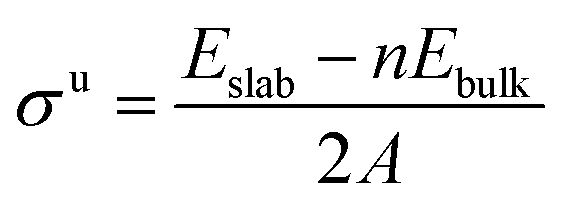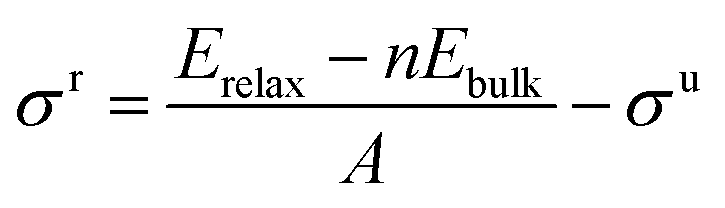How bulk and surface properties of Ti4SiC3, V4SiC3, Nb4SiC3 and Zr4SiC3 tune reactivity: a computational study†
Received
11th January 2021
, Accepted 5th February 2021
First published on 5th February 2021
Abstract
We present several in silico insights into the MAX-phase of early transition metal silicon carbides and explore how these affect carbon dioxide hydrogenation. Periodic density functional methodology is applied to models of Ti4SiC3, V4SiC3, Nb4SiC3 and Zr4SiC3. We find that silicon and carbon terminations are unstable, with sintering occurring in vacuum and significant reconstruction taking place under an oxidising environment. In contrast, the metal terminated surfaces are highly stable and very active towards CO2 reduction. However, we predict that under reaction conditions these surfaces are likely to be oxidised. These results are compared to studies on comparable materials and we predict optimal values for hydrogen evolution and CO2 reduction.
Introduction
The H-(Hägg)phase of over 100 ternary carbides/nitrides was first identified in the 1960s.1,2 This class of material is also given the annotation “MAX”-phase, an abbreviation of the general formula of these 2D materials ‘Mn+1AXn’, where M denotes an early transition metal and A/X represent an A group element and carbon/nitrogen, respectively.3–5 Importantly, these initial studies also found that these materials possess a novel mixture of chemical properties, including the high thermo-/electrical-conductivity of a metal and a resistance to thermo/oxidative shocks more typical of a ceramic.1,6–10 Owing to this set of novel properties these materials are also called “metallic ceramics”.11 Interestingly, this same mixture of properties is also present in the closely related monocarbide materials which in recent years have received a lot of interest from the catalytic community owing to their activity in many important industrial processes, including CO oxidation,12,13 various hydrogenation14 and the water–gas shift15,16 reactions. Importantly, for our work as well as the theme of this Faraday Discussions meeting, these carbides have also been shown effectively to hydrogenate CO2 to useful chemicals.17–20
Structurally, the (0001) basal plane of silicon carbides strongly resembles the pristine (111) facets of early transition metal carbides (TMCs), which are the facets that have been shown to be most efficient at catalysing the hydrogenation of CO2.17,21 Indeed, detailed in silico investigations by our group clearly indicate that the metal terminated (111) surfaces of the equivalent TMCs to the MAX-phase silicon carbides investigated here are extremely active toward CO2 activation.22 Expectations for the performance of these materials has also been informed by recent modelling work into another closely related class of material, i.e. the MXene-phase of early transition metal carbides, which also show a remarkable selectivity for carbon capture/utilisation.23–25 Again, the (0001) facet of these materials shows a remarkably similar topology to the metal terminated facet of silicon carbides. However, the MAXene stoichiometry of M2C leads to the production of two metal terminations, instead of the facets with one metal termination and one termination by either carbon or silicon, as seen in the silicon carbides discussed in this work. The choice of the transition metal components was partly informed by these studies and partly by a systematic screening of the bulk and surface properties of a diverse array of carbides.26 The latter suggested that the early transition metals had the most promising combination of surface properties for the catalysis of CO2 reduction by hydrogen. Therefore, this paper presents the results from a computational study into the bulk and catalytic properties of Ti4SiC3, V4SiC3, Nb4SiC3 and Zr4SiC3 and compares these properties to those already obtained for TiC, VC, NbC and ZrC.
Methodology
Unless otherwise stated, all energies were obtained using the standard Perdew–Burke–Ernzerhof (PBE) functional27 in combination with the Vienna Ab initio Simulation (VASP) Package.28–30 Benchmark studies, using formation energies for these materials with both RPBE45 and PBEsol,46 have produced consistent values for all methods which validates our choice of methodology. Such a protocol has been shown to be sufficient for replicating experimental trends for both surface properties and reactivities of closely related carbide26 and MXene materials.25 Plane-wave basis sets are applied to valence electrons with core potentials produced using projected augmented wave methodology (PAW)31 to describe the core electrons of each element. Long-range dispersion interactions were introduced via the D3 method.32,33 Bulk structures for α-Ti4SiC3 (ref. 7) and α-Nb4SiC3 (ref. 34) were taken from the Inorganic Structure Database (ICSD),35 whilst the MAX-phase of α-V4SiC3 and α-Zr4SiC3 were created by in silico modification of the former structures. The lattice parameters and the internal coordinates of each material were then fully optimized for all degrees of freedom and the (0001) basal plane was cut between the metal–carbon and metal–silicon layers using the METADISE code.36 Since the materials consist of alternating layers of metal and either carbon or silicon, only two possible slabs can be constructed in this plane: both express one metal layer, with one slab also terminated by carbon and the other slab terminated by the corresponding silicon surface. This sequence is due to the stoichiometry of these materials with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of metal to either carbon or silicon. The primitive cell of each silicon carbide was replicated into a 2 × 2 × 2 simulation cell to give slabs with nine atoms in each layer and 16 atomic layers along the z-axis to give a total of 144 atoms per slab. Above each termination was placed 15 Å of vacuum. A fine Monkhorst–Pack grid with a 5 × 5 × 5 k-point mesh was applied to all bulk calculations, with surface properties being determined using a 5 × 5 × 1 mesh. In all cases, energies were converged to within 520 eV and a threshold of 0.01 eV Å−1 was used for geometry optimisations. Self-consistency cycles (SCF) were converged to within 10−5 eV and the Blöchl smearing method was applied for higher accuracy.37 In all cases, half of each slab (the top 8 layers) was allowed to relax fully, whilst the bottom 8 layers were fixed to maintain their bulk-optimised positions. Spin polarization was also enabled to account for the magnetic moments in the materials and electron transfer during catalysis.
:1 ratio of metal to either carbon or silicon. The primitive cell of each silicon carbide was replicated into a 2 × 2 × 2 simulation cell to give slabs with nine atoms in each layer and 16 atomic layers along the z-axis to give a total of 144 atoms per slab. Above each termination was placed 15 Å of vacuum. A fine Monkhorst–Pack grid with a 5 × 5 × 5 k-point mesh was applied to all bulk calculations, with surface properties being determined using a 5 × 5 × 1 mesh. In all cases, energies were converged to within 520 eV and a threshold of 0.01 eV Å−1 was used for geometry optimisations. Self-consistency cycles (SCF) were converged to within 10−5 eV and the Blöchl smearing method was applied for higher accuracy.37 In all cases, half of each slab (the top 8 layers) was allowed to relax fully, whilst the bottom 8 layers were fixed to maintain their bulk-optimised positions. Spin polarization was also enabled to account for the magnetic moments in the materials and electron transfer during catalysis.
Adsorption energies for H2O, H2, O2, OH and CO2 are defined as:
| | | Eads = E(slab/molec) − [E(slab) + E(molec)], | (1) |
where the sum of the energies of the pristine slab and gas phase adsorbate(s) are subtracted from the total energy of the minimum energy structure of the slab with adsorbates. Vibrational frequencies, calculated with the finite difference method, confirmed the global minima for all absorbed species.
Results
Bulk properties
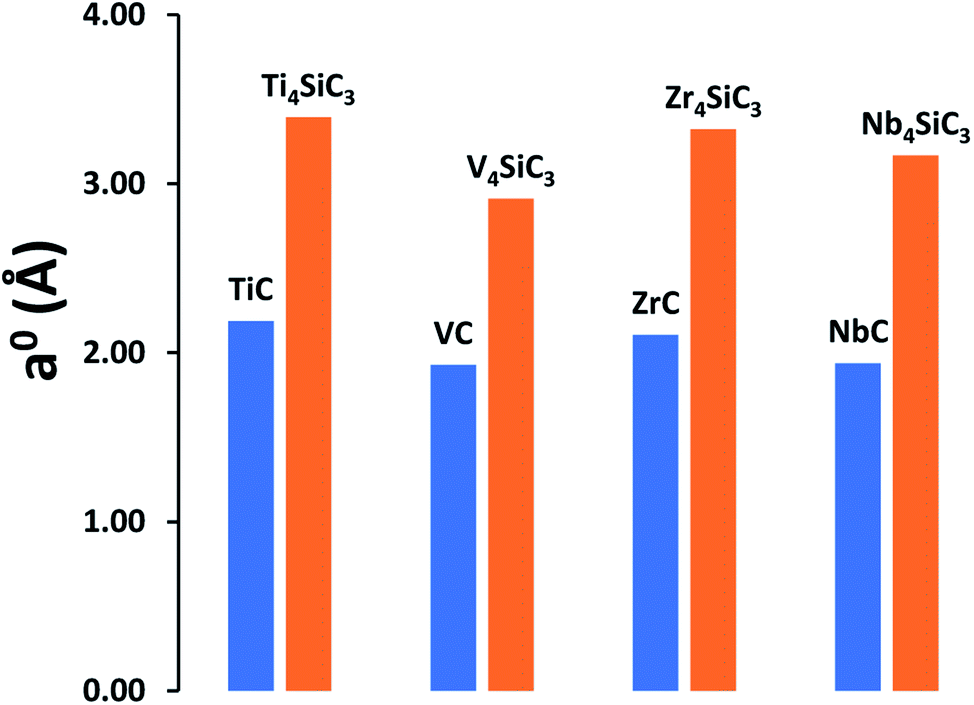
MAX-phase silicon carbide crystal structures for α-Ti4SiC3 (ref. 7) and α-Nb4SiC3 (ref. 34) were taken from the Inorganic Structure Database (ICSD).35 Since no structures were available for α-V4SiC3 and α-Zr4SiC3, they were created in silico by exchanging the transition metal component and running full optimisations of both the lattice parameters and atomic coordinates. For a meaningful comparison, the same bulk references for the individual elements were used as previously reported for the carbides,26 with the addition of an elemental silicon reference.38 Our results show that the addition of interstitial silicon layers into the carbide lattice has a relatively small effect on formation energies (see Table 1) with no consistent trend observed, although silicon addition does appear to have a very large effect on the relative lattice constants (Fig. 1), which is unsurprising since at 210 pm silicon has the largest atomic radius of any of the elements in the materials under investigation. This effect is most apparent when examining the relative surface areas of the carbide (111) and silicon carbide (0001) facets, which have the same number of atomic sites in both facets, where the relative values are as follows: Ti4SiC3 (149.04 Å2) vs. TiC (139.79 Å2); V4SiC3 (132.30 Å2) vs. VC (125.09 Å2); Zr4SiC3 (175.33 Å2) vs. ZrC (162.36 Å2); and Nb4SiC3 (157.15 Å2) vs. NbC (149.99 Å2).
Table 1 Energies of formation (OfE0) for each silicon carbide compared to the corresponding monocarbide material (with the same metal component). All values are normalised to eV per atom and compared with the experimental heat of formation
|
|
OfE0 |
OfE0 |
OfE0 |
OfH0 |
|
PBE.
PBEsol.
RPBE.
Experimental values.
|
| TiC |
−0.830 |
−0.915 |
−0.836 |
−0.951 (ref. 39) |
| Ti4SiC3 |
−0.863 |
−0.933 |
−0.848 |
| VC |
−0.439 |
−0.558 |
−0.425 |
−0.524 (ref. 39) |
| V4SiC3 |
−0.518 |
−0.363 |
−0.501 |
| ZrC |
−0.950 |
−1.010 |
−0.933 |
−0.959 (ref. 39) |
| Zr4SiC3 |
−0.937 |
−0.996 |
−0.915 |
| NbC |
−0.551 |
−0.622 |
−0.613 |
−0.676 (ref. 39) |
| Nb4SiC3 |
−0.563 |
−0.632 |
−0.562 |
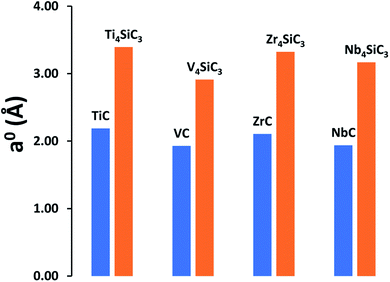 |
| | Fig. 1 Comparison of lattice constants (a0) for both the monocarbides and their comparable silicon carbides. All results are calculated using the PBE functional. | |
Density of states
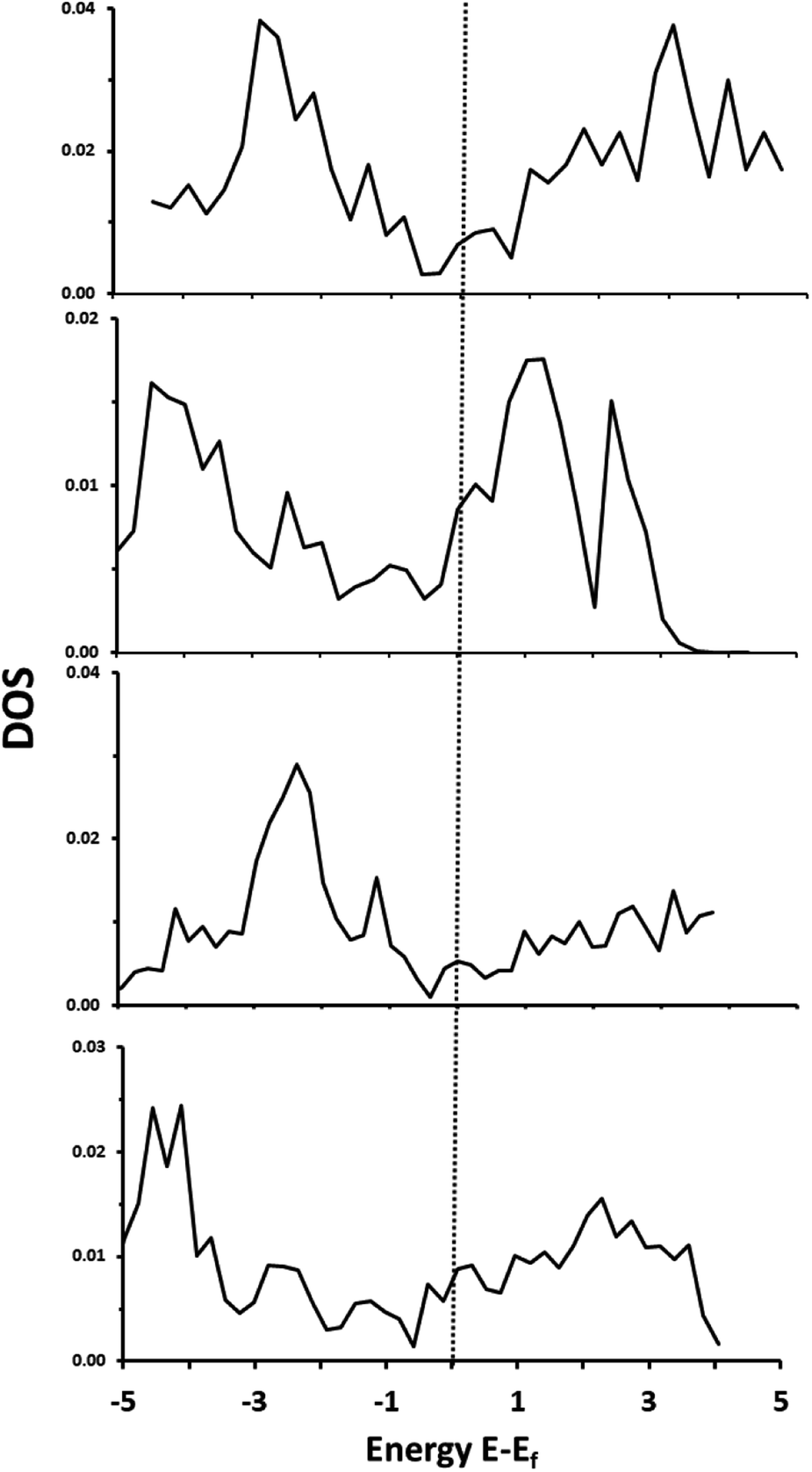
Total density of states plots for each of the materials are shown in Fig. 2. Importantly, all four materials are clearly metallic in nature, with some bands crossing the Fermi level.
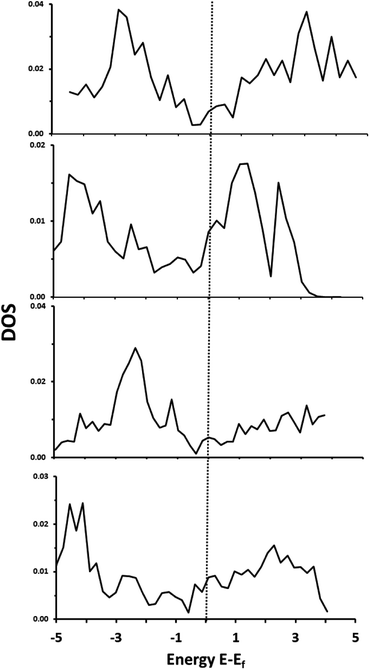 |
| | Fig. 2 Total DOS of: α-Ti4SiC3 (top), α-V4SiC3 (second from top), α-Zr4SiC3 (second from bottom), α-Nb4SiC3 (bottom). | |
Decomposition of these states indicates that there is a high level of hybridization between 3d or 4d orbitals of the metal with both the 2p states of the carbon and 3s orbitals of the silicon. All these materials, therefore, show considerable amounts of M–C and M–Si covalency. These same general properties were also reported previously for the related monocarbide materials,26 and our results replicate previously reported electronic structure analysis for α-Nb4SiC3.34 There is also the same apparent increase in peak intensity in the group 5 over the group 6 metals that in carbides has been associated with a decrease in the strength of the M–C bond and an increase in ionic character.40 This shift is primarily due to an increase in the number of valence d-electrons and also causes a negative shift in the position of the d-band centre in relation to the Fermi level. Finally, the density of states in the conduction band is much higher in the silicon-containing carbides with d2 metals (i.e. the metals in group 4 with the valence electrons d2, s2) than for the d3 which could indicate greater redox potential in the former.
To a large extent, catalytic activity as well as other surface properties, such as the stability of different facets, can be linked to surface energies (σ), and work functions (Φ).41 We have calculated these properties for each silicon carbide by cleaving along the (0001) plane and allowing the uppermost half of the slab to relax; depictions of the morphology of each relaxed surface can be found in Fig. S1 (see ESI†). Interestingly, major reconstruction (leading to surface-mediated formation of elemental silicon) is observed when the silicon-terminated surfaces of the 3d silicon carbides are relaxed, which indicates that the M–Si bond broken is far weaker than the new Si–Si bonds formed. Relaxed surface energies (σ) were calculated using eqn (2) and (3). A denotes the area of each surface, where Eslab signifies the total surface energy and n denotes the number of unit cells used for each slab. When calculating the energy to create two unrelaxed surfaces (eqn (2)) 2 is added to the denominators.
| | 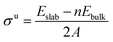 | (2) |
| | 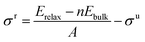 | (3) |
When only half the slab is relaxed, the relaxation component of the surface energy (σ) must be determined, as shown in eqn (3); here the area of only one surface is included in the denominator and this energy is subtracted from that of the unrelaxed surfaces. The computed σ for various facets of both the silicon-containing carbides reported here and their corresponding monocarbide equivalents are shown in Table 2. Previous work by our group showed that relaxing only the surface layers of the (111) facets of the corresponding monocarbides was critically important in both explaining the energetic differences between the two very different facets and to maintain the constraints provided by the bulk properties.26 From the values in Table 2, it is clear that surface energies for the carbon-terminated slabs in both the monocarbide and silicon carbide materials are very high.
Table 2 Relaxed surface energies (σ) and surface areas (a2) for each possible termination of the four silicon containing carbides compared to values for the (001) and metal terminated (111) surfaces of the corresponding monocarbides.26 All surface energies are given in J m−2 with the corresponding areas shown in Å2
|
|
SiC |
SiC |
SiC |
C |
C |
C |
|
σ
2
|
σ
2
|
,
a
2
|
σ
2
|
a
2
|
σ
2
|
a
2
|
σ
2
|
σ
2
|
,
a
2
|
|
Metal silicon facets, metal layer relaxed.
Metal silicon facets, silicon layer relaxed.
Metal carbon facets, carbon layer relaxed.
(001) monocarbide facet.
(111) metal terminated carbide facet.
(111) carbon terminated facet.
|
| Ti |
2.822 |
3.095 |
139.79 |
6.337 |
139.79 |
2.190 |
79.89 |
5.615 |
5.891 |
149.04 |
| V |
3.127 |
3.853 |
125.09 |
5.116 |
125.09 |
1.927 |
70.53 |
4.742 |
4.938 |
132.30 |
| Zr |
2.641 |
2.891 |
162.36 |
5.962 |
162.36 |
2.106 |
102.63 |
5.043 |
5.086 |
175.33 |
| Nb |
3.012 |
2.104 |
149.99 |
5.507 |
149.99 |
1.937 |
79.87 |
4.709 |
4.347 |
157.15 |
The silicon carbide (0001) slabs with both silicon and metal terminations have much lower surface energies, closer to the dominant (001) surface of the monocarbides than to the (111) facets. This result is perhaps unsurprising since in the presence of absorbates the carbon terminated (111) facet of early transition metal carbides has been shown to be extremely unstable and therefore would only be expected as a meta-stable phase under vacuum conditions.22,42 Fig. S1† shows that there is a major reconstruction of both the silicon and carbon terminated surfaces upon relaxation. To attempt to see if this major surface reconstruction also occurs in an oxygen-rich environment, we have also added a monolayer of eight oxygen atoms to each pristine termination; however, upon optimisation, various partially oxidised silicon and carbon species were formed in all cases (see Fig. S2, ESI†). Owing to their potential instability and relatively high surface energies, it is clear that the carbon and silicon terminated (0001) facets of these silicon carbides will be unstable under reaction conditions and that the surface morphology of the catalysts will be either dominated by pristine metal surfaces or by sintered coke/elemental silicon on top of a metal surface support. Therefore, unless otherwise stated, the remainder of this study will focus on the activity of the metal terminated (0001) facets.
Reactivity on the pristine surfaces
Hydrogen adsorption.
We attempted to identify barriers for hydrogen adsorption by performing geometry scans that sequentially lower physically adsorbed hydrogen from the vacuum to the silicon carbide surfaces. To this end, each hydrogen molecule was fixed in the z-direction and sequentially lowered towards the surface, but allowing full relaxation along all other degrees of freedom. The resulting energies of these scans are shown in Table S3 (see ESI†). However, whilst no local minima in such stepwise pathways to adsorption were found, the subsequent chemical adsorption step was shown to be a barrierless and extremely exothermic process. Since the same barrierless adsorption mechanism has been reported previously for the metal terminated (111) surfaces of the corresponding monocarbides,42 we will continue our study by examining hydrogen loading effects on these facets.
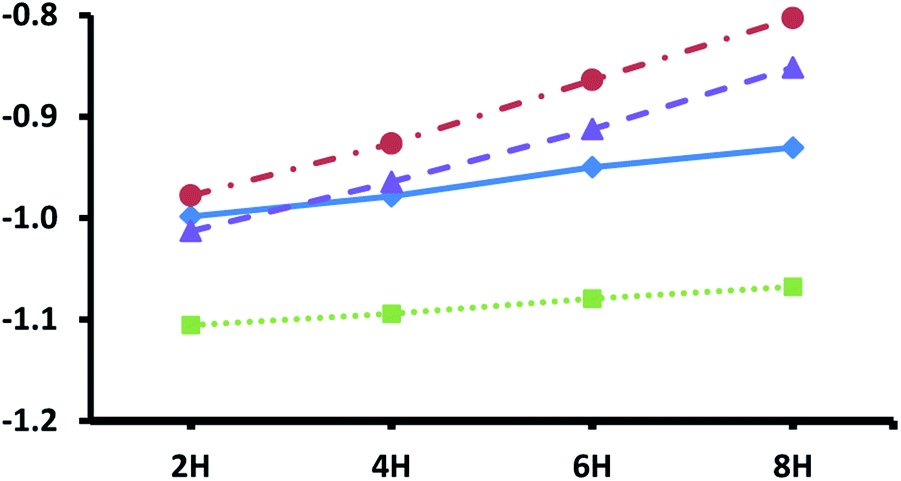
Fig. 3 shows the adsorption energies for the most exothermic adsorption modes of hydrogen under various coverages on the pristine metal terminated surfaces of the four silicon carbides considered in this study. The trends reported here are very similar to those reported previously for the metal terminated (111) facets of the corresponding monocarbides,42 where it was demonstrated that the decrease in adsorption energies with higher loading of hydrogen is primarily due to electronic effects, whereby electron donation into the conduction band of the carbides increases the surface work function. The same phenomenon is observed here, with a steeper slope obtained for the silicon carbides with group 5 transition metals owing to their increased number of electrons in the d-bands exacerbating this effect. Unfortunately, whilst smaller than those observed for the metal terminated (111) surfaces of the monocarbides (almost certainly due in part to the higher surface energies of those facets) the hydrogen adsorption energies here may still be too large at even the highest loadings for the adsorbed hydrogen to be useful for efficient CO2 reduction. This prediction is informed by our previous work, where very similar hydrogen adsorption energies on the (111) surfaces of the corresponding monocarbides makes those facets less than ideal for hydrogenation processes under reaction conditions.42
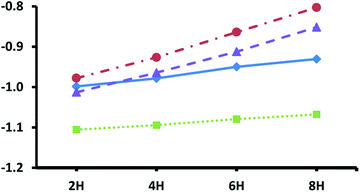 |
| | Fig. 3 Adsorption energies for different hydrogen loadings on top of pristine metal terminated surfaces of four MAX-phase silicon carbides. Values are shown for: Ti4SiC3 blue diamonds (solid trend line), V4SiC3 brown circles (dotted and dashed trend line), Zr4SiC3 green squares (dotted trend line) and Nb4SiC3 purple triangles (dashed trend line). Energies are given in eV per atom with reference to gas phase molecular hydrogen. Number of hydrogen atoms added is given along the x-axis in multiples of two (i.e. indicating increasing molecules of H2 added). | |
Carbon dioxide adsorption.
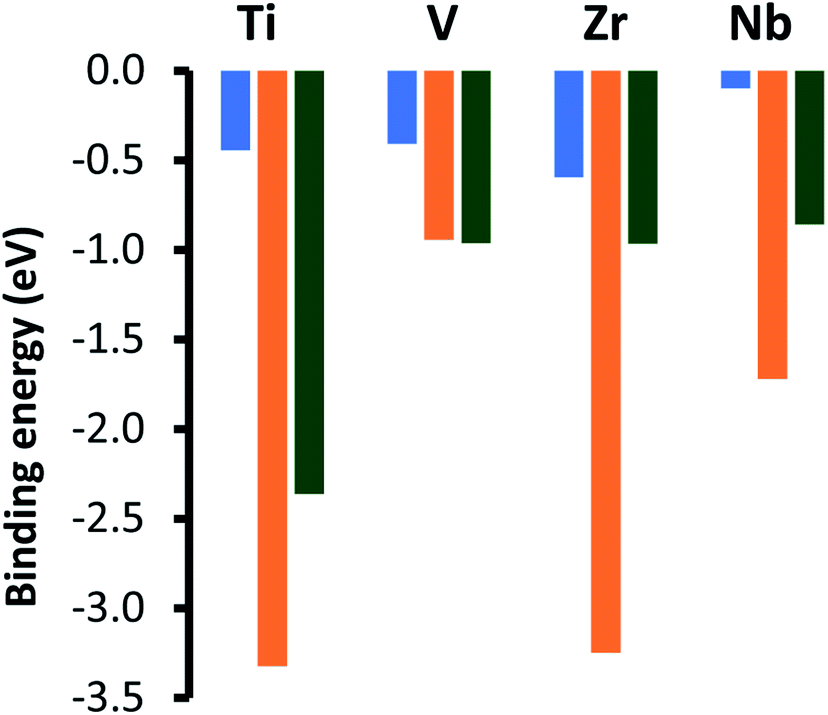
The adsorption energies for the lowest energy chemical adsorption modes for CO2 on top of the metal terminated (0001) facets of the four MAX-phase catalysts are shown by the orange bars in Fig. 4 and are considerably more exothermic than the energies previously reported for the monocarbide surfaces22 for all materials except for V4SiC3, which is calculated to lead to a very similar adsorption energy to that of the metal terminated (111) surface of VC. The values that we present here correlate more closely to values obtained for MAXenes with M2C stoichiometry, for which adsorption energies of −3.69 for Ti2C and −3.16 for Zr2C were calculated.43 Important geometric and electronic information about the lowest energy adsorption modes for carbon dioxide on top of each silicon carbide surface is given in Fig. S5, ESI.† These data show that whilst the average metal oxygen bond distance between the surface and adsorbate is modulated almost completely by the ideal M–C bond distance the amount of charge transfer (and corresponding elongation of the C–O bond length) varies depending on the periodic position of the parent transition metal. Importantly, group IV metals (Ti/V) see an increase of ∼0.6 in number of electrons transferred to the CO2 upon adsorption from the values observed for their V metal equivalents (Zr/Nb), which corresponds to both greater activation of the adsorbate and a more exothermic adsorption energy. Additionally, modelling of MAXenes with a M3C2 stoichiometry and d3 transition metals predicted much smaller adsorption energies of −2.19 for V3C2 and −2.35 for Nb3C2.23 Whilst high carbon dioxide capture on the pristine facets is more reminiscent of the MAXene-phase than the metal terminated (111) facets of the fcc-monocarbides, the adsorption energies are still slightly smaller than those on the MAXenes. Such high energies in the MAXenes are explained by the absence of a free (pristine) metal surface under either synthesis or reaction conditions. Indeed, all these materials are capped by a terminating layer of either OH, O, F or H monolayers.44 Therefore, the remainder of this paper will focus on the effects of monolayer formation on the metal facet of MAX-phase silicon carbides.
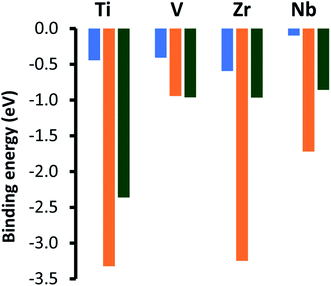 |
| | Fig. 4 Adsorption energies for chemically adsorbed carbon dioxide on the metal terminated (0001) basal plane of the four silicon carbides, shown by the orange bars. These values are compared to adsorption energies for the comparable (001) (blue bar) and metal terminated (111) (green bar) surfaces of the comparable monocarbide catalysts [ref. 22]. | |
Atomic monolayer formation and reactivity
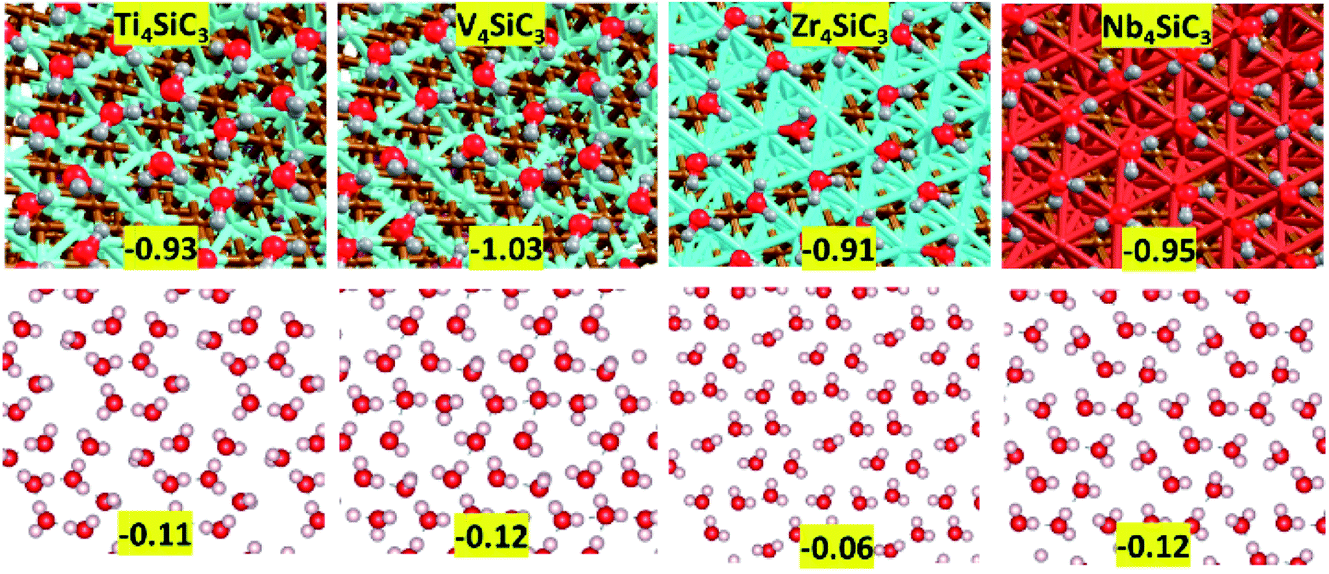
First, we consider solvation of the surfaces in an aqueous environment. The energy per molecule of eight water molecules adsorbed on each surface is shown in Fig. 5 and there is little variation between the values for each material, which range between −1.03 for V4SiC3 to −0.91 for Zr4SiC3. To create each layer, water molecules were initially placed in the most exothermic binding motifs (corresponding to the highly coordinated hollow sites) after which other molecules were sequentially added to fill each surface, whilst every attempt was made to maximise hydrogen-bonding networks between molecules. The extent to which this was possible was largely determined by surface structure, as is also shown in Fig. 5, where the bottom panels show the hydrogen-bonding energy per molecule, obtained after the surface was removed and a single point energy calculation was performed. These results show that for each silicon carbide surface, except for Zr4SiC3 (where the network is much weaker), the surface-mediated hydrogen-bonding networks maintain approximately half the energy of each hydrogen-bond formed by water under atmospheric conditions. The anomalous value for Zr4SiC3 is possibly due to the larger distances between binding sites caused by the larger surface area.
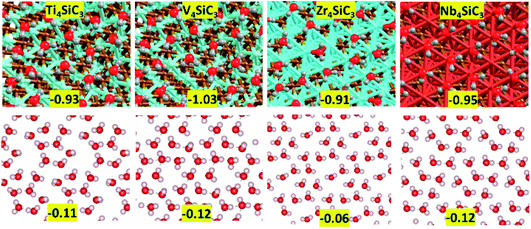 |
| | Fig. 5 Water monolayers on top of exposed metal terminated (0001) basal plane of silicon carbides. (top) Adsorption energy in eV per H2O for eight water molecules adsorbed on each surface. (bottom) Hydrogen-bonding energy in eV per H2O for the water layer without the surface interactions. | |
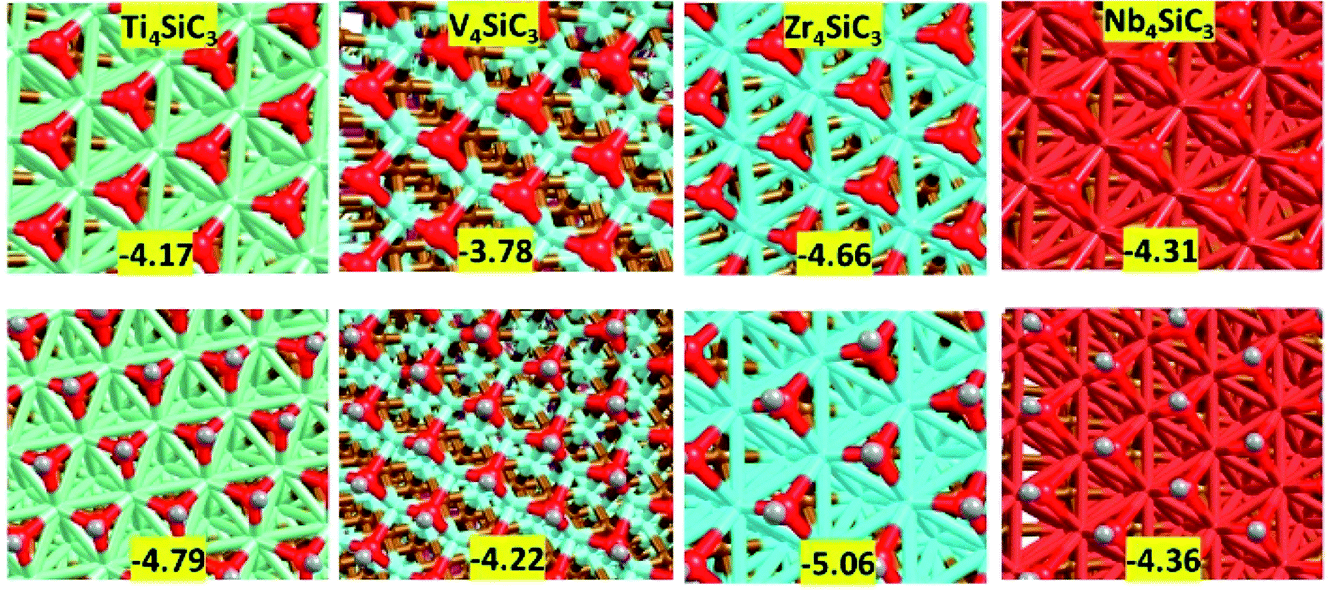
Finally, we examined surface modifications under oxidising environments. Fig. 6 shows the morphology and energetics of a fully oxidised or hydroxylated metal terminated (0001) surface for each catalyst. The references for the surface formation energies are triplet oxygen and singlet hydrogen molecules in the gas phase. Importantly, the oxide and hydroxide layers on this surface are formed without major modification to the rest of the slab which contrasts strongly with the results obtained using the carbon and silicon terminations (see ESI Fig. S2†). The addition of other adlayers has an extremely exothermically favourable stabilising effect that again is in excellent agreement with previous experimental results from the related MAXene-phases.44 More interestingly, by subtracting the energies for the formation of the oxide layers from those of the hydroxyl layers, we observe a large reduction in the hydrogen adsorption values: Ti4SiC3 pristine (−0.93 eV per H) vs. oxidised (−0.62 eV per H); V4SiC3 pristine (−0.80 eV per H) vs. oxidised (−0.44 eV per H); Zr4SiC3 pristine (−1.07 eV per H) vs. oxidised (−0.40 eV per H); and Nb4SiC3 pristine (−0.85 eV per H) vs. oxidised (−0.05 eV per H). These latter energies are interesting, because they are close to the values determined via modelling of chemical potentials to be optimal for the CO2 hydrogenation reaction over the monocarbide materials.22 However, it is still unknown whether these hydroxylated surfaces will also maintain their other catalytic properties that make silicon carbide MAX-phases such exciting candidates for CCC and CCU processes.
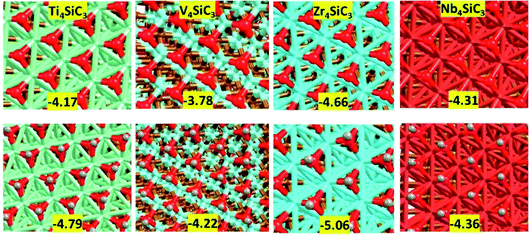 |
| | Fig. 6 Models of oxidised surfaces of the four metal terminated silicon carbides. (top) Oxygen monolayer of nine oxygen atoms with energies given in eV per Oatom. (bottom) Hydroxylated surfaces with nine –OH groups per surface with values given in eV per OHmolecule. | |
Conclusions
The MAX-phases of early transition metal silicon carbides represent an interesting class of materials, owing to both their novel mix of metallic, covalent and ionic properties, as well as their structural similarities to related redox catalysts for carbon utilisation reactions. Our results demonstrate that the silicon and carbon terminations of these materials are unstable, with strong silicon–silicon and carbon–carbon bonds causing sintering and coking respectively, when undercoordinated atomic layers are exposed to the vacuum. In the presence of oxygen these terminations lead to the formation of partially oxidised silicon- and carbon-containing species as well as massive surface reconstruction. However, these materials can also terminate at an extremely stable metal (0001) facet that is very active towards CO2 adsorption and activation. Unfortunately, these facets adsorb hydrogen far too strongly for them to be useful for the hydrogenation of carbon dioxide, although further calculations suggest that oxidised surfaces may have the ideal properties for this process. Finally, whilst these materials appear to show many promising characteristics as potential CCU catalysts, much more experimental and theoretical work is required to study their activity and selectivity towards the production of industrially useful chemicals.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was funded as part of an EPSRC low carbon fuels grant (EP/N009533/1). Computing facilities for this work were provided by ARCCA at Cardiff University, HPC Wales, and through our membership of the UK’s Materials Chemistry Consortium (MCC). The UK Catalysis Hub is thanked for resources and support provided via membership of the UK Catalysis Hub Consortium and funded by EPSRC (Grants EP/R026815/1, EP/K014854/1, and EP/M013219/1). The MCC is funded by EPSRC (EP/F067496). All data created as part of this study are openly available at: http://doi/10.17035/d.2021.0128671491.
Notes and references
- M. W. Barsoum, The MN+1AXN phases: a new class of solids, Prog. Solid State Chem., 2000, 28, 201–281 Search PubMed.
- W. Jeitschko, H. Nowotny and F. Benesovsky, Kohlenstoffhaltige ternäre Verbindungen (H-Phase), Monatsh. Chem., 1963, 94, 672–676 Search PubMed.
- Z. M. Sun, Progress in research and development on MAX phases: a family of layered ternary compounds, Int. Mater. Rev., 2011, 56, 143–166 Search PubMed.
- M. W. Barsoum and T. El-Raghy, New phases: carbide and nitride materials unique ternary ceramics turn out to be surprisingly soft and machinable, yet also heat-tolerant, strong and lightweight, Am. Sci., 2001, 89, 334–343 Search PubMed.
- W. Jeitschko and H. Nowotny, Die Kristallstruktur von Ti3SiC2-ein neuer Komplexcarbid-Typ, Monatsh. Chem., 1967, 98, 329–337 Search PubMed.
- P. Eklund, M. Beckers, U. Jansson, H. Högberg and L. Hultman, Thin Solid Films, 2010, 518, 1851–1878 Search PubMed.
- E. H. Kisi, J. A. A. Crossley, S. Myhra and M. W. Barsoum, Structure and crystal chemistry of Ti3SiC2, J. Phys. Chem. Solids, 1998, 59, 1437–1443 Search PubMed.
- T. El-Raghy, A. Zavaliangos, M. W. Barsoum and S. R. Kalidindi, Damage Mechanisms around Hardness Indentations in Ti3SiC2, J. Am. Ceram. Soc., 2005, 80, 513–516 Search PubMed.
- M. W. Barsoum, T. El-Raghy and L. U. J. T. Ogbuji, Oxidation of Ti3SiC2 in air, J. Electrochem. Soc., 1997, 144, 2508–2516 Search PubMed.
- T. El-Raghy and M. W. Barsoum, Diffusion kinetics of the carburization and silicidation of Ti3SiC2, J. Appl. Phys., 1998, 83, 112–119 Search PubMed.
- Z. M. Sun, H. Hashimoto, Z. F. Zhang, S. L. Yang and S. Tada, Synthesis and Characterization of a Metallic Ceramic Material–Ti3SiC2, Mater. Trans., 2006, 47, 170–174 Search PubMed.
- L. K. Ono, D. Sudfeld and B. Roldan Cuenya, In situ gas-phase catalytic properties of TiC-supported size-selected gold nanoparticles synthesized by diblock copolymer encapsulation, Surf. Sci., 2006, 600, 5041–5050 Search PubMed.
- K.-Z. Qi, G.-C. Wang and W.-J. Zheng, A first-principles study of CO hydrogenation into methane on molybdenum carbides catalysts, Surf. Sci., 2013, 614, 53–63 Search PubMed.
- P. M. Patterson, T. K. Das and B. H. Davis, Carbon monoxide hydrogenation over molybdenum and tungsten carbides, Appl. Catal., A, 2003, 251, 449–455 Search PubMed.
- P. Lui and J. A. Rodriguez, Water-Gas-Shift Reaction on Molybdenum Carbide Surfaces: Essential Role of the Oxycarbide, J. Phys. Chem. B, 2006, 110, 19418–19425 Search PubMed.
- N. M. Schweitzer, J. A. Schaidle, O. K. Ezekoye, X. Pan, S. Linic and L. T. Thompson, High Activity Carbide Supported Catalysts for Water Gas Shift, J. Am. Chem. Soc., 2011, 133, 2378–2381 Search PubMed.
- M. D. Porosoff, S. Kattel, W. Li, P. Liu and J. G. Chen, Identifying trends and descriptors for selective CO2 conversion to CO over transition metal carbides, Chem. Commun., 2015, 51, 6988–6991 Search PubMed.
- C. Kunkel, F. Viñes and F. Illas, Transition metal carbides as novel materials for CO2 capture, storage, and activation, Energy Environ. Sci., 2016, 9, 141–144 Search PubMed.
- S. Posada-Pérez, F. Viñes, P. J. Ramirez, A. B. Vidal, J. A. Rodriguez and F. Illas, The bending machine: CO2 activation and hydrogenation on δ-MoC(001) and β-Mo2C(001) surfaces, Phys. Chem. Chem. Phys., 2014, 16, 14912–14921 Search PubMed.
- M. D. Porosoff, X. Yang, J. A. Boscoboinik and J. G. Chen, Molybdenum Carbide as Alternative Catalysts to Precious Metals for Highly Selective Reduction of CO2 to CO, Angew. Chem., Int. Ed., 2014, 53, 6705–6709 Search PubMed.
- C. Kunkel, F. Viñes and F. Illas, Transition metal carbides as novel materials for CO2 capture, storage, and activation, Energy Environ. Sci., 2016, 9, 141–144 Search PubMed.
- M. G. Quesne, A. Roldan, N. H. de Leeuw and C. R. A. Catlow, Carbon dioxide and water co-adsorption on the low-index surfaces of TiC, VC, ZrC and NbC: a DFT study, Phys. Chem. Chem. Phys., 2019, 21, 10750–10760 Search PubMed.
- Á. Morales-García, M. Mayans-Llorach, F. Viñes and F. Illas, Thickness biased capture of CO2 on carbide MXenes, Phys. Chem. Chem. Phys., 2019, 21, 23136–23142 Search PubMed.
- M. T. Aswathi, N. Kuriakose, K. Mondal and P. Ghosh, CO2 capture, activation and dissociation on the Ti2C surface and Ti2C MXene: the role of surface structure, Phys. Chem. Chem. Phys., 2020, 22, 14599–14612 Search PubMed.
- H. Prats, H. Mcaloone, F. Viñes and F. Illas, Ultra-high selectivity biogas upgrading through porous MXenes, J. Mater. Chem. A, 2020, 8, 12296–12300 Search PubMed.
- M. G. Quesne, A. Roldan, N. H. De Leeuw and C. R. A. Catlow, Bulk and surface properties of metal carbides: implications for catalysis, Phys. Chem. Chem. Phys., 2018, 20, 6905–6916 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 Search PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 Search PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 Search PubMed.
- G. Kresse and J. Hafner,
Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 Search PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 Search PubMed.
- S. Grimme, Semiempirical GGA‐type density functional constructed with a long‐range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 Search PubMed.
- C. Li, J. Kuo, B. Wang, Y. Li and R. Wang, A new layer compound Nb4SiC3 predicted from first-principles theory, J. Phys. D: Appl. Phys., 2009, 42, 075404 Search PubMed.
- M. Hellenbrandt, The Inorganic Crystal Structure Database (ICSD)—Present and Future, Crystallogr. Rev., 2004, 10, 17–22 Search PubMed.
- G. W. Watson, E. T. Kelsey, N. H. de Leeuw, D. J. Harris and S. C. Parker, Atomistic simulation of dislocations, surfaces and interfaces in MgO, J. Chem. Soc., Faraday Trans., 1996, 92, 433 Search PubMed.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Improved tetrahedron method for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 Search PubMed.
-
D. M. Többens, N. Stüßer, K. Knorr, H. M. Mayer and G. Lampert, in Materials Science Forum, Trans Tech Publications Ltd, 2001, vol. 378–381, pp. 288–293 Search PubMed.
- S. R. Shatynski, The Thermochemistry of Transition Metal Carbides, Oxid. Met., 1979, 13, 105–118 Search PubMed.
- H. H. Hwu and J. G. Chen, Surface Chemistry of Transition Metal Carbides, Chem. Rev., 2005, 105, 185–212 Search PubMed.
- H. W. Hugosson, O. Eriksson, U. Jansson, A. V. Ruban, P. Souvatzis and I. A. Abrikosov, Surface energies and work functions of the transition metal carbides, Surf. Sci., 2004, 557, 243–254 Search PubMed.
- F. Silveri, M. G. Quesne, A. Roldan, N. H. De Leeuw and C. R. A. Catlow, Hydrogen adsorption on transition metal carbides: a DFT study, Phys. Chem. Chem. Phys., 2019, 21, 5335–5343 Search PubMed.
- Á. Morales-García, A. Fernández-Fernández, F. Viñes and F. Illas, CO2 abatement using two-dimensional MXene carbides, J. Mater. Chem. A, 2018, 6, 3381–3385 Search PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Two-Dimensional Materials: 25th Anniversary Article: MXenes: A New Family of Two-Dimensional Materials (Adv. Mater. 7/2014), Adv. Mater., 2014, 26, 982 Search PubMed.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(11), 7413–7421 Search PubMed.
-
J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Restoring the Density-Gradient Expansion for Exchange, in Solids and Surfaces, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1fd00004g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
C. Richard A.
Catlow
*ab,
C. Richard A.
Catlow