 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonolithic metal–organic frameworks for carbon dioxide separation†
David G.
Madden‡
 a,
Robin
Babu‡
a,
Ceren
Çamur
a,
Nakul
Rampal
a,
Joaquin
Silvestre-Albero
b,
Teresa
Curtin
c and
David
Fairen-Jimenez
*a
a,
Robin
Babu‡
a,
Ceren
Çamur
a,
Nakul
Rampal
a,
Joaquin
Silvestre-Albero
b,
Teresa
Curtin
c and
David
Fairen-Jimenez
*a
aAdsorption & Advanced Materials Laboratory (A2ML), Department of Chemical Engineering & Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge, CB3 0AS, UK. E-mail: df334@cam.ac.uk
bLaboratorio de Materiales Avanzados, Departamento de Química Inorgánica, Universidad de Alicante, San Vicente del Raspeig, E-03690, Spain
cBernal Institute, Department of Chemical Sciences, University of Limerick, Limerick, V94 T9PX, Republic of Ireland
First published on 26th March 2021
Abstract
Carbon dioxide (CO2) is both a primary contributor to global warming and a major industrial impurity. Traditional approaches to carbon capture involve corrosive and energy-intensive processes such as liquid amine absorption. Although adsorptive separation has long been a promising alternative to traditional processes, up to this point there has been a lack of appropriate adsorbents capable of capturing CO2 whilst maintaining low regeneration energies. In the context of CO2 capture, metal–organic frameworks (MOFs) have gained much attention in the past two decades as potential materials. Their tuneable nature allows for precise control over the pore size and chemistry, which allows for the tailoring of their properties for the selective adsorption of CO2. While many candidate materials exist, the amount of research into material shaping for use in industrial processes has been limited. Traditional shaping strategies such as pelletisation involve the use of binders and/or mechanical processes, which can have a detrimental impact on the adsorption properties of the resulting materials or can result in low-density structures with low volumetric adsorption capacities. Herein, we demonstrate the use of a series of monolithic MOFs (monoUiO-66, monoUiO-66-NH2 & monoHKUST-1) for use in gas separation processes.
Introduction
Anthropogenic emissions of carbon dioxide (CO2) are acknowledged to be a significant risk to the global climate. The atmospheric CO2 concentration has surpassed 400 ppm on several occasions since 2013, which represents an increase of over 100 ppm compared to pre-industrial revolution levels.1 Carbon capture will be a crucial technology in achieving carbon neutrality by 2050.2 While clean technologies such as wind and solar power will play a major role in energy provision over the coming decades, increasing energy demands dictate that fossil fuels will remain a key component of the global energy system into the second half of this century.3 Additionally, CO2 represents a significant impurity in industrial processes and its removal from important gases such as methane (CH4) can help improve the overall quality of pipeline grade natural gas (NG). The upgrading of alternative forms of CH4 such as biogas and landfill gas to produce biomethane is also an incredibly attractive source of renewable NG that, once purified, can be pumped directly into the national grid. The development of next-generation carbon capture and sequestration (CCS) technologies in the coming decades will be imperative in the fight again global climate change.To date, CCS has been hampered by high costs (>100 US$ per t CO2 captured) and the techno-economic uncertainties of liquid amine-based technologies.4 Liquid amine-based CCS technologies have been around for over half a century. However, liquid amine chemical capture relies upon chemical reactions and is energy intensive, therefore reducing the overall efficiency of a power plant by up to 40%.5 Additionally, liquid amines are volatile and prone to foaming, leading to the corrosion of industrial equipment. Liquid amines are thus not economically viable and offer little room for innovation. With the advent of the 2015 Paris agreement,6 there has been a political shift towards reducing CO2 emissions globally. In particular, Canada recently announced a direct tax on carbon emissions of at least 10 C$ per t, rising by 10 C$ per t per year until it reaches 50 C$ per t by 2022.7 At 50 C$ per t, this will significantly improve the competitiveness of CCS technologies. Similar ambitious CO2 emission reduction targets have been set in Europe and, right now, a new approach to CCS technologies is required, since renewable and new energy technologies alone will be unable to sustain the growing demand for energy in Europe. This requires a new paradigm for CCS technologies that will fundamentally improve the environmental footprint and cost effectiveness of CSS technologies.
As an alternative to traditional amine-based technologies, solid adsorbents represent a viable alternative for the next generation of low-temperature CCS technologies. To date, much of the research into solid adsorbent based CO2 capture has focused on traditional porous materials such as zeolites, activated carbons and amine-modified silicas.7–11 While significant progress has been made, there are many drawbacks to these materials. In the case of zeolites and activated carbons, these materials lack the tunability and chemical functionalities to improve important process parameters such as chemical interactions, selectivity and hydrophobicity, leading to high CO2 capture costs as a result of their low working capacities and high regeneration costs. Similarly, amine-modified adsorbents rely on similar capture mechanisms to liquid amines and require elevated temperatures (>100 °C) for adsorbent regeneration. Besides, gas constituents such as NOx, SOx and CO2 itself can negatively impact amine-modified solids by poisoning the chemisorbent and deactivating the amine adsorption sites.12–14 Furthermore, amine-modified materials are sometimes subject to thermal and oxidative degradation.15,16
As an alternative to traditional porous materials, metal–organic frameworks (MOFs)17,18 or porous coordination polymers (PCPs)19 represent a broad class of materials that have received a great deal of attention over the past two decades. MOFs are composed of metal ions or clusters, commonly referred to as nodes, bridged by organic ligands and in some cases organic and inorganic pillars, to form various structures and networks. There are currently ca. 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MOF20,21 structures included in the Cambridge Structural Database – MOF subset vs. ca. 1000 silicas and zeolites; their tuneable nature enables the precise control of the material design at the molecular level. Using crystal engineering and reticular chemistry approaches, it is possible to tailor the pore size and chemistry by the rational selection of the organic ligand, functional group, metal ion and activation method. MOFs have already displayed exceptional performance for a wide array of applications including gas storage,22–24 catalysis25,26 and drug delivery.27–30 Their performance for gas separation has also been widely studied, with MOFs having benchmark physisorptive performances for numerous processes including carbon capture,9 CO2 direct air capture (DAC),31–33 C2/C3/C4 separation34–36 and natural gas processing.37–39 The tunability of MOFs and the vast array of platforms available gives MOFs great promise for revolutionising industrial processes in the coming decades.
000 MOF20,21 structures included in the Cambridge Structural Database – MOF subset vs. ca. 1000 silicas and zeolites; their tuneable nature enables the precise control of the material design at the molecular level. Using crystal engineering and reticular chemistry approaches, it is possible to tailor the pore size and chemistry by the rational selection of the organic ligand, functional group, metal ion and activation method. MOFs have already displayed exceptional performance for a wide array of applications including gas storage,22–24 catalysis25,26 and drug delivery.27–30 Their performance for gas separation has also been widely studied, with MOFs having benchmark physisorptive performances for numerous processes including carbon capture,9 CO2 direct air capture (DAC),31–33 C2/C3/C4 separation34–36 and natural gas processing.37–39 The tunability of MOFs and the vast array of platforms available gives MOFs great promise for revolutionising industrial processes in the coming decades.
Despite their potential, the lack of a suitable method for production scale-up and shaping has thus far been a barrier to maximising the potential of MOFs for numerous applications. Their synthesis traditionally relies on solution-based methods, i.e. layering or solvothermal synthesis, both of which are time-consuming and require large amounts of solvent. Mechanochemical synthesis has recently garnered attention for adsorbent scale-up,22–29 where the synthesis can be conducted at scale using continuous processes such as twin-screw extrusion (TSE).30–36 While the scale-up of MOFs has been well-studied, these processes generally lead to powdered materials that require post-synthetic shaping. Indeed, the shaping of MOF powders into bulk samples with desired sizes, shapes, densities and mechanical stabilities is a critical step for their industrial deployment, as it is required to minimise pressure drops and to increase the volumetric adsorption capacity in adsorption columns (Fig. 1).22 To date, mechanical shaping using binders has already been widely utilised for shaping MOFs.33,38–43 While mechanical shaping is relatively simple and fast, the resulting materials often display two major issues. On one hand, extruded materials where low mechanical pressures are applied display low bulk densities due to the presence of large void spaces. Conversely, in powder pressing, the delicate crystalline structures of MOFs are prone to collapsing under high mechanical pressures, leading to crumple zones of amorphous material.20 These amorphous phases can give way to pellets with high bulk densities but with large reductions in the overall porosity. The development of strategies that can address the issues of powder processing whilst maintaining the gas separation performance is critical for the real-world applications of solid adsorbents.
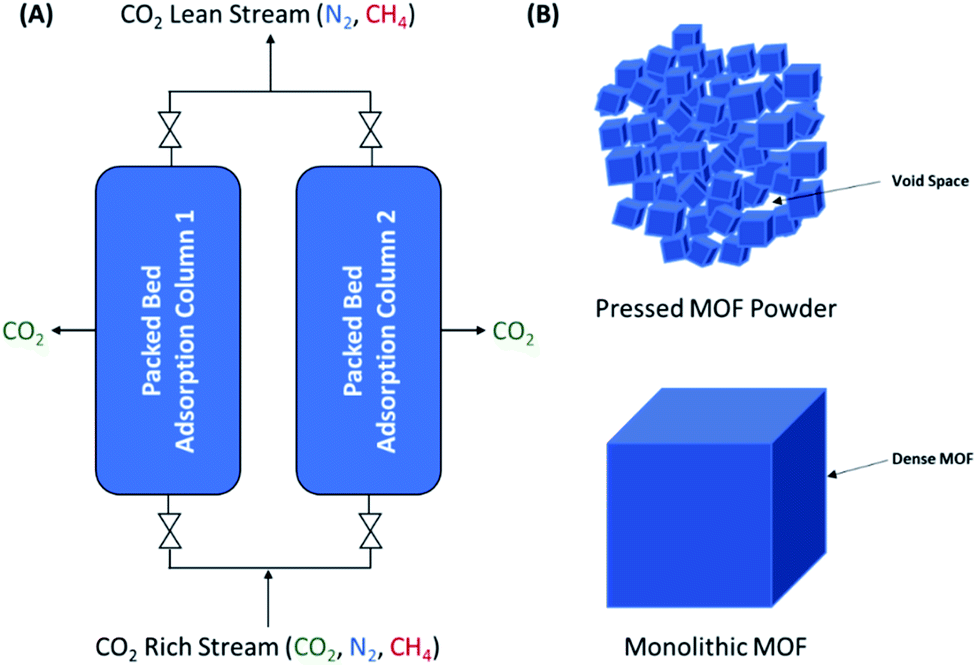 | ||
| Fig. 1 (A) Schematic of a dual-column pressure swing adsorption/temperature swing adsorption (PSA/TSA) system with MOF packed beds. (B) Representation of the abundant void spaces amongst pressed MOF particles compared with a densified monolithic MOF. | ||
In contrast to traditional shaping, self-shaping methods can effectively circumvent the issues related to the extrusion and high-pressure pressing of MOFs. Self-shaping can eliminate the need for additives and/or the use of mechanical presses or extruders. These unique methodologies hold promise for reducing the performance-related issues of MOF shaping whilst simultaneously reducing the cost for shaped MOF production. So far, there have been a limited number of reports on self-shaping MOFs.44–52 In early self-shaping MOFs, researchers utilised precursor MOF gels to form self-shaped materials via syneresis when the MOF gel was dried under ambient conditions, while elevated drying temperatures gave way to powder formation.53–56 These materials displayed similar properties to xerogels, displaying large volumes of hierarchical porosity and low bulk densities. The development of MOF xerogels gave way to the development of the first monolithic MOFs (monoMOFs). Similar to previously reported monolithic gels, monoMOFs have been formed via a sol–gel synthesis approach which has offered a viable alternative to traditional MOF shaping processes. monoMOFs enable the synthesis of high-density, mechanically and chemically stable, centimeter-scale shaped materials, which retain their porosity during synthesis. The first such report on monoMOF synthesis came with the development of monoZIF-8.60 The transparent, glassy-looking material displayed a high BET area (SBET = 1423 m2 g−1) and a density of ρb = 1.05 g cm−3 (single crystal ρb = 0.95 g cm−3).
The sol–gel synthesis approach in MOFs was subsequently extended to other classical MOFs such as HKUST-1 and UiO-66.49,50 The remarkable physical properties displayed by monoHKUST-1 (ρb = 1.06 g cm−3 and SBET = 1288 m2 g−1) resulted in an outstanding volumetric methane uptake capacity of 261 cm3 (STP) cm−3 (65 bar, 298 K). This was found to substantially exceed the previously reported results for pelletised HKUST-1 compacted under a range of pressures and effectively rendered it as the first material to reach the DOE target for NG storage.61 Recently, the formation of monoUiO-66 was achieved by varying the sol–gel drying conditions employed during synthesis.49 The bulk physical properties of monoUiO-66 were tuned with a high level of experimental control, resulting in materials with bulk densities varying between 0.43 and 1.05 g cm−3 (single crystal ρb = 1.20 g cm−3). The inclusion of mesoporosity and its resultant alteration of the adsorptive properties of the MOF yielded outstanding improvements in the methane working capacity of monoUiO-66 (261 cm3 (STP) cm−3, 5–100 bar, 298 K). This demonstrated that unprecedented levels of synthetic control can be exerted on local structures of monoMOFs, enabling the enhancement of the gas adsorption properties beyond those of purely microporous materials. While the field of monoMOFs is relatively new, these materials have shown potential as viable alternatives to traditional shaping methods to produce high-density materials for industrial use.
In this contribution, we examine the gas separation performance of monoMOFs in comparison to their powdered variants under both gravimetric and volumetric conditions. The performances of all of the materials were then evaluated using single-component isotherms, gravimetric gas uptake and mixed gas dynamic breakthrough experiments. We used single-component isotherms to determine important parameters such as the gas uptake and mixed gas selectivity values, while we used gravimetric uptake experiments to determine the gas uptake kinetics. Finally, we analysed these materials for mixed gas separation in gas streams associated with carbon capture (15/85 v/v CO2/N2, dry and 74% relative humidity) and natural gas/biogas upgrading (50/50 v/v CO2/CH4). This work represents the first demonstration of the use of monoMOFs for gas separation applications.
Results & discussion
Synthesis, characterisation and physicochemical properties
Monolithic and powdered variants of HKUST-1, UiO-66 and UiO-66-NH2 were initially synthesised via previously reported methodologies.57–59,62–64 The crystallinities and thermal stabilities of the pristine samples were investigated using powder X-ray diffraction (PXRD) and thermogravimetric analysis (TGA), respectively. The experimental PXRD patterns were found to match the calculated PXRD patterns for each material. The PXRD patterns for the monolithic samples display Scherrer line broadening, caused by the non-convergence of the diffraction line in nano-size particles (Fig. S1–S3†). Besides, monolithic HKUST-1, UiO-66 and UiO-66-NH2 were found to be thermally stable up to 300 °C, 400 °C and 280 °C, respectively (Fig. S4†), which is consistent with previous reports for these materials.Further characterisation of the synthesised materials was performed using 77 K N2 adsorption isotherms (Fig. 2) and Hg porosimetry (Table 1) to analyse the porosity and bulk densities, respectively, of the powdered and monolithic materials. Table 1 displays the BET areas (SBET) calculated using our extended Rouquerol’s criteria using our BETSI protocol (Fig. S8–S13†),65 as well as the total (Vtot) pore volumes and bulk densities (ρbulk) of each material synthesised. All of the materials were fully activated by heating under vacuum before performing the porosimetry experiments. The experimental BET areas calculated for the powdered materials were consistent with those previously reported for HKUST-1, UiO-66 and UiO-66-NH2.62–64 The BET areas of the monoMOFs were also consistent with previous reports.57,58 For both the powdered and monolithic variants, the N2 isotherms displayed high gas uptake below 0.1 P/Po, indicating extensive microporosity within the samples. N2 uptake was also observed at higher relative pressures (>0.8 P/Po) for the Zr-MOFs, indicating the presence of mesoporosity. monoUiO-66-NH2 displayed a relatively large mesoporous step, while the lower N2 uptake above 0.8 P/Po for monoUiO-66 indicates a relatively low amount of mesoporosity. This mesoporosity has been previously observed for monoZr-MOFs and can be attributed to the void spaces between the crystallites and UiO-type material defects.57
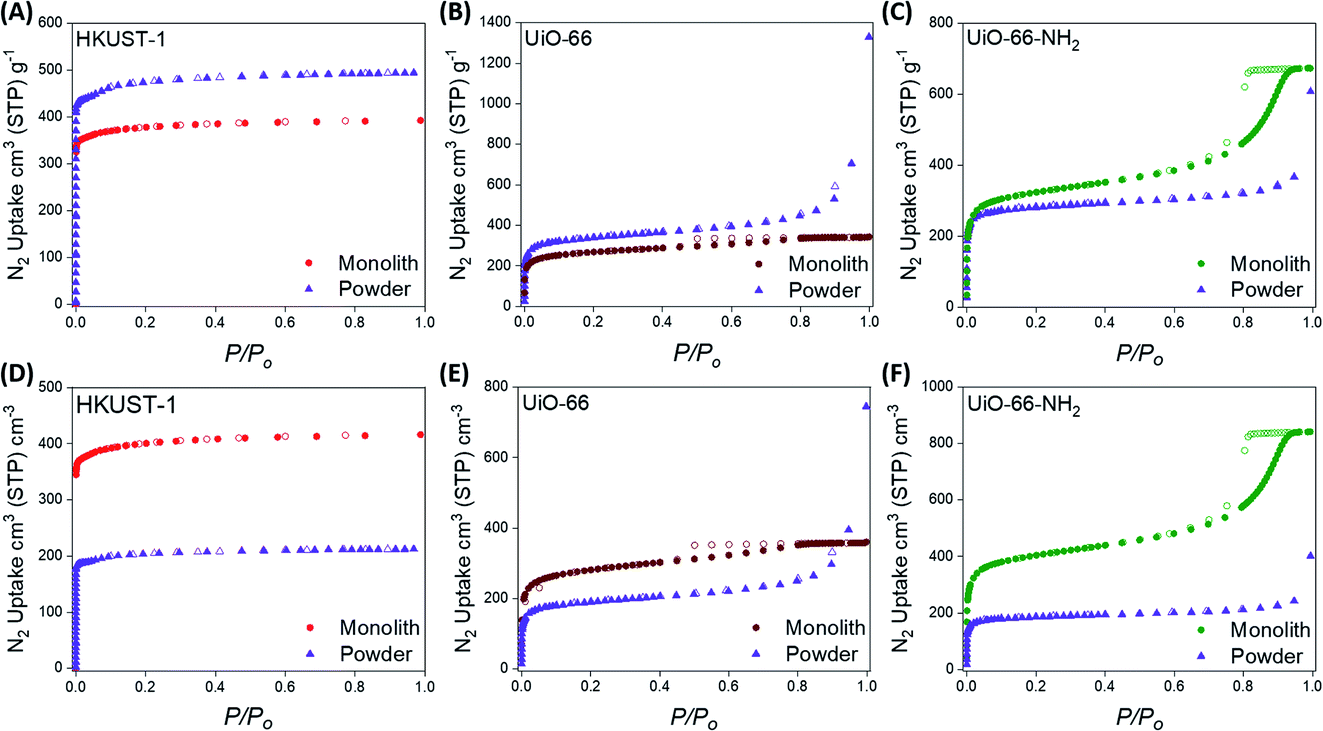 | ||
| Fig. 2 Gravimetric (A–C) and volumetric (D–F) N2 adsorption isotherms at 77 K for the monolithic and powdered HKUST-1, UiO-66 and UiO-66-NH2 materials. Closed symbols represent adsorption while open symbols represent desorption. | ||
| Materials | S BET m2 g−1 | V Tot cm3 g−1 | ρ bulk g cm−3 | ρ crystal g cm−3 | S BET m2 cm−1 | V Tot cm3 cm−3 | Single component gas adsorption | Dynamic breakthrough studies | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CO2 uptake (0.15 bar) cm3 g−1 (cm3 cm−3) | CO2 uptake (0.5 bar) cm3 g−1 (cm3 cm−3) | S CN (0.15 bar) | S CM (0.5 bar) | CO2 uptake 15/85 CO2/N2 cm3 g−1 (cm3 cm−3) | CO2 uptake 50/50 CO2/CH4 cm3 g−1 (cm3 cm−3) | |||||||
| a Obtained at P/Po = 0.98. b Quantified using Hg porosimetry. c Quantified using ideal adsorption solution theory (IAST).66 Physical properties for pellHKUST-1 obtained from the study by Peng et al.67 d Physical properties for pellUiO-66 and pellUiO-66-NH2 obtained from the study by Dhainaut et al.68 | ||||||||||||
| monoHKUST-1 | 1512 | 0.634 | 1.060 | 0.883 | 1603 | 0.672 | 18.5 (19.6) | 53.8 (57.0) | 23 | 12 | 21.3 (22.6) | 53.3 (56.5) |
| powdHKUST-1 | 1871 | 1.290 | 0.500 | 0.883 | 936 | 0.645 | 23.8 (11.9) | 76.2 (38.1) | — | — | 24.7 (12.4) | 61.7 (30.8) |
| pellHKUST-1c | 1340 | 0.570 | 0.824 | 0.883 | 1102 | 0.470 | — | — | — | — | — | — |
| monoUiO-66 | 1015 | 0.530 | 1.050 | 1.237 | 1066 | 0.557 | 14.9 (15.6) | 33.1 (34.8) | 28 | 36 | 15.2 (16.0) | 42.0 (44.1) |
| powdUiO-66 | 1288 | 2.050 | 0.560 | 1.237 | 721 | 1.148 | 18.1 (10.1) | 39.9 (22.3) | — | — | 17.9 (10.0) | 44.0 (24.6) |
| pellUiO-66d | 1459 | 0.540 | 0.430 | 1.237 | 627 | 0.232 | — | — | — | — | — | — |
| monoUiO-66-NH2 | 1226 | 1.040 | 1.250 | 1.246 | 1533 | 1.300 | 15.1 (18.9) | 34.9 (43.6) | 30 | 54 | 16.0 (20.0) | 36.2 (45.2) |
| powdUiO-66-NH2 | 1094 | 0.941 | 0.660 | 1.246 | 722 | 0.621 | 17.2 (11.4) | 39.7 (26.2) | — | — | 17.6 (11.6) | 38.6 (25.5) |
| pellUiO-66-NH2d | 625 | 0.250 | 0.930 | 1.246 | 581 | 0.234 | — | — | — | — | — | — |
To investigate the bulk density properties of the synthesised materials, we performed Hg porosimetry on the monolithic and powdered MOF materials (Table 1). We have shown previously that monoMOFs display higher bulk densities than powders and pelletised materials due to the exceptional control and close packing of the primary particles (i.e. crystallites) during the sol–gel synthesis. In turn, this leads to materials that maintain their porosity and performances and overcomes the limitations of traditional shaping techniques (i.e. pore collapse or low density due to high or low pressure, respectively, during the compression or pore blockage due to the use of binders). The bulk densities observed for monoHKUST-1 (ρbulk = 1.06 g cm−3), monoUiO-66 (ρbulk = 1.05 g cm−3), and monoUiO-66-NH2 (ρbulk = 1.25 g cm−3) were comparable to previous reports.57,58 In contrast, the bulk densities of the powdered materials were significantly lower for HKUST-1 (ρbulk = 0.5 g cm−3), UiO-66 (ρbulk = 0.56 g cm−3) and UiO-66-NH2 (ρbulk = 0.66 g cm−3). The lower density can be attributed to the poor packing, leading to large amounts of void space in the powder samples. When the bulk density is taken into account to calculate the volumetric BET area and volumetric total pore volume of each material (Table 1), the monolithic materials display significantly higher values compared to the powdered variants. In terms of performance, the monolithic materials display volumetric BET areas which are 79%, 48% and 150% higher for HKUST-1, UiO-66 and UiO-66-NH2, respectively, compared to their powdered variants.
Single-component gas adsorption isotherms and kinetic studies
The exceptional physical properties of the monoMOFs prompted us to examine their CO2 adsorption and separation performances. We collected CO2, N2 and CH4 single-component isotherms at 298 and 273 K for all monolithic materials (Fig. 3 & S14–S16†). We also collected CO2 single-component isotherms at 298 K for the powdered variants for comparison (Fig. 3 & S17–S19†). The CO2 uptake values at 298 K and 1 bar observed for monoHKUST-1, monoUiO-66 and monoUiO-66-NH2 were found to be 4.2, 2.2 and 2.1 mmol g−1, respectively. The CO2 uptake values for the monolithic MOFs were found to be consistent with values reported for the powdered variants of each material in the literature and in the NIST/ARPA-E Database of Novel and Emerging Adsorbent Materials.9,69,70 Similarly, the lower uptake values for N2 and CH4 gases observed for the monoliths were also consistent with previous reports for powdered variants of each MOF material.9,69,70 Although the powdered materials display higher gravimetric CO2 adsorption performances compared to the monolithic materials for each MOF variant, this trend is once again reversed when the bulk density is used to calculate the volumetric CO2 adsorption performance (Fig. 3). In this case, the monolithic variants display a superior volumetric performance. While many studies report the gravimetric CO2 uptake performances of MOF materials, the volumetric performances of MOFs are often reported based on the crystal densities of the MOFs as opposed to the experimental bulk densities. To the best of our knowledge, the volumetric CO2 uptake value for monoHKUST-1 of 99.7 cm3 cm−3 at 1 bar represents a new benchmark for volumetric performance under these conditions for MOF materials.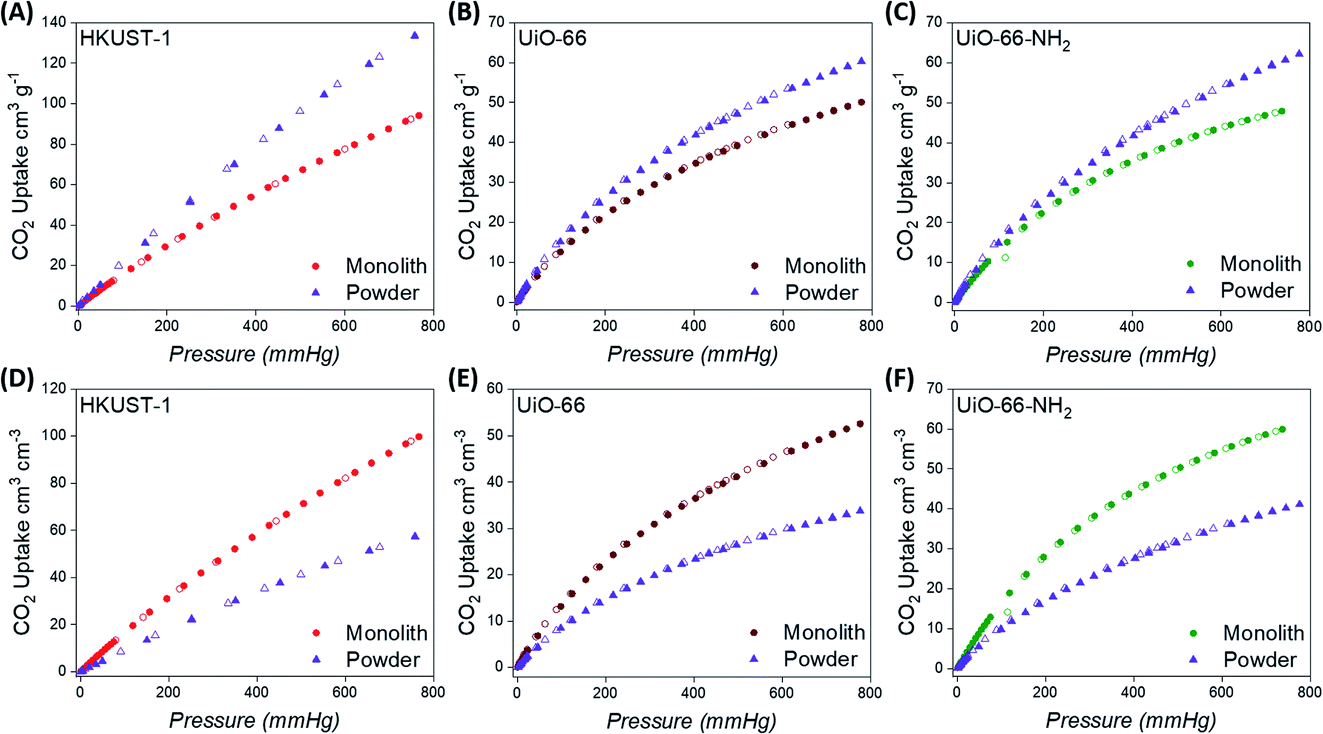 | ||
| Fig. 3 Gravimetric (A–C) and volumetric (D–F) CO2 adsorption isotherms at 298 K for monolithic and powdered HKUST-1, UiO-66 and UiO-66-NH2 materials. Closed symbols represent adsorption while open symbols represent desorption. | ||
To examine the effect of MOF shaping on the adsorbent–adsorbate interactions, we collected variable temperature gas adsorption isotherms to determine the CO2 isosteric heats of adsorption (Qst) for the monoMOF materials. To obtain the CO2 adsorption energies for these compounds, we fitted the CO2 adsorption data at 273 and 298 K using the virial equation (Fig. S20–S22†), calculating the Qst using the Clausius–Clapeyron equation (Fig. S23†). monoHKUST-1 displays a CO2Qst of ca. 25 kJ mol−1, while monoUiO-66 and monoUiO-66-NH2 display values of ca. 25 and 37 kJ mol−1, respectively, similar to previous reports for the powdered variants of each MOF material.69,70 The higher Qst displayed by monoUiO-66-NH2 can be attributed to the higher electrostatic contribution of the amino group which, in turn, leads to stronger adsorbent–adsorbate interactions.
To estimate the CO2/N2 (SCN) and CO2/CH4 (SCM) selectivities of the materials, we first fitted the adsorption isotherms to a dual-site Langmuir–Freundlich (DSLF) model (Tables S1–S3†) and then we used the ideal adsorbed solution theory (IAST).66 We estimated the selectivities under the relevant conditions for carbon capture and natural gas purification (CO2 mole fractions of 0.15 and 0.5, respectively) for all monolithic MOF materials (Fig. S24–S26†). For the CO2 separations associated with carbon capture, monoHKUST-1, monoUiO-66 and monoUiO-66-NH2 exhibited IAST SCN values (at 1 bar and 298 K) of 23, 28 and 30, respectively. For the CO2 separations associated with natural gas purification, monoHKUST-1, monoUiO-66 and monoUiO-66-NH2 exhibited IAST SCM values (at 1 bar and 298 K) of 12, 36 and 54, respectively. The high SCN and SCM values exhibited by the monolithic MOFs suggest that they have potential for use in gas separations relevant to post combustion carbon capture (15/85 v/v CO2/N2) and natural gas purification (50/50 v/v CO2/CH4).
We examined further the CO2 adsorption performances of the monolithic materials using kinetic studies on pristine samples of monoHKUST-1, monoUiO-66 and monoUiO-66-NH2 (Fig. S30–S32†). We exposed the activated samples of the monolithic MOFs to a constant 20 ml min−1 flow of 1.0 bar CO2 at 308 K, while constantly recording the weight change. We found that, despite having shaped the MOFs into larger bodies than the microcrystalline powder, the CO2 uptake kinetics were consistent with previous reports,32,33 with monoHKUST-1, monoUiO-66 and monoUiO-66-NH2 achieving 90% saturation loading in 60, 80 and 40 min, respectively. This demonstrates negligible loss in terms of the gas adsorption kinetics between the powdered and monolithic materials, something that is critical when evaluating the utility of monolithic materials for use in gas separation applications.
Dynamic mixed gas breakthrough studies
At this point, we examined the gas separation performances of the monoMOFs using experimental breakthrough studies on the pristine samples of powdered and monolithic variants of monoHKUST-1, monoUiO-66 and monoUiO-66-NH2. We examined gas mixtures associated with post-combustion carbon capture (15/85 v/v CO2/N2, dry and 74% relative humidity) and natural gas purification (50/50 v/v CO2/CH4) at room temperature (Fig. 4, 5 & S33–S41†). All of the materials examined under both dry and moist conditions achieved efficient CO2/N2 and CO2/CH4 separation. N2 and CH4 gases eluted through the bed immediately, whereas CO2 was retained in the adsorbent bed. For the 15/85 v/v CO2/N2 gas mixture, monoHKUST-1 and the powdered variant achieved CO2 uptake capacities of 21.3 and 24.7 cm3 g−1, respectively, under dry conditions (Table 1). The CO2 uptake value for monoHKUST-1 was in agreement with the IAST predicted value (Fig. S27†). Importantly, when the bulk densities of the monolithic and powdered variants of HKUST-1 were taken into account, the monoHKUST-1 material displayed a volumetric CO2 uptake of 22.6 cm3 cm−3, which is nearly double that of the HKUST-1 packed powder at 12.4 cm3 cm−3. Similar trends were observed for the volumetric CO2 uptake performance of the UiO-66 (monolith = 16.0 cm3 cm−3; powder = 10.0 cm3 cm−3) and UiO-66-NH2 (monolith = 20.0 cm3 cm−3; powder = 11.6 cm3 cm−3) materials for the 15/85 v/v CO2/N2 gas mixture (Table 1 & Fig. S34, S35†). When we exposed the materials to a humid (ca. 74% relative humidity) 15/85 v/v CO2/N2 gas stream, both the packed powder and monolithic MOF materials displayed a ca. 40% drop in performance compared to the dry gas mixture (Fig. S36–S38†). The drop in performance is attributed to the competitive adsorption between CO2 and H2O molecules.9,32,33 Interestingly, despite this reduction in performance, the monoMOFs exhibited nearly double the volumetric CO2 uptake performance under humid conditions compared to the powders for each of the MOF variants studied. | ||
| Fig. 4 Gravimetric (A–C) and volumetric (D–F) CO2 breakthrough curves for a 15/85 v/v CO2/N2 gas stream at 298 K for monolithic and powdered HKUST-1, UiO-66 and UiO-66-NH2 materials. | ||
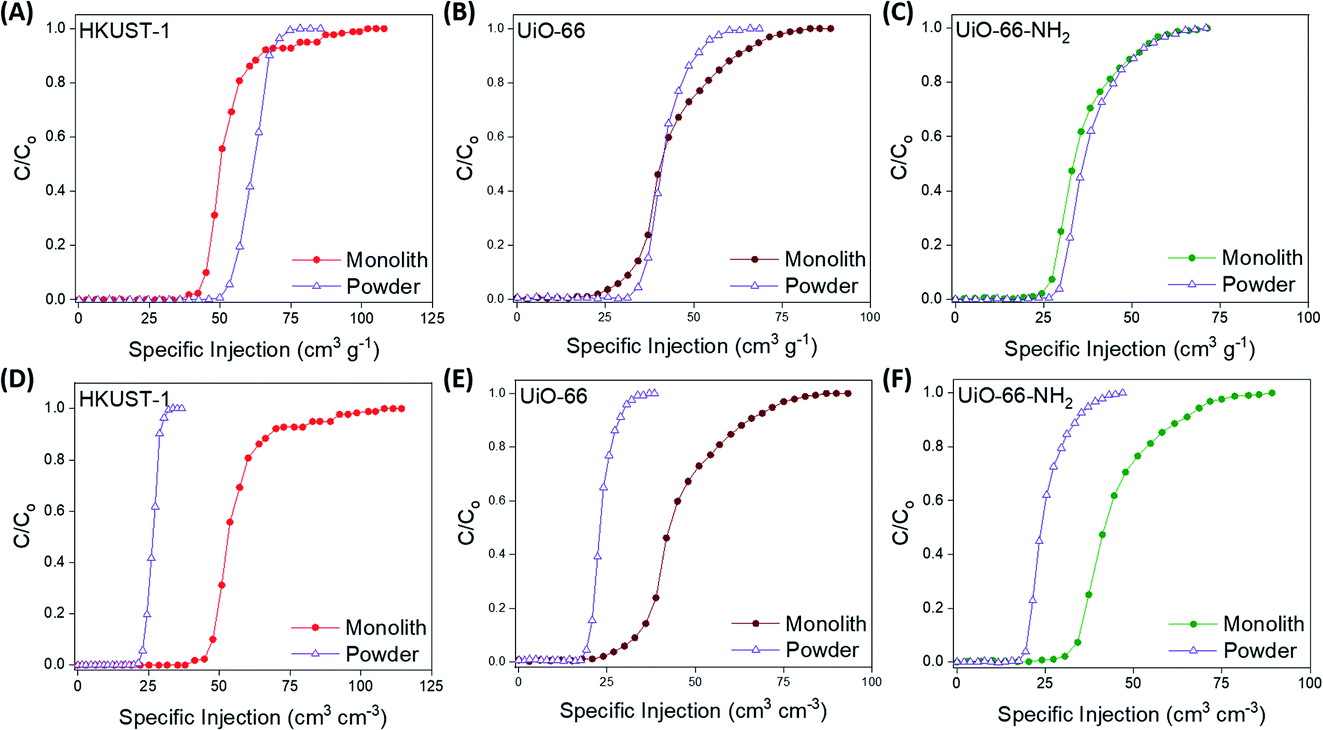 | ||
| Fig. 5 Gravimetric (A–C) and volumetric (D–F) CO2 breakthrough curves for a 50/50 v/v CO2/CH4 gas stream at 298 K for monolithic and powdered HKUST-1, UiO-66 and UiO-66-NH2 materials. | ||
When we examined the synthesized materials with the 50/50 v/v CO2/CH4 gas mixture, monoHKUST-1, monoUiO-66 and monoUiO-66-NH2 displayed exceptional volumetric CO2 uptake values of 56.5, 44.1 and 45.2 cm3 cm−3, respectively (Table 1 & Fig. S39–S41†). The gravimetric performances of each material were in agreement with the IAST predictions (Fig. S27–S29†). Once again, the monolithic materials significantly outperformed the packed powders in terms of the volumetric CO2 uptake performance in the breakthrough studies, with each monolithic variant adsorbing nearly double the amount of CO2 per cm−3 compared to the powdered materials. The CO2/CH4 separation performances of monoHKUST-1, monoUiO-66 and monoUiO-66-NH2 were comparable to a number of benchmark MOFs such as TIFSIX-3-Ni, NbOFFIVE-1-Ni and TIFSIX-2-Cu-i, with only materials such as Mg-MOF-74 and UTSA-16 displaying superior gravimetric CO2 uptakes under similar conditions.37–39,71 While many MOFs demonstrate benchmark performances for gravimetric CO2 capture, their performances do not translate well to volumetric performance. Many studies rely on theoretical crystal densities when calculating volumetric performances, which are often not achievable during traditional MOF shaping and densification processes such as extrusion and pressing due to mechanical degradation and pore collapse.38,50,54 Again, to the best of our knowledge, the CO2 removal performance of monoHKUST-1 represents the highest CO2 removal performance achieved by any adsorbent after successful pelletisation and shaping under these conditions. Finally, we carried out recyclability tests on monoHKUST-1, monoUiO-66 and monoUiO-66-NH2 for the 50/50 v/v CO2/CH4 gas mixture (Fig. S42†). In order to examine the recyclability of the materials, we heated the adsorbents to 120 °C under a helium flow between tests. The monoMOFs were stable under dry conditions, displaying negligible reductions in performance over five successive adsorption/desorption cycles.
Conclusions
Adsorption based gas separation processes have shown huge potential for important industrial processes such as carbon capture and gas purification. The deployment of porous adsorbents in these processes, however, has been hampered by a lack of suitable shaping processes which allow high density materials while maintaining gas sorption performance. In conclusion, we have demonstrated that monoMOFs display superior volumetric gas separation performances compared to packed powder materials. Single-component gas adsorption isotherms suggest that all six materials examined herein are efficient at removing CO2 from CO2/N2 and CO2/CH4 gas mixtures. However, when the bulk density of each material is evaluated to determine the volumetric performances in both gas adsorption isotherms and dynamic breakthrough studies, monoMOFs exhibit superior CO2 separation performances under all conditions. monoMOFs display similar kinetics to their powdered variants, suggesting that kinetic limitations do not exist after monoMOF synthesis. While many benchmark MOFs display exceptional gravimetric CO2 adsorption performances, these rarely translate to volumetric CO2 adsorption due to issues regarding MOF shaping and densification. Many reports on MOF materials rely on theoretical crystal structure densities when reporting volumetric performances, which rarely translate to experimental bulk densities upon powder processing and pelletisation. While high-density monoMOFs have previously demonstrated benchmark performances for gas storage applications, this report represents the first demonstration of the gas separation performances of monoMOFs. This work further illustrates the potential of this unique class of materials for a myriad of commercially relevant gas separation applications and paves the way for the development of next generation monoMOFs with superior physical properties and enhanced gas adsorption performance.Conflicts of interest
D. F.-J. has financial interests in the start-up company immaterial, which is seeking to commercialise metal–organic frameworks.Acknowledgements
D. F.-J. thanks the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (NanoMOFdeli), ERC-2016-COG 726380 and Innovate UK (104384) and EPSRC IAA. JSA would like to acknowledge the financial support from MINECO (PID2019-108453GB-C21).References
- R. Monastersky, Nature, 2013, 497, 13–14 CrossRef CAS PubMed.
- M. Hulme, Nat. Clim. Change, 2016, 6, 222 CrossRef.
- M. Odenberger and F. Johnsson, Int. J. Greenhouse Gas Control, 2010, 4, 327–340 CrossRef CAS.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross and J. P. Hallett, Energy Environ. Sci., 2014, 7, 130–189 RSC.
- K. Goto, K. Yogo and T. Higashii, Appl. Energy, 2013, 111, 710–720 CrossRef CAS.
- J. Rogelj, M. den Elzen, N. Hohne, T. Fransen, H. Fekete, H. Winkler, R. Schaeffer, F. Sha, K. Riahi and M. Meinshausen, Nature, 2016, 534, 631–639 CrossRef CAS PubMed.
- T. Guardian, Canada will tax carbon emissions to meet Paris climate agreement targets, https://www.theguardian.com/world/2016/oct/03/canada-carbon-emissions-tax-paris-climate-agreement, accessed 23/01/2017, 2017 Search PubMed.
- A. Goeppert, H. Zhang, M. Czaun, R. B. May, G. K. S. Prakash, G. A. Olah and S. R. Narayanan, ChemSusChem, 2014, 7, 1386–1397 CrossRef CAS.
- J. A. Mason, T. M. McDonald, T. H. Bae, J. E. Bachman, K. Sumida, J. J. Dutton, S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2015, 137, 4787–4803 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS.
- D. M. D’Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef.
- A. P. Hallenbeck and J. R. Kitchin, Ind. Eng. Chem. Res., 2013, 52, 10788–10794 CrossRef CAS.
- A. Sayari, A. Heydari-Gorji and Y. Yang, J. Am. Chem. Soc., 2012, 134, 13834–13842 CrossRef CAS.
- A. Sayari, Y. Belmabkhout and E. Da’na, Langmuir, 2012, 28, 4241–4247 CrossRef CAS PubMed.
- A. Heydari-Gorji and A. Sayari, Ind. Eng. Chem. Res., 2012, 51, 6887–6894 CrossRef CAS.
- A. Heydari-Gorji, Y. Belmabkhout and A. Sayari, Microporous Mesoporous Mater., 2011, 145, 146–149 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- L. R. MacGillivray, Metal–organic frameworks: design and application, John Wiley & Sons, 2010 Search PubMed.
- S. Kitagawa, R. Kitaura and S. i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS.
- P. Z. Moghadam, S. M. J. Rogge, A. Li, C.-M. Chow, J. Wieme, N. Moharrami, M. Aragones-Anglada, G. Conduit, D. A. Gomez-Gualdron, V. Van Speybroeck and D. Fairen-Jimenez, Matter, 2019, 1, 219–234 CrossRef.
- P. Z. Moghadam, A. Li, X.-W. Liu, R. Bueno-Perez, S.-D. Wang, S. B. Wiggin, P. A. Wood and D. Fairen-Jimenez, Chem. Sci., 2020, 11, 8373–8387 RSC.
- B. M. Connolly, D. G. Madden, A. E. H. Wheatley and D. Fairen-Jimenez, J. Am. Chem. Soc., 2020, 142, 8541–8549 CrossRef CAS PubMed.
- K. V. Kumar, K. Preuss, M. M. Titirici and F. Rodriguez-Reinoso, Chem. Rev., 2017, 117, 1796–1825 CrossRef CAS PubMed.
- B. Li, H.-M. Wen, W. Zhou, J. Q. Xu and B. Chen, Chem, 2016, 1, 557–580 CAS.
- Q. Wang and D. Astruc, Chem. Rev., 2020, 120, 1438–1511 CrossRef CAS.
- J. P. Mehta, T. Tian, Z. Zeng, G. Divitini, B. M. Connolly, P. A. Midgley, J.-C. Tan, D. Fairen-Jimenez and A. E. H. Wheatley, Adv. Funct. Mater., 2018, 28, 1705588 CrossRef.
- J. W. Osterrieth and D. Fairen-Jimenez, Biotechnol. J., 2020, 2000005 Search PubMed.
- I. A. Lazaro and R. S. Forgan, Coord. Chem. Rev., 2019, 380, 230–259 CrossRef.
- M. X. Wu and Y. W. Yang, Adv. Mater., 2017, 29, 1606134 CrossRef PubMed.
- C. Orellana-Tavra, E. F. Baxter, T. Tian, T. D. Bennett, N. K. Slater, A. K. Cheetham and D. Fairen-Jimenez, Chem. Commun., 2015, 51, 13878–13881 RSC.
- S. Mukherjee, N. Sikdar, D. O’Nolan, D. M. Franz, V. Gascon, A. Kumar, N. Kumar, H. S. Scott, D. G. Madden, P. E. Kruger, B. Space and M. J. Zaworotko, Sci. Adv., 2019, 5, eaax9171 CrossRef CAS.
- D. G. Madden, H. S. Scott, A. Kumar, K. J. Chen, R. Sanii, A. Bajpai, M. Lusi, T. Curtin, J. J. Perry and M. J. Zaworotko, Philos. Trans. R. Soc. London, Ser. A, 2017, 375, 20160025 CrossRef.
- A. Kumar, D. G. Madden, M. Lusi, K. J. Chen, E. A. Daniels, T. Curtin, J. J. t. Perry and M. J. Zaworotko, Angew. Chem., Int. Ed., 2015, 54, 14372–14377 CrossRef CAS PubMed.
- K. J. Chen, D. G. Madden, S. Mukherjee, T. Pham, K. A. Forrest, A. Kumar, B. Space, J. Kong, Q. Y. Zhang and M. J. Zaworotko, Science, 2019, 366, 241–246 CrossRef CAS PubMed.
- A. Cadiau, K. Adil, P. M. Bhatt, Y. Belmabkhout and M. Eddaoudi, Science, 2016, 353, 137–140 CrossRef CAS PubMed.
- G. Liu, V. Chernikova, Y. Liu, K. Zhang, Y. Belmabkhout, O. Shekhah, C. Zhang, S. Yi, M. Eddaoudi and W. J. Koros, Nat. Mater., 2018, 17, 283–289 CrossRef CAS.
- D. G. Madden, D. O’Nolan, K. J. Chen, C. Hua, A. Kumar, T. Pham, K. A. Forrest, B. Space, J. J. Perry, M. Khraisheh and M. J. Zaworotko, Chem. Commun., 2019, 55, 3219–3222 RSC.
- Y. Belmabkhout, P. M. Bhatt, K. Adil, R. S. Pillai, A. Cadiau, A. Shkurenko, G. Maurin, G. Liu, W. J. Koros and M. Eddaoudi, Nat. Energy, 2018, 3, 1059–1066 CrossRef CAS.
- D. Britt, H. Furukawa, B. Wang, T. G. Glover and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20637 CrossRef CAS PubMed.
- A. Pichon and S. L. James, CrystEngComm, 2008, 10, 1839–1847 RSC.
- T. Friščić, D. G. Reid, I. Halasz, R. S. Stein, R. E. Dinnebier and M. J. Duer, Angew. Chem., Int. Ed., 2010, 122, 724–727 CrossRef.
- K. Užarević, T. C. Wang, S.-Y. Moon, A. M. Fidelli, J. T. Hupp, O. K. Farha and T. Friščić, Chem. Commun., 2016, 52, 2133–2136 RSC.
- P. A. Julien, K. Užarević, A. D. Katsenis, S. A. Kimber, T. Wang, O. K. Farha, Y. Zhang, J. Casaban, L. S. Germann and M. Etter, J. Am. Chem. Soc., 2016, 138, 2929–2932 CrossRef CAS.
- M. Rubio-Martinez, C. Avci-Camur, A. W. Thornton, I. Imaz, D. Maspoch and M. R. Hill, Chem. Soc. Rev., 2017, 46, 3453–3480 RSC.
- D. E. Crawford and J. Casaban, Adv. Mater., 2016, 28, 5747–5754 CrossRef CAS.
- J. Stojaković, B. S. Farris and L. R. MacGillivray, Chem. Commun., 2012, 48, 7958–7960 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- R. P. Ribeiro, C. L. Antunes, A. U. Garate, A. F. Portela, M. G. Plaza, J. P. Mota and I. A. Esteves, Microporous Mesoporous Mater., 2019, 275, 111–121 CrossRef CAS.
- J. Cousin-Saint-Remi, S. Van der Perre, T. Segato, M.-P. Delplancke, S. Goderis, H. Terryn, G. Baron and J. Denayer, ACS Appl. Mater. Interfaces, 2019, 11, 13694–13703 CrossRef CAS.
- A. I. Spjelkavik, S. Divekar, T. Didriksen and R. Blom, Chem.–Eur. J., 2014, 20, 8973–8978 CAS.
- M. Tagliabue, C. Rizzo, R. Millini, P. D. Dietzel, R. Blom and S. Zanardi, J. Porous Mater., 2011, 18, 289–296 CrossRef CAS.
- Y. Hara, K. Kanamori and K. Nakanishi, Angew. Chem., Int. Ed., 2019, 131, 19223–19229 CrossRef.
- M. R. Lohe, M. Rose and S. Kaskel, Chem. Commun., 2009, 6056–6058 RSC.
- S. M. Vilela, P. Salcedo-Abraira, L. Micheron, E. L. Solla, P. G. Yot and P. Horcajada, Chem. Commun., 2018, 54, 13088–13091 RSC.
- B. Bueken, N. Van Velthoven, T. Willhammar, T. Stassin, I. Stassen, D. A. Keen, G. V. Baron, J. F. Denayer, R. Ameloot and S. Bals, Chem. Sci., 2017, 8, 3939–3948 RSC.
- J.-W. Ye, X. Zhou, Y. Wang, R.-K. Huang, H.-L. Zhou, X.-N. Cheng, Y. Ma and J.-P. Zhang, Sci. China Mater., 2018, 61, 424–428 CrossRef CAS.
- B. M. Connolly, M. Aragones-Anglada, J. Gandara-Loe, N. A. Danaf, D. C. Lamb, J. P. Mehta, D. Vulpe, S. Wuttke, J. Silvestre-Albero, P. Z. Moghadam, A. E. H. Wheatley and D. Fairen-Jimenez, Nat. Commun., 2019, 10, 2345 CrossRef CAS PubMed.
- T. Tian, Z. Zeng, D. Vulpe, M. E. Casco, G. Divitini, P. A. Midgley, J. Silvestre-Albero, J. C. Tan, P. Z. Moghadam and D. Fairen-Jimenez, Nat. Mater., 2018, 17, 174–179 CrossRef CAS.
- J. P. Mehta, T. Tian, Z. Zeng, G. Divitini, B. M. Connolly, P. A. Midgley, J.-C. Tan, D. Fairen-Jimenez and A. E. H. Wheatley, Adv. Funct. Mater., 2018, 28, 1705588 CrossRef.
- T. Tian, J. Velazquez-Garcia, T. D. Bennett and D. Fairen-Jimenez, J. Mater. Chem. A, 2015, 3, 2999–3005 RSC.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, J. Am. Chem. Soc., 2013, 135, 11887–11894 CrossRef CAS PubMed.
- F. Raganati, V. Gargiulo, P. Ammendola, M. Alfe and R. Chirone, Chem. Eng. J., 2014, 239, 75–86 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- J. Osterrieth, J. Rampersad, D. G. Madden, N. Rampal, L. Skoric and B. Connolly, et al. , ChemRxiv, 2021 DOI:10.26434/chemrxiv.14291644.v2.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121–127 CrossRef CAS.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, J. Am. Chem. Soc., 2013, 135(32), 11887–11894 CrossRef CAS.
- J. Dhainaut, C. Avci-Camur, J. Troyano, A. Legrand, J. Canivet, I. Imaz, D. Maspoch, H. Reinsch and D. Farrusseng, CrystEngComm, 2017, 19, 4211–4218 RSC.
- G. E. Cmarik, M. Kim, S. M. Cohen and K. S. Walton, Langmuir, 2012, 28, 15606–15613 CrossRef CAS PubMed.
- D. Siderius, V. Shen, R. Johnson III and R. van Zee, https://adsorbents.nist.gov/, accessed, 2018, 3.
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1fd00017a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |