatic,10,41 and the electron diffraction (ED) patterns were recorded by a Timepix hybrid detector QTPX-262k (512 × 512 pixels, pixel size 55 μm, Amsterdam Sci. Ins.). A tomography holder was used for the data collection, which could tilt from −70° to +70° in the TEM. The area used for cRED data collection was about 1.0 μm in diameter, as defined by a selected area aperture. To minimize electron beam damage of the crystal and maximize the data quality, data was collected at a low electron dose condition. Ten cRED datasets were collected on the ZIF-EC1 crystals, covering a tilt range from 42.34° to 122.71°. Two different rotation speeds were used for the datasets, and the data collection time ranges from 77.7 s to 239.5 s. The detailed conditions for acquiring each dataset can be found in Table 1.
Table 1 Selected parameters and data information of ten cRED datasets collected on ZIF-EC1 crystals (200 kV, λ = 0.02508 Å)
| Dataset no. |
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
| Group |
A
|
A
|
A
|
A
|
B
|
B
|
C
|
C
|
D
|
D
|
| Rotation range (°) |
122.71 |
103.77 |
97.20 |
87.15 |
81.89 |
108.92 |
102.05 |
94.07 |
44.31 |
42.34 |
| Tilting rate (° s−1) |
1.13 |
1.13 |
1.13 |
1.13 |
0.45 |
0.45 |
0.45 |
0.45 |
0.45 |
0.45 |
| Collection time (s) |
108.3 |
91.7 |
86.1 |
77.7 |
181.0 |
239.5 |
223.7 |
208.5 |
97.7 |
93.6 |
| Resolution (Å) |
0.70 |
0.78 |
0.75 |
0.74 |
0.76 |
0.78 |
0.93 |
0.92 |
1.21 |
1.00 |
| Completeness (%) |
44.5 |
47.1 |
55.0 |
58.3 |
52.9 |
71.6 |
62.9 |
32.5 |
40.8 |
24.1 |
cRED data processing and structural analysis
The X-ray crystallography software package XDS42 was used for data processing to determine the unit cell and space group, index the diffraction spots and estimate integrated diffraction intensities. XSCALE, which is part of the XDS package, was then applied for data merging. The SHELX software package43 was used for structural analysis, where SHELXT was used for structure solution and SHELXL for structure refinement. Atomic scattering factors for electrons based on neutral atoms were used. All atoms were refined anisotropically. While the model obtained from each individual dataset could be refined, for easy comparison, the same starting SHELXL input file was used for all the structure refinements presented here. EADP were applied on two carbon atoms (C5 and C20). Planarity (FLAT) and distance restraints (DFIX and DANG) were applied to the 2-methylimidazolate (mIm−) groups to maintain a reasonable geometry. At the end, the extinction parameter EXTI44 was applied in the refinement. The crystallographic details can be found in Table 2.
Table 2 Refinement details of the merged datasets
| Dataset no. |
1
|
M_A1
|
M_A2
|
M_A3
|
5
|
M_B
|
7
|
M_C
|
M_BC
|
M_ABC
|
M_ABCD
|
| Datasets used for merging |
N/A |
1 & 2 |
1–3
|
1–4
|
N/A |
5 & 6 |
N/A |
7 & 8 |
5–8
|
1–8
|
1–10
|
|
The structural model refined against dataset M_ABC was used as the reference to calculate the ADRA values.
|
| Completeness (%) |
44.5 |
71.5 |
75.5 |
81.4 |
53.5 |
85.0 |
62.9 |
63.3 |
87.0 |
89.1 |
92.0 |
| Resolution (Å) |
0.78 |
0.78 |
0.78 |
0.78 |
0.78 |
0.78 |
0.93 |
0.93 |
0.78 |
0.78 |
0.78 |
| No. of reflections (total) |
3935 |
9224 |
14352 |
19372 |
6502 |
16354 |
4958 |
7456 |
23817 |
43219 |
46079 |
| No. of reflections (all unique) |
2189 |
3615 |
3848 |
4654 |
3132 |
4859 |
2088 |
2138 |
4976 |
5093 |
5262 |
| No. of reflections (I > 2 sigma(I)) |
1495 |
2420 |
2845 |
3306 |
1874 |
3263 |
1221 |
1409 |
3345 |
3933 |
3669 |
|
R
1 (I > 2 sigma(I)) |
0.174 |
0.179 |
0.178 |
0.181 |
0.178 |
0.182 |
0.153 |
0.150 |
0.170 |
0.173 |
0.181 |
|
R
1 (all reflections) |
0.204 |
0.213 |
0.204 |
0.211 |
0.219 |
0.211 |
0.193 |
0.181 |
0.201 |
0.193 |
0.250 |
| GooF |
1.564 |
1.486 |
1.542 |
1.544 |
1.870 |
1.442 |
1.321 |
1.409 |
1.450 |
1.676 |
1.530 |
| ADRA (Å)a |
0.035 |
0.027 |
0.018 |
0.015 |
0.038 |
0.015 |
0.036 |
0.034 |
0.010 |
0 |
0.012 |
Electrochemical application
All electrochemical tests are evaluated by a three-electrode system (the counter electrode and the reference electrode are graphite rods and saturated Ag/AgCl electrodes, respectively) on the CHI 660E electrochemical workstation at 30 °C. The working electrode is a rotating disk electrode (RDE) with a diameter of 5 mm (0.196 cm2). The CV test is carried out in 0.1 M KOH saturated with Ar or O2 (scan rate is 50 mV s−1). The linear sweep voltammetry (LSV) test is carried out in 0.1 M KOH saturated with O2 (scan rate is 5 mV s−1). The catalyst ink for the ORR tests was prepared as follows: 4 mg ZIF-EC1(Zn,Co) and 1 mg carbon nanotubes was weighed and dispersed in the mixed solution containing 450 μL isopropanol, 40 μL deionized water and 10 μL Nafion D520 aqueous solution (5 wt%). The mixed solution was treated by ultrasound for 1 h to form a homogeneous suspension. Then 15 μL catalyst solution was taken from the pipette and uniformly dripped onto the rotating disc electrode (RDE). After drying, it was used for electrocatalytic ORR performance evaluation.
Results and discussion
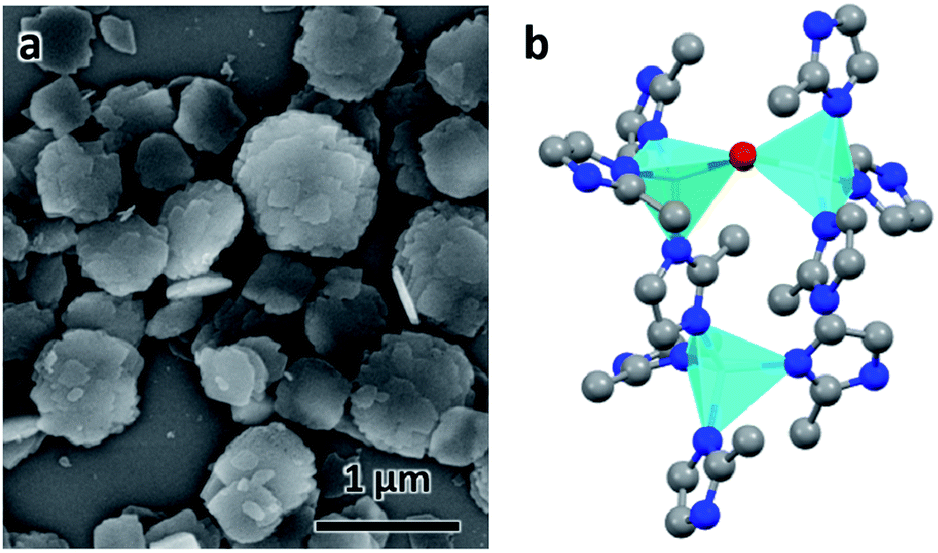
ZIF-EC1 crystallizes in a monoclinic system with a plate-like morphology (Fig. 1a). With chemical formula Zn3(mIm)5(OH), ZIF-EC1 is composed of ZnN4 mononuclear clusters and binuclear Zn2N6(OH) clusters, where Zn(II) cations link to mIm− ions (Fig. 1b). Ten cRED datasets collected on the ZIF-EC1 crystals were chosen for this study (Table 1), among which eight (no. 1–8) are good datasets with high resolution (0.70–0.93 Å) and another two (no. 9–10) with lower resolution (1.00–1.21 Å). The 3D reciprocal lattices reconstructed from datasets 1–8 are presented in Fig. 2, where the missing cones appear at different locations. As ZIF-EC1 is crystallized in a monoclinic system, the individual 3DED datasets have completeness ranging from 24.1–71.6% after the reflections were indexed using X-ray crystallography software package XDS42 (Table 1). The structure could be solved and refined from each of the datasets 1–8.
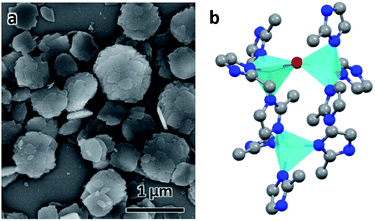 |
| | Fig. 1 (a) SEM image of ZIF-EC1 crystals. (b) The structural model of ZIF-EC1. Cyan tetrahedra: Zn atoms; red spheres: O atoms; blue spheres: N atoms; grey spheres: C atoms. | |
 |
| | Fig. 2 3D reciprocal lattices of ZIF-EC1 reconstructed from datasets 1–8, (a–h) respectively. They are viewed along the same direction, with data completeness of 44.5%, 47.1%, 55.0%, 58.3%, 52.9%, 71.6%, 62.9%, and 32.5%, respectively. The different data cover different parts of the reciprocal space. | |
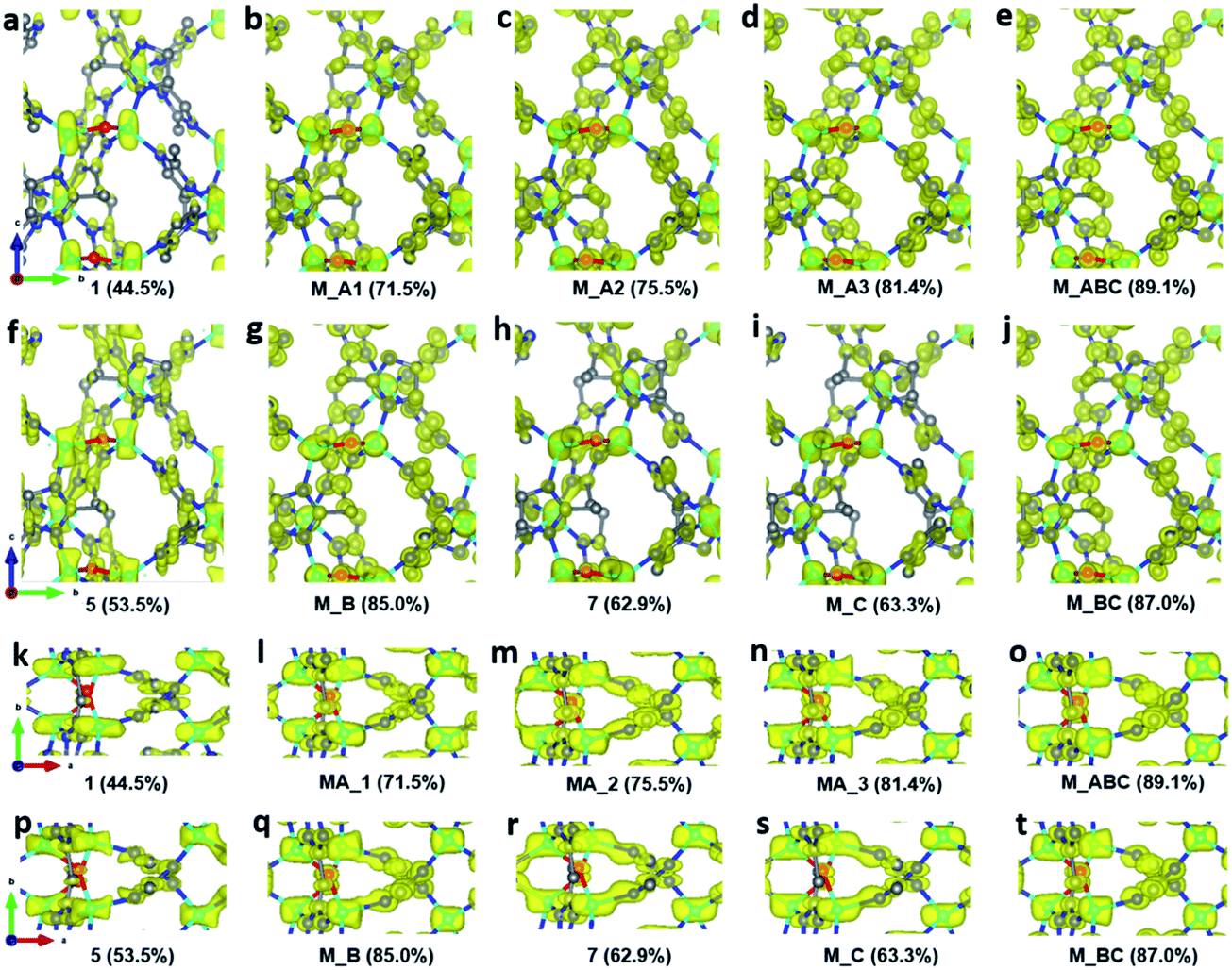
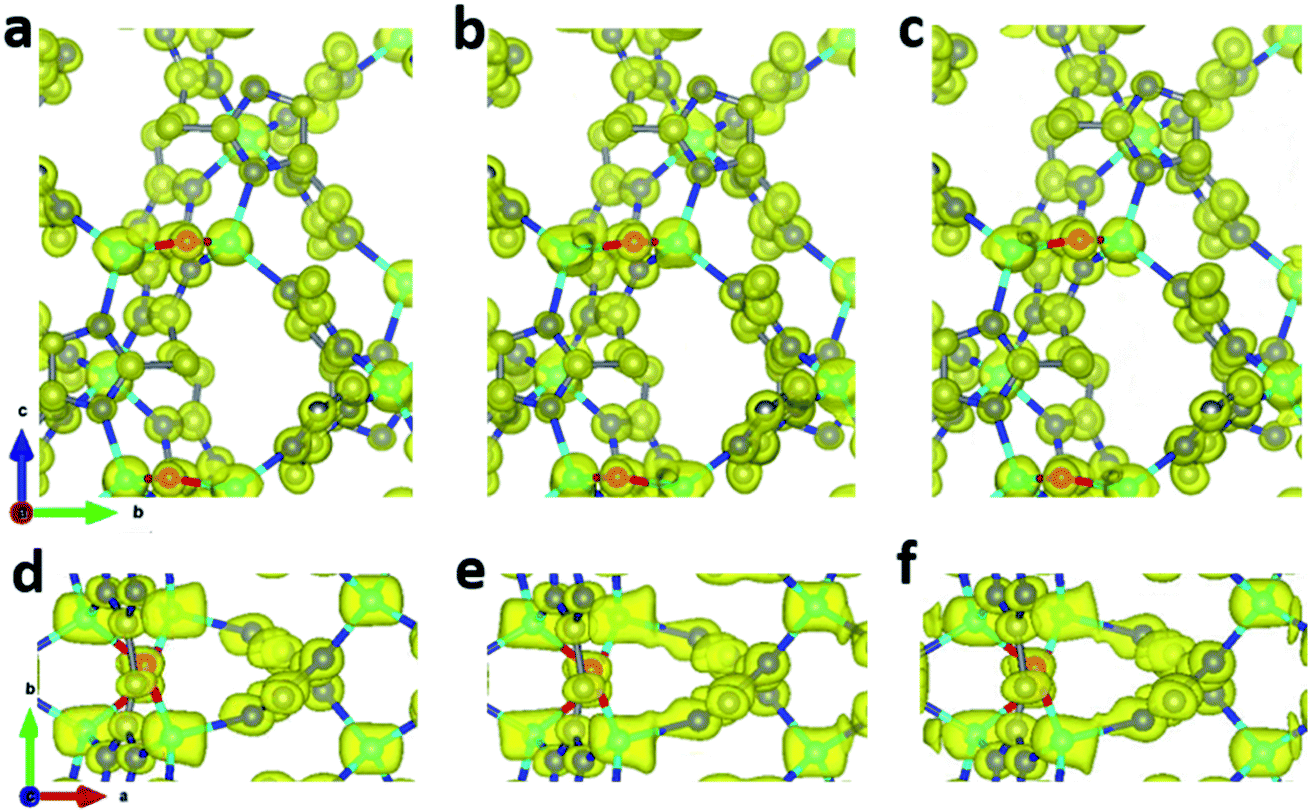
To improve the overall data quality, XSCALE was applied for data merging. We performed systematic studies by merging a different number of datasets to study the influence of data completeness on the structural analysis of ZIF-EC1. We divided the ten datasets into four groups, collected under different tilting rates and/or with different resolutions. Group A includes four datasets 1–4 collected using a high tilting rate (1.13° s−1), all with high resolution (0.70–0.78 Å). The remaining datasets 5–10 were collected using a low tilting rate (0.45° s−1). The groups were in accordance with the data resolution: datasets 5–6 with 0.76–0.78 Å resolution belong to group B, datasets 7–8 with 0.92–0.93 Å resolution to group C, and datasets 9–10 with 1.00–1.21 Å resolution to group D. We first merged datasets in group A, which has completeness ranging from 44.5% to 58.3%. The data completeness was improved steadily from 44.5% of the single dataset 1 to 71.5, 75.5, and 81.4% for combining two (M_A1, merged dataset A1), three (M_A2), and four datasets (M_A3), respectively (Table 2). To investigate the relationship between data completeness and the structural model, we refined the structural model of ZIF-EC1 against the data merged from different numbers of cRED datasets. While the number of unique reflections increased significantly, from 2189 for a single dataset 1 to 4154 by merging four datasets, the R1 values are very similar, varying between 0.204 and 0.213. This indicates that all datasets have similar degrees in consistence with the structural models. The improvement can be easily visualized in the corresponding electrostatic potential maps (Fig. 3a–d and k–n). For those with high data completeness (75.5% for M_A2 and 81.4% for MA_3), the peaks appear more spherical with similar peak heights for the same atom types (Fig. 3d and n). In contrast, data with low completeness (44.5% for 1 and 62.6% for M_A1) resulted in either missing peaks with large variations in peak heights (Fig. 3a and k) or severe peak elongation (Fig. 3a, b, k and l). Because the electrostatic potential map is the basis of a structural model, distortion of the map leads to reduced reliability and accuracy of the atom coordinates and atomic displacement parameters. Therefore, even though a structural model can be obtained from a single dataset, it is vital to improve data completeness.
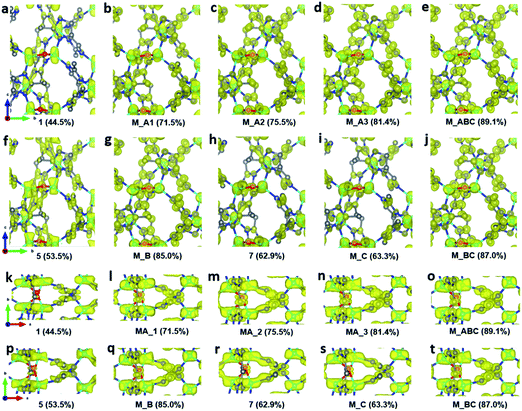 |
| | Fig. 3 Electrostatic potential maps calculated based on datasets (a and k) 1, (b and l) MA_1, (c and m) MA_2, (d and n) MA_3, (e and o) M_ABC, (f and p) 5, (g and q) M_B, (h and r) 7, (i and s) M_C, and (j and t) M_BC, viewed along (a–j) the a-axis and (k–t) the c-axis. The refined structural model is superimposed in the map and used for calculating the structure factor phases. Missing peaks are observed on the maps constructed from data with low completeness. Peaks are more spherical with the increase of completeness. All electrostatic potential maps are drawn at the same contour level, which is the 2σ level of M_ABC dataset, and is equal to an isosurface level diso = 0.811. Atom color codes: grey, C; blue, N; red, O; cyan, Zn. | |
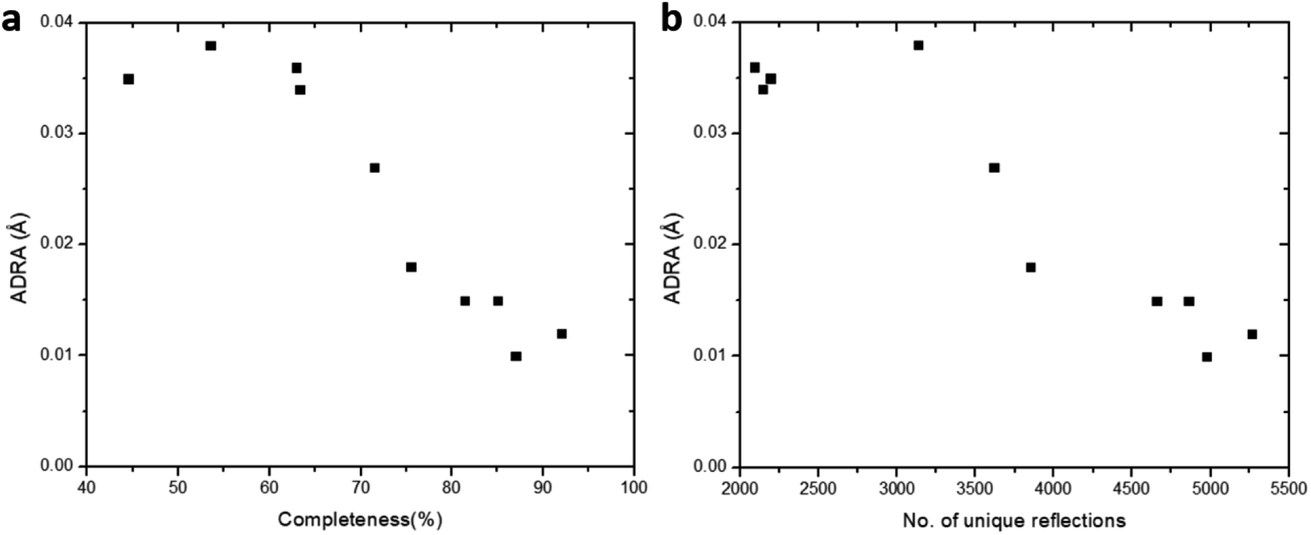
Furthermore, we applied data merging on datasets collected using a low tilting rate (groups B and C). The completeness increased significantly from 53.5% for single dataset 5 to 85.0% upon merging datasets 5 and 6 (M_B). However, there is little increase in data completeness for group C, when datasets 7 and 8 are merged (from 62.9% for 7 to 63.3% for M_B). The electrostatic potential maps present a significant improvement from single dataset 5 to M_B as the completeness is increased from 53.5 to 85.0% (Fig. 3f, g, p, and q). On the contrary, there is very limited improvement in the electrostatic potential maps from single dataset 7 to M_C, because the completeness was only increased by 0.4% (Fig. 3h, i, r, and s). Notably, merging the four datasets 5–8 in groups B and C (M_BC) led to improved completeness (87.0%), and the electrostatic potential map (Fig. 3j and t). Further merging all the aforementioned datasets 1–8 (M_ABC) resulted in the highest completeness (89.1%), and thus the best electrostatic potential map with most spherical peaks (Fig. 3e and o). The structural model refined against the dataset M_ABC is therefore used as a reference model, to which the atomic coordinates from the structural models obtained from datasets M_A3, M_B, M_C and M_BC were compared. It shows that with a low completeness, e.g. 63.3% for M_C, the corresponding structural model resulted in higher deviations on atomic positions, compared to those from other datasets with high completeness (Table 3). We further calculated the average deviation from reference atoms (ADRA)45 values to compare the different models (Table 2). The ADRA values decreased successively from 0.035 Å to 0.015 Å by merging more and more datasets in group A (M_A3). Improving completeness from 53.5 to 85.0% in group B (M_B) also led to decreasing ADRA values from 0.038 Å to 0.015 Å. On the other hand, the ADRA values showed little improvement by merging datasets 7 and 8 in group C (M_C), varying from 0.036 to 0.034 Å, with a completeness of 62.9 and 63.3%, respectively. Moreover, merging the four datasets 5–8 in groups B and C (87.0%, M_BC) led to a reduced ADRA value (0.010 Å). Fig. 4 shows a clear trend that the ADRA values are reduced with the increase of the data completeness as well as the increase in the number of unique reflections.
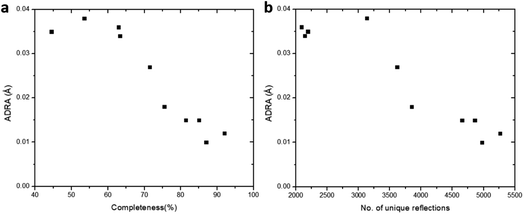 |
| | Fig. 4 The effect of (a) data completeness and (b) number of total unique reflections on the ADRA values. The structural model refined against the merged data from the eight cRED datasets with the highest CCI was used as the reference model. The ADRA values are reduced with the increase of the data completeness. | |
Table 3 Deviations of the atomic coordination. The structural model obtained from M_ABC is set as the reference
| Atoms |
M_A3
|
M_B
|
M_C
|
M_BC
|
| Zn1 |
0.004(3) |
0.005(4) |
0.028(7) |
0.005(3) |
| Zn2 |
0.002(4) |
0.005(5) |
0.031(8) |
0.003(4) |
| Zn3 |
0.004(5) |
0.006(4) |
0.015(7) |
0.006(4) |
| N1 |
0.008(15) |
0.014(12) |
0.030(21) |
0.014(10) |
| N2 |
0.019(17) |
0.014(10) |
0.014(14) |
0.013(13) |
| N3 |
0.019(17) |
0.027(11) |
0.014(17) |
0.021(12) |
| N4 |
0.001(17) |
0.014(14) |
0.051(19) |
0.006(13) |
| N5 |
0.007(12) |
0.013(14) |
0.045(20) |
0.003(11) |
| N6 |
0.012(13) |
0.017(11) |
0.048(21) |
0.008(11) |
| N7 |
0.012(10) |
0.010(14) |
0.069(17) |
0.015(12) |
| N8 |
0.011(15) |
0.015(15) |
0.037(16) |
0.005(16) |
| N9 |
0.008(13) |
0.008(15) |
0.040(18) |
0.005(11) |
| N10 |
0.019(16) |
0.015(10) |
0.033(16) |
0.006(11) |
| O1 |
0.025(13) |
0.015(17) |
0.048(22) |
0.010(18) |
| C1 |
0.012(11) |
0.024(14) |
0.040(18) |
0.004(11) |
| C2 |
0.013(17) |
0.006(12) |
0.024(17) |
0.004(12) |
| C3 |
0.020(13) |
0.020(9) |
0.033(21) |
0.020(10) |
| C4 |
0.007(13) |
0.003(14) |
0.037(23) |
0.010(15) |
| C5 |
0.009(17) |
0.019(13) |
0.017(28) |
0.015(13) |
| C6 |
0.030(19) |
0.033(17) |
0.041(20) |
0.018(16) |
| C7 |
0.032(19) |
0.031(14) |
0.053(17) |
0.027(18) |
| C8 |
0.034(15) |
0.004(14) |
0.073(29) |
0.019(13) |
| C9 |
0.025(16) |
0.003(18) |
0.030(18) |
0.007(17) |
| C10 |
0.015(15) |
0.015(14) |
0.041(21) |
0.010(12) |
| C11 |
0.007(20) |
0.016(15) |
0.025(21) |
0.013(14) |
| C12 |
0.010(16) |
0.017(18) |
0.022(19) |
0.008(16) |
| C13 |
0.022(17) |
0.012(15) |
0.017(16) |
0.014(14) |
| C14 |
0.029(16) |
0.009(14) |
0.034(26) |
0.008(14) |
| C15 |
0.019(20) |
0.045(14) |
0.030(23) |
0.015(13) |
| C16 |
0.007(14) |
0.006(18) |
0.016(28) |
0.005(19) |
| C17 |
0.008(21) |
0.006(14) |
0.030(20) |
0.005(13) |
| C18 |
0.019(17) |
0.027(21) |
0.028(7) |
0.008(20) |
| C19 |
0.025(21) |
0.015(20) |
0.031(8) |
0.012(19) |
| C20 |
0.022(16) |
0.027(16) |
0.015(7) |
0.006(14) |
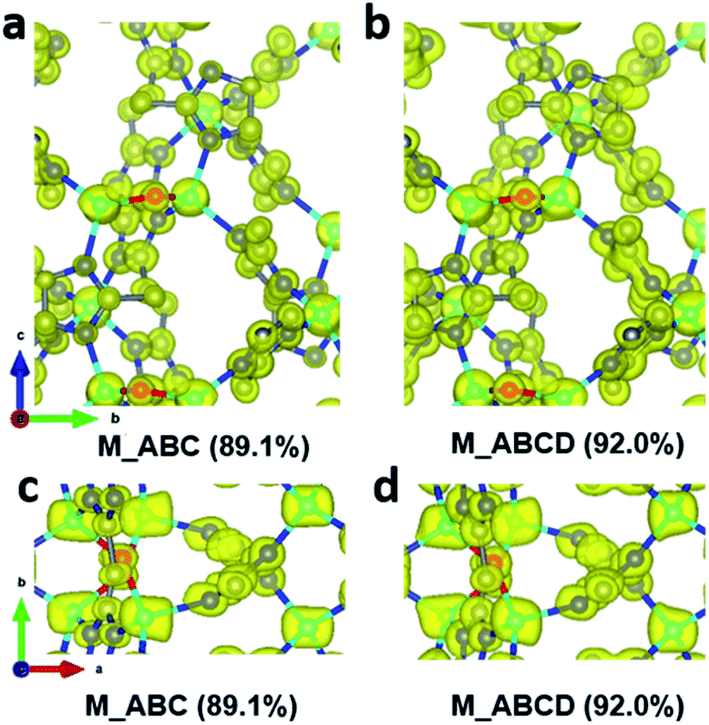
During data merging, correlation coefficients of the common reflection intensities (CCI) between two datasets are calculated by XSCALE, which indicate the degree of intensity similarity between the datasets (Table 4). The correlation among the datasets 1–8 are very high, as indicated by the CCI values (>0.95). In contrast, datasets 9 and 10 are rather poorly correlated with the other datasets with the CCI values are mostly below 0.80. Combining the additional datasets 9 and 10 to the merged dataset M_ABC increased the data completeness from 89.1% to 92.0%. However, this led to a significant increase of the R1 values for all reflections, from 0.193 to 0.250. On the other hand, very little change is observed in the electrostatic potential maps (Fig. 5), and the ADRA remains at a low value. This indicates that the structural models are very similar, and the increased R1 value mainly results from merging poorly correlated datasets. Therefore, there would be little benefit to add more datasets with low intensity correlations, after reasonable data completeness has already been reached.
Table 4 The CCI values between two datasets
| Dataset no. |
Dataset no. |
No. of common reflections |
CCI |
|
Dataset no. |
Dataset no. |
No. of common reflections |
CCI |
|
1
|
2
|
2077 |
0.973 |
|
3
|
8
|
1181 |
0.979 |
|
1
|
3
|
1572 |
0.967 |
|
4
|
8
|
944 |
0.983 |
|
2
|
3
|
1323 |
0.964 |
|
5
|
8
|
870 |
0.985 |
|
1
|
4
|
2904 |
0.986 |
|
6
|
8
|
1305 |
0.982 |
|
2
|
4
|
1783 |
0.975 |
|
7
|
8
|
867 |
0.982 |
|
3
|
4
|
1274 |
0.981 |
|
1
|
9
|
536 |
0.695 |
|
1
|
5
|
2096 |
0.972 |
|
2
|
9
|
398 |
0.660 |
|
2
|
5
|
1018 |
0.961 |
|
3
|
9
|
359 |
0.783 |
|
3
|
5
|
1220 |
0.991 |
|
4
|
9
|
432 |
0.617 |
|
4
|
5
|
1620 |
0.988 |
|
5
|
9
|
221 |
0.684 |
|
1
|
6
|
2463 |
0.953 |
|
6
|
9
|
346 |
0.456 |
|
2
|
6
|
1663 |
0.978 |
|
7
|
9
|
193 |
0.927 |
|
3
|
6
|
1606 |
0.952 |
|
8
|
9
|
218 |
0.772 |
|
4
|
6
|
1999 |
0.981 |
|
1
|
10
|
580 |
0.826 |
|
5
|
6
|
1962 |
0.962 |
|
2
|
10
|
508 |
0.775 |
|
1
|
7
|
684 |
0.961 |
|
3
|
10
|
592 |
0.856 |
|
2
|
7
|
898 |
0.977 |
|
4
|
10
|
477 |
0.556 |
|
3
|
7
|
934 |
0.995 |
|
5
|
10
|
410 |
0.722 |
|
4
|
7
|
504 |
0.954 |
|
6
|
10
|
475 |
0.748 |
|
5
|
7
|
484 |
0.990 |
|
7
|
10
|
312 |
0.369 |
|
6
|
7
|
747 |
0.975 |
|
8
|
10
|
406 |
0.449 |
|
1
|
8
|
1317 |
0.967 |
|
9
|
10
|
141 |
0.948 |
|
2
|
8
|
1642 |
0.974 |
|
|
|
|
|
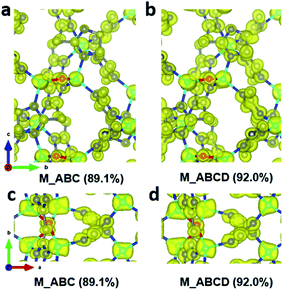 |
| | Fig. 5 Electrostatic potential maps calculated based on datasets (a and c) M_ABC, and (b and d) M_ABCD viewed along (a and b) the a-axis and (c and d) the c-axis. The refined structural model is superimposed in the map and used for calculating the structure factor phases. All electrostatic potential maps are drawn at the same contour level, which is the 2σ level of the M_ABC dataset, and is equal to an isosurface level diso = 0.811. Atom color codes: grey, C; blue, N; red, O; cyan, Zn. | |
Due to the development of a continuous rotation setup for collecting cRED data, software for X-ray crystallography can be easily used to process 3DED data. Thus, we investigate different algorithms of X-ray crystallography for data merging and evaluate the data quality. XSCALE, XPREP46 and AIMLESS47 are among the other programs that are commonly applied for merging datasets. We chose datasets 1–8 for merging because they have high correlation between each other. Using different algorithms for scaling and merging data, XSCALE, XPREP, and AIMLESS resulted in slightly different completeness of 89.1, 85.2, and 90.4%, respectively (Table 5). The R1 values are also similar (0.192–0.213), and the electrostatic potential maps exhibit well defined and spherical peaks (Fig. 6). Although using kinematical assumption, this indicates that different merging algorithms in X-ray crystallography programs can be used to process cRED data for high data completeness.
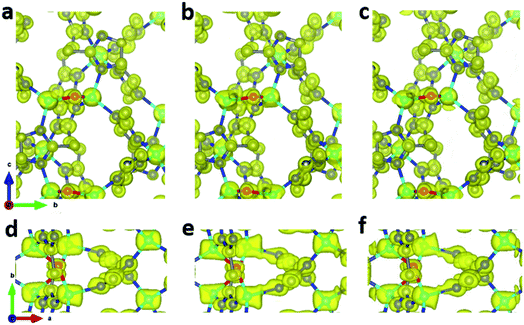 |
| | Fig. 6 Electrostatic potential maps calculated based on datasets 1–8 merged by (a and d) XSCALE, (b and e XPREP, and (c and f) AIMLESS. The refined structural model is superimposed in the map and used for calculating the structure factor phases. All electrostatic potential maps are drawn at the same contour level, which is the 2σ level of M_ABC dataset, and equals to an isosurface level diso = 0.811. Atom color codes: grey: C; blue: N; red: O; cyan: Zn. | |
Table 5 Refinement details for merging datasets 1–8 using different methods
| Software used for merging |
XSCALE |
XPREP |
AIMLESS |
| No. of reflections (all unique) |
5093 |
5046 |
5116 |
| Completeness (%) |
89.1 |
85.2 |
90.4 |
| Resolution (Å) |
0.78 |
0.78 |
0.78 |
| No. of reflections (I > 2 sigma(I)) |
3933 |
3964 |
3879 |
|
R
1 (I > 2 sigma(I)) |
0.172 |
0.193 |
0.187 |
|
R
1 (all reflections) |
0.192 |
0.210 |
0.213 |
| GooF |
1.676 |
1.956 |
1.536 |
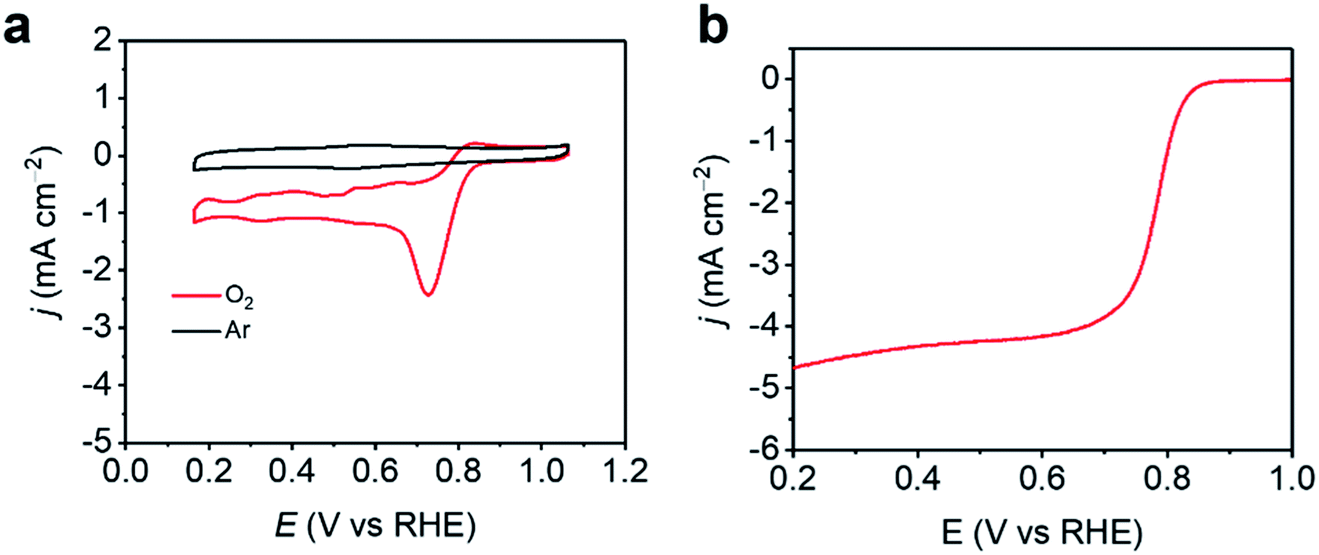
As 3DED revealed the accurate structure of ZIF-EC1, ZIF-EC1 has been utilized as a precursor to generate carbon materials for catalyzing electrochemical reactions due to its high density of metal ions.40 Here, we added Co(II) ions in the synthesis to further take advantage of the highly dense structure of ZIF-EC1, and use its pristine form for electrocatalytic oxygen reduction reaction (ORR). The electroactivity of ZIF-EC1(Zn,Co) was tested in 0.1 M KOH solution using the rotating disk electrode (RDE). The cyclic voltammetry (CV) data of ZIF-EC1(Zn,Co) showed that a strong electrocatalytic oxygen reduction peak near 0.74 V was observed in O2-saturated solution, while there was no reduction peak in an Ar-saturated solution (Fig. 7). Linear sweep voltammetry (LSV) further confirmed the good catalytic performance of ZIF-EC1, showing an onset potential (Eonset) of 0.85 V and a half-wave potential (E1/2) of 0.78 V. In addition, the electrocatalytic ORR limiting current density can reach 4.7 mA cm−2. The efficiency of ZIF-EC1(Zn,Co) for ORR is among one of the best MOF electrocatalysts in comparison with the others.48–51
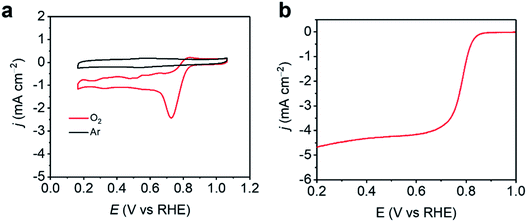 |
| | Fig. 7 (a) CVs of ZIF-EC1(Zn,Co) catalyst in 0.1 M KOH in an Ar/O2-saturated solution. (b) LSV curves of ZIF-EC1(Zn,Co) catalyst in 0.1 M KOH in an O2-saturated solution at 1600 rpm. | |
Conclusions
From the studies of the influence of data completeness on the structural analysis of ZIF-EC1, we show that the structure could be solved and refined to a high precision from cRED data with different completeness, even as low as 44.5% collected at various resolutions (0.70–0.93 Å). Importantly, we demonstrate that the completeness of 3DED data can be improved by merging data obtained from a series of individual crystals. The increased data completeness led to an improved structural model. In addition, the increased data completeness also resulted in decreasing of the ADRA values. More importantly, peaks corresponding to atom positions appear more spherical in the electrostatic potential maps, with similar peak heights for the same atom types. On the other hand, a low data completeness results in a large uncertainty and variations in peak heights and elongation of the peaks. A completeness of >75% already gave a low ADRA value (<0.018 Å) and good electrostatic potential maps. The structural model may be further improved by merging more datasets, but only when the datasets with high intensity correlations (i.e. high CCI values) are used. Data merging using different algorithms implemented in X-ray crystallographic software XSCALE, XPREP, and AIMLESS gave similar refinement results, showing the high quality of cRED data. Finally, we show that ZIF-EC1(Zn,Co) is an efficient electrocatalyst for ORR. With improved data quality, we anticipate that more structural details can be uncovered, which could promote discovery of novel properties of MOFs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Swedish Research Council (VR, 2016-04625, Z. H and 2017-04321, X. Z.), the Swedish Research Council Formas (2020-00831, Z. H.), and the National Natural Science Foundation of China (Grant No. 21975148, and 21601118).
Notes and references
- M. Gemmi, E. Mugnaioli, T. E. Gorelik, U. Kolb, L. Palatinus, P. Boullay, S. Hovmöller and J. P. Abrahams, ACS Cent. Sci., 2019, 5, 1315–1329 CrossRef CAS.
- Z. Huang, E. S. Grape, J. Li, A. K. Inge and X. Zou, Coord. Chem. Rev., 2021, 427, 213583 CrossRef CAS.
- Z. Huang, T. Willhammar and X. Zou, Chem. Sci., 2021, 12, 1206–1219 RSC.
- Z. Huang, M. Ge, F. Carraro, C. Doonan, P. Falcaro and X. Zou, Faraday Discuss., 2021, 225, 118–132 RSC.
- U. Kolb, T. Gorelik, C. Kübel, M. T. Otten and D. Hubert, Ultramicroscopy, 2007, 107, 507–513 CrossRef CAS PubMed.
- P. Boullay, L. Palatinus and N. Barrier, Inorg. Chem., 2013, 52, 6127–6135 CrossRef CAS PubMed.
- D. Zhang, P. Oleynikov, S. Hovmöller and X. Zou, Z. Kristallogr., 2010, 225, 94–102 CrossRef CAS.
- W. Wan, J. Sun, J. Su, S. Hovmöller and X. Zou, J. Appl. Crystallogr., 2013, 46, 1863–1873 CrossRef CAS.
- M. Gemmi and P. Oleynikov, Z. Kristallogr. - Cryst. Mater., 2013, 228, 51–58 CrossRef CAS.
- M. O. Cichocka, J. Ångström, B. Wang, X. Zou and S. Smeets, J. Appl. Crystallogr., 2018, 51, 1652–1661 CrossRef CAS.
- M. Gemmi, M. G. I. La Placa, A. S. Galanis, E. F. Rauch and S. Nicolopoulos, J. Appl. Crystallogr., 2015, 48, 718–727 CrossRef CAS.
- S. Plana-Ruiz, Y. Krysiak, J. Portillo, E. Alig, S. Estradé, F. Peiró and U. Kolb, Ultramicroscopy, 2020, 211, 112951 CrossRef CAS PubMed.
- B. L. Nannenga, D. Shi, A. G. W. Leslie and T. Gonen, Nat. Methods, 2014, 11, 927–930 CrossRef CAS PubMed.
- I. Nederlof, E. van Genderen, Y.-W. Li and J. P. Abrahams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1223–1230 CrossRef CAS.
- J. Jiang, J. L. Jorda, J. Yu, L. A. Baumes, E. Mugnaioli, M. J. Diaz-Cabanas, U. Kolb and A. Corma, Science, 2011, 333, 1131–1134 CrossRef CAS PubMed.
- T. Willhammar, A. W. Burton, Y. Yun, J. Sun, M. Afeworki, K. G. Strohmaier, H. Vroman and X. Zou, J. Am. Chem. Soc., 2014, 136, 13570–13573 CrossRef CAS PubMed.
- J. Simancas, R. Simancas, P. J. Bereciartua, J. L. Jorda, F. Rey, A. Corma, S. Nicolopoulos, P. Pratim Das, M. Gemmi and E. Mugnaioli, J. Am. Chem. Soc., 2016, 138, 10116–10119 CrossRef CAS.
- P. Guo, J. Shin, A. G. Greenaway, J. G. Min, J. Su, H. J. Choi, L. Liu, P. A. Cox, S. B. Hong, P. A. Wright and X. Zou, Nature, 2015, 524, 74–78 CrossRef CAS PubMed.
- L. A. Villaescusa, J. Li, Z. Gao, J. Sun and M. A. Camblor, Angew. Chem., 2020, 132, 11379–11382 CrossRef.
- Z. Huang, S. Seo, J. Shin, B. Wang, R. G. Bell, S. B. Hong and X. Zou, Nat. Commun., 2020, 11, 3762 CrossRef.
- Y. Yun, W. Wan, F. Rabbani, J. Su, H. Xu, S. Hovmöller, M. Johnsson and X. Zou, J. Appl. Crystallogr., 2014, 47, 2048–2054 CrossRef CAS.
- E. Buixaderas, M. Kempa, V. Bovtun, C. Kadlec, M. Savinov, F. Borodavka, P. Vaněk, G. Steciuk, L. Palatinus and J. Dec, Phys. Rev. Mater., 2018, 2, 124402 CrossRef CAS.
- A. E. Lanza, M. Gemmi, L. Bindi, E. Mugnaioli and W. H. Paar, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 711–716 CrossRef CAS PubMed.
- J. Li, C. Lin, Y. Min, Y. Yuan, G. Li, S. Yang, P. Manuel, J. Lin and J. Sun, J. Am. Chem. Soc., 2019, 141, 4990–4996 CrossRef CAS PubMed.
- S. Samuha, E. Mugnaioli, B. Grushko, U. Kolb and L. Meshi, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 999–1005 CrossRef CAS.
- O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- M. Feyand, E. Mugnaioli, F. Vermoortele, B. Bueken, J. M. Dieterich, T. Reimer, U. Kolb, D. de Vos and N. Stock, Angew. Chem., Int. Ed., 2012, 51, 10373–10376 CrossRef CAS PubMed.
- N. Portolés-Gil, A. Lanza, N. Aliaga-Alcalde, J. A. Ayllón, M. Gemmi, E. Mugnaioli, A. M. López-Periago and C. Domingo, ACS Sustainable Chem. Eng., 2018, 6, 12309–12319 CrossRef.
- S. Yuan, J.-S. Qin, H.-Q. Xu, J. Su, D. Rossi, Y. Chen, L. Zhang, C. Lollar, Q. Wang, H.-L. Jiang, D. H. Son, H. Xu, Z. Huang, X. Zou and H.-C. Zhou, ACS Cent. Sci., 2018, 4, 105–111 CrossRef CAS PubMed.
- T. Rhauderwiek, H. Zhao, P. Hirschle, M. Döblinger, B. Bueken, H. Reinsch, D. D. Vos, S. Wuttke, U. Kolb and N. Stock, Chem. Sci., 2018, 9, 5467–5478 RSC.
- B. Wang, T. Rhauderwiek, A. K. Inge, H. Xu, T. Yang, Z. Huang, N. Stock and X. Zou, Chem.–Eur. J., 2018, 24, 17429–17433 CrossRef CAS PubMed.
- E. S. Grape, J. G. Flores, T. Hidalgo, E. Martínez-Ahumada, A. Gutiérrez-Alejandre, A. Hautier, D. R. Williams, M. O’Keeffe, L. Öhrström, T. Willhammar, P. Horcajada, I. A. Ibarra and A. K. Inge, J. Am. Chem. Soc., 2020, 142, 16795–16804 CrossRef CAS PubMed.
- T. He, Z. Huang, S. Yuan, X.-L. Lv, X.-J. Kong, X. Zou, H.-C. Zhou and J.-R. Li, J. Am. Chem. Soc., 2020, 142, 13491–13499 CrossRef CAS PubMed.
- M. O. Cichocka, Z. Liang, D. Feng, S. Back, S. Siahrostami, X. Wang, L. Samperisi, Y. Sun, H. Xu, N. Hedin, H. Zheng, X. Zou, H.-C. Zhou and Z. Huang, J. Am. Chem. Soc., 2020, 142, 15386–15395 CrossRef CAS.
- F. Carraro, M. d. J. Velásquez-Hernández, E. Astria, W. Liang, L. Twight, C. Parise, M. Ge, Z. Huang, R. Ricco, X. Zou, L. Villanova, C. O. Kappe, C. Doonan and P. Falcaro, Chem. Sci., 2020, 11, 3397–3404 RSC.
- J.-H. Dou, M. Q. Arguilla, Y. Luo, J. Li, W. Zhang, L. Sun, J. L. Mancuso, L. Yang, T. Chen, L. R. Parent, G. Skorupskii, N. J. Libretto, C. Sun, M. C. Yang, P. V. Dip, E. J. Brignole, J. T. Miller, J. Kong, C. H. Hendon, J. Sun and M. Dincă, Nat. Mater., 2021, 20, 222–228 CrossRef CAS PubMed.
- J. T. C. Wennmacher, C. Zaubitzer, T. Li, Y. K. Bahk, J. Wang, J. A. van Bokhoven and T. Gruene, Nat. Commun., 2019, 10, 3316 CrossRef PubMed.
- M. Ge, Y. Wang, F. Carraro, W. Liang, M. Roostaeinia, S. Siahrostami, D. M. Proserpio, C. Doonan, P. Falcaro, H. Zheng, X. Zou and Z. Huang, Angew. Chem., Int. Ed., 2021, 60, 11391–11397 CrossRef CAS PubMed.
- S. Smeets and W. Wan, J. Appl. Crystallogr., 2017, 50, 885–892 CrossRef CAS.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
-
A. C. Larson, Crystallography Computing, ed. F. R. Ahmed, Copenhagen, 1970 Search PubMed.
- L. Palatinus, C. A. Corrêa, G. Steciuk, D. Jacob, P. Roussel, P. Boullay, M. Klementová, M. Gemmi, J. Kopeček, M. C. Domeneghetti, F. Cámara and V. Petříček, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 740–751 CrossRef CAS.
-
G. M. Sheldrick, XPREP, Version 2008/2, Bruker AXS Inc., Madison, USA, 2008 Search PubMed.
- P. R. Evans and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1204–1214 CrossRef CAS PubMed.
- E. M. Miner, T. Fukushima, D. Sheberla, L. Sun, Y. Surendranath and M. Dincă, Nat. Commun., 2016, 7, 10942 CrossRef CAS PubMed.
- W. Cheng, X. Zhao, H. Su, F. Tang, W. Che, H. Zhang and Q. Liu, Nat. Energy, 2019, 4, 115–122 CrossRef CAS.
- E. M. Miner, S. Gul, N. D. Ricke, E. Pastor, J. Yano, V. K. Yachandra, T. Van Voorhis and M. Dincă, ACS Catal., 2017, 7, 7726–7731 CrossRef CAS.
- H. Zhong, K. H. Ly, M. Wang, Y. Krupskaya, X. Han, J. Zhang, J. Zhang, V. Kataev, B. Büchner, I. M. Weidinger, S. Kaskel, P. Liu, M. Chen, R. Dong and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 10677–10682 CrossRef CAS PubMed.
Footnote |
| † CCDC The crystallographic data for the datasets 1, M_A1, M_A2, M_A3, 5, M_B, 7, M_C, M_BC, M_ABC and M_ABCD have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition number CCDC 2063943–2063953. For crystallographic data in CIF or other electronic format see DOI: 10.1039/d1fd00020a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Taimin
Yang
a,
Yanzhi
Wang
b,
Francesco
Carraro
a,
Taimin
Yang
a,
Yanzhi
Wang
b,
Francesco
Carraro