 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into air pollution chemistry and sulphate formation from nitrous acid (HONO) measurements during haze events in Beijing†
William J.
Bloss
 *a,
Louisa
Kramer‡
a,
Leigh R.
Crilley§
a,
Tuan
Vu¶
a,
Roy M.
Harrison
a,
Zongbo
Shi
a,
James D.
Lee
b,
Freya A.
Squires
b,
Lisa K.
Whalley
c,
Eloise
Slater
c,
Robert
Woodward-Massey||
c,
Chunxiang
Ye||
c,
Dwayne E.
Heard
c,
Shengrui
Tong
d,
Siqi
Hou
d,
Yele
Sun
e,
Jingsha
Xu
a,
Lianfang
Wei
e and
Pingqing
Fu
f
*a,
Louisa
Kramer‡
a,
Leigh R.
Crilley§
a,
Tuan
Vu¶
a,
Roy M.
Harrison
a,
Zongbo
Shi
a,
James D.
Lee
b,
Freya A.
Squires
b,
Lisa K.
Whalley
c,
Eloise
Slater
c,
Robert
Woodward-Massey||
c,
Chunxiang
Ye||
c,
Dwayne E.
Heard
c,
Shengrui
Tong
d,
Siqi
Hou
d,
Yele
Sun
e,
Jingsha
Xu
a,
Lianfang
Wei
e and
Pingqing
Fu
f
aSchool of Geography, Earth and Environmental Sciences, University of Birmingham, UK. E-mail: w.j.bloss@bham.ac.uk
bWolfson Atmospheric Chemistry Laboratories, Department of Chemistry, University of York, UK
cSchool of Chemistry, University of Leeds, UK
dInstitute of Chemistry, Chinese Academy of Sciences, Beijing, China
eState Key Laboratory of Atmospheric Boundary Layer Physics and Atmospheric Chemistry, Institute of Atmospheric Physics, Chinese Academy of Sciences, Beijing, China
fInstitute of Surface-Earth System Science, Tianjin University, Tianjin 300072, China
First published on 21st September 2020
Abstract
Wintertime urban air pollution in many global megacities is characterised by episodic rapid increase in particulate matter concentrations associated with elevated relative humidity – so-called haze episodes, which have become characteristic of cities such as Beijing. Atmospheric chemistry within haze combines gas- and condensed-phase chemical processes, leading to the growth in secondary species such as sulphate aerosols. Here, we integrate observations of reactive gas phase species (HONO, OH, NOx) and time-resolved aerosol composition, to explore observational constraints on the mechanisms responsible for sulphate growth during the onset of haze events. We show that HONO abundance is dominated by established fast gas-phase photochemistry, but the consideration of the additional formation potentially associated with condensed-phase oxidation of S species by aqueous NO2 leading to NO2− production and hence HONO release, improves agreement between observed and calculated gas-phase HONO levels. This conclusion is highly dependent upon aerosol pH, ionic strength and particularly the parameterisation employed for S(IV) oxidation kinetics, for which an upper limit is derived.
Background
Atmospheric chemical processing leads to the removal of primary pollutants and determines the rate of production of secondary species, including ozone and many components of aerosol particles. Gas-phase processing is driven by a small number of key oxidants, of which the OH radical is predominant for the majority of species during daytime, including many hydrocarbons and inorganic gas-phase species such as SO2 and NO2. Gas- and condensed-phase oxidation chemistry are coupled across the two phases of aerosol, with a wide range of aqueous oxidants driving the particle phase oxidation of species such as SO2, including NO2 itself.In many urban areas, particulate matter (PM, particularly PM2.5) is a key regulated air pollutant, whose abundance drives substantial adverse human health impacts, in addition to affecting visibility, radiation transfer and amenity. PM is formed of a complex chemical mixture including both directly emitted (primary) and chemically produced (secondary) components; understanding the relationship between these underpins the accurate identification of direct and indirect PM sources, formulating the most effective and efficient clean air policy.
This challenge is emphasised by the air pollution climatology of Beijing and the North China Plain, where seasonal haze events characterised by episodic PM2.5 excursions to 500 μg m−3 and above, impact the health and wellbeing of around 400 million people. Sulphate is known to be a major component of the fine size fraction of Beijing haze. The rate of formation of sulphate aerosol from SO2, driven by chemical oxidation, is observed to increase as PM2.5 levels rise, and recent studies into haze formation have found that the rate of production of sulphate aerosol is significantly accelerated in megacities such as Beijing.1 New chemical mechanisms have been proposed, notably including the aqueous phase oxidation of SO2 by NO2, if aerosol pH is sufficiently high. A by-product of this mechanism is expected to be the production of gas-phase HONO.
Here, we analyse in situ observations collected during the UK–Chinese Air Pollution and Human Health (APHH) campaign in central Beijing during haze episodes in November–December 2016, which constrain the gas-phase production and removal of HONO, and the observed rate of formation of sulphate. We identify constraints from the in situ observations upon potential chemical SO2 oxidation mechanisms contributing to haze formation.
Sulphate aerosol formation in haze
Secondary SO4 formation is driven by a number of chemical mechanisms. Globally, the dominant process is the gas-phase reaction of SO2 with OH radicals, leading to the formation of HSO3 intermediates, which rapidly react with O2 forming SO3, which is converted to H2SO4 through reaction with water vapour. The recently characterized rapid reaction between SO2 and criegee intermediates formed through alkene ozonolysis,2 is unlikely to significantly impact boundary layer SO2 oxidation due to the rapid removal of criegee intermediates by water vapour and the water vapour dimer.3,4 SO2 may also partition to aqueous aerosol particles, where a series of equilibria partition between SO2·H2O (at low pH), HSO3− (pH 2–7) or SO32− under basic conditions. In the aqueous phase, SO2 processing is driven by H2O2 (when pH is near neutral) and O3 (of increasing importance under more acidic conditions). Transition metals (Fe, Mn) can also catalyse O2 reactions leading to condensed phase sulphate formation. During the onset of haze events, sulphate formation rates of the order of tens of μg per m3 per hour are observed.1 Very recently, Wang et al.5 reported the potential for HONO-mediated SO2-to-SO4 oxidation in Beijing cloud/fog, with associated N2O formation.However, current air quality models fail to reproduce elevated sulphate levels in winter haze. Several mechanisms for S(IV) oxidation are still under consideration to account for the underestimation of sulphate concentration between model simulations and field observations, including reactive nitrogen chemistry involving the oxidation of S(IV) by NO2,6–10 underestimation of the transient metals catalyzed pathway,11,12 the possible oxidation on the surface of acid droplets by O2,13 or the otherwise dominant role of S(IV) + H2O2 pathway.14 Although these pathways can improve the agreement between sulphate simulation and measurement, there remains a lack of independent constraints on modelling results, especially the potentially dominant role of the NO2–S reaction. A tagging methodology with triple oxygen isotopes has been applied as a proxy to constrain the relative contributions of the individual oxidation pathways of S(IV),12,15 with results showing that the NO2–S reaction is highly dependent on pH and dominant under high-pH conditions, however, a very substantial uncertainty resulted from the assumed activity coefficient between the aerosol liquid phase ionic strength and parameterisation of the kinetic reaction rate.
The reaction between NO2, partitioned into the aqueous phase, with HSO3− remains a potentially dominant driver of sulphate formation under haze conditions, forming SO42− at rates of 1–7 μg m−3 h−1:6,8
| 2NO2(aq) + HSO3(aq)− + H2O(aq) → 3H(aq)+ + 2NO2(aq)− + SO4(aq)2− | (1) |
This is in contrast to tropospheric cloud processing of sulphur species, which is dominated by the H2O2 and O3 reactions, with NO2 processing relatively unimportant.16 The mechanism has the potential to drive a positive feedback in haze formation, where increased sulphate production leads to increased aerosol water and hence SO2 uptake and oxidation, under conditions of high NO2.6 Of particular note here, the condensed phase nitrite is expected to be released from acidic aerosol to the atmosphere as HONO.8
The ionic composition of the aqueous aerosol is critically important in calculating the importance of the NO2 mechanism – ionic strength (which impacts the rate constants for the S(VI) oxidation reactions) and aerosol pH. Elevated pH enhances SO2 solubility (higher effective Henry’s law constant); consequently chemical factors to maintain elevated aerosol pH (5–7) are required, and have been attributed to high levels of ammonia emissions/ammonium in the North China Plain,6,8 However, subsequent analyses have concluded that the highest observed NH3 levels are insufficient to raise pH levels above 4.5 under haze conditions, and hence it is unlikely that NO2-mediated oxidation of SO2 is a major sulphate formation route in Beijing haze, compared with O3 and transition-metal mediated oxidation.11
Boundary layer HONO photochemistry
The basic gas-phase chemistry of HONO combines formation through the homogenous termolecular OH + NO reaction (R2), and reaction of HONO with OH (R3) and relatively rapid solar photolysis (R4), where the photolysis frequency (1.7 × 10−3 s−1 at SZA = 40°) corresponds to a typical mid-latitude local solar noon lifetime of around 10 min.| OH + NO + M → HONO + M | (2) |
| HONO + OH → H2O + NO2 | (3) |
| HONO + hν → OH + NO | (4) |
HONO photolysis (R4) is an important, and frequently the dominant, source of OH radicals in the continental boundary layer, accounting for 40% of OH formation in central London,17 over 80% of OH production at a rural site in Colorado,18 and most recently an average of 83% of OH production in wintertime Beijing19 – hence, HONO abundance and formation mechanisms are tightly coupled to the production of OH and rate of in situ gas-phase oxidation for many atmospheric pollutants.
HONO levels may be predicted by reactions (R2)–(R4), assuming a photochemical steady state (see below), which may then be compared with observed concentrations to ascertain the presence of additional HONO sources. This exercise has been undertaken a number of times, leading to a range of additional HONO sources being identified – which are frequently significantly larger than the homogeneous OH + NO reaction. Examples include source strengths exceeding reaction (R2) by factor 13 in a European forest,20 30 at Hyytiälä21 and a factor of 19 in rural China.22 In performing such analyses, it is important to note the timescales for HONO are rather longer than those of OH, hence analysis in highly spatially heterogeneous environments can be challenging as noted by Lee et al.23 and Crilley et al.24
In general terms, such sources may be combined as an additional source term, SHONO, if HONO may be placed into the steady state:
| SHONO → HONO | (5) |
| d[HONO]/dt = k2[OH][NO] + SHONO − k3[OH][HONO] − j4[HONO] | (E1) |
A number of mechanisms for further atmospheric HONO emission and production have been suggested. Direct emission sources include vehicle exhausts (with HONO emissions equivalent to 1.24%, and 0.85% of NOx emissions recently derived for the Hong Kong and UK vehicle fleets, respectively),25,26 biocrusts,27 and microbial soil emissions.28 Heterogeneous HONO sources include dark reactions between NO2 and water (e.g. Finlayson-Pitts et al.29) on ground and aerosol surfaces, photolysis of nitrophenols, and photoenhanced conversion of NO2 to HONO in the presence of organic chromophores.30 Substantial work has been undertaken and the reader is directed to the reviews of Kleffman31 and Spataro and Ianniello.32
The UK–Chinese Air Pollution and Human Health – Beijing experiment (APHH-Beijing), was funded by the UK Natural Environment Research Council and the National Science Foundation of China. APHH-Beijing brought together well over 100 instruments to study boundary layer atmospheric composition and haze formation at the Institute of Atmospheric Physics (IAP), Chinese Academy of Sciences in central Beijing in November–December 2016 and spring–summer 2017.
Here, we combine co-located measurements from the APHH study, alongside numerical model calculations, to explore potential constraints on the sulphate formation mechanisms under haze conditions in Beijing. We compare the abundance of HONO with potentially determinant parameters including PM, meteorological factors and aerosol composition (including SO4). We calculate the HONO concentrations which would be predicted in ambient air from established gas-phase chemistry (reactions (2)–(4)), and the rate of HONO formation from the aerosol-mediated NO2/SO2 oxidation process (reaction (1)). We compare predicted HONO levels with those observed, and discuss the quantitative agreement in the context of different approaches to parameterising the NO2-enhanced SO2 oxidation kinetics. Implications for air quality are discussed.
APHH-Beijing measurements
Measurements were acquired during the APHH-Beijing winter experiment, conducted from 10 November–10 December 2016 at the Institute of Atmospheric Physics (IAP), Chinese Academy of Sciences in central Beijing 39°58′33′′ N, 116°22′41′′ E, between the third and fourth North ring roads. The site is in a residential area, with a number of substantial highways ca. 150 m away to the south, north and west. On the site itself, an open grassed area is surrounded by 2–3 storey buildings, with larger structures (residential blocks) some 200–300 m away. A canal runs past the north perimeter of the site, with a park (including conifer pine trees) further to the west. Given recent work to relocate industry from central Beijing, the dominant local emission sources are anticipated to be traffic and domestic activities, with possible contributions from light industry, for NOx. For SO2, sources are anticipated to be regional, industry and power/heating-related combustion (including coal) around the city periphery. For full details of the site, instruments and campaign overview, including a detailed meteorological timeseries, see Shi et al.33OH radicals were measured via laser-induced fluorescence, using the fluorescence assay by gas expansion (FAGE) methodology. Full details of the instrument operation and calibration are given in Slater et al.19 NO was measured using chemiluminescence employing a Thermo TE42i NOx monitor, calibrated to an NPL α-standard. Photolysis frequencies (jHONO) were measured at ground level using a calibrated spectral radiometer. HONO was measured using the LOPAP (Long Path Optical Absorption Photometry) approach. The LOPAP is a wet chemical technique and has been described in detail in Heland et al.34 and Kleffmann et al.35 Briefly, a stripping coil entrains gas-phase HONO into an acidic solution where it is derivatized into an azo dye, detected by long path absorption at 550 nm, and calibrated against an aqueous phase nitrite standard. The LOPAP was operated and calibrated according to the standard procedures described in Kleffmann and Wiesen.36 A detailed account of the APHH-Beijing HONO dataset, including an intercomparison of HONO measurements by LOPAP, broadband cavity absorption spectrometry, chemical ionisation mass spectrometry and proton transfer reaction mass spectrometry is given in Crilley et al.37 All data were acquired at ground level (inlet heights = 5 m).
Aerosol data for sulphate mass concentration were taken from AMS (aerosol mass spectrometer) measurements; these therefore correspond to the fine fraction (PM1) of the particulate matter present. The sulphate concentrations determined by AMS data were compared with the results obtained by 24 hour high-volume PM2.5 filter samples collected at the IAP site during December 2016, and analysed in the laboratory by ion chromatography.38 The two datasets were found to be within 20% of each other (r2 = 0.97) and the AMS data were used without further adjustment. PM2.5 mass concentrations were derived by TEOM-FDMS, and were recorded at the top of a two-storey building (about 10 m above ground level). Data were averaged to hourly mean mixing ratios using in situ temperature and pressure measurements. The period 2–10 December was selected as this encompassed several periods of haze, with rapid and dramatic PM2.5 growth reaching nearly 500 μg m−3 (haze periods typically identified as PM2.5 > 75 μg m−3), and maximised overlap between the operational periods of the various instruments used – during this date range, 65% of the necessary measurements were present (see Fig. 1), limited primarily by the OH and HONO datasets. Conditions during this period were typical of the wider APHH winter campaign (see Shi et al.33 for climatological details).
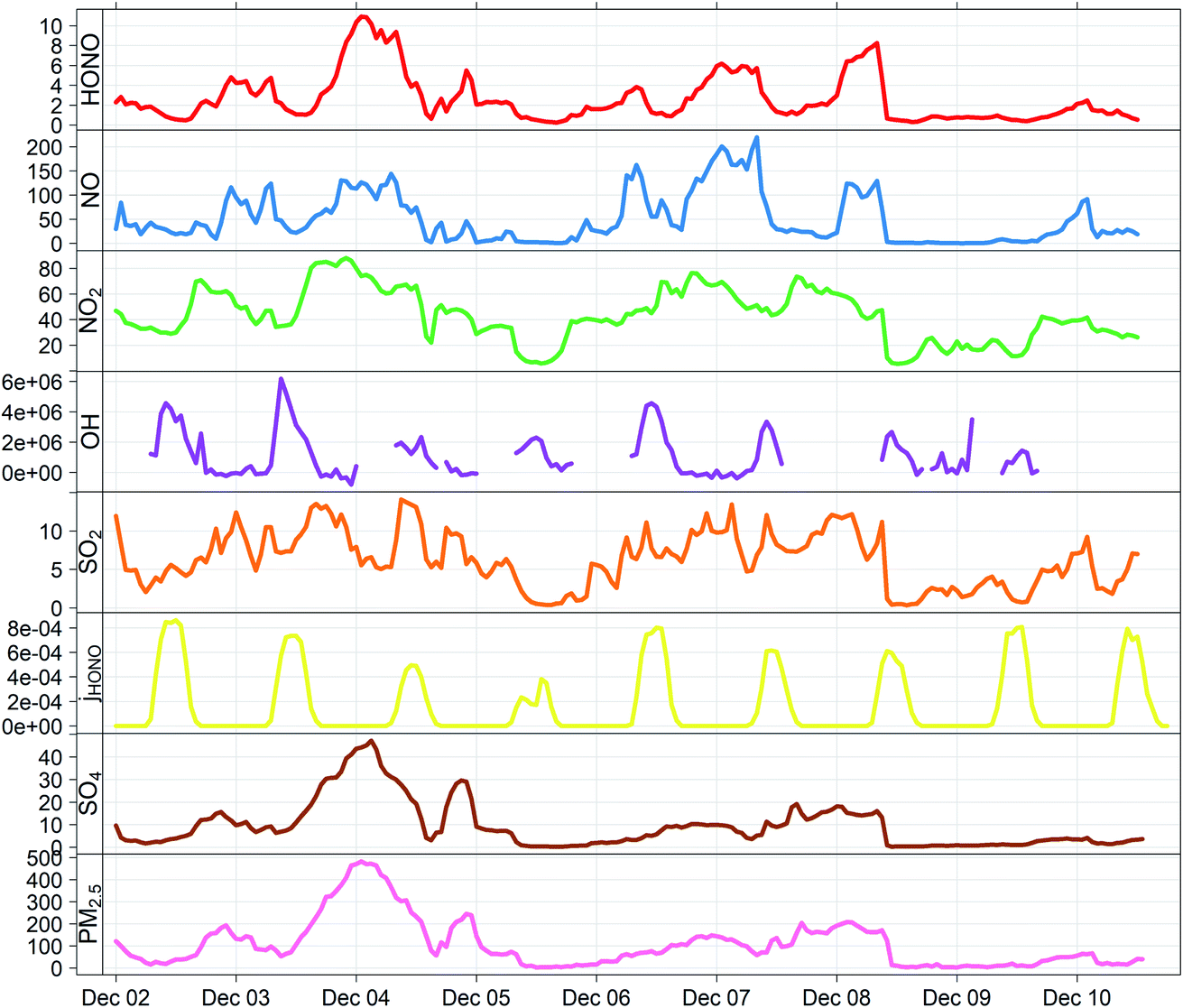 | ||
| Fig. 1 Measured timeseries for HONO (ppb), NO (ppb), NO2 (ppb), OH (molec cm−3), SO2 (ppb), jHONO (s−1), SO4 (AMS; μg m−3) and PM2.5 (TEOM FDMS; μg m−3) at the IAP tower site during the APHH Beijing winter campaign, 2016. | ||
Analysis of in situ observations
Fig. 1 shows the characteristic pattern of haze event onset observed previously, and associated with increases in many air pollutant species and intermediates; daily mean mixing ratios/concentrations are given in ESI Tables ST1 and ST2.† There is a positive correlation between HONO and sulphate abundance, indicating commonality in their mechanisms of formation/removal. In general, HONO abundance is frequently closely coupled with that of NO2 (through heterogeneous NO2-to-HONO conversion chemistry, and/or co-emission of both species), and variations in the HONO/NO2 ratio can be used to characterise this relationship. We find HONO levels are correlated with the abundance of PM2.5 (r2 = 0.67) and of aerosol sulphate (r2 = 0.59), but the HONO/NO2 ratio is much less strongly related to PM2.5 abundance (r2 = 0.27), indicating that the interaction between particulate matter and HONO may be more strongly related to sulphate chemistry, than NO2-to-HONO heterogeneous conversion. The HONO–SO4 relationship does not show any dependence upon SO2, but higher levels of both species are strongly associated with higher RH (Fig. 2), indicating a potential association with aqueous content, such as the condensed phase SO2 oxidation mechanisms outlined above.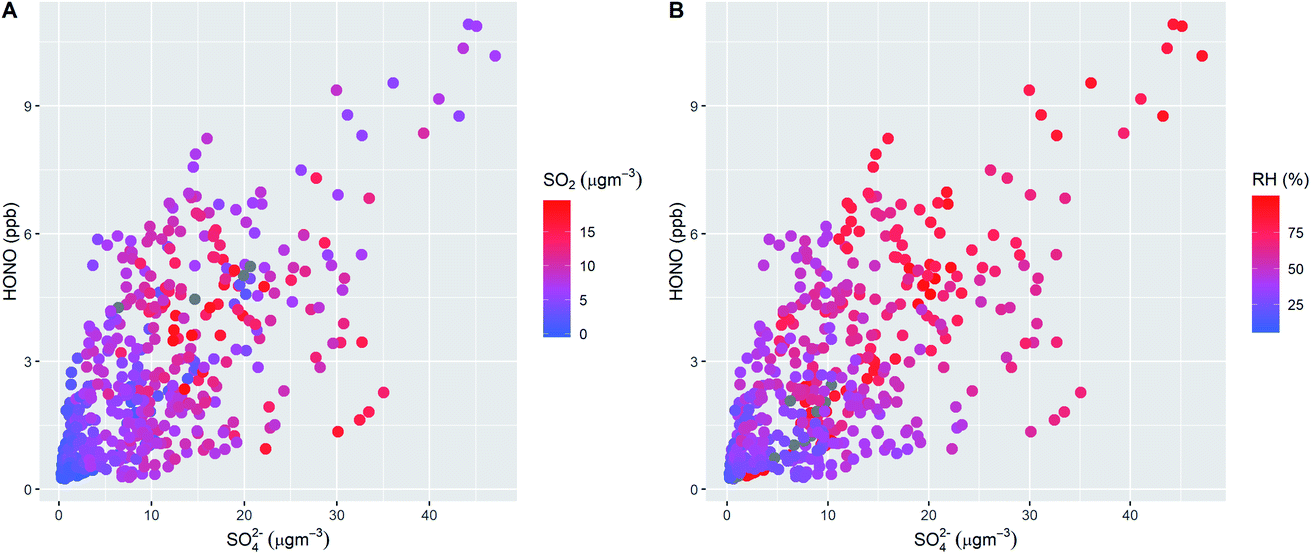 | ||
| Fig. 2 Relationship between HONO and SO4 concentrations, coloured by SO2 abundance (panel A) and by relative humidity (RH, panel B), during the 2–10 December focus period. The data show the limited impact of SO2, but the highest levels of both HONO and SO4 are associated with elevated humidity. | ||
The measurement data coverage during the APHH-Beijing campaign at IAP provides direct constraints on the major (gas-phase, photochemical) processes forming and removing ambient nitrous acid, including measurements of concentrations of OH, NO, HONO and meteorological/photolysis parameters. We averaged data to an hourly time resolution, and evaluated the consecutive changes in concentration of HONO (i.e. d[HONO]/dt, defined simply as the difference in consecutive hourly mean measurements of HONO abundance), and rate of the chemical reactions forming and removing HONO over the equivalent time periods (E1).
Fig. 3 shows the derived timeseries for d[HONO]/dt, and the individual components of HONO formation and removal, focusing upon the case study period of 2–10 December. The dominance of the conventional gas-phase chemistry is apparent, with HONO formation dominated by the OH + NO reaction, and removal by photolysis, with a small to negligible contribution from the OH + HONO reaction.
 | ||
| Fig. 3 Timeseries of the observationally-derived components of in situ HONO formation and destruction: reactions of OH + NO (R2), OH + HONO (R3), HONO + hν(R4), plus the observed rate of change of HONO, d[HONO]/dt. | ||
Discounting potential additional sources of HONO (i.e., assuming SHONO = 0), we can calculate the HONO abundance that would be predicted assuming the steady state applies. We limit the analysis to daylight only (here, hourly-averaged data where the HONO lifetime is less than 60 min), corresponding to jHONO > 2.78 × 10−4 s−1, and where all measurements are available (HONO, OH, NO, SO2, photolysis parameters, aerosol composition) – this condition reduces the available data to 44 hourly averages, 23% of the total over the focus period (the principal reduction vs. the coverage noted above arises due to the restriction to daylight hours). The predicted HONO concentration is compared with that observed in Fig. 4. The PSS-calculated (red triangles) and observed (blue circles) HONO mixing ratios are in relatively good agreement, indicating limited immediate need for additional production terms. A clearer picture is obtained from the regression analysis (Fig. 5), comparing the PSS-calculated and observed HONO mixing ratios (red triangles). While the data are scattered, overall the observed and calculated mixing ratios are similar in overall magnitude, without evidence for orders-of-magnitude error. Regression analysis derives gradient and intercept values of 0.90 ± 0.38 and (0.59 ± 0.84) ppb respectively (±2σ), with an r2 coefficient of 0.35. HONO measurements performed by multiple independent techniques during the APHH-Beijing study have been compared by Crilley et al.,37 who showed that the LOPAP-derived HONO data had a detection limit of around 35 ppt (i.e. very much smaller than the scatter of the observations), and that all instruments (using different techniques) were extremely well correlated with each other (r > 0.97), but with variations in slope (i.e. absolute accuracy) of 0.61–0.88. Therefore the scatter in the observed data in Fig. 4 does not reflect measurement precision (although the observations may collectively be biased high or low, depending on the absolute accuracy), and rather reflects changes in the actual observed concentrations, in turn reflecting variations in the atmospheric source and sink-terms as integrated over the HONO lifetime.
 | ||
| Fig. 4 Timeseries of observed daytime HONO mixing ratios (blue circles), steady state calculated HONO mixing ratios – HONO PSS, as derived from application of eqn (E1) – (red triangles) and model calculated HONO (see text; green squares). | ||
 | ||
| Fig. 5 Correlation between observed HONO and PSS-predicted mixing ratios (red triangles), regression line (red dashes), and model calculated HONO levels (see text; green squares) and associated regression (green dots). | ||
The data can also be analysed to quantify HONO formation (or removal) mechanisms not accounted for by the basic chemistry (reactions (R2)–(R4)) through application of (E1) alongside the observed d[HONO]/dt to derive SHONO, however the inferred net source for HONO, SHONO, is very scattered and displays no correlation with SO2 or SO4 abundance (r2 = 0.04 and 0.02 respectively), or with the SO2-normalised rate of SO4 formation (r2 = 0.05) – not shown. A crude estimation of the rate of formation of HONO (technically, NO2−) which would be associated with SO2 oxidation (Psulphate) can be obtained from the observed rate of formation of sulphate, d[SO4]/dt, multiplied by a factor of 2 for the stoichiometry of NO2− formation:
| Psulphate = 2 × d[SO4]/dt | (E2) |
Critically, in addition to disregarding other (non–NO2–mediated) mechanisms for SO2 oxidation, this estimate for HONO formation associated with SO4 production neglects the effect of dynamics, i.e. advection of more (or less) polluted airmasses across the study site – leading to negative as well as positive values (Fig. S1†). Nonetheless it is instructive to compare the resulting daytime Psulphate values, which ranged from 0.2 to 2.84 ppb h−1, to those predicted for the OH + NO reaction (Fig. 3) – the Psulphate values are nearly one order of magnitude smaller (and likely significant overestimates during periods of haze growth, due to pollution advection).
Model quantification of SO2 oxidation pathways
To more comprehensively quantify the relative importance of different SO2 oxidation mechanisms and the interaction with HONO formation, a zero-dimensional numerical model constrained to the observed conditions was used to calculate the gas- and condensed-phase SO2 oxidation rates for the focus period. The model framework involved a series of processes, consisting of gas-phase reaction of SO2 with OH, absorption equilibria of gaseous species, ionization, aqueous phase chemical reactions of S(IV), and the kinetics of mass transport. The model assumed that air infuses into the aerosol water and then participates in chemical reactions. The condensed-phase oxidation of S(IV) by H2O2, O3, oxygen catalyzed by Fe3+/Mn2+ or NO2 in the aerosol water were explicitly represented. The influence of primary sulphate emission and regional transportation were disregarded – the focus is on in situ chemical production only. The kinetics of sulphate formation and its dependence on ionic strength of aerosol water were implemented following Cheng et al.6 and He et al.15 Concentrations of Fe3+ and Mn2+ were pH-dependent and followed the precipitation equilibria of Fe(OH)3 and Mn(OH)2.The Nested Air Quality Prediction Model System (NAQPMS) developed by the Institute of Atmospheric Physics, Chinese Academy of Sciences (IAP/CAS) was adopted to simulate hourly concentrations of OH free radicals and H2O2 during the field campaign.39 NAQPMS incorporated the Carbon Bond Mechanism version Z (CBM-Z)40 to simulate gas-phase reactions. The modeled OH and H2O2 concentrations were used in the estimation of sulphate formation from the gas-phase reaction of OH with SO2 and the aqueous-phase reaction of H2O2 with HSO3−, and (in the case of OH) to evaluate the model oxidant field calculation. The observed aerosol composition, gaseous NH3 and meteorological conditions were input into the ISORROPIA-II thermodynamic equilibrium model41 to estimate the aerosol water content (AWC), aerosol water pH, and ionic strength (Table ST2†). The model was run in forward mode and predicted the phase partitioning of a K+/Ca2+/Mg2+/NH4+/Na+/SO42−/NO3−/Cl−/H2O in thermodynamic equilibrium with gas-phase precursors.
By way of validation, the modelled and observed OH levels19 as a sensitive test of the integrated NAQPMS oxidation (VOC, NOx and O3 chemistry), are compared in Fig. S2.† Overall, agreement was good, with the model showing good skill in reproducing the diurnal behaviour of OH. The absolute modelled OH levels were within 15% of those observed on 4 of the 6 days considered, but exceeded those observed (by up to a factor of 2) on the remaining 2 days (interestingly, those characterised by the lowest NO levels).
The modelled rate of formation of SO4 was determined for OH (gas phase), H2O2, O3, transition metals (Fe, Mn catalysed O2 reaction) and NO2 driven processes. Ionic strength is a key factor in such calculations: sulphate production rates were calculated for three cases: (A) neglecting the impact of ionic strength; (B) considering the impact of ionic strength upon the solubility of SO2 and the dissociation of H2SO3 (ref. 42) and upon the kinetics of the H2O2, O3 and transition metal reactions; and (C) also considering the impact of ionic strength upon the rate constant for the NO2 + S(IV) reaction, following the parameterisations of Cheng et al.6 We compared these approaches as there is a lack of experimental data for the influence of ionic strength upon the NO2–S reaction, and the calculated aerosol ionic strength during severe haze events reaches 60 M (when RH > 40% and PM2.5 > 75 μg m−3) hence extrapolations from the conditions of dilute solutions may not be warranted.
Discounting ionic strength effects (i.e., case A), SO4 formation averaged over the whole 12 November–10 December 2016 campaign period is found to be dominated by gas-phase OH chemistry (51%), followed by condensed phase H2O2 and transition metal reactions, with NO2 reaction responsible for 1.6%. Considering ionic strength effects other than NO2 kinetics (case B), oxidation of SO2 (g) by OH drives 81% of SO4 formation, with 18% arising from H2O2 and 0.6% for reaction with NO2 – however the average and maximum rates of SO4 formation calculated were 0.036 and 0.29 μg m−3 h−1 respectively, much lower than those observed. Inclusion of the ionic strength parameterisation for the NO2 + S kinetics (case C) increases the calculated rate of SO4 formation by two orders of magnitude (a factor of 96), with the calculated contribution of the NO2 pathway for SO4 formation increasing to 98% overall, as a consequence of the very strong dependence or the rate constant upon ionic strength, highlighting this critical uncertainty in this aspect of the kinetics parameterisation. The average and maximum rates of SO4 formation calculated for case C were 3.5 and 12 μg m−3 h−1, respectively, comparable to the behaviour observed (Fig. 1 and S3†).
The timeseries for the modelled SO4 formation rates for each chemical mechanism under case (B), and for the NO2-driven processing under case (C), are shown in Fig. 6 for the 2–7 December focus period – note the different y axis scale used for the latter data series (see also Fig. S4†). The significant increase in the overall rate of formation of SO4 for case (C) is in much closer accord with the rates of SO4 formation derived from the observed timeseries (Fig. 1), than for cases (A) and (B).
 | ||
| Fig. 6 Modelled SO4 formation rates (converted to ppb h−1) through reaction of SO2 with OH (orange line), condensed phase reactions (all case B) with H2O2 (grey), O3 (yellow), transition metals (blue) and NO2 (green). Dashed line, second y axis: NO2 reaction considering impacts of ionic strength on NO2 + S(IV) reaction kinetics (i.e. case C). See Fig. S5 (ESI†) for expansion of the H2O2, O3 and TMI timeseries. | ||
Considering the rate of production of NO2−, as an estimate to the HONO formation rate associated with NO2/SO2 oxidation, it is clear that the non-ionic-strength enhanced kinetics (case (A) and (B) – the latter as show in Fig. 6) are too small to make any appreciable impact upon HONO production, compared with the dominant OH + NO reaction flux. Calculated HONO formation from case (C), dominated by the ionic-strength-dependent reaction with NO2, was added to the homogeneous OH + NO production to determine the impact upon calculated steady state HONO concentrations, according to eqn (E3), and as shown in Fig. 4 and 5 as HONO (+Model).
| [HONO] = {k1[OH][NO] + [Modelled SO2 + NO2 rate]}/{k2[OH][HONO] + j3[HONO]} | (E3) |
Addition of the SO2 + NO2 derived HONO flux increases the HONO levels by 27% on average, and modestly improves agreement between the calculated and observed HONO levels (r2 = 0.98). The observed and PSS-calculated HONO levels are compared as a function of SO4 in Fig. S3† – while higher levels of both are associated with more polluted conditions (more PM2.5, more SO4), no bias between the observed and PSS-calculated levels with respect to SO4 is apparent, indicating this factor is not dominant. By applying a scale factor to the model-derived HONO production component, it can be shown that the rate of SO2 oxidation by NO2, as simulated here, is consistent with the observed HONO within the scatter of the data, but would cease to be so if the ionic strength enhancement factor were to be significantly increased – an upper limit (corresponding to a regression gradient of 1.5) of a factor of 4.5 fold higher may be estimated.
Conclusions
Nitrous acid was measured in the urban atmosphere of Beijing, at mixing ratios of up to 10 ppb (significantly higher than many other cities, e.g. 1.6 ppb for London17). HONO abundance, alongside that of many air pollutant species, was correlated with the formation of episodic haze events, associated with rapid growth in PM2.5 under elevated RH conditions, including increases in sulphate concentration. The HONO abundance was correlated with that of SO4, and with RH. The HONO/NO2 ratio, a marker for heterogeneous interconversion of reactive nitrogen oxides to HONO through dark- and photo-enhanced reaction processes, was not correlated with aerosol abundance, indicating heterogeneous formation does not dominate HONO production. Rather, during daytime, fast conventional gas-phase chemistry – the OH + NO reaction (2), and HONO photolysis (4) – dominates the HONO abundance in wintertime Beijing, and considering these processes (and the minor HONO + OH reaction (R3)), permits alone good quantitative agreement (regression coefficient = 0.90 ± 0.38) between measured and calculated photostationary steady state HONO levels.Combination of observationally-constrained rates of HONO formation and loss through reactions (2)–(4), with the calculated rates of HONO production through heterogeneous mechanisms including H2O2, O3, transition metal and S–NO2 chemistry showed that, discounting the effects of ionic strength, the condensed phase mechanisms made a negligible contribution to HONO formation, while SO2 oxidation was dominated by gas-phase OH chemistry, with the S–NO2 reaction contributing 1.6% of the total. Inclusion of the ionic strength effects, including parameterisation of the NO2–S(IV) kinetics following Cheng et al.,6 led to calculated condensed-phase nitrite production equivalent to increases in the steady state HONO levels of 27%, modestly increasing agreement with the HONO levels observed. Under this scenario, the NO2–S processing dominates SO4 formation. Considering the constraint the HONO levels place upon the additional production attributable to the NO2–S reaction, an upper limit to the maximum possible enhancement of the ionic-strength-dependent kinetics of a factor of 4.5 is estimated.
This work demonstrates the potential, and limitations, for coordinated observations of gaseous and particulate atmospheric composition to constrain fundamental processes which are extremely challenging to evaluate accurately in the laboratory or field – in this case, aerosol liquid water pH and ionic strength kinetic behaviour under very strongly non-ideal conditions. A key limitation of the data which are available here is their fixed measurement location: a lagrangian approach, allowing parameters to be evaluated along the trajectory of an evolving airmass at higher altitude and remote from possible surface influence may facilitate greater insight.
Conflicts of interest
There are no conflicts of interest.Acknowledgements
This work was funded by the UK Natural Environment Research Council (NERC), the Medical Research Council and Natural Science Foundation of China under the framework of the Newton Innovation Fund (NE/N007190/1 and NE/N007077/1), and by the UK NERC through the project Sources of Nitrous Acid in the Atmospheric Boundary Layer (SNAABL, NE/M013405/1). We acknowledge the support from Zifa Wang and Jie Li for hosting the APHH-Beijing campaign at IAP, and thank the whole APHH team for their support throughout the project campaigns in Beijing and beyond.References
- B. Zheng, Q. Zhang, Y. Zhang, K. He, K. Wang, G. Zheng, F. Duan, Y. Ma and T. Kimoto, Atmos. Chem. Phys., 2015, 15, 2031–2049 CrossRef CAS.
- O. Welz, J. D. Savee, D. L. Osborn, S. S. Vasu, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Science, 2012, 335, 204 CrossRef CAS.
- T. Berndt, T. Jokinen, M. Sipilä, R. L. Mauldin III, H. Herrmann, F. Stratmann, H. Junninen and M. Kulmala, Atmos. Environ., 2014, 89, 603 CrossRef CAS.
- M. J. Newland, A. R. Rickard, L. Vereecken, A. Munoz, M. Rodenas and W. J. Bloss, Atmos. Chem. Phys., 2015, 15, 9521 CrossRef CAS.
- J. Wang, J. Li, J. Ye, J. Zhao, Y. Wu, J. Hu, D. Liu, D. Nie, F. Shen, X. Huang, D. D. Huang, D. Ji, X. Sun, W. Xu, J. Guo, S. Song, Y. Qin, P. Liu, J. R. Turner, H. C. Lee, S. Hwang, H. Liao, S. T. Martin, Q. Zhang, M. Chen, Y. Sun, X. Ge and D. J. Jacob, Nat. Commun., 2020, 11, 2844 CrossRef CAS.
- Y. Cheng, G. Zheng, C. Wei, Q. Mu, B. Zheng, Z. Wang, M. Gao, Q. Zhang, K. He, G. Carmichael, U. Poschl and H. Su, Sci. Adv., 2016, 2, e1601530 CrossRef.
- M. Gao, G. R. Carmichael, Y. Wang, D. Ji, Z. Liu and Z. Wang, Front. Environ. Sci. Eng., 2016, 10, 16 CrossRef.
- G. Wang, R. Zhang, M. E. Gomez, L. Yang, M. L. Zamora, M. Hu, Y. Lin, J. Peng, S. Guo and J. Meng, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13630 CrossRef CAS.
- Y. Xie, A. Ding, W. Nie, H. Mao, X. Qi, X. Huang, Z. Xu, V. M. Kerminen, T. Petäjä, X. Chi, A. Virkkula, M. Boy, L. Xue, J. Guo, J. Sun, X. Yang, M. Kulmala and C. Fu, J. Geophys. Res., 2015, 120, 12679–12694 CAS.
- R. Zhang, G. Wang, S. Guo, M. L. Zamora, Q. Ying, Y. Lin, W. Wany, M. Hu and Y. Wang, Chem. Rev., 2015, 115, 3803 CrossRef CAS.
- H. Guo, R. J. Weber and A. Nenes, Sci. Rep., 2017, 7, 12109 CrossRef.
- J. Shao, Q. Chen, Y. Wang, X. Lu, P. He, Y. Sun, V. Shah, R. V. Martin, S. Philip, S. Song, Y. Zhao, Z. Xie, L. Zhang and B. Alexander, Atmos. Chem. Phys., 2019, 19, 6107–6123 CrossRef CAS.
- H.-M. Hung and M. R. Hoffmann, Environ. Sci. Technol., 2015, 49, 13768 CrossRef CAS.
- C. Ye, P. Liu, Z. Ma, C. Xue, C. Zhang, Y. Zhang, J. Liu, C. Liu, X. Sun and Y. Mu, Environ. Sci. Technol. Lett., 2018, 5, 757–763 CrossRef CAS.
- P. He, B. Alexander, L. Geng, X. Chi, S. Fan, H. Zhan, H. Kang, G. Zheng, Y. Cheng, H. Su, C. Liu and Z. Xie, Atmos. Chem. Phys., 2018, 8, 5515 CrossRef.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, Inc., Hoboken, New Jersey, 3rd edn, 2016 Search PubMed.
- J. D. Lee, L. K. Whalley, D. E. Heard, D. Stone, R. E. Dunmore, J. F. Hamilton, D. E. Young, J. D. Allan, S. Laufs and J. Kleffmann, Atmos. Chem. Phys., 2016, 16, 2747 CrossRef CAS.
- S. Kim, T. C. VandenBoer, C. J. Young, T. P. Riedel, J. A. Thornton, B. Swarthout, B. Sive, B. Lerner, J. B. Gilman, C. Warneke, J. M. Roberts, A. Guenther, N. L. Wagner, W. P. Dubé, E. Williams and S. S. Brown, J. Geophys. Res., 2014, 119, 6886–6896 CAS.
- E. J. Slater, L. K. Whalley, R. Woodward-Massey, C. Ye, J. D Lee, F. Squires, J. R. Hopkins, R. E. Dunmore, M. Shaw, J. F. Hamilton, A. C. Lewis, L. R. Crilley, L. Kramer, W. Bloss, T. Vu, Y. Sun, W. Xu, S. Yue, L. Ren, W. J. F. Acton, C. N. Hewitt, X. Wang, P. Fu and D. E. Heard, Atmos. Chem. Phys. Discuss., 2020 DOI:10.5194/acp-2020-362.
- J. Kleffman, T. Gavriloaiei, A. Hofzumahaus, F. Holland, R. Koppmann, L. Rupp, E. Schosser, M. Siese and A. Wahner, Geophys. Res. Lett., 2005, 32, L05818 CrossRef.
- R. Oswald, M. Ermel, K. Hens, A. Novelli, H. G. Ouwersloot, P. Paasonen, T. Petäjä, M. Sipilä, P. Keronen, J. Bäck, R. Königstedt, Z. Hosaynali Beygi, H. Fischer, B. Bohn, D. Kubistin, H. Harder, M. Martinez, J. Williams, T. Hoffmann, I. Trebs and M. Sörgel, Atmos. Chem. Phys., 2015, 15, 799 CrossRef.
- X. Li, T. Brauers, R. Häseler, B. Bohn, H. Fuchs, A. Hofzumahaus, F. Holland, S. Lou, K. D. Lu, F. Rohrer, M. Hu, L. M. Zeng, Y. H. Zhang, R. M. Garland, H. Su, A. Nowak, A. Wiedensohler, N. Takegawa, M. Shao and A. Wahner, Atmos. Chem. Phys., 2012, 12, 1497 CrossRef.
- B. H. Lee, E. C. Wood, S. C. Herndon, B. L. Lefer, W. T. Luke, W. H. Brune, D. D. Nelson, M. S. Zahniser and J. W. Munger, J. Geophys. Res., 2013, 118, 12274–12281 CAS.
- L. R. Crilley, L. Kramer, F. D. Pope, L. K. Whalley, D. R. Dryer, D. E. Heard, J. D. Lee, C. Reed and W. J. Bloss, Faraday Discuss., 2016, 189, 191 RSC.
- Y. Liang, Q. Zha, W. Wang, L. Cui, K. H. Lui, K. F. Ho, Z. Wang, S. C. Lee and T. Wang, J. Air Waste Manage. Assoc., 2017, 67, 797 CrossRef CAS.
- L. J. Kramer, L. R. Crilley, T. J. Adams, S. M. Ball, F. D. Pope and W. J. Bloss, Atmos. Chem. Phys., 2020, 20, 5231 CrossRef CAS.
- B. Weber, D. Wu, A. Tamm, N. Ruckteschler, E. Rodríguez-Caballero, J. Steinkamp, H. Meusel, W. Elbert, T. Behrendt, M. Sörgel, Y. Cheng, P. J. Crutzen, H. Su and U. Pöschl, Proc. Natl. Acad. Sci. U. S. A., 2015, 15, 15384 CrossRef.
- H. Su, Y. Cheng, R. Oswald, T. Behrendt, I. Trebs, F. X. Meixner, M. O. Andreae, P. Cheng, Y. Zhang and U. Pöschl, Science, 2011, 333, 1616 CrossRef CAS.
- B. J. Finlayson-Pitts, L. M. Wingen, A. L. Sumner, D. Syomin and K. A. Ramazan, Phys. Chem. Chem. Phys., 2003, 5, 223 RSC.
- C. George, R. S. Strekowski, J. Kleffmann, K. Stemmler and M. Ammann, Faraday Discuss., 2005, 130, 195 RSC.
- J. Kleffmann, ChemPhysChem, 2007, 8, 1137 CrossRef CAS.
- F. Spataro and A. Ianniello, J. Air Waste Manage. Assoc., 2014, 64, 1232 CrossRef CAS.
- Z. Shi, T. Vu, S. Kotthaus, R. M. Harrison, S. Grimmond, S. Yue, T. Zhu, J. Lee, Y. Han, M. Demuzere, R. E. Dunmore, L. Ren, D. Liu, Y. Wang, O. Wild, J. Allan, W. J. Acton, J. Barlow, B. Barratt, D. Beddows, W. J. Bloss, G. Calzolai, D. Carruthers, D. C. Carslaw, Q. Chan, L. Chatzidiakou, Y. Chen, L. Crilley, H. Coe, T. Dai, R. Doherty, F. Duan, P. Fu, B. Ge, M. Ge, D. Guan, J. F. Hamilton, K. He, M. Heal, D. Heard, C. N. Hewitt, M. Hollaway, M. Hu, D. Ji, X. Jiang, R. Jones, M. Kalberer, F. J. Kelly, L. Kramer, B. Langford, C. Lin, A. C. Lewis, J. Li, W. Li, H. Liu, J. Liu, M. Loh, K. Lu, F. Lucarelli, G. Mann, G. McFiggans, M. R. Miller, G. Mills, P. Monks, E. Nemitz, B. Ouyang, P. I. Palmer, C. Percival, O. Popoola, C. Reeves, A. R. Rickard, L. Shao, G. Shi, D. Spracklen, D. Stevenson, Y. Sun, Z. Sun, S. Tao, S. Tong, Q. Wang, W. Wang, X. Wang, X. Wang, Z. Wang, L. Wei, L. Whalley, X. Wu, Z. Wu, P. Xie, F. Yang, Q. Zhang, Y. Zhang, Y. Zhang and M. Zheng, Atmos. Chem. Phys., 2019, 19, 7519 CrossRef CAS.
- J. Heland, J. Kleffmann, R. Kurtenbach and P. Wiesen, Environ. Sci. Technol., 2001, 35, 3207 CrossRef CAS.
- J. Kleffmann, J. Heland, R. Kurtenbach, J. C. Lorzer and P. Wiesen, Environ. Sci. Pollut. Res., 2002, 9, 48 CrossRef.
- J. Kleffmann and P. Wiesen, Atmos. Chem. Phys., 2005, 5, 77 CrossRef CAS.
- L. R. Crilley, L. J. Kramer, B. Ouyang, J. Duan, W. Zhang, S. Tong, M. Ge, K. Tang, M. Qin, P. Xie, M. D. Shaw, A. C. Lewis, A. Mehra, T. J. Bannan, S. D. Worrall, M. Priestley, A. Bacak, H. Coe, J. Allan, C. J. Percival, O. A. M. Popoola, R. L. Jones and W. J. Bloss, Atmos. Meas. Tech., 2019, 12, 6449 CrossRef CAS.
- J. Xu, S. Song, R. M. Harrison, C. Song, L. Wei, Q. Zhang, Y. Sun, L. Lei, C. Zhang, X. Yao, D. Chen, W. Li, M. Wu, H. Tian, L. Luo, S. Tong, W. Li, J. Wang, G. Shi, Y. Huangfu, Y. Tian, B. Ge, S. Su, C. Peng, Y. Chen, F. Yang, A. Mihajlidi-Zelić, D. Đorđević, S. J. Swift, I. Andrews, J. F. Hamilton, Y. Sun, A. Kramawijaya, J. Han, S. Saksakulkrai, C. Baldo, S. Hou, F. Zheng, K. R. Daellenbach, C. Yan, Y. Liu, M. Kulmala, P. Fu and Z. Shi, Atmos. Meas. Tech., 2020 DOI:10.5194/amt-2020-156.
- H. Du, J. Li, X. Chen, Z. Wang, Y. Sun, P. Fu, J. Li, J. Gau and Y. Wei, Atmos. Chem. Phys., 2019, 19, 9351 CrossRef CAS.
- R. A. Zaveri and L. K. Peters, J. Geophys. Res.: Atmos., 1999, D23, 30387 CrossRef.
- C. Fountoukis and A. Nenes, Atmos. Chem. Phys., 2007, 7, 4639 CrossRef CAS.
- F. J. Millero, Mar. Chem., 1989, 28, 1 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0fd00100g |
| ‡ Currently at Ricardo Energy & Environment, Harwell, Oxfordshire, OX11 0QR, UK. |
| § Currently at the Department of Chemistry, York University, Toronto, ON, Canada. |
| ¶ Currently at the School of Public Health, Imperial College London, UK. |
| || Currently at the College of Environmental Sciences and Engineering, Peking University, Beijing, 100871, China. |
| This journal is © The Royal Society of Chemistry 2021 |