 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A high-throughput screening of metal–organic framework based membranes for biogas upgrading†
Joseph
Glover‡
 and
Elena
Besley
*
and
Elena
Besley
*
School of Chemistry, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: Elena.Besley@nottingham.ac.uk
First published on 28th April 2021
Abstract
Applications of biomethane as a source of renewable energy and transport fuel rely heavily on successful implementation of purification methods capable of removing undesirable impurities from biogas and increasing its calorific content. Metal–organic frameworks (MOFs) are competitive candidates for biogas upgrading due to a versatile range of attractive physical and chemical properties which can be utilised in membrane materials. In this work, we present a high-throughput computational screening methodology for efficient identification of MOF structures with promising gas separation performance. The proposed screening strategy is based on initial structural analysis and predictions of the single-component permeation of CO2, CH4 and H2S from adsorption and diffusion calculations at infinite dilution. The identified top performing candidates are subject to further analysis of their gas separation performance at the operating conditions of 10 bar and 298 K, using grand canonical Monte Carlo and equilibrium molecular dynamics simulations on equimolar CO2/CH4 and H2S/CH4 mixtures. The Henry constant for the adsorption of H2O was also calculated to determine the hydrophobicity of MOF structures, as the presence of H2O often leads to membrane instability and performance limitations. For the considered gas mixtures, the top MOF candidates exhibit superior separation capabilities over polymer-, zeolite-, and mixed matrix-based membranes as indicated by the predicted values of selectivity and permeability. The proposed screening protocol offers a powerful tool for the rational design of novel MOFs for biogas upgrading.
1 Introduction
Agricultural, industrial and municipal wastes are some of the many feedstocks which, in the absence of O2, can be broken down to form biogas – a mixture of gases composed of CH4 (50–65%), CO2 (35–50%) and small amounts of trace gases such as H2S, N2, H2 and NH3.1 The composition of biogas is largely dependent on the waste substrate and the conditions under which substrate digestion occurs.2 Raw biogas has a high calorific content between 5000–7000 kcal m−3, making it ideal for heat and electricity generation.3 Through separation of its components, biogas can be upgraded to provide an enriched source of CH4, further improving its calorific content for utilisation in the transport sector. This process is essential as high levels of CO2 lower the calorific content of the fuel. In addition, small concentrations of H2S (>50 ppm) have been shown to corrode metal, making raw biogas unsuitable for standard combustion engines.4In industry, there are many techniques that can be employed to upgrade biogas including absorption, adsorption and cryogenic distillation methods. Absorption methods involve the capture of impurities by dissolving them in a solvent at elevated pressure, or by reacting them with an inorganic/amine based solution.5 Adsorption methods make use of molecular sieves, activated carbon and zeolites to separate out biogas due to differences in affinity between the biogas components and the adsorbent bed. The contaminants are usually adsorbed at high pressure from the biogas stream, leaving CH4 to pass over the adsorbent bed. The bed can then be regenerated by reducing the pressure which desorbs the contaminants. These are the core principles underlying pressure-swing adsorption methods used to separate gaseous mixtures.6 Similar principles can also be employed using changes in other stimuli such as temperature. Finally, as each biogas component condenses at different temperatures and pressures, one can use distillation methods to separate contaminants.7
These methods can provide high CH4 purity typically between 90–99%, however, they can be quite energy intensive, suffering from high investment, operation and/or maintenance costs.8 One of the most promising and cheaper alternatives is to use membrane materials to separate gas components through differences in permeation. Membranes can be synthesised from various materials, including organic polymers, molecular sieves, palladium alloys, amorphous silicas, ceramics and zeolites.9 Polymer based membranes currently dominate the membrane market as they are cheap, easily scalable, can be fabricated into hollow fibers or sheets and have been more extensively studied. However, they suffer from short lifetime, low thermal and chemical stability, and low selectivity.10 Furthermore, the work of Robeson revealed a trade-off relationship between selectivity and permeability in polymer membranes which limits their overall separation performance.11,12 Inorganic based membranes tend to produce higher selectivity and possess greater thermal and chemical stability than polymer membranes making them suitable for separations that require harsh conditions.13 However, they typically have higher production costs and can have reproducibility issues when attempting to create uniform, defect-free membranes.14 While a lot of progress has been made in the development of synthesis methods for existing membranes,15–17 the discovery of new materials which can rival the separation performance of existing membranes is also a key area of research.18–20 One example of these materials are metal–organic frameworks (MOFs): a rapidly growing hybrid class of porous materials that are self-assembled from inorganic clusters (known as secondary building units) and organic ligands. Due to the diverse nature of their building blocks, MOFs can offer a versatile range of attractive physical and chemical properties such as high porosity and large surface area.21 Furthermore, these properties are highly tuneable by choosing appropriate building blocks, making MOFs competitive over traditional porous materials. As such, MOFs have been extensively studied for a wide array of applications including gas storage,22 separation,23 drug delivery,24 catalysis,25 and sensing.26
When fabricating MOF thin films or membranes, a number of different strategies may be employed, such as direct growth, layer-by-layer growth, seeded growth or chemical solution deposition.27,28 The development of these synthetic routes has led to the construction of continuous membranes for many different applications. An example of these applications is gas separations, including CO2/CH4 separation found in biogas upgrading. In addition to pure MOF membranes, the development of composite materials such as mixed matrix membranes (MMM) has been used as a strategy to combine the high separation performance of many zeolitic and MOF membranes with the low processing costs of polymer membranes.1,29–34 An overview of the CO2/CH4 separation performance for membranes previously reported in the literature is presented later in the paper.
Many experimental studies demonstrate the promising application of MOFs to biogas upgrading. However, they only cover a small fraction of the total number of MOFs that have been synthesised. In 2017, Moghadam and co-workers developed a series of criteria which estimates close to 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MOF-like structures within the Cambridge Structural Database (CSD).35 While this creates many opportunities for application-driven research, it is clearly not feasible to synthesise all of these structures. In such scenarios, high-throughput computational studies can be useful for accurately identifying top performing MOFs for a particular application. Furthermore, they can provide insight and relationships elucidating the link between structural properties and the performance of a MOF. In the literature, there are a number of excellent high-throughput studies which investigate the CO2 capture and separation properties of both experimental and hypothetical MOF adsorbents.36–40 In contrast, there are far fewer studies which examine the membrane separation potential of MOFs, particularly focusing on CO2/CH4. This may be attributed to the higher computational cost of simulating gas permeability which requires both adsorption and diffusion calculations. In an early study, Erucar and co-workers tested several permeation models to compare predicted and experimental permeabilities in MMMs. With the best model, they screened a modest number of MOF-based MMMs, identifying trends between the choice of MOF filler particles and its effect on CO2/CH4 selectivity and CO2 permeability.41 Jiang and co-workers screened a large database containing hypothetical MOF membranes for CO2/CH4 and N2/CH4 separations. They identified MOFs with CO2/CH4 membrane selectivities exceeding 100 and CO2 permeabilities greater than 104 barrer (1 barrer = 3.35 × 10−16 mol m−2 s−1 Pa−1). The top candidates were further examined for a ternary CO2/CH4/N2 separation at 10 bar and 298 K.42 Recently, Altintas, Erucar and Keskin screened MOF membranes to identify top performing MOFs that could be used as filler particles in MMMs and found that MOF-based MMMs have much higher CO2 permeability and CO2/CH4 selectivity than the pure polymers.43
000 MOF-like structures within the Cambridge Structural Database (CSD).35 While this creates many opportunities for application-driven research, it is clearly not feasible to synthesise all of these structures. In such scenarios, high-throughput computational studies can be useful for accurately identifying top performing MOFs for a particular application. Furthermore, they can provide insight and relationships elucidating the link between structural properties and the performance of a MOF. In the literature, there are a number of excellent high-throughput studies which investigate the CO2 capture and separation properties of both experimental and hypothetical MOF adsorbents.36–40 In contrast, there are far fewer studies which examine the membrane separation potential of MOFs, particularly focusing on CO2/CH4. This may be attributed to the higher computational cost of simulating gas permeability which requires both adsorption and diffusion calculations. In an early study, Erucar and co-workers tested several permeation models to compare predicted and experimental permeabilities in MMMs. With the best model, they screened a modest number of MOF-based MMMs, identifying trends between the choice of MOF filler particles and its effect on CO2/CH4 selectivity and CO2 permeability.41 Jiang and co-workers screened a large database containing hypothetical MOF membranes for CO2/CH4 and N2/CH4 separations. They identified MOFs with CO2/CH4 membrane selectivities exceeding 100 and CO2 permeabilities greater than 104 barrer (1 barrer = 3.35 × 10−16 mol m−2 s−1 Pa−1). The top candidates were further examined for a ternary CO2/CH4/N2 separation at 10 bar and 298 K.42 Recently, Altintas, Erucar and Keskin screened MOF membranes to identify top performing MOFs that could be used as filler particles in MMMs and found that MOF-based MMMs have much higher CO2 permeability and CO2/CH4 selectivity than the pure polymers.43
High-throughput studies which identify the “best” materials for separating CO2 from CH4 can provide a great deal of insight into biogas upgrading. However, for the production of high purity CH4, it is also important to be able to selectively remove the smaller quantities of H2S present in biogas streams. Additionally, many biogas streams contain water vapour which may potentially affect the separation performance and stability of MOF membranes.44 In this work, we apply a multi-stage screening methodology to focus on CO2/CH4 and H2S/CH4 separations across a range of hydrophobic MOF membranes from the CSD database. Top selected candidates encompass high selectivity and permeability for CO2 and H2S which is essential for the biogas upgrading process.
Additionally, a set of membranes with improved moisture stability due to their enhanced hydrophobic character has been identified. The initial filtering of porous candidates is based on the differences in their geometric properties. Next, a time efficient estimation of the ideal CO2/CH4 and H2S/CH4 selectivity and permeability is carried out at infinite dilution conditions. This is achieved by calculating the Henry coefficients using a grand canonical Monte Carlo (GCMC) approach and the diffusion coefficients extracted from equilibrium molecular dynamics (EMD) calculations. We also explore how different input parameters, used to calculate partial charges via the extended charge equilibration (EQeq) scheme,45 can affect the physical nature and the overall separation properties of MOFs. The top hydrophobic MOFs found above the Robeson upper bound are then subjected to calculations of the performance prediction under working conditions. We perform equimolar CO2/CH4 and H2S/CH4 GCMC and EMD simulations to predict mixture selectivity and permeability at 298 K and 10 bar. The best selected membranes are compared to literature zeolites, MOFs and MMMs to assess their overall performance and viability for biogas upgrading. Finally, we examine the structural properties of 1183 hydrophobic membranes that lie above the Robeson limit. This enables us to draw conclusions about the relationships which lead to high separation performance, providing useful insight into the design of future membranes.
2 Computational details
A set of geometric criteria, detailed in the ESI,† is applied to the CSD MOF data set (67675 structures) to remove structures with non-accessible surface area and pore diameters smaller than the individual components of biogas. Solvent molecules are removed from each structure using a python script readily available from the literature.35 In this work, a diverse set of 7909 MOF structures are found to be suitable for high-throughput screening. The RASPA software package46 is employed to calculate the self-diffusivity (D0), Henry constant (K0) and heat of adsorption (Q0st) of CO2, H2S, CH4 and H2O at infinite dilution. K0 is determined using the Widom insertion method at 298 K, using a total of 100000 cycles.47 For calculations of D0, EMD simulations in the NVT ensemble are employed. A time step of 1 fs is used, and the number of equilibration and production cycles are set to 10000 and 100000, respectively. Heating is controlled using a Nosé–Hoover thermostat.47 To mimic infinite dilution, adsorbate–adsorbate interactions are neglected and only framework–adsorbate interactions are considered. The statistical accuracy of infinite dilution conditions is improved by using 30 gas molecules per simulation. The initial positions of the gas molecules are determined using 1000 Monte Carlo initialization cycles. The mean square displacement (MSD) of the gas molecules is computed and the self-diffusivity is obtained using the gradient of the MSD via Einstein’s relation.47 Based on previous work, we exclude materials where D0 < 10−8 cm2 s−1 as these values approach the limit of accurate description of molecular diffusion by EMD simulations.48 The gas permeability for each component i at infinite dilution (P0i) is calculated from the product of self-diffusivity and the Henry constant, i.e. P0i = D0i × K0i. The ratios K0i/K0j, D0i/D0j and P0i/P0j of two components are then used to determine the ideal adsorption (S0ads,i/j), diffusion (S0diff,i/j) and membrane selectivity (S0mem,i/j), respectively.
A hydrophobic subset of MOFs is identified by comparing 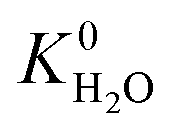 of each membrane against the hydrophobic ZIF-8 structure (
of each membrane against the hydrophobic ZIF-8 structure (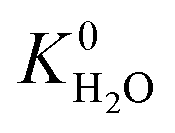 = 5.00 × 10−6 mol kg−1 Pa−1). A membrane is considered hydrophobic if the value of
= 5.00 × 10−6 mol kg−1 Pa−1). A membrane is considered hydrophobic if the value of 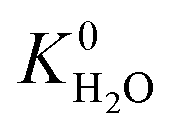 is smaller or equal to that of ZIF-8. This approach has been used previously to identify hydrophobic MOFs for hydrocarbon separations and toxic industrial chemical capture.49,50 Only hydrophobic membranes above the 2008 Robeson bound12 that are selective to both H2S and CO2 and possess
is smaller or equal to that of ZIF-8. This approach has been used previously to identify hydrophobic MOFs for hydrocarbon separations and toxic industrial chemical capture.49,50 Only hydrophobic membranes above the 2008 Robeson bound12 that are selective to both H2S and CO2 and possess 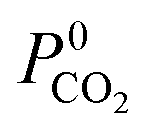 and
and 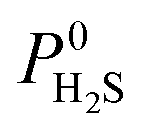 greater than 2.5 × 107 barrer are considered in the final phase of simulations where their performance at working conditions of 10 bar and 298 K is analysed. The value of 2.5 × 107 barrer is chosen to reduce the total number of structures and does not reflect a particular target permeability. For GCMC simulations, the number of initialization and production cycles is set to 10000 each (20000 total). Gas phase fugacities are calculated using the Peng–Robinson equation of state.51 Due to the high cost of the EMD simulations, the LAMMPS software package is used to enable parallelisation of the calculations.52 The initial states of each EMD simulation are determined from the loadings predicted in the GCMC simulations. In contrast to calculations at infinite dilution, both host–adsorbate and adsorbate–adsorbate interactions are included. A time step of 1 fs is employed with simulations running for a total of 1 ns (50% equilibration). Atomic coordinates for each adsorbate are recorded every 5 fs and are used to calculate the MSD and self-diffusivity via Einstein’s relation. A minimum of 5 trajectories are used to compute the mixture self-diffusivity of each component. The gas permeability in the binary mixture is re-evaluated using
greater than 2.5 × 107 barrer are considered in the final phase of simulations where their performance at working conditions of 10 bar and 298 K is analysed. The value of 2.5 × 107 barrer is chosen to reduce the total number of structures and does not reflect a particular target permeability. For GCMC simulations, the number of initialization and production cycles is set to 10000 each (20000 total). Gas phase fugacities are calculated using the Peng–Robinson equation of state.51 Due to the high cost of the EMD simulations, the LAMMPS software package is used to enable parallelisation of the calculations.52 The initial states of each EMD simulation are determined from the loadings predicted in the GCMC simulations. In contrast to calculations at infinite dilution, both host–adsorbate and adsorbate–adsorbate interactions are included. A time step of 1 fs is employed with simulations running for a total of 1 ns (50% equilibration). Atomic coordinates for each adsorbate are recorded every 5 fs and are used to calculate the MSD and self-diffusivity via Einstein’s relation. A minimum of 5 trajectories are used to compute the mixture self-diffusivity of each component. The gas permeability in the binary mixture is re-evaluated using 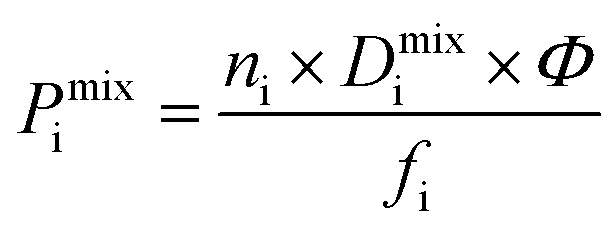 , where Pmixi, ni, Dmixi and fi are the gas permeability, uptake, self-diffusion coefficient and fugacity of component i, respectively, and Φ is the fractional pore volume of the membrane.53 The membrane selectivity (Smixmem,i/j) can then be calculated from the ratio of the gas permeabilities for components i and j of the binary mixture as Pmixi/Pmixj.
, where Pmixi, ni, Dmixi and fi are the gas permeability, uptake, self-diffusion coefficient and fugacity of component i, respectively, and Φ is the fractional pore volume of the membrane.53 The membrane selectivity (Smixmem,i/j) can then be calculated from the ratio of the gas permeabilities for components i and j of the binary mixture as Pmixi/Pmixj.
2.1 Force-field description and validation
The force-fields used to describe intermolecular interactions are formed from a Lennard-Jones (LJ) potential with a cut-off of 12.8 Å. Long-range coulombic interactions are handled using the Ewald summation technique.54 Cross-terms are calculated using the Lorentz–Berthelot mixing rules.55,56 Frameworks are assumed to be rigid with atoms fixed in their crystallographic positions and modelled using the Universal Force-Field (UFF).57 Atomic partial charges are assigned to framework atoms using the EQeq scheme available within the RASPA software package.45 A list of oxidation states used in EQeq calculations can be found in the ESI.† CO2 and CH4 molecules are represented by rigid 3-site and single united-atom models, respectively, using LJ parameters and partial charges from the TraPPE force-field.58,59 H2O is represented by a rigid 4-site model using the TIP4P-Ew model60 and H2S is represented by a rigid 4-site model using parameters derived by Kristóf and co-workers.61 A full list of force-field parameters may be found in Tables S1 and S2.† For MOFs, so called “off-the-shelf” force-fields such as UFF are commonly employed to study adsorption and diffusion properties. These are typically combined with force-fields designed specifically for adsorbates to reproduce thermophysical properties, such as the TraPPE and TIP force-fields. This computational setup has been used extensively over the past 15 years and has been shown to give good agreement across a wide range of adsorbates and MOFs.62–64 Furthermore, they have been previously validated for reproducing experimental membrane properties in a variety of CO2 and CH4 based separations.65,66 In addition to these previous studies, we have also performed similar calculations which show good agreement between experimental and simulated values for permeability of CO2 and CH4 (Fig. 1 – top).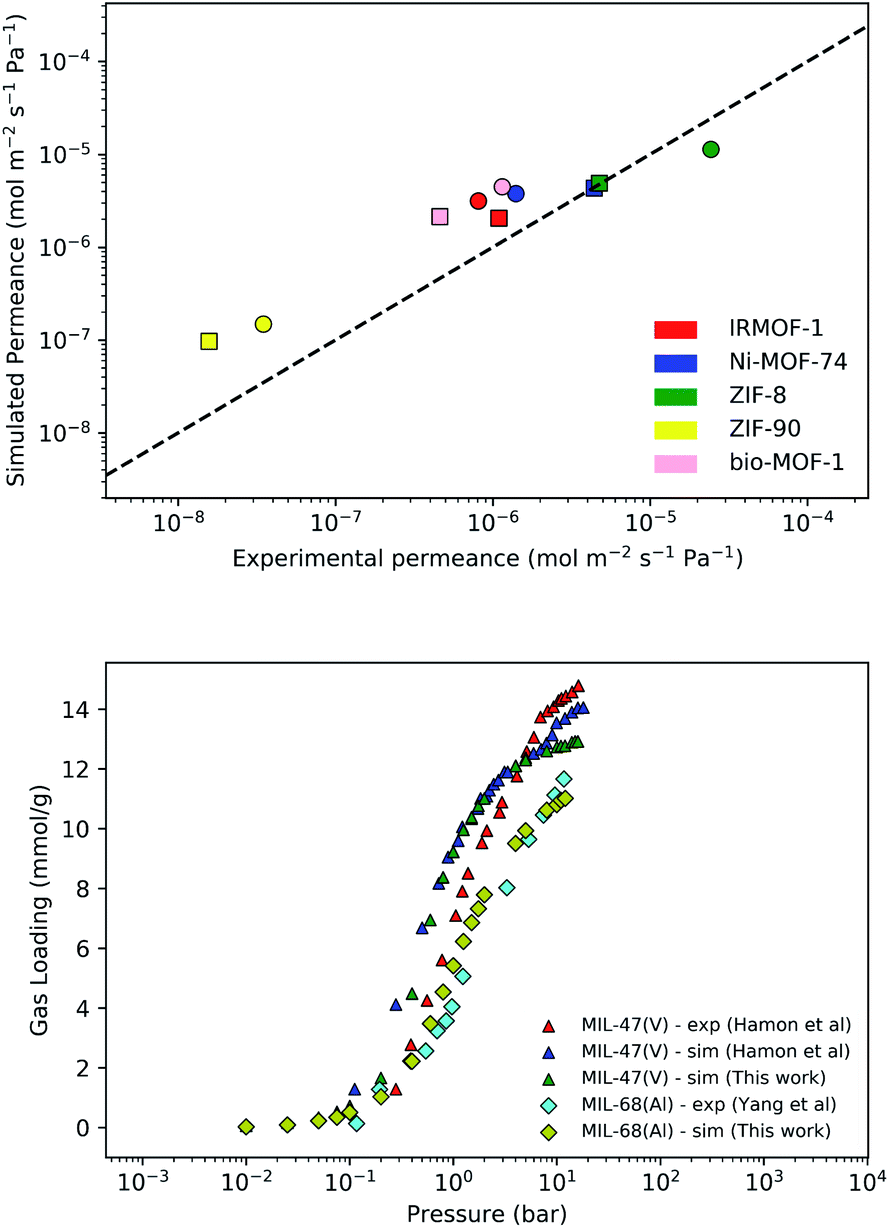 | ||
| Fig. 1 (Top) Comparison of simulated and experimental values of permeance for CO2 (circle symbols) and CH4 (square symbols) in selected MOF membranes. Experimental conditions may be found in Table S3 of the ESI;† (Bottom) Comparison of simulated and experimental uptakes of H2S in MIL-47 and MIL-68 MOFs at 303 K. Experimental data taken from Hamon et al.67 and Yang et al.68 | ||
Studies of H2S adsorption and separation are less documented than for other small molecules such as CO2 and CH4.69 In many MOFs, the formation of strong metal–sulphur bonds can lead to poor regeneration of the empty host or even displacement of linkers causing loss of permanent porosity. Despite these issues, a small number of MOFs from the MIL family display H2S adsorption isotherms over large pressure ranges, which are suitable for validation of the H2S model. Our results on MIL-47(V) indicate good agreement with experimental results (Fig. 1 – bottom) and similar agreement to simulations using a 3-site H2S model employed by Hamon and co-workers.67 Good agreement between theory and experiment is also achieved for MIL-68(Al).68 An evaluation test for H2S permeability over a greater range of MOF membranes is not feasible with the current availability of experimental data.
A perfect, rigid, solvent- and defect-free MOF crystal structure is assumed in all calculations, thus providing an upper limit of permeation properties achievable in these materials. Whilst this approximation has been used ubiquitously in the literature, some membranes may be sensitive to guest activation leading to structural changes. Additional chemical and mechanical stability tests of the candidates identified in a high-throughput screening may be also required.
One limitation of the force-fields used in this work is that strong interactions between adsorbates and open metal sites are not captured, leading to under-prediction of the separation performance in these materials. Studies that have attempted to parametrise force-fields using quantum chemical calculations have successfully shown that this method can correctly capture host–guest interactions around open metal sites.70,71 While this approach has been used at the high-throughput scale previously, it is a very costly process which can only target a small subset of MOFs possessing similar structural motifs (e.g. metal paddlewheels).72,73 Finally, a number of materials, e.g. some MOFs containing open-metal sites, are omitted from this screening due to poor separation performance which may arise from limitations in the employed models. As our aim is to target a range of MOFs with a diverse set of structural properties, general force-fields such as UFF will give the best compromise of accuracy and cost.
3 Results and discussion
3.1 Accuracy in evaluation of partial charges on metal sites
In the early stages of the screening, the distribution of partial charges were evaluated for 7909 MOFs using the EQeq method. Unlike the original Qeq method reported by Rappe et al.,74 this method uses a neutral oxidation state Taylor expansion for ligand atoms, and a higher oxidation state Taylor expansion for metals. In some systems, the choice of Taylor expansion can be the difference between convergence to a physical set of partial charges or an unphysical set. In previous work, Ongari and co-workers compared the partial charges obtained from different charge equilibration methods against high quality density functional theory-derived DDEC charges for a large set of MOFs.75 They showed that smaller deviations between the Qeq methods and DDEC charges were obtained when using a higher oxidation state for the metal centres present in the MOFs. Furthermore, using the data from DDEC charge distributions, they showed that by imposing an upper and lower atomic charge limit of +3 and −2, respectively, fewer structures containing unphysical charges were obtained from using the higher oxidation state Taylor expansion. In this work, MOFs with physical partial charges are obtained by applying two sets of criteria. Firstly, MOFs containing atoms with a partial charge greater than +3.5 or less than −2 were removed. This criterion is closely based on the condition proposed by Ongari and co-workers, however, we find that a slight buffer added to the upper limit is useful for structures with highly charged metal centres containing Zr(IV). We find that this criterion alone is sufficient for identifying metals with unphysical charges but less so for ligand atoms such as carbon or hydrogen (Fig. S1 and S2 of the ESI†). Therefore, we apply a stricter charge criteria on C and H atoms to only allow MOFs with C and H charges between +1 and −1. A total of 800 MOFs are removed from the screening leaving 7109 MOFs to be taken forward into the first phase of high-throughput screening.To demonstrate how the choice of Taylor expansion may influence the results, Fig. 2 compares Henry coefficients for CO2 and H2S (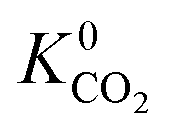 and
and 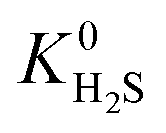 ) in MOFs for which a converged EQeq partial charge evaluation was achieved using both neutral and higher oxidation states of the metal atoms. For both CO2 and H2S, using the higher oxidation state Taylor expansion (x-axis) provides a narrower and more chemically meaningful spread of values for Henry coefficients. This is because the neutral oxidation state expansion has a higher propensity to provide structures with unphysical charges.75 When using the neutral oxidation state Taylor expansion, we analyse the charge distributions in 76 MOFs which possess a
) in MOFs for which a converged EQeq partial charge evaluation was achieved using both neutral and higher oxidation states of the metal atoms. For both CO2 and H2S, using the higher oxidation state Taylor expansion (x-axis) provides a narrower and more chemically meaningful spread of values for Henry coefficients. This is because the neutral oxidation state expansion has a higher propensity to provide structures with unphysical charges.75 When using the neutral oxidation state Taylor expansion, we analyse the charge distributions in 76 MOFs which possess a 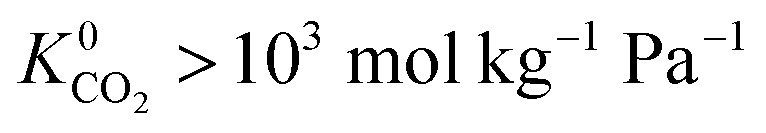 , (i.e. any value greater than the largest
, (i.e. any value greater than the largest 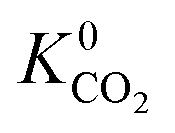 predicted from using the higher oxidation state expansion). We find that 50% of these MOFs contain Li, Na or K sites with charges ranging from +1.34 to +6.75, and 30% contain Nd sites with charges ranging from +2.89 to +46.4. Consequently, the unphysical Coulomb interactions between CO2 and these metals centres lead to large overestimation of
predicted from using the higher oxidation state expansion). We find that 50% of these MOFs contain Li, Na or K sites with charges ranging from +1.34 to +6.75, and 30% contain Nd sites with charges ranging from +2.89 to +46.4. Consequently, the unphysical Coulomb interactions between CO2 and these metals centres lead to large overestimation of 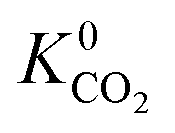 . Using the higher oxidation state expansion, the Li, Na and K atoms in these MOFs have charges ranging from +0.55 to +0.71. Additionally, the Nd charges ranging between +1.45 and +2.46.
. Using the higher oxidation state expansion, the Li, Na and K atoms in these MOFs have charges ranging from +0.55 to +0.71. Additionally, the Nd charges ranging between +1.45 and +2.46.
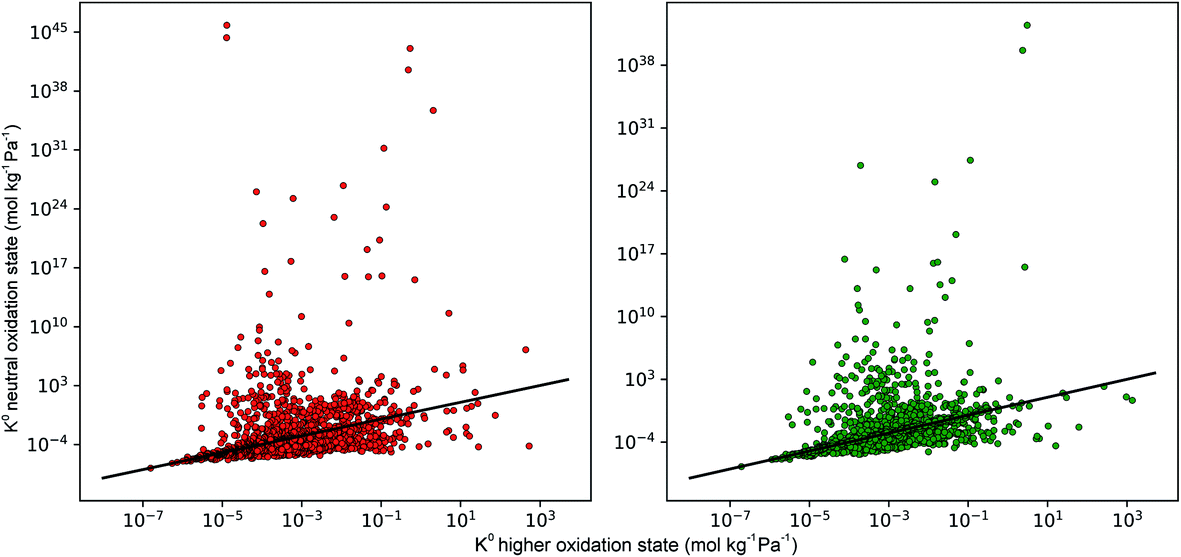 | ||
| Fig. 2 Comparison of the Henry coefficients for CO2 (red) and H2S (green) in MOFs, predicted by using different sets of partial charges on metal sites. The y-axis corresponds to partial charges calculated using a neutral oxidation state on metal atoms, while the x-axis corresponds to partial charges calculated using a higher oxidation state on metal atoms. A full list of oxidation states may be found in the ESI.† | ||
A total of 4418 MOFs (68%) have a larger 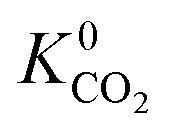 when using the higher oxidation state expansion instead of the neutral oxidation state expansion (Fig. 2 left). However, in 81% of these MOFs, this increase in
when using the higher oxidation state expansion instead of the neutral oxidation state expansion (Fig. 2 left). However, in 81% of these MOFs, this increase in 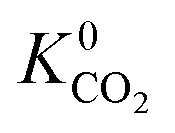 is relatively small (1 to 10 times larger). Although this change is small, it is worth noting that the adsorption selectivity obtained from the ratio of the CO2 and CH4 Henry constants can be directly influenced by switching from one Taylor expansion to another. Similar trends are also observed for H2S with 4013 MOFs (62%) possessing a larger
is relatively small (1 to 10 times larger). Although this change is small, it is worth noting that the adsorption selectivity obtained from the ratio of the CO2 and CH4 Henry constants can be directly influenced by switching from one Taylor expansion to another. Similar trends are also observed for H2S with 4013 MOFs (62%) possessing a larger 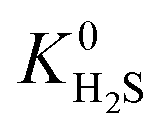 when using the higher oxidation state expansion. In 88% of these MOFs,
when using the higher oxidation state expansion. In 88% of these MOFs, 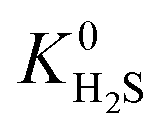 is 1 to 10 times larger compared to that calculated using the neutral oxidation state expansion. In general, these results demonstrate the importance of using a higher oxidation state Taylor expansion for metal atoms for generating a more accurate and physical set of partial charges.
is 1 to 10 times larger compared to that calculated using the neutral oxidation state expansion. In general, these results demonstrate the importance of using a higher oxidation state Taylor expansion for metal atoms for generating a more accurate and physical set of partial charges.
3.2 Permeability at infinite dilution
Within the solution-diffusion model, the process of gas permeation across a membrane can be broken down into three steps: adsorption onto the membrane surface, diffusion through the membrane pore network, and desorption from the membrane surface. Therefore, it is expected that differences in host–guest and guest–guest interactions for different biogas components will strongly influence the rate at which a component permeates through the membrane. For an initial insight, calculations at very low concentrations (in the Henry region) are useful for comparing differences in permeation as these are solely influenced by the strength of host–guest interactions. Fig. 3 compares the diffusion coefficients and permeability of 6768 MOF membranes for CH4, CO2 and H2S at infinite dilution conditions. We note to the reader that from the initial 7109 structures, 341 membranes are excluded as they possess diffusion coefficients smaller than 10−8 cm2 s−1.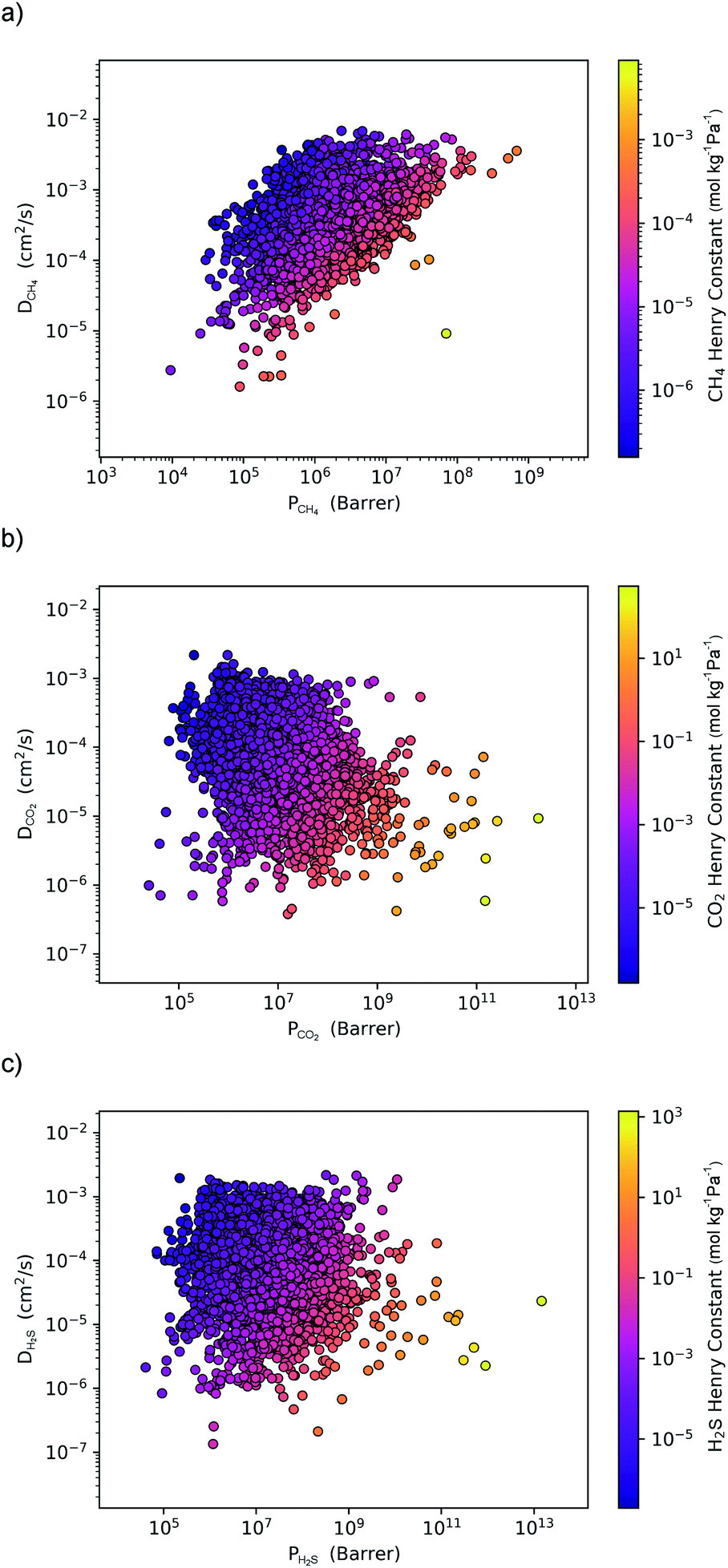 | ||
| Fig. 3 A comparison of the diffusion coefficient versus the permeability under infinite dilution conditions for (a) CH4, (b) CO2 and (c) H2S. Each point represents a different MOF membrane and the colour corresponds to the magnitude of the Henry constant. | ||
For CH4 gas, there appears to be a positive correlation in which structures possessing large 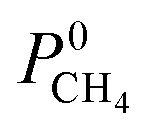 are likely to possess large
are likely to possess large 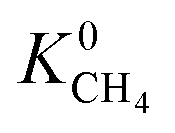 and
and 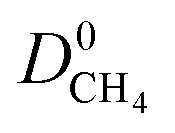 (Fig. 3a). For 5264 membranes,
(Fig. 3a). For 5264 membranes, 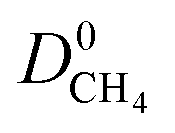 lies between 10−3 and 10−4 cm2 s−1, resulting in fast rates of diffusion which we attribute to weak CH4–MOF interactions. Although
lies between 10−3 and 10−4 cm2 s−1, resulting in fast rates of diffusion which we attribute to weak CH4–MOF interactions. Although 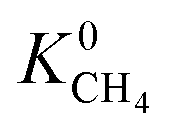 values range between 8.91 × 10−3 mol kg−1 Pa−1 and 1.58 × 10−7 mol kg−1 Pa−1, 81% of MOFs possess
values range between 8.91 × 10−3 mol kg−1 Pa−1 and 1.58 × 10−7 mol kg−1 Pa−1, 81% of MOFs possess 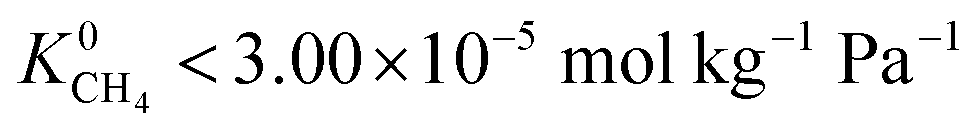 also indicating weak CH4–MOF interactions. Unlike CH4 gas, both CO2 and H2S interact with MOF membranes through a combination of van der Waals and electrostatic interactions. Therefore, on average, we expect CO2–MOF and H2S–MOF interactions to be stronger than CH4–MOF interactions. We see evidence of this in the range of
also indicating weak CH4–MOF interactions. Unlike CH4 gas, both CO2 and H2S interact with MOF membranes through a combination of van der Waals and electrostatic interactions. Therefore, on average, we expect CO2–MOF and H2S–MOF interactions to be stronger than CH4–MOF interactions. We see evidence of this in the range of 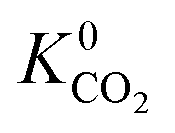 and
and 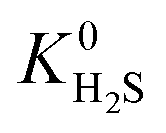 values which extend further than
values which extend further than 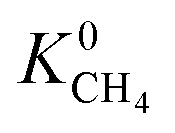 to values as large as 103 mol kg−1 Pa−1 (Fig. 3b and c). Furthermore, 76% of MOFs possess a
to values as large as 103 mol kg−1 Pa−1 (Fig. 3b and c). Furthermore, 76% of MOFs possess a 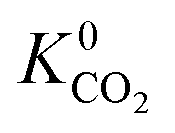 between 10−3 mol kg−1 Pa−1 and 10−5 mol kg−1 Pa−1 (70% for
between 10−3 mol kg−1 Pa−1 and 10−5 mol kg−1 Pa−1 (70% for 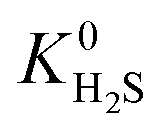 ). Therefore, on average these gases diffuse slower than CH4 through each MOF membrane. While 21% of MOFs possess
). Therefore, on average these gases diffuse slower than CH4 through each MOF membrane. While 21% of MOFs possess 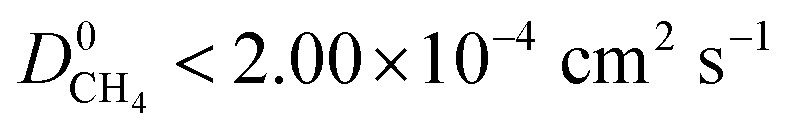 , this increases to 63% and 66% when considering
, this increases to 63% and 66% when considering 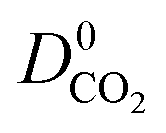 and
and 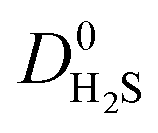 . The combination of these factors lead to a more negative looking correlation as presented in Fig. 3b and c.
. The combination of these factors lead to a more negative looking correlation as presented in Fig. 3b and c.
For the 6768 MOFs included in our study, the values for the heat of adsorption, 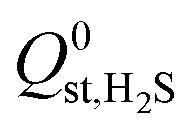 and
and 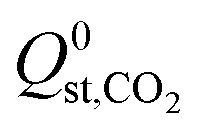 , are smaller than −25 kJ mol−1 in 67% of cases. While these membranes are expected to strongly adsorb the main contaminants in biogas, the presence of water vapour in the biogas mixture may affect the stability of these membranes. In addition, water may act as a competitive adsorbate, resulting in different separation properties. For these MOFs, 70% have
, are smaller than −25 kJ mol−1 in 67% of cases. While these membranes are expected to strongly adsorb the main contaminants in biogas, the presence of water vapour in the biogas mixture may affect the stability of these membranes. In addition, water may act as a competitive adsorbate, resulting in different separation properties. For these MOFs, 70% have 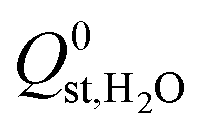 greater than
greater than 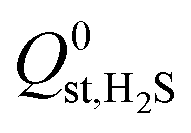 or
or 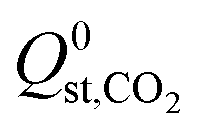 . Therefore, a hydrophobicity criterion was employed to identify promising membranes which are expected to exhibit improved hydrolytic stability. This criterion reduced the data set to 1685 MOF membranes with a significantly lower average value for
. Therefore, a hydrophobicity criterion was employed to identify promising membranes which are expected to exhibit improved hydrolytic stability. This criterion reduced the data set to 1685 MOF membranes with a significantly lower average value for 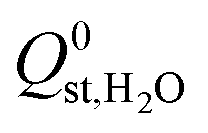 (the average
(the average 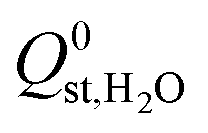 decreased from −47.1 kJ mol−1 to −16.1 kJ mol−1, as shown in Fig. S5 of the ESI†). Furthermore, less than 1% of the hydrophobic MOF data set possess
decreased from −47.1 kJ mol−1 to −16.1 kJ mol−1, as shown in Fig. S5 of the ESI†). Furthermore, less than 1% of the hydrophobic MOF data set possess 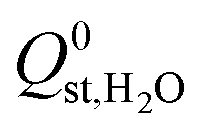 values that are greater than
values that are greater than  ,
, 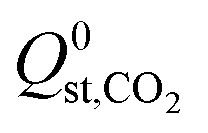 or
or 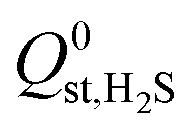 , compared to 59% in the original set of 6768 MOFs.
, compared to 59% in the original set of 6768 MOFs.
The permeability at infinite dilution for CH4, CO2 and H2S was determined for the reduced set of 1685 membranes. To assess the CO2/CH4 and H2S/CH4 separation performance of these membranes, Robeson plots were constructed, as shown in Fig. 4. For CO2/CH4, the 1991 and 2008 Robeson upper bounds11,12 are included to illustrate the performance of these MOFs relative to some of the best available organic polymer membranes. 1499 MOFs are found to be more selective for CO2 gas with 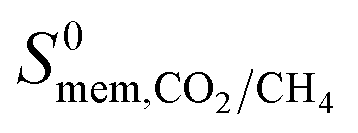 ranging between 1 and 26.9 and
ranging between 1 and 26.9 and 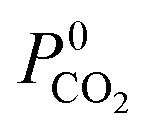 values ranging between 4.10 × 104 barrer and 1.56 × 108 barrer. As seen in this study, MOFs have the potential to overcome the Robeson upper bound limit corresponding to the selectivity–permeability trade-off problem found in organic polymer membranes. Highlighted in red and yellow in Fig. 4 are a total of 1183 MOFs that exceed the 2008 Robeson limit for CO2/CH4 separation. For H2S/CH4 separation, only 14 MOFs were found to be more selective for CH4 than H2S. The remaining 1671 possess
values ranging between 4.10 × 104 barrer and 1.56 × 108 barrer. As seen in this study, MOFs have the potential to overcome the Robeson upper bound limit corresponding to the selectivity–permeability trade-off problem found in organic polymer membranes. Highlighted in red and yellow in Fig. 4 are a total of 1183 MOFs that exceed the 2008 Robeson limit for CO2/CH4 separation. For H2S/CH4 separation, only 14 MOFs were found to be more selective for CH4 than H2S. The remaining 1671 possess 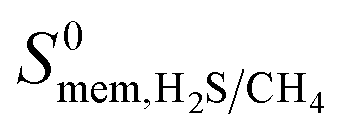 and
and 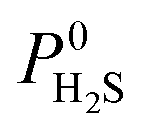 ranging from 1.03 to 82.3 and from 7.23 × 104 to 1.98 × 109 barrer, respectively. Due to the limited availability of H2S permeability data, an upper limit for the H2S/CH4 separation is not established. Therefore, the structures highlighted in red and yellow in Fig. 4b are the same as those shown in Fig. 4a and are highlighted for comparative purposes.
ranging from 1.03 to 82.3 and from 7.23 × 104 to 1.98 × 109 barrer, respectively. Due to the limited availability of H2S permeability data, an upper limit for the H2S/CH4 separation is not established. Therefore, the structures highlighted in red and yellow in Fig. 4b are the same as those shown in Fig. 4a and are highlighted for comparative purposes.
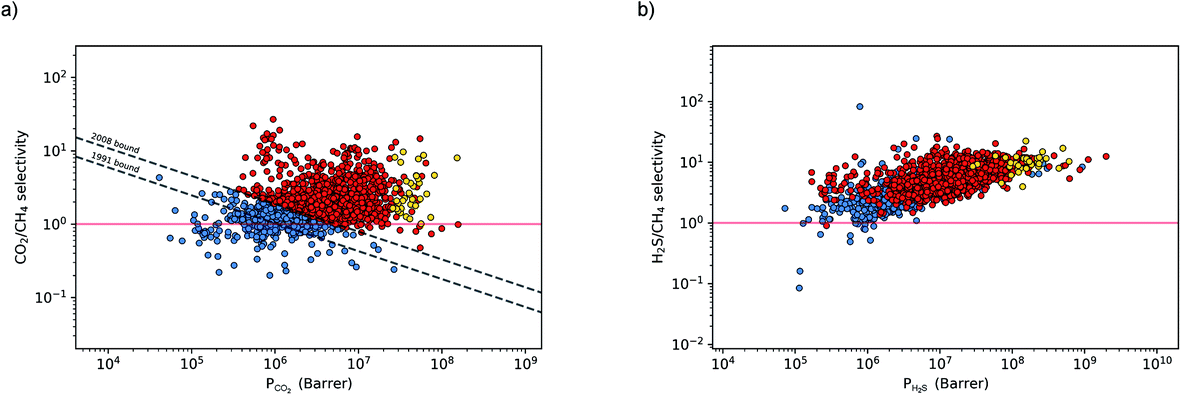 | ||
| Fig. 4 Robeson trade-off plots for CO2/CH4 (a) and H2S/CH4 (b) ideal separations in 1685 MOFs. The CO2/CH4 Robeson 1991 and 2008 limits are used to compare performance of each MOF to some of the best polymer membranes. 1183 MOFs lie above the 2008 limit (red and yellow points) and 35 (yellow points) are used in the study of binary mixture separations at working conditions of 10 bar and 298 K. | ||
Due to the large computational cost associated with modelling molecular dynamics at high pressure, we focus our study on MOF candidates that show the most promise at infinite dilution conditions. Hence, we consider MOF structures above the upper bound that are selective to CO2 and H2S with 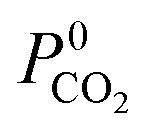 and
and 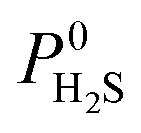 greater than 2.50 × 107 barrer, leaving a total of 42 structures. Seven of these structures (CSD reference codes: CAFSUY, CAFTAF, COMDOY, CUMDIY, LODPOL, OHAKEO and SETQAL) are reported to collapse in response to activation or possess degrees of structural flexibility when exposed to different stimuli.76–81 Therefore, the rigid lattice approximation used in our calculations will not provide an accurate representation of their structure used under working conditions. These MOFs were removed from the screening leaving a total of 35 unique MOFs represented by yellow circles in Fig. 4.
greater than 2.50 × 107 barrer, leaving a total of 42 structures. Seven of these structures (CSD reference codes: CAFSUY, CAFTAF, COMDOY, CUMDIY, LODPOL, OHAKEO and SETQAL) are reported to collapse in response to activation or possess degrees of structural flexibility when exposed to different stimuli.76–81 Therefore, the rigid lattice approximation used in our calculations will not provide an accurate representation of their structure used under working conditions. These MOFs were removed from the screening leaving a total of 35 unique MOFs represented by yellow circles in Fig. 4.
3.3 Separations at working conditions
For the promising membrane candidates, we employ GCMC and EMD simulations to study equimolar binary mixture separations of CO2/CH4 and H2S/CH4 at 10 bar and 298 K. Unlike calculations at infinite dilution, these simulations include the role of adsorbate–adsorbate interactions in biogas separation at operating conditions. One MOF structure (REFCODE: HONCIY) was removed from the screening as an accurate estimation of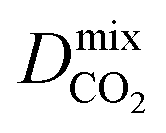 and
and 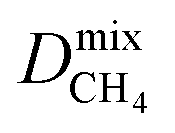 was not possible within the simulation timescale.
was not possible within the simulation timescale.
A large decrease in gas permeability 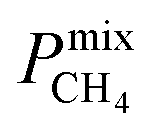 ,
, 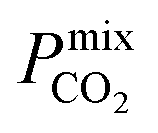 and
and 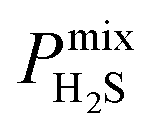 is observed at working conditions, with values ranging between 101 and 105 barrer (Fig. 5). For 22 of the 34 MOFs, the values of both
is observed at working conditions, with values ranging between 101 and 105 barrer (Fig. 5). For 22 of the 34 MOFs, the values of both 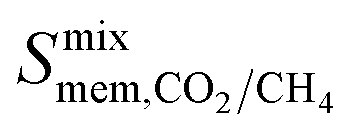 and
and 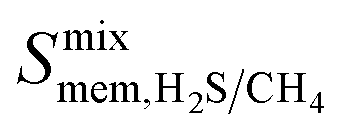 are greater than their ideal selectivity at infinite dilution. With the inclusion of guest–guest interactions and increased gas loading under working conditions, the rate of diffusion for each component in the gas mixture is reduced leading to slower permeation rates. Furthermore, each component now competes against one another for adsorption sites which is not captured in the ideal calculations at infinite dilution. Under working conditions, the separation performance of 20 MOFs deteriorates such that they now lie below the 1991 and 2008 Robeson bounds (Fig. 5 – top). Of the remaining 14 MOFs, 6 lie between the 1991 and 2008 bounds and 8 lie above both bounds. These materials possess
are greater than their ideal selectivity at infinite dilution. With the inclusion of guest–guest interactions and increased gas loading under working conditions, the rate of diffusion for each component in the gas mixture is reduced leading to slower permeation rates. Furthermore, each component now competes against one another for adsorption sites which is not captured in the ideal calculations at infinite dilution. Under working conditions, the separation performance of 20 MOFs deteriorates such that they now lie below the 1991 and 2008 Robeson bounds (Fig. 5 – top). Of the remaining 14 MOFs, 6 lie between the 1991 and 2008 bounds and 8 lie above both bounds. These materials possess 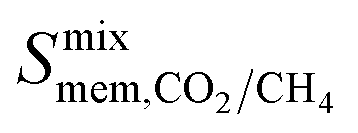 between 3.09 and 12.5,
between 3.09 and 12.5, 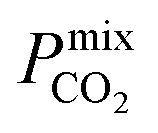 between 2.87 × 104 barrer and 1.24 × 105 barrer,
between 2.87 × 104 barrer and 1.24 × 105 barrer, 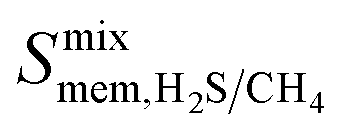 between 25.0 and 123 and
between 25.0 and 123 and 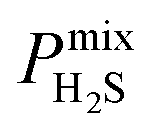 between 2.72 × 103 barrer and 1.03 × 105 barrer.
between 2.72 × 103 barrer and 1.03 × 105 barrer.
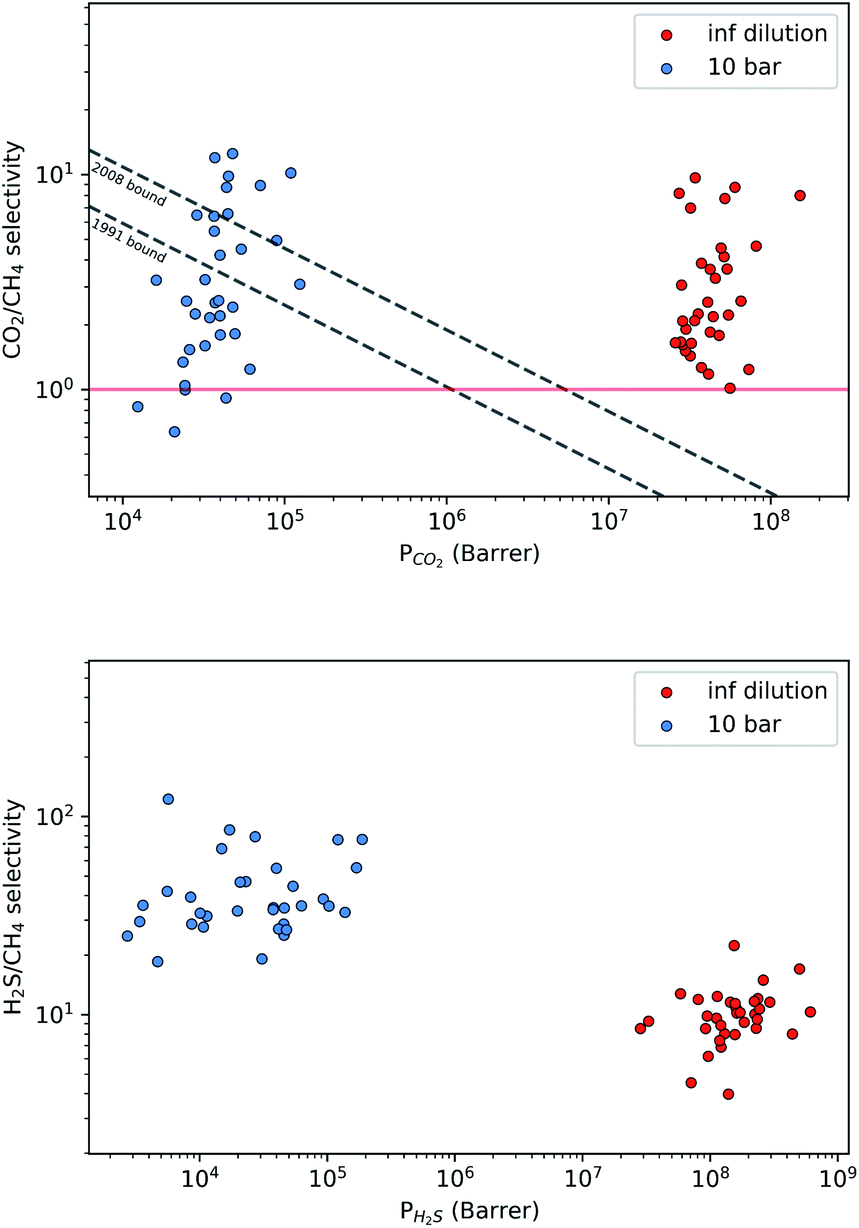 | ||
| Fig. 5 Robeson trade-off plots comparing the CO2/CH4 (top) and H2S/CH4 (bottom) separation performance of 34 MOFs under infinite dilution (red) and at working conditions of 10 bar and 298 K (blue). | ||
3.4 Top candidates
In order to rank the performance of each MOF based on the values of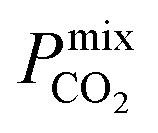 ,
, 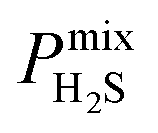 ,
, 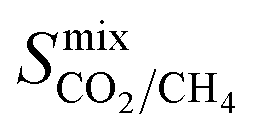 , and
, and 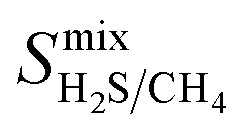 , a scoring system has to be generated by weighting each of these parameters. This is because much larger concentrations of CO2 need to be removed from a biogas stream than H2S, which may affect the rankings of one MOF to another. However, the list of top structures is strongly correlated with the type and size of the applied weighting constraints, which in some cases identified top structures that fall below the Robeson upper bounds. Furthermore, it is possible that a weighting system of such design may introduce bias into the selection of the top candidates. One possible solution to this problem is to convert each property into a descriptor that is passed through a machine-learning algorithm. This may generate rankings in a more unbiased manner based on the descriptors and problem alone, however this lies outside the scope of this publication. For the remainder of this discussion, the top 8 MOFs are referred to as the top candidates identified by our high-throughput screening and correspond to those which exceed the 2008 CO2/CH4 Robeson limit (Fig. 6). While this does not give a quantitative rank for each material, it does highlight a set of MOF membranes that tend to perform better than other candidates in terms of permeability and selectivity. Void analysis of the top candidates indicates that they all possess very similar structural characteristics such as one-dimensional pore channels (see ESI†). The geometric and physical properties summarised in Table 1 indicate that in these materials, the largest cavity diameter (LCD) is typically smaller than 6 Å, accessible surface area (ASA) ranges from 164 to 1018 m2 g−1, and void fraction (VF) is typically less than 0.5. Of the top 8 candidates, the MOF with CSD reference code QOKCID has the smallest LCD resulting in the lowest gravimetric and volumetric storage capacities for CO2 and H2S. In contrast, the RUVBER MOF exhibits the largest LCD with capacity for more CO2 and H2S storage than most top candidates. Pores with large diameters typically possess a greater proportion of sites with weak host–guest interactions, affecting their selectivity for contaminants. Despite this, RUVBER is still able to selectively adsorb both CO2 and H2S over CH4 with large Smixads values of 8.94 and 26.4 for CO2/CH4 and H2S/CH4 separations, respectively.
, a scoring system has to be generated by weighting each of these parameters. This is because much larger concentrations of CO2 need to be removed from a biogas stream than H2S, which may affect the rankings of one MOF to another. However, the list of top structures is strongly correlated with the type and size of the applied weighting constraints, which in some cases identified top structures that fall below the Robeson upper bounds. Furthermore, it is possible that a weighting system of such design may introduce bias into the selection of the top candidates. One possible solution to this problem is to convert each property into a descriptor that is passed through a machine-learning algorithm. This may generate rankings in a more unbiased manner based on the descriptors and problem alone, however this lies outside the scope of this publication. For the remainder of this discussion, the top 8 MOFs are referred to as the top candidates identified by our high-throughput screening and correspond to those which exceed the 2008 CO2/CH4 Robeson limit (Fig. 6). While this does not give a quantitative rank for each material, it does highlight a set of MOF membranes that tend to perform better than other candidates in terms of permeability and selectivity. Void analysis of the top candidates indicates that they all possess very similar structural characteristics such as one-dimensional pore channels (see ESI†). The geometric and physical properties summarised in Table 1 indicate that in these materials, the largest cavity diameter (LCD) is typically smaller than 6 Å, accessible surface area (ASA) ranges from 164 to 1018 m2 g−1, and void fraction (VF) is typically less than 0.5. Of the top 8 candidates, the MOF with CSD reference code QOKCID has the smallest LCD resulting in the lowest gravimetric and volumetric storage capacities for CO2 and H2S. In contrast, the RUVBER MOF exhibits the largest LCD with capacity for more CO2 and H2S storage than most top candidates. Pores with large diameters typically possess a greater proportion of sites with weak host–guest interactions, affecting their selectivity for contaminants. Despite this, RUVBER is still able to selectively adsorb both CO2 and H2S over CH4 with large Smixads values of 8.94 and 26.4 for CO2/CH4 and H2S/CH4 separations, respectively.
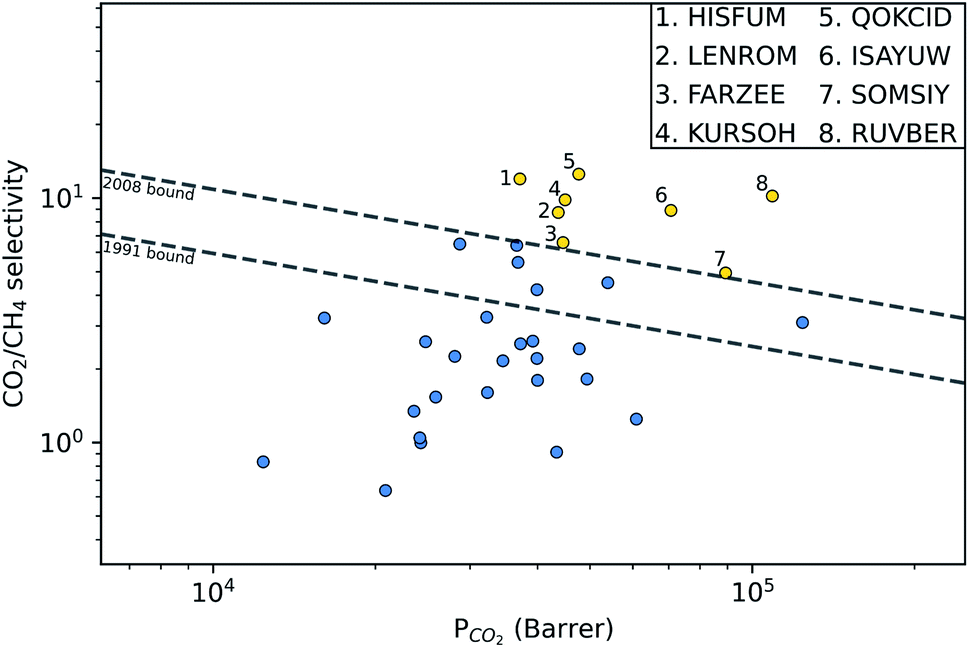 | ||
| Fig. 6 Robeson trade-off plots at 298 K comparing the CO2/CH4 separation performance of 34 MOFs. The top 8 candidates identified in the proposed screening are highlighted in yellow and are labelled with their corresponding CSD reference code. | ||
| REFCODE | PLD (Å) | LCD (Å) | ASA (m2 g−1) | VF | n CO2 (mol kg−1) | n H2S (mol kg−1) |
|
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| × 10−6 (cm2 s−1) | ||||||||||||||
a PLD = pore-limiting diameter; LCD = largest cavity diameter; ASA = accessible surface area; VF = void fraction; ni = loading of adsorbate i obtained from 50:50 binary mixture GCMC; Dmixi = diffusion coefficient of adsorbate i; 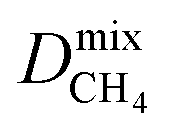 I is derived from CO2/CH4 simulations, and I is derived from CO2/CH4 simulations, and 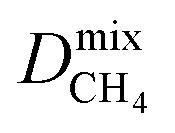 II is derived from H2S/CH4 simulations; Pmixi = permeability of adsorbate i; Smixi/j = membrane selectivity for adsorbates i and j in the binary mixture. II is derived from H2S/CH4 simulations; Pmixi = permeability of adsorbate i; Smixi/j = membrane selectivity for adsorbates i and j in the binary mixture.
|
||||||||||||||
| FARZEE | 4.11 | 4.36 | 418 | 0.32 | 2.84 | 4.04 | 18.1 | 34.3 | 10.5 | 17.6 | 44582 |
6.56 | 37661 |
33.8 |
| HISFUM | 3.81 | 4.53 | 225 | 0.22 | 2.39 | 2.56 | 16.6 | 27.5 | 8.52 | 6.83 | 37063 |
11.9 | 20790 |
46.6 |
| ISAYUW | 4.32 | 4.59 | 376 | 0.31 | 2.80 | 3.46 | 28.4 | 32.5 | 19.9 | 22.7 | 70681 |
8.89 | 62670 |
35.3 |
| KURSOH | 4.60 | 5.22 | 674 | 0.43 | 4.55 | 3.61 | 13.2 | 11.6 | 3.67 | 4.31 | 45013 |
9.83 | 10136 |
32.7 |
| LENROM | 4.28 | 4.83 | 411 | 0.31 | 3.32 | 2.44 | 14.8 | 13.5 | 2.56 | 0.787 | 43673 |
8.72 | 5694 | 123 |
| QOKCID | 3.84 | 4.28 | 164 | 0.21 | 1.55 | 1.55 | 28.5 | 43.8 | 23.3 | 23.6 | 47688 |
12.5 | 39939 |
54.9 |
| RUVBER | 5.68 | 6.05 | 1018 | 0.51 | 5.58 | 6.05 | 27.1 | 24.7 | 10.8 | 11.3 | 108500 |
10.1 | 48046 |
26.8 |
| SOMSIY | 5.71 | 6.00 | 984 | 0.44 | 5.60 | 5.15 | 19.5 | 33.8 | 23.9 | 27.0 | 89295 |
4.93 | 103161 |
35.4 |
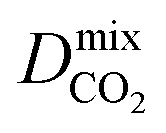
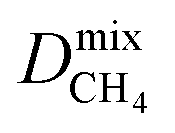
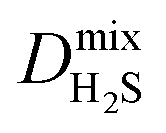
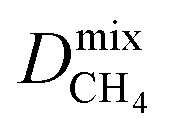
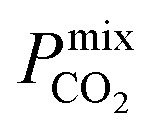
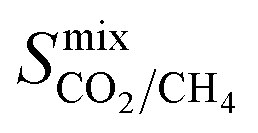
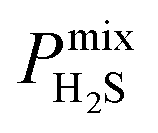
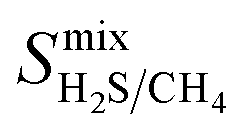
Diffusion coefficients 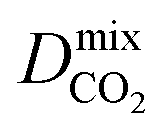 ,
, 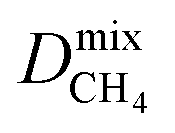 and
and 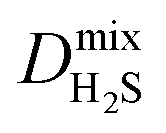 of the top candidates range from 4.38 × 10−5 cm2 s−1 to 7.87 × 10−7 cm2 s−1. The average diffusion selectivity Smixdiff for each binary mixture is fairly close to unity, ranging between 0.53 and 1.14 for CO2/CH4 mixtures and between 0.60 and 3.25 for H2S/CH4 mixtures. In the CO2/CH4 mixtures, CO2 molecules interact more strongly with the framework than CH4 resulting in a reduced degree of freedom that is generally reflected in their smaller self-diffusion coefficients. In the H2S/CH4 mixtures, the large gas loadings and high
of the top candidates range from 4.38 × 10−5 cm2 s−1 to 7.87 × 10−7 cm2 s−1. The average diffusion selectivity Smixdiff for each binary mixture is fairly close to unity, ranging between 0.53 and 1.14 for CO2/CH4 mixtures and between 0.60 and 3.25 for H2S/CH4 mixtures. In the CO2/CH4 mixtures, CO2 molecules interact more strongly with the framework than CH4 resulting in a reduced degree of freedom that is generally reflected in their smaller self-diffusion coefficients. In the H2S/CH4 mixtures, the large gas loadings and high 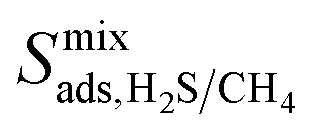 result in each CH4 molecule being surrounded by a large number of contaminant molecules. This strongly influences the diffusion rate of CH4 such that both H2S and CH4 molecules possess similar rates of diffusion through the pore channels. In general,
result in each CH4 molecule being surrounded by a large number of contaminant molecules. This strongly influences the diffusion rate of CH4 such that both H2S and CH4 molecules possess similar rates of diffusion through the pore channels. In general, 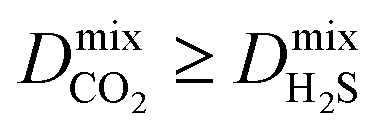 because each MOF possesses a lower
because each MOF possesses a lower 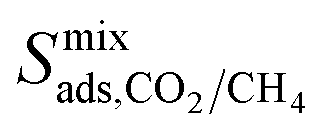 value than
value than 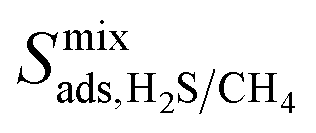 . Consequently, a larger concentration of CH4 is found in the pores during the CO2/CH4 MD simulations which can collide with nearby CO2 molecules to help increase their diffusion rate.
. Consequently, a larger concentration of CH4 is found in the pores during the CO2/CH4 MD simulations which can collide with nearby CO2 molecules to help increase their diffusion rate.
We next compare the top MOF candidates to those of other porous membranes reported in the literature, which include pure zeolite-, MOF- and ZIF-based membranes, in addition to a number of novel MMMs (Table 2). Note that many studies report CO2 separations over a range of conditions which may not be exactly the same as those used in biogas upgrading. However, these studies are still useful for evaluating the separation performance of the top MOF candidates identified in the proposed screening. Some of the largest values of Smixmem were reported by Carreon84 and Shi85 for SAPO-34 membranes, and many of the reported MOF- and ZIF-based membranes possess 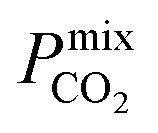 between 104 barrer and 105 barrer and Smixmem ranging between 1 and 25. For many MOFs, Smixmem appears to improve in composite MMMs at the cost of lowering the value of
between 104 barrer and 105 barrer and Smixmem ranging between 1 and 25. For many MOFs, Smixmem appears to improve in composite MMMs at the cost of lowering the value of 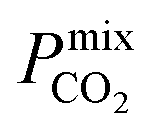 which typically ranges from 10s to 1000s of barrer. However, it is clear from the studies reported in Table 2 that changes to the polymer and MOF loading can significantly alter the permeability, resulting in an additional level of control over the permeation properties. In comparison, it appears that the top MOFs membranes identified in this work generally possess larger Smixmem and
which typically ranges from 10s to 1000s of barrer. However, it is clear from the studies reported in Table 2 that changes to the polymer and MOF loading can significantly alter the permeability, resulting in an additional level of control over the permeation properties. In comparison, it appears that the top MOFs membranes identified in this work generally possess larger Smixmem and 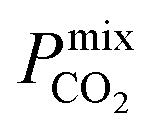 values. Furthermore, as these materials lie above the Robeson limit, their separation performance is competitive with traditional polymer membranes. As previously mentioned, our simulations assume a perfect, defect-free crystal structure, in which the values of
values. Furthermore, as these materials lie above the Robeson limit, their separation performance is competitive with traditional polymer membranes. As previously mentioned, our simulations assume a perfect, defect-free crystal structure, in which the values of 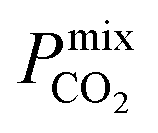 and Smixmem represent an upper performance limit. Despite this, the top MOFs in this work appear to show promise in separating biogas contaminants. Further insight which assesses the top membranes as filler particles in MMMs would be beneficial to establish whether their separation performance can be enhanced further. However, this consideration goes beyond the scope of the current study.
and Smixmem represent an upper performance limit. Despite this, the top MOFs in this work appear to show promise in separating biogas contaminants. Further insight which assesses the top membranes as filler particles in MMMs would be beneficial to establish whether their separation performance can be enhanced further. However, this consideration goes beyond the scope of the current study.
| Material | CO2 permeability (barrer) | CO2/CH4 membrane selectivity | Feed/permeate pressure (bar) | Temp. (K) | Ref. |
|---|---|---|---|---|---|
| H-ZSM-5 | 1.91 × 103 | 5.50 | 2.70/1.38 | 300 | 82 |
| Cu2(bza)4(pyz) | 4.48 × 103 | 25.0 | 1.50/1.00 | 293 | 83 |
| SAPO-34 | 4.03 × 104 | 171 | 2.24/0.84 | 295 | 84 |
| SAPO-34 | 4.84 × 104 | 186 | 2.40/1.00 | 295 | 85 |
| HKUST-1 | 5.00 × 104 | 1.70 | 1.00/1.00 | 298 | 86 |
| ZIF-8 | 3.63 × 105 | 5.10 | 1.38/0.99 | 295 | 87 |
| Bio-MOF-1 | 5.14 × 104 | 2.50 | 1.38/0.99 | 298 | 88 |
| ZIF-90 (modified with APTES ligand) | 7.78 × 102 | 4.70 | 1.00/1.00 | 498 | 89 |
| Ni-MOF-74 | 1.04 × 105 | 0.316 | 1.00/1.00 | 298 | 90 |
| ZIF-69 | 1.22 × 104 | 4.60 | 1.00/1.00 | 298 | 91 |
| 20 wt% ZIF-8@Matrimid®5218 | 16.6 | 35.8 | 4.00/0.05 | 503 | 92 |
| 20 wt% SAPO-34@PU | 28.7 | 25.6 | 12.0 (feed) | 298 | 93 |
| 40 wt% NOTT-300(Al)@Pebax®1657 | 395 | 36.3 | 10 bar (feed) | 298 | 94 |
| 10 wt% ZIF-8@(10:90) PIM-1/6FDA-DAM |
2891 | 26.6 | 3.00/1.00 | 308 | 95 |
| 20 wt% NH2-MIL-53(Ti)@PSF | 29.3 | 29.5 | 3.00 (feed) | 303 | 96 |
| FARZEE | 4.46 × 104 | 6.56 | 10.0/vacuum | 298 | This work |
| HISFUM | 3.71 × 104 | 11.9 | 10.0/vacuum | 298 | This work |
| ISAYUW | 7.07 × 104 | 8.89 | 10.0/vacuum | 298 | This work |
| KURSOH | 4.50 × 104 | 9.83 | 10.0/vacuum | 298 | This work |
| LENROM | 4.37 × 104 | 8.72 | 10.0/vacuum | 298 | This work |
| QOKCID | 4.77 × 104 | 12.5 | 10.0/vacuum | 298 | This work |
| RUVBER | 1.09 × 105 | 10.1 | 10.0/vacuum | 298 | This work |
| SOMSIY | 8.93 × 104 | 4.93 | 10.0/vacuum | 298 | This work |
3.5 Structure–performance relationships
One of the main advantages of working with large sets of structures is the ability to investigate the relationships between their performance and physical properties, which are crucial in the rational design of future materials. Generally, the separation performance for a class of materials cannot be linked to an individual structural, physical or chemical property; it requires understanding of a complex interplay between all of them. Fig. 4 shows that 1183 MOFs can surpass the selectivity–permeability trade-off relationship described by Robeson’s upper bound. By comparing the structural properties of the MOFs considered in this study, we aim to answer three closely linked questions: (1) are there any common properties shared by these 1183 structures? (2) How do these properties account for the observed separation performance? And (3) are there any relationships between the structural properties and the separation performance that will allow us to construct design principles for future MOFs?Histogram distributions shown in Fig. 7 for different structural properties of the 1183 MOFs indicate that many of these contain small pore sizes, with 80% of the structures having a pore-limiting diameter (PLD) less than 6 Å and 81% possessing LCD below 8 Å. The ultra-microporous nature of many of these materials also leads to small ASAs with 68% found to possess an ASA less than 1250 m2 g−1. Of the remaining membranes that possess an ASA greater than 1250 m2 g−1, only 42% possess an LCD greater than 8 Å. Large pores with high surface area can make it more difficult to separate mixtures through differences in binding affinity, making them undesirable for these types of separation. The porosity distribution in Fig. 7a indicates that MOFs with low VF appear more frequently, with 42% having a VF < 0.4 and 61% < 0.5. Analysis of the metals in each structure indicates that 29% of the structures are constructed from zinc-based nodes, forming the most common metal ahead of copper (14%), cadmium (14%) and cobalt (11%). Furthermore, these four metals were exclusively found in 7 of the 10 top MOFs. In a recent study by Altintas and co-workers, it was shown that these four metals also frequently appear in the top membranes predicted for CH4/H2 separations.65 Finally, the density distribution in Fig. 7f shows that 64% of MOFs above the upper bound have densities between 1 g cm−3 and 1.5 g cm−3. In summary, our calculations at infinite dilution suggest that MOFs with a PLD between 3.8 Å and 6 Å, LCD between 4 Å and 8 Å, ASA less than 1250 m2 g−1, VF between 0.3 and 0.5 and density between 1 g cm−3 and 1.5 g cm−3 are likely to appear above the Robeson upper bound and exhibit desirable separation performance.
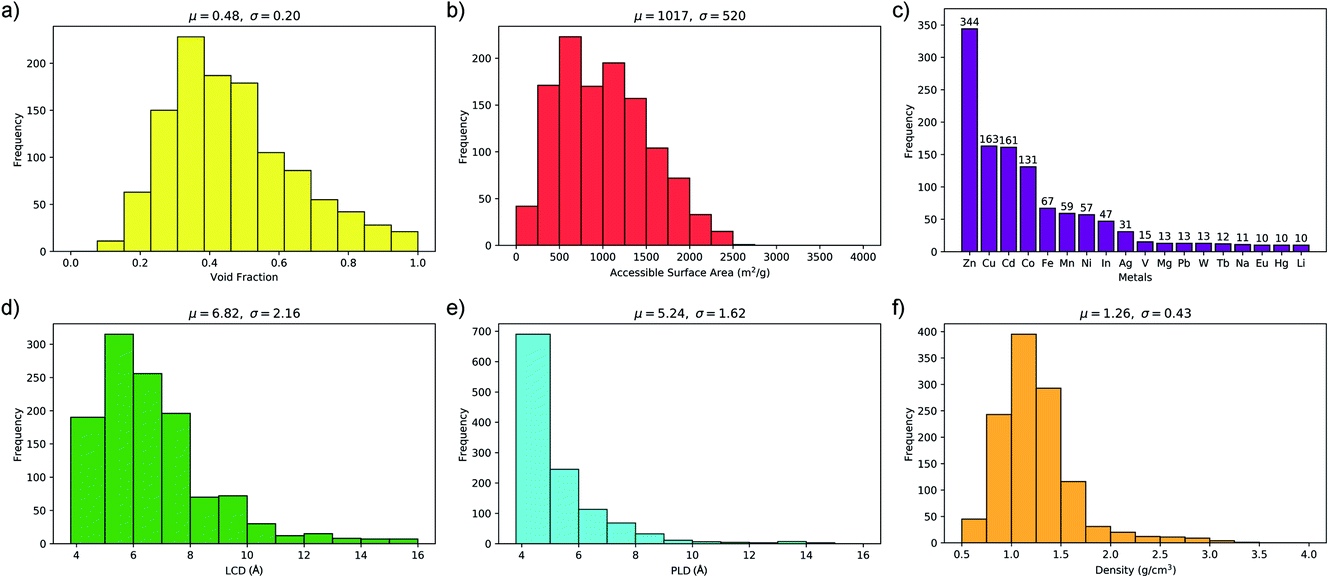 | ||
| Fig. 7 Distributions of (a) void fraction, (b) accessible surface area, (c) metals, (d) largest cavity diameter, (e) pore-limiting diameter, (f) density in the 1183 MOFs above the Robeson upper bound (μ = mean, σ = standard deviation). | ||
It is useful to compare these results with the structural property distributions from low-performing materials that lie below the 2008 Robeson bound. If structures below the upper bound also have similar distributions, then this suggests that these properties are not of significance and do not correlate with high performance. Interestingly, it is found that only 10% of membranes below the upper bound possess similar structural properties which all lie in the same ranges that were summarised at the end of the previous paragraph. Two of the most significant differences appear to be the PLD and LCD. In the low-performing subset of membranes, 42% possess a PLD smaller than 6 Å, and 43% have a LCD smaller than 8 Å. In contrast, the percentage increases to 80% and 81%, respectively, in membranes above the upper bound. In addition, only 29% of membranes below the upper bound possess a VF smaller than 0.5, in contrast to the 61% of structures above the upper bound. Finally, 32% of membranes below the upper bound have a density between 1 and 1.5 g cm−3, in contrast to 64% of membranes above the upper bound. These results suggest that the structure–performance relationships described in this study are significant and may provide target structural properties for designing future membranes with excellent separation performance.
Calculations of the heats of adsorption (Q0st) can provide useful insight into a MOF’s affinity for different gas molecules. These can be directly linked to the strength of host–guest interactions which are related to structural properties such as pore diameter (PLD, LCD) and porosity (VF). The Q0st distributions for the MOFs above and below the 2008 Robeson upper bound are presented in Fig. 8. For CH4, CO2 and H2S, the distributions representing MOFs above the upper bound (red) lie further to the left than those below the upper bound (blue), implying that, in general, there are stronger host–guest interactions present in the MOFs above the upper bound. This is also reflected in the average values of 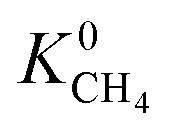 ,
, 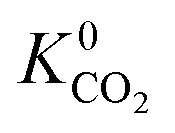 and
and 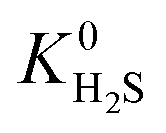 which were a factor of 3.03, 3.90 and 4.73 larger for MOFs above the upper bound, respectively. From these results, we expect to see a reduction in the rate of diffusion for each biogas component. For the MOFs below the upper bound, we found that the average
which were a factor of 3.03, 3.90 and 4.73 larger for MOFs above the upper bound, respectively. From these results, we expect to see a reduction in the rate of diffusion for each biogas component. For the MOFs below the upper bound, we found that the average 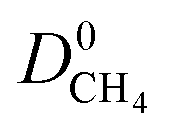 ,
, 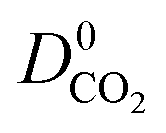 and
and 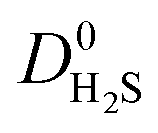 were a factor of 1.87, 1.42 and 1.64 times larger than the MOFs above the upper bound. As permeability is calculated from the product of the solubility and diffusion properties of the material, we can see that the changes to K0i outweigh the changes to D0i and therefore, permeability and selectivity become larger for the MOFs above the upper bound.
were a factor of 1.87, 1.42 and 1.64 times larger than the MOFs above the upper bound. As permeability is calculated from the product of the solubility and diffusion properties of the material, we can see that the changes to K0i outweigh the changes to D0i and therefore, permeability and selectivity become larger for the MOFs above the upper bound.
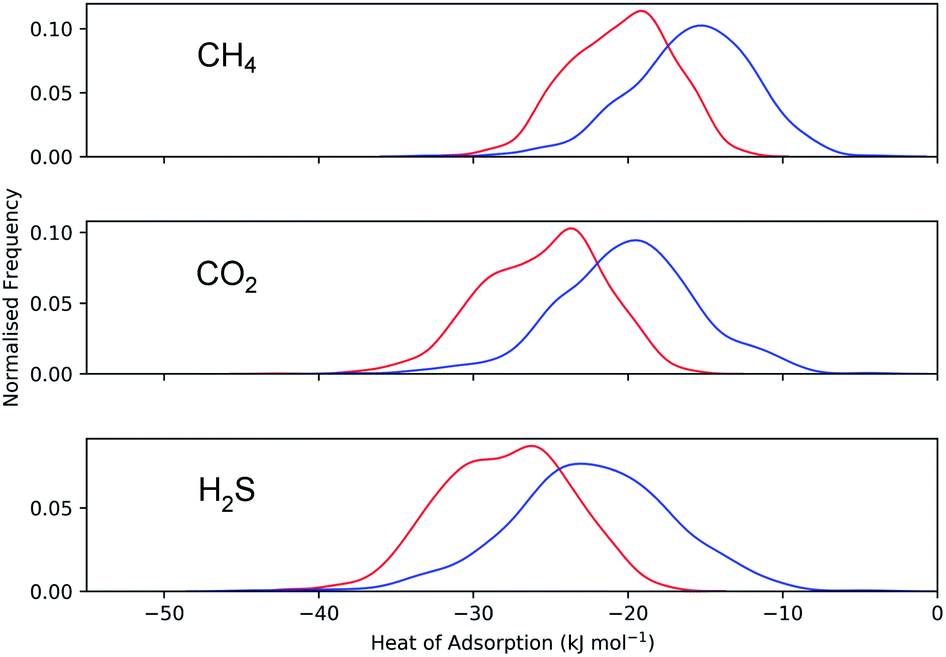 | ||
| Fig. 8 Distribution of Q0st for CH4, CO2 and H2S in the MOFs above (red) and below (blue) the 2008 CO2/CH4 Robeson limit. | ||
4 Conclusions
In this work, a multi-stage screening methodology was designed to identify the most promising porous MOFs for biogas upgrading. The Cambridge structural database was pre-screened using a set of geometric criteria in order to remove structures with non-accessible surface area and pore diameters smaller than the individual components of biogas. The initial phase of screening predicted the single-component permeation of CO2, CH4 and H2S from adsorption and diffusion calculations at infinite dilution. In addition, Henry constants for the adsorption of H2O were calculated for each structure to determine the relative levels of hydrophobicity. Only the top hydrophobic MOF structures which exceed the Robeson upper bound (these are assumed to be the least affected by co-adsorption of H2O molecules within the biogas stream) were considered for the final phase of screening. For these materials, mixture selectivity and permeability at 298 K and 10 bar were predicted from equimolar CO2/CH4 and H2S/CH4 simulations. The top 8 MOFs identified in this work possess selectivity and permeability that rivals that of polymer-, zeolite-, ZIF- and MMM-based membranes. For the top candidates, the predicted values of CO2 permeability range between 3.71 × 104 barrer and 1.09 × 105 barrer, CO2 selectivity between 4.93 and 11.9, H2S permeability between 5.69 × 103 barrer and 1.03 × 105 barrer and H2S selectivity between 32.7 and 123 at the operating conditions of 10 bar and 298 K. In addition, the structural properties of the 1183 hydrophobic membranes with membrane selectivity values above the Robeson limit were examined. Structural property histograms indicated that MOF membranes with PLDs between 3.8 Å and 6 Å, LCDs between 4 Å and 8 Å, ASAs less than 1250 m2 g−1, VF between 0.3 and 0.5 and density between 1 g cm−3 and 1.5 g cm−3 appear the most frequently.To verify the significance of these properties, a direct comparison was made against the structural property distributions from low-performing materials found below the 2008 Robeson bound. It was found that 42% of low-performing materials possess PLDs and LCDs smaller than 6 Å, and 8 Å, respectively, which increases to 80% and 81% for membranes above the upper bound. In addition, 29% of low-performing materials possess a VF smaller than 0.5, in contrast to the 61% of membranes above the upper bound. A similar result was also noted when comparing the density distributions. This suggests that the structural properties that are frequently observed in the membranes above the upper bound are significant and contribute to the superior separation performance of these membranes. Therefore, these results may influence the future design of novel MOFs for biogas upgrading.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for access to the University of Nottingham’s Augusta HPC service. E. B. acknowledges a Royal Society Wolfson Fellowship for financial support.References
- X. Y. Chen, H. Vinh-Thang, A. A. Ramirez, D. Rodrigue and S. Kaliaguine, RSC Adv., 2015, 5, 24399–24448 RSC.
- S. Rasi, A. Veijanen and J. Rintala, Energy, 2007, 32, 1375–1380 CrossRef CAS.
- M. Harasimowicz, P. Orluk, G. Zakrzewska-Trznadel and A. G. Chmielewski, J. Hazard. Mater., 2007, 144, 698–702 CrossRef CAS PubMed.
- D. P. Gosh, Wet H2S cracking problem in oil refinery processes – Material selection and operation control issues, Tri-Service Corrosion Conference, Denver, Colorado, USA, 2007 Search PubMed.
- E. Ryckebosch, M. Drouillon and H. Vervaeren, Biomass Bioenergy, 2011, 35, 1633–1645 CrossRef CAS.
- D. Ruthven, S. Farooq and K. Knaebel, Pressure Swing Adsorption, VCH Publishers, New York, 1994 Search PubMed.
- A. M. Yousef, W. M. El-Maghlany, Y. A. Eldrainy and A. Attia, Energy, 2018, 156, 328–351 CrossRef CAS.
- University of Vienna, Biogas to biomethane technology review, Task 3.1.1, Institute of Chemical Engineering, Research Divison Thermal Process Engineering and Simulation, Intelligent Energy Europe Report, 2012 Search PubMed.
- S. Basu, A. L. Khan, A. Cano-Odena, C. Liu and I. F. J. Vankelecom, Chem. Soc. Rev., 2010, 39, 750–768 RSC.
- M. Shah, M. C. McCarthy, S. Sachdeva, A. K. Lee and H.-K. Jeong, Ind. Eng. Chem. Res., 2012, 51, 2179–2199 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- E. Adatoz, A. K. Avci and S. Keskin, Sep. Purif. Technol., 2015, 152, 207–237 CrossRef CAS.
- P. S. Goh and A. F. Ismail, Desalination, 2018, 434, 60–80 CrossRef CAS.
- N. Rangnekar, N. Mittal, B. Elyassi, J. Caro and M. Tsapatsis, Chem. Soc. Rev., 2015, 44, 7128–7154 RSC.
- J. Gascon, F. Kapteijn, B. Zornoza, V. Sebastián, C. Casado and J. Coronas, Chem. Mater., 2012, 24, 2829–2844 CrossRef CAS.
- K. Xie, Q. Fu, G. G. Qiao and P. A. Webley, J. Membr. Sci., 2019, 572, 38–60 CrossRef CAS.
- S. R. Venna and M. A. Carreon, Chem. Eng. Sci., 2015, 124, 3–19 CrossRef CAS.
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303–307 CrossRef CAS PubMed.
- M. Vinoba, M. Bhagiyalakshmi, Y. Alqaheem, A. A. Alomair, A. Pérez and M. S. Rana, Sep. Purif. Technol., 2017, 188, 431–450 CrossRef CAS.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H.-C. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef PubMed.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- X. Zhao, Y. Wang, D.-S. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef PubMed.
- S. Rojas, A. Arenas-Vivo and P. Horcajada, Coord. Chem. Rev., 2019, 388, 202–226 CrossRef CAS.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepúlveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, F. X. Llabrés i Xamena, V. Van Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- H. Wang, W. P. Lustig and J. Li, Chem. Soc. Rev., 2018, 47, 4729–4756 RSC.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- S. Qiu, M. Xue and G. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC.
- D. Bastani, N. Esmaeili and M. Asadollahi, J. Ind. Eng. Chem., 2013, 19, 375–393 CrossRef CAS.
- G. Liu, V. Chernikova, Y. Liu, K. Zhang, Y. Belmabkhout, O. Shekhah, C. Zhang, S. Yi, M. Eddaoudi and W. J. Koros, Nat. Mater., 2018, 17, 283–289 CrossRef CAS PubMed.
- H. B. Tanh Jeazet, S. Sorribas, J. M. Román-Marín, B. Zornoza, C. Téllez, J. Coronas and C. Janiak, Eur. J. Inorg. Chem., 2016, 4363–4367 CrossRef CAS.
- M. R. Khdhayyer, E. Esposito, A. Fuoco, M. Monteleone, L. Giorno, J. C. Jansen, M. P. Attfield and P. M. Budd, Sep. Purif. Technol., 2017, 173, 304–313 CrossRef CAS.
- S. Basu, A. Cano-Odena and I. F. J. Vankelecom, Sep. Purif. Technol., 2011, 81, 31–40 CrossRef CAS.
- G. Dong, H. Li and V. Chen, J. Mater. Chem. A, 2013, 1, 4610–4630 RSC.
- P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Chem. Mater., 2017, 29, 2618–2625 CrossRef CAS.
- D. Wu, Q. Yang, C. Zhong, D. Liu, H. Huang, W. Zhang and G. Maurin, Langmuir, 2012, 28, 12094–12099 CrossRef CAS PubMed.
- Z. Qiao, K. Zhang and J. Jiang, J. Mater. Chem. A, 2016, 4, 2105–2114 RSC.
- Z. Li, G. Xiao, Q. Yang, Y. Xiao and C. Zhong, Chem. Eng. Sci., 2014, 120, 59–66 CrossRef CAS.
- C. E. Wilmer, O. K. Farha, Y.-S. Bae, J. T. Hupp and R. Q. Snurr, Energy Environ. Sci., 2012, 5, 9849–9856 RSC.
- E. Haldoupis, S. Nair and D. S. Sholl, J. Am. Chem. Soc., 2012, 134, 4313–4323 CrossRef CAS PubMed.
- I. Erucar and S. Keskin, Ind. Eng. Chem. Res., 2011, 50, 12606–12616 CrossRef CAS.
- Z. Qiao, C. Peng, J. Zhou and J. Jiang, J. Mater. Chem. A, 2016, 4, 15904–15912 RSC.
- C. Altintas and S. Keskin, ACS Sustainable Chem. Eng., 2019, 7, 2739–2750 CrossRef CAS PubMed.
- C. Wang, X. Liu, N. Keser Demir, J. P. Chen and K. Li, Chem. Soc. Rev., 2016, 45, 5107–5134 RSC.
- C. E. Wilmer, K. C. Kim and R. Q. Snurr, J. Phys. Chem. Lett., 2012, 3, 2506–2511 CrossRef CAS PubMed.
- D. Dubbeldam, S. Calero, D. E. Ellis and R. Q. Snurr, Mol. Simul., 2016, 42, 81–101 CrossRef CAS.
- D. Frenkel and B. Smit, Understanding Molecular Simulation, Academic Press, San Diego, 2nd edn, 2002 Search PubMed.
- H. Daglar and S. Keskin, J. Phys. Chem. C, 2018, 122, 17347–17357 CrossRef CAS PubMed.
- Z. Qiao, Q. Xu and J. Jiang, J. Mater. Chem. A, 2018, 6, 18898–18905 RSC.
- P. Z. Moghadam, D. Fairen-Jimenez and R. Q. Snurr, J. Mater. Chem. A, 2016, 4, 529–536 RSC.
- D.-Y. Peng and D. B. Robinson, Ind. Eng. Chem. Fundam., 1976, 15, 59–64 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- R. Krishna and J. M. van Baten, J. Membr. Sci., 2010, 360, 323–333 CrossRef CAS.
- P. P. Ewald, Ann. Phys., 1921, 369, 253–287 CrossRef.
- H. A. Lorentz, Ann. Phys., 1881, 248, 127–136 CrossRef.
- D. Berthelot, C. R. Hebd. Seances Acad. Sci., 1898, 126, 1703–1855 Search PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 1998, 102, 2569–2577 CrossRef CAS.
- J. J. Potoff and J. I. Siepmann, AIChE J., 2001, 47, 1676–1682 CrossRef CAS.
- H. W. Horn, W. C. Swope, J. W. Pitera, J. D. Madura, T. J. Dick, G. L. Hura and T. Head-Gordon, J. Chem. Phys., 2004, 120, 9665–9678 CrossRef CAS PubMed.
- T. Kristóf and J. Liszi, J. Phys. Chem. B, 1997, 101, 5480–5483 CrossRef.
- J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef CAS PubMed.
- Y. J. Colón and R. Q. Snurr, Chem. Soc. Rev., 2014, 43, 5735–5749 RSC.
- R. B. Getman, Y.-S. Bae, C. E. Wilmer and R. Q. Snurr, Chem. Rev., 2012, 112, 703–723 CrossRef CAS PubMed.
- C. Altintas, G. Avci, H. Daglar, E. Gulcay, I. Erucar and S. Keskin, J. Mater. Chem. A, 2018, 6, 5836–5847 RSC.
- G. Avci, S. Velioglu and S. Keskin, ACS Appl. Mater. Interfaces, 2018, 10, 33693–33706 CrossRef CAS PubMed.
- L. Hamon, C. Serre, T. Devic, T. Loiseau, F. Millange, G. Férey and G. D. Weireld, J. Am. Chem. Soc., 2009, 131, 8775–8777 CrossRef CAS PubMed.
- Q. Yang, S. Vaesen, M. Vishnuvarthan, F. Ragon, C. Serre, A. Vimont, M. Daturi, G. De Weireld and G. Maurin, J. Mater. Chem., 2012, 22, 10210–10220 RSC.
- M. S. Shah, M. Tsapatsis and J. I. Siepmann, Chem. Rev., 2017, 117, 9755–9803 CrossRef CAS PubMed.
- L. Chen, C. A. Morrison and T. Düren, J. Phys. Chem. C, 2012, 116, 18899–18909 CrossRef CAS.
- M. Fischer, J. R. B. Gomes and M. Jorge, Mol. Simul., 2014, 40, 537–556 CrossRef CAS.
- C. Zhang, L. Wang, G. Maurin and Q. Yang, AIChE J., 2018, 64, 4089–4096 CrossRef CAS.
- A. R. Kulkarni and D. S. Sholl, J. Phys. Chem. C, 2016, 120, 23044–23054 CrossRef CAS.
- A. K. Rappe and W. A. Goddard, J. Phys. Chem., 1991, 95, 3358–3363 CrossRef CAS.
- D. Ongari, P. G. Boyd, O. Kadioglu, A. K. Mace, S. Keskin and B. Smit, J. Chem. Theory Comput., 2019, 15, 382–401 CrossRef CAS PubMed.
- Y.-B. Dong, M. D. Smith, R. C. Layland and H.-C. zur Loye, Chem. Mater., 2000, 12, 1156–1161 CrossRef CAS.
- M. Vougo-Zanda, J. Huang, E. Anokhina, X. Wang and A. J. Jacobson, Inorg. Chem., 2008, 47, 11535–11542 CrossRef CAS PubMed.
- J. P. S. Mowat, J. A. Groves, M. T. Wharmby, S. R. Miller, Y. Li, P. Lightfoot and P. A. Wright, J. Solid State Chem., 2009, 182, 2769–2778 CrossRef CAS.
- J. H. Park, W. R. Lee, Y. Kim, H. J. Lee, D. W. Ryu, W. J. Phang and C. S. Hong, Cryst. Growth Des., 2014, 14, 699–704 CrossRef CAS.
- A. Aijaz, E. Barea and P. K. Bharadwaj, Cryst. Growth Des., 2009, 9, 4480–4486 CrossRef CAS.
- R. El Osta, A. Carlin-Sinclair, N. Guillou, R. I. Walton, F. Vermoortele, M. Maes, D. de Vos and F. Millange, Chem. Mater., 2012, 24, 2781–2791 CrossRef CAS.
- J. C. Poshusta, R. D. Noble and J. L. Falconer, J. Membr. Sci., 1999, 160, 115–125 CrossRef CAS.
- S. Takamizawa, Y. Takasaki and R. Miyake, J. Am. Chem. Soc., 2010, 132, 2862–2863 CrossRef CAS PubMed.
- M. A. Carreon, S. Li, J. L. Falconer and R. D. Noble, J. Am. Chem. Soc., 2008, 130, 5412–5413 CrossRef CAS PubMed.
- H. Shi, New J. Chem., 2014, 38, 5276–5278 RSC.
- H. Guo, G. Zhu, I. J. Hewitt and S. Qiu, J. Am. Chem. Soc., 2009, 131, 1646–1647 CrossRef CAS PubMed.
- S. R. Venna and M. A. Carreon, J. Am. Chem. Soc., 2010, 132, 76–78 CrossRef CAS PubMed.
- J. A. Bohrman and M. A. Carreon, Chem. Commun., 2012, 48, 5130–5132 RSC.
- A. Huang, Q. Liu, N. Wang and J. Caro, Microporous Mesoporous Mater., 2014, 192, 18–22 CrossRef CAS.
- D.-J. Lee, Q. Li, H. Kim and K. Lee, Microporous Mesoporous Mater., 2012, 163, 169–177 CrossRef CAS.
- Y. Liu, G. Zeng, Y. Pan and Z. Lai, J. Membr. Sci., 2011, 379, 46–51 CrossRef CAS.
- Q. Song, S. K. Nataraj, M. V. Roussenova, J. C. Tan, D. J. Hughes, W. Li, P. Bourgoin, M. A. Alam, A. K. Cheetham, S. A. Al-Muhtaseb and E. Sivaniah, Energy Environ. Sci., 2012, 5, 8359–8369 RSC.
- G. Sodeifian, M. Raji, M. Asghari, M. Rezakazemi and A. Dashti, Chin. J. Chem. Eng., 2019, 27, 322–334 CrossRef CAS.
- N. Habib, Z. Shamair, N. Tara, A.-S. Nizami, F. H. Akhtar, N. M. Ahmad, M. A. Gilani, M. R. Bilad and A. L. Khan, Sep. Purif. Technol., 2020, 234, 116101 CrossRef CAS.
- J. Sánchez-Laínez, A. Pardillos-Ruiz, M. Carta, R. Malpass-Evans, N. B. McKeown, C. Téllez and J. Coronas, Sep. Purif. Technol., 2019, 224, 456–462 CrossRef.
- X. Guo, H. Huang, Y. Ban, Q. Yang, Y. Xiao, Y. Li, W. Yang and C. Zhong, J. Membr. Sci., 2015, 478, 130–139 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1fd00005e |
| ‡ Current address: School of Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK. |
| This journal is © The Royal Society of Chemistry 2021 |