 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInclusion of viologen cations leads to switchable metal–organic frameworks†
Laura K.
Cadman
,
Mary F.
Mahon
and
Andrew D.
Burrows
 *
*
Department of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: a.d.burrows@bath.ac.uk
First published on 19th August 2020
Abstract
The reactions of Zn(NO3)2·6H2O with the polycarboxylic acids 1,3-benzenedicarboxylic acid (H2mbdc), 1,4-benzenedicarboxylic acid (H2bdc), 1,3,5-benzenetricarboxylic acid (H3btc) and 4,4′-biphenyldicarboxylic acid (H2bpdc) in the presence of methyl viologen iodide ([MV]I2) in DMF gave anionic frameworks with methyl viologen species incorporated as counter-ions. When the reactions were carried out at 120 °C, the blue products [MV][Zn3(mbdc)4] (1-ht), [MV]0.44[H2MV]0.36[NMe2H2]0.4[Zn3(bdc)4]·0.6DMF (2-ht), [MV]0.5[Zn(btc)]·DMF (4-ht) and [MV][Zn4(bpdc)5]·8DMF·10H2O (5-ht) were formed, and these were shown to contain the radical cation [MV]˙+. In contrast, the same reactions carried out at 85 °C gave orange isostructural compounds containing the dication [MV]2+. Similar observations were made for reactions with ethyl viologen bromide. The compounds 1-ht, 2-ht and 4-ht contain similar framework topologies to analogues in which NMe2H2+ is the included cation. In contrast, 5-ht is based on a previously unreported interpenetrated network. Compound 2-ht contains the protonated species [H2MV]2+ in addition to [MV]˙+ and the crystal structure shows that the two rings in the former are staggered with respect to each other. This species is believed to form under the reaction conditions employed in the synthesis and the formation of [H2MV]2+ is suppressed by using an alternative approach in which methyl viologen is formed in situ from viologen diacetic acid. In the bdc-containing products, the radical cation is rapidly oxidised to the dication on exposure to air, as witnessed by the colour change from blue to orange. This change is reversed either by heating to 120 °C or exposure to UV radiation, both under nitrogen. This is in contrast to observations with the mbdc and btc analogues 1-ht and 4-ht, as in these compounds the blue colour persists for weeks. The difference can be related to the structures, with the channels present in 2-ht allowing oxygen to reach the radical cations.
Introduction
One of the most significant features of metal–organic frameworks (MOFs) is their relative ease of functionalisation.1 Taken together with the concept of isoreticular synthesis,2 this provides a powerful means of controlling pore environments, which in turn allows the properties of the materials to be tuned. MOFs can be functionalised to some extent by the use of pre-functionalised linkers in the synthesis or, more generally, by post-synthetic modification techniques.3,4 A less well-studied means of controlling the properties of a MOF is by inclusion of an appropriate guest molecule within the pores.5Generally, as-synthesised MOFs contain solvent molecules and/or unreacted starting materials within the pores of the structure. These guests can often be removed through an activation process to give a guest-free framework, and many studies have concentrated on the use of these activated porous materials to adsorb gaseous species such as hydrogen6 or carbon dioxide.7 Increasingly, researchers are looking to include a wider range of molecules as potential guests within porous frameworks, including organic dyes,8 drug molecules,9 reaction intermediates10 and products.11
Host–guest complexes can also give rise to properties that are native to neither the MOF nor the guest.5,12 Allendorf and co-workers reported the inclusion of tetracyanoquinodimethane (TCNQ) into [Cu3(btc)2] (HKUST-1).13 This afforded a charge transfer material that displays significant electrical conductivity. Neither HKUST-1 nor TCNQ are conductive in isolation and it is thought that the binding of the TCNQ molecules to copper centres in the HKUST-1 framework leads to a conductive channel through the MOF.
In addition to host–guest systems, the inclusion of particular counter-ions into charged frameworks can lead to the MOF material displaying emergent properties. For example, viologen-containing frameworks have been studied for their thermochromic and photochromic properties. Viologen (N,N′-disubstituted bipyridinium) species undergo reversible redox reactions that are typically accompanied by distinct colour changes.14 As such, these species have been studied as redox indicators in biological systems and they have more recently gained attention for their potential uses in electrochromic displays and photochemical applications.15
The vast majority of studies on viologens have been conducted in the solution phase. Here, they exhibit three redox states, the dication V2+, radical cation V˙+ and neutral V0, with the transformations between these shown in Scheme 1.
 | ||
| Scheme 1 Redox transformations undertaken by viologen species. | ||
The first reduction, from V2+ to V˙+, is reversible and can be cycled multiple times. However, the second reduction, from V˙+ to V0, generally has limited reversibility due to the insoluble nature of the V0 product.14 These redox reactions have been studied predominantly through electrochemistry techniques, although chemically reduced reactions have also been reported.16
Each redox step is associated with a distinct colour change. For example, methyl viologen dichloride, [MV]Cl2, contains R = Me and is a yellow solid in its dication form, MV2+. It can undergo a reduction to give the deep blue radical cation, MV˙+, whereas the neutral MV0 compound has been reported to be yellow/orange in a solution of acetonitrile and heptane.16 Viologen radicals react rapidly with molecular oxygen to regenerate the dication species. As this reaction is accompanied by a colour change, it has been proposed as a process that might be employed for detection of low concentrations of dissolved oxygen in solution.17 Viologen solutions have also demonstrated reduction of V2+ to V˙+ through thermal or photosensitised methods.18 This process has been demonstrated through heating or UV irradiation of viologens in alkaline aqueous or alcoholic solutions in the absence of air.16
While the study of the photochromic behaviour of viologens in solution is well documented, reports of solid state viologen photochromism are less common. Crystal packing influences the success of viologen photochromism in the solid state, with geometric restraints making photoinduced electron transfer between the donor and the viologen acceptor moieties difficult to achieve.19
Examples of both the inclusion of viologens as ligands or as counter-ions in the pores of charged MOFs have been reported. For example, Fu and co-workers reported the use of the viologen-based ligand N-(3-carboxyphenyl)-4,4-bipyridinium (cpbpy) and 1,3-benzenedicarboxylate (mbdc) in [Cd(cpbpy)(mbdc)]·H2O.20 Crystals of the product undergo a colour change from yellow to blue upon exposure to sunlight, indicating the formation of the radical viologen species.
A methyl viologen MOF with the formula [MV][Zn3(mbdc)4] has recently been reported from the reaction of Zn(NO3)2·6H2O with H2mbdc and viologen diacetic acid dichloride, which hydrolyses to generate methyl viologen cations during the synthesis.21 [MV][Zn3(mbdc)4] displays both photochromic and thermochromic behaviours, changing from yellow/orange to blue upon light irradiation or heating to above 120 °C. A short N⋯O contact (3.256(5) Å) was reported to lead to charge transfer between the framework and the MV2+ cation, reducing the viologen to its blue radical cation form. Once generated, the radical species is stable in air for several weeks, forming a long-lived charge separated state. This behaviour contrasts with that in solution, in which the methyl viologen radical cation rapidly oxidises to MV2+ in air.
We were interested in determining whether viologen-containing MOFs could be formed in a more rational manner through direct synthesis in the presence of the viologen species, rather than via decomposition of viologen diacetic acid dichloride. To this end, the zinc-based anionic frameworks [Zn3(bdc)4]2−, [Zn(btc)]− (btc = 1,3,5-benzenetricarboxylate) and [Zn3(bpdc)4]2− (4,4′-biphenyldicarboxylate) were selected for study alongside [Zn3(mbdc)4]2−, with methyl viologen diiodide, [MV]I2, and ethyl viologen dibromide, [EV]Br2, chosen as viologen sources.
Results and discussion
Direct synthesis of [MV][Zn3(mbdc)4]
In order to determine whether [MV][Zn3(mbdc)4] could be prepared directly, rather than via viologen diacetic acid dichloride, the reaction between Zn(NO3)2·6H2O, H2mbdc and [MV]I2 was carried out in DMF at both 85 °C and 120 °C. The lower temperature was selected to mirror the conditions used in the synthesis of [MV][Zn3(mbdc)4] from viologen diacetic acid, whereas the higher temperature was chosen as the reported temperature at which [MV][Zn3(mbdc)4] forms methyl viologen radical cations.The reaction carried out at 85 °C yielded a mixture of yellow and blue crystals (1-lt), while the reaction at 120 °C gave a product containing only blue crystals (1-ht). Powder X-ray diffraction analysis of 1-lt and 1-ht (Fig. S1†) showed similar peak positions and relative intensities both to each other and to the pattern simulated from the reported crystal structure of [MV][Zn3(mbdc)4].21 This confirms that direct synthesis does indeed give the same framework as the reaction with viologen diacetic acid dichloride. Furthermore, from the colours of the products, it can be postulated that 1-lt contains some crystals containing methyl viologen in the yellow dicationic form (MV2+) and others containing methyl viologen in the blue radical cationic form (MV˙+), whereas 1-ht only contains crystals with MV˙+ radical cations. The similarity of the powder X-ray diffraction patterns of 1-lt and 1-ht provides good evidence that the oxidation state of the methyl viologen cation within the framework, MV2+ or MV˙+, has no effect on the gross structure. The blue colour of 1-ht persists when it is left in air at ambient temperature for at least three months, consistent with the previously reported data.21
1H NMR spectra of DCl-digested samples of 1-lt and 1-ht (Fig. S2†) show the presence of D2mbdc (δ/ppm 7.61t, 8.11d and 8.42s) and MV2+ (δ/ppm 4.43s, 8.73d and 9.26d), with the orange-yellow colour of the solution confirming that digestion occurs alongside oxidation of the radical cation. Analysis of the integrals reveals that both 1-lt and 1-ht contain mbdc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MV ratios of 4:1, which is consistent with both compounds having the formula [MV][Zn3(mbdc)4]. Conversion of MV2+ to MV˙+ occurs through electron transfer from the framework to the methyl viologen cation, so no change in formula is needed despite the change in cation charge.21 Thermogravimetric analysis of the product found no included solvent molecules to be present within the structure (Fig. S3†).
:MV ratios of 4:1, which is consistent with both compounds having the formula [MV][Zn3(mbdc)4]. Conversion of MV2+ to MV˙+ occurs through electron transfer from the framework to the methyl viologen cation, so no change in formula is needed despite the change in cation charge.21 Thermogravimetric analysis of the product found no included solvent molecules to be present within the structure (Fig. S3†).
Reaction of zinc(II) nitrate and H2bdc with [MV]I2
The reported synthesis for [NMe2H2]2[Zn3(bdc)4]·DMF·H2O22 involves the reaction of zinc nitrate with H2bdc in DMF, with the addition of [NMe2H2]Cl used to template the formation of the ionic product, though dimethylammonium cations can also be derived from the solvent. The reaction of Zn(NO3)2·6H2O with H2bdc and [MV]I2 was carried out in a similar manner. The reaction was conducted at both 85 °C and 120 °C, over 48 h, to determine the effect of reaction temperature on product formation and the methyl viologen form. The reactions yielded a mixture of blue and yellow crystals (2-lt) from the reaction at 85 °C and blue crystals (2-ht) from the reaction at 120 °C. As with 1-lt and 1-ht, this suggests that the lower temperature gives a product containing both MV2+ and MV˙+, whereas the higher temperature gives only MV˙+.Powder X-ray diffraction analysis (Fig. S4†) revealed that 2-lt and 2-ht have similar gross features to one another and contain peaks at similar positions to those observed in the powder diffraction pattern simulated from the crystal structure of [NMe2H2]2[Zn3(bdc)4]·DMF·H2O.22 Some additional peaks are present in the experimental powder patterns that may result from the presence of the larger and more rigid methyl viologen cations.
The methyl viologen radicals within 2-lt and 2-ht were notably less stable in air than those in 1-ht, with the blue crystals turning orange (2-ht′) within 5–10 minutes of air exposure. The MV˙+ species could be regenerated by either heating the orange crystals to 120 °C in oxygen-free conditions or by UV irradiation in the absence of oxygen. Under either of these conditions the orange crystals undergo a colour change to blue, which is reversed upon exposure to air. These photochromic and thermochromic conversions are summarised in Scheme 2.
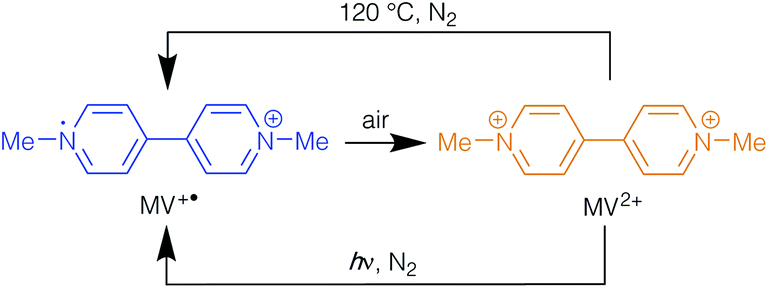 | ||
| Scheme 2 Transformations of the methyl viologen cation in 2-ht. | ||
Electron paramagnetic resonance (EPR) spectroscopy was carried out on solid-state samples of both 2-ht and 2-ht′. The EPR spectrum of the blue crystals of 2-ht is shown in Fig. 1. The signal is indicative of an organic radical species, confirming that the included methyl viologen counter-ions are present as MV˙+. The EPR spectrum of the orange crystals of 2-ht′ shows no signal, in contrast, which confirms that there are no radicals present after exposure to air, consistent with the oxidation of MV˙+ to MV2+.
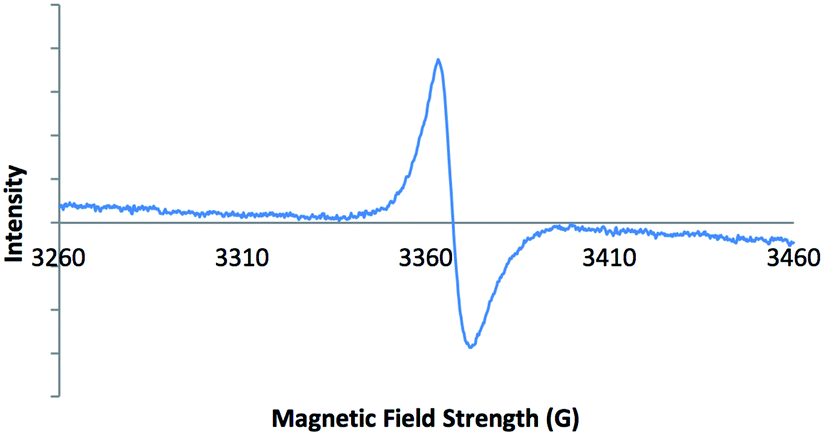 | ||
| Fig. 1 EPR spectrum of 2-ht, confirming the presence of a radical species. | ||
As with 1-lt and 1-ht, samples of 2-ht turned orange on digestion in DCl/D2O/DMSO-d6, consistent with the conversion of MV˙+ to MV2+. The 1H NMR spectrum (Fig. S6†) showed the expected presence of D2bdc (δ/ppm 8.02s) and MV2+ (δ/ppm 4.44s, 8.78d and 9.31d), but also another organic species with a similar symmetry to MV2+ (δ/ppm 4.30s, 8.01(sh), 8.93d), the spectrum of which was partly obscured by the strong D2bdc signal. Recording the 1H NMR spectrum in D2O/DCl in the absence of DMSO-d6 (Fig. S7†) reduced the intensity of the D2bdc resonance due to its poor solubility in DMSO, allowing the feature at δ 8.01 ppm to be resolved into a doublet. 1H–1H COSY and NOESY spectra of the samples digested in D2O/DCl/DMSO-d6 (Fig. S8 and S9†) confirmed that the resonances at δ 4.30, 8.01 and 8.93 ppm were from the same compound.
Crystals of 2-ht were suitable for single crystal X-ray analysis, and data collection was carried out at 150 K. The blue colour of the compound was retained throughout the data collection. The asymmetric unit contains two zinc atoms, Zn(1) at full occupancy and Zn(2) with site occupancy of 50%, one complete and two half bdc ligands, one fractional occupancy DMF molecule and half of a methyl viologen cation with a site occupancy of 80%.
Zn(1) displays distorted tetrahedral geometry, whilst Zn(2) lies on an inversion centre and displays distorted octahedral geometry. The SBUs of the framework are formed from three zinc atoms and eight carboxylate groups, and are linked together into sheets through the benzene rings attached to six of the carboxylates (Fig. 2a). These sheets are further linked into a three-dimensional net through bdc linkers coordinated to Zn(1) (Fig. 2b). There are channels present within the structure, which contain the included solvent molecules and cations. The network formed by 2-ht is similar to that previously reported for [NMe2H2]2[Zn3(bdc)4]·DMF·H2O.22
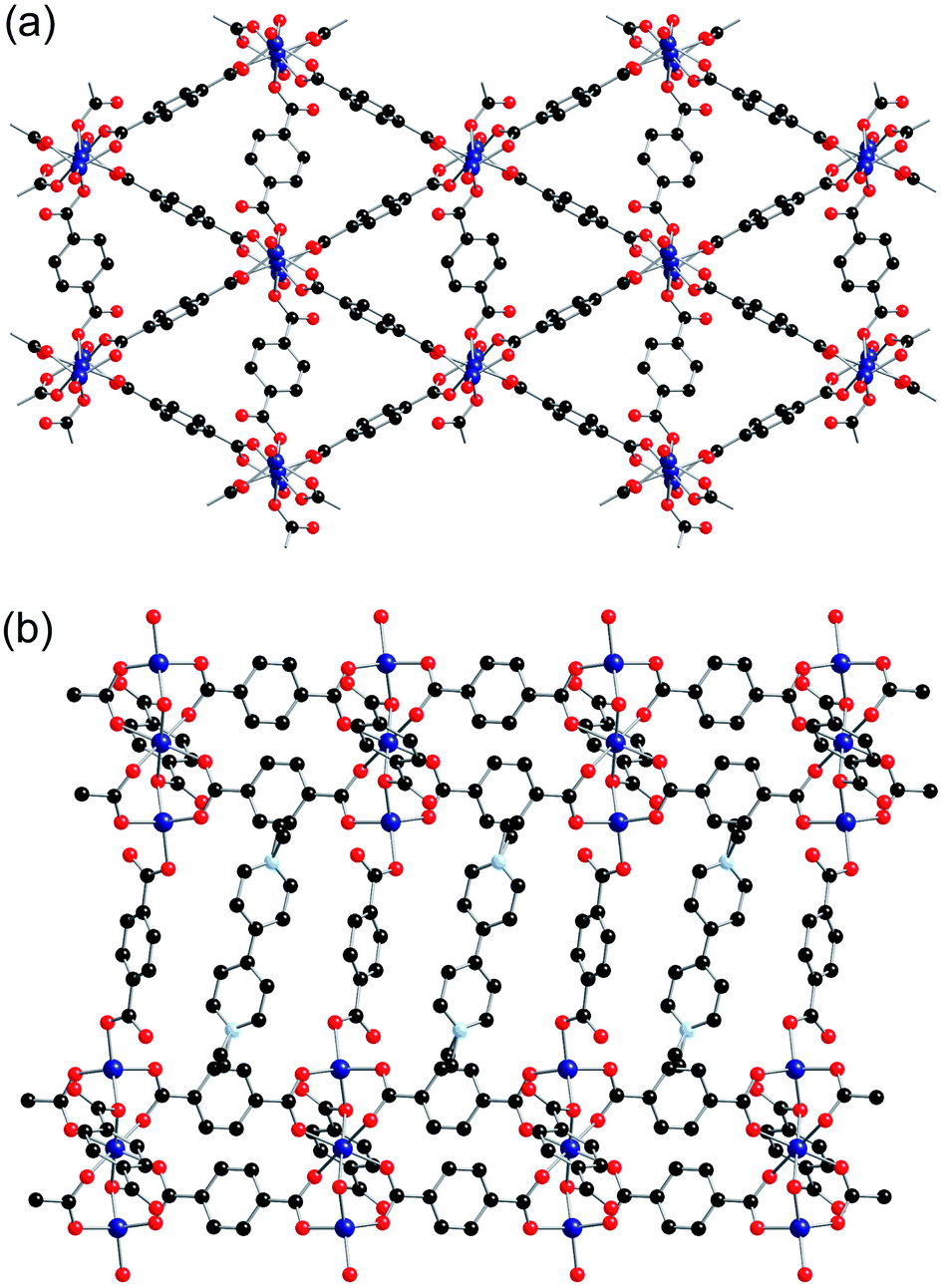 | ||
| Fig. 2 The structure of 2-ht, showing (a) the two-dimensional Zn3(bdc)3 sheets and (b) linking of the sheets into a 3D structure through additional bridging bdc linkers, showing the positions of the cations between layers. Hydrogen atoms are omitted for clarity. | ||
Although the electron density of the methyl viologen cation is smeared, it is notable that the methyl group attached to N(1) is disordered over two positions, C(22) and C(22A). One of these is co-planar with the pyridyl ring to which it is attached, while the other is notably out of the plane. We have therefore interpreted the cation as a mixture of MV˙+ and the doubly-protonated ion [H2MV]2+. The presence of the two cations in 2-ht is consistent with the 1H NMR spectrum of the digested compound, and the spectrum was used to attribute site occupancies for C(22) and C(22A) as 44% and 36%, respectively. Due to the smearing of the electron density, restraints on the atomic displacement parameters and the N–C and C–C distances were required for the MV˙+/H2MV2+ moieties. Overall, this led to a formula for 2-ht of [MV]0.44[H2MV]0.36[NMe2H2]0.4[Zn3(bdc)4]·0.6DMF, with the dimethylammonium ions required for charge balance.
A comparison of the structures of 2-ht and [MV][Zn3(mbdc)4] reveals a clear difference in the methyl viologen geometry. The MV˙+ cations present in [MV][Zn3(mbdc)4] adopt a planar geometry (Fig. 3a), whereas in the disordered MV˙+/H2MV2+ cations of 2-ht the pyridyl rings are twisted with respect to one another, with an interplanar angle of 80° (Fig. 3b).
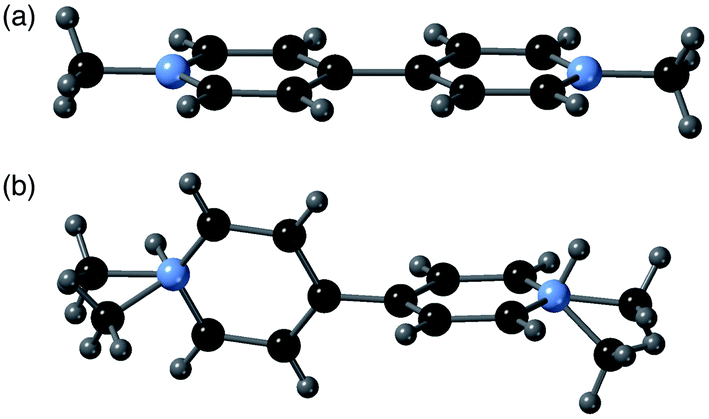 | ||
| Fig. 3 (a) The planar MV˙+ cations in [MV][Zn3(mbdc)4] (1-ht), and (b) the disordered MV˙+/H2MV2+ cations in [MV]0.44[H2MV]0.36[NMe2H2]0.4[Zn3(bdc)4]·2H2O·1.2DMF (2-ht). | ||
MV2+ dications are known to display a twisted geometry, but MV˙+ radical cations are usually planar in their geometry, allowing for stabilisation of the radical across both rings. Compound 2-ht is unusual in that it contains a radical species, unambiguously identified through colour and EPR spectroscopy, but containing a twisted geometry. 1-ht contains square channels in which the MV˙+ cations lie parallel to the mbdc ligands in the framework, oriented into the channels and stacked end-to-end. In 2-ht, the pores are narrower and rectangular in shape, and the methyl viologen cations stack with the rings parallel to one another in a face-to-face arrangement. The narrower pores of the [Zn3(bdc)4]2− framework together with the stacking arrangement of the MV˙+/H2MV2+ cations suggest that steric interactions may play a role in the observed ring twist.
A non-coordinated oxygen atom from a carboxylate group is directed towards the nitrogen atom on the methyl viologen in 2-ht, in a similar manner to that observed in [MV][Zn3(mbdc)4].21 The O⋯N distance is 3.635(9) Å, within the expected value for a charge transfer donor–acceptor pair. This contact is likely to facilitate electron transfer between the framework and the MV2+ dication.
The H2MV2+ cation, observed in the structure of 2-ht and the 1H NMR spectrum of the digested MOF, has not been previously reported to the best of our knowledge, though the singly protonated species, HMV+, is hypothesised to form when MV˙+ radicals are heated in acidic conditions,23 and the non-protonated species MV0 is well-established. H2bdc is an acidic source, which may account for the formation of H2MV2+ during the synthesis.
Reaction of zinc(II) nitrate and H2bdc with viologen diacetic acid dichloride
In order to ascertain whether the in situ generation of methyl viologen cations would lead to a product that did not contain H2MV2+ ions, the reaction between Zn(NO3)2·6H2O, H2bdc and viologen diacetic acid dichloride was investigated. The reaction was carried out in DMF at both 85 °C and 120 °C for 48 h, and the reactions generated orange crystals (3-lt) and blue crystals (3-ht), respectively.The PXRD patterns of 3-lt and 3-ht both show peaks in similar positions to those of 2-ht (Fig. S11†), though in the case of 3-ht extra peaks suggest the presence of an additional phase. The 1H NMR spectrum of 3-lt, digested in DCl/D2O/DMSO-d6, shows the presence of D2bdc (δ/ppm 8.02s) and MV2+ (δ/ppm 4.45s, 8.80d, 9.33d), but no evidence for the formation of H2MV2+ (Fig. S12†). This confirms that by generating methyl viologen in situ, the presence of H2MV2+ can be prevented. Analysis of the integrals in the spectrum of digested 3-lt gives a D2bdc:MV ratio of 83:17, leading to a formula of [MV]0.8[NMe2H2]0.4[Zn3(bdc)4], with the dimethylammonium cations required for charge balance. A comparison with 2-ht shows a similar total viologen content, with 0.8 viologen moieties per unit formula. On heating or UV exposure, 3-lt turns blue, with the expected formation of MV˙+.
It can therefore be concluded that methyl viologen salts of [Zn3(bdc)4]2− can be formed either through the reaction of zinc(II), H2bdc and viologen diacetic acid dichloride or through direct synthesis using [MV]I2, though some conversion of the methyl viologen to H2MV2+ occurs under the latter conditions.
Synthesis and characterisation of [MV]0.5[Zn(btc)]·DMF
[NMe2H2][Zn(btc)]·DMF has been prepared previously by combining zinc(II) acetate with 1,3,5-benzenetricarboxylic acid (H3btc) in DMF at 140 °C over 48 h.24 The SBU contains two zinc atoms that are bridged by two bidentate carboxylate groups. Two further carboxylate groups coordinate in a unidentate manner to each zinc centre. The SBUs are linked together by the btc ligands to give a three-dimensional network that contains one-dimensional rectangular channels.The synthesis of the methyl viologen derivative of [Zn(btc)]− was carried out by combining Zn(NO3)2·6H2O with H3btc and [MV]I2 in DMF at 120 °C over 48 h. The reaction yielded blue crystals (4-ht), suggesting the presence of methyl viologen in its radical cation form. The synthesis was also carried out by heating the same starting materials at 85 °C for 48 h and a microcrystalline, orange product (4-lt) was obtained, typically indicative of the structure containing methyl viologen in its dicationic form.
Both products were analysed by powder X-ray diffraction (Fig. S13†). The X-ray powder diffraction pattern for 4-lt contained only a few low intensity peaks against high background noise, suggesting poor crystallinity, and no further analysis was undertaken on this material. The X-ray powder diffraction pattern for 4-ht showed better crystallinity, and revealed there were some features in common with the pattern simulated from the X-ray crystal structure of [NMe2H2][Zn(btc)]·DMF. However, there were significant differences between the two patterns, suggesting structural differences between the two compounds.
The EPR spectrum of 4-ht confirmed the presence of methyl viologen radical cations (Fig. S15†). The 1H NMR spectrum of acid-digested 4-ht (Fig. S16†) revealed the presence of both D3btc (δ/ppm 8.62s) and methyl viologen (δ/ppm 4.45s, 8.81d, 9.33d). The solution was orange in colour, indicating that digestion had, as expected, converted MV˙+ to MV2+.
A blue crystal of 4-ht was analysed by single crystal X-ray crystallography. The asymmetric unit consists of one zinc centre, one btc ligand, one DMF molecule, disordered over two positions, and half a methyl viologen cation, which lies proximate to an inversion centre. This gives a formula for 4-ht as [MV]0.5[Zn(btc)]·DMF.
The btc ligand coordinates through each of the three carboxylate groups to four different zinc centres. Two of the carboxylate groups are monodentate, whereas the third binds to two zinc centres through both oxygen atoms. There was no credible crystallographic evidence for the presence of hydrogen atoms on non-coordinated oxygen atoms. The zinc centre is in a distorted tetrahedral coordination environment, coordinating to four carboxylate groups from four different btc ligands. The SBU consists of a dimeric zinc unit bridged by two carboxylate groups, and these are linked together by the btc linkers to form chains running along the a-axis, which are themselves linked into a three-dimensional network containing channels along the a-axis (Fig. 4). The channels are occupied by methyl viologen cations, which lie in a near-perpendicular arrangement to the btc ligands of the framework. The methyl viologens stack down the channels, alternating between layers of DMF solvent molecules.
 | ||
| Fig. 4 The structure of 4-ht, showing (a) Zn2(btc)2 chains and (b) the linking of the chains to form a 3D structure, showing the position of the cations. Hydrogen atoms and included DMF molecules have been omitted for clarity. | ||
One of the non-coordinated oxygen atoms from the btc ligand is directed towards the nitrogen atom of the methyl viologen. The O⋯N distance is 2.972(4) Å, which suggests a charge transfer interaction between the framework and methyl viologen, in a similar manner to that observed in 2-ht.
The [MV]0.5[Zn(btc)]·DMF structure displays a similar framework connectivity to that observed in [NMe2H2][Zn(btc)]·DMF. Both systems crystallise in the monoclinic space group P21/n but display some differences in their unit cell parameters, which is consistent with the differences in the X-ray powder diffraction patterns observed between the two species. A clear difference between the two structures can be seen in the pore geometries, with those in 4-ht more square (Fig. 5a) than those in [NMe2H2][Zn(btc)]·DMF, which are more rectangular (Fig. 5b). This difference in pore geometry is related to a compression of the Zn⋯Zn distances in the dimeric SBUs of 4-ht compared to those of [NMe2H2][Zn(btc)]·DMF. The Zn⋯Zn distance across the SBU of [NMe2H2][Zn(btc)]·DMF is 4.0133(7) Å, whilst in 4-ht the zinc atoms are offset and have a reduced Zn⋯Zn distance of 3.8250(7) Å. This distortion enables the larger MV˙+ ions to be accommodated in the channels.
 | ||
| Fig. 5 The network structures in (a) 4-ht and (b) [NMe2H2][Zn(btc)]·DMF, with included solvent molecules and cations removed for clarity, and the dimeric secondary building units of (c) 4-ht and (d) [NMe2H2][Zn(btc)]·DMF. | ||
Crystals of 4-ht retain their blue colour when exposed to air for at least six weeks. Similar stability of the methyl viologen radical was observed for 1-ht, which contains 1,3-benzenedicarboxylate.
Synthesis and characterisation of [MV][Zn4(bpdc)5]·8DMF·10H2O
The previously reported MOF [NMe2H2]2[Zn3(bpdc)4]·5DMF (bpdc = 4,4′-biphenyldicarboxylate)25 is isoreticular to [NMe2H2]2[Zn3(bdc)4]·DMF·H2O, though the longer length of the linkers leads to this structure being interpenetrated. [NMe2H2]2[Zn3(bpdc)4]·5DMF was synthesised in the reaction between Zn(NO3)2·6H2O, H2bpdc and [NMe2H2]Cl in DMF at 120 °C for 48 h. Undertaking the reaction using [MV]I2 in place of [NMe2H2]Cl gave blue crystals of 5-ht, whereas when the reaction was carried out at 85 °C, orange-yellow crystals of 5-lt were produced. Both 5-ht and 5-lt gave very different X-ray powder diffraction patterns (Fig. S18†) from that simulated for the single crystal structure of [NMe2H2]2[Zn3(bpdc)4]·5DMF, suggesting the formation of structurally different products. The pattern for 5-lt shows some similarity with that for 5-ht, but the lower intensity of the peaks and higher background noise suggests lower crystallinity, and as a consequence the remaining analyses were carried out only on 5-ht.In common with many of the other compounds reported herein, 5-ht changes colour from blue to orange on digestion in DCl/D2O/DMSO-d6, consistent with the conversion of MV˙+ into MV2+. The 1H NMR spectrum (Fig. S20†) showed the presence of D2bpdc (δ/ppm 7.76d, 8.03d) and MV2+ (δ/ppm 4.45s, 8.80d, 9.32d), with the integrals revealing a bpdc:MV ratio of 88:12.
A crystal of 5-ht was suitable for single crystal X-ray diffraction analysis and the asymmetric unit was found to contain eight zinc atoms, ten bpdc ligands, sixteen DMF solvent molecules and two methyl viologen cations. The water content could not be resolved in the crystal structure and was treated with PLATON SQUEEZE. A value of twenty water molecules per asymmetric unit was determined using this solvent mask algorithm, giving a formula for 5-ht of [MV][Zn4(bpdc)5]·8DMF·10H2O.
All eight of the independent zinc atoms in the asymmetric unit are four coordinate, displaying distorted tetrahedral geometry. Pairs of zinc atoms are bridged by three bidentate carboxylate groups of bpdc ligands to form dimeric SBUs and are further coordinated by two axially bound monodentate carboxylate groups to give Zn2(O2C)5 SBUs (Fig. 6a). Six of the ten bpdc ligands within the asymmetric unit coordinate to four different zinc atoms employing all of the oxygen atoms, whilst the remaining four bpdc ligands coordinate through two oxygen atoms to two zinc atoms. There is no evidence for the presence of hydrogen atoms on the non-coordinated carboxylate oxygen atoms.
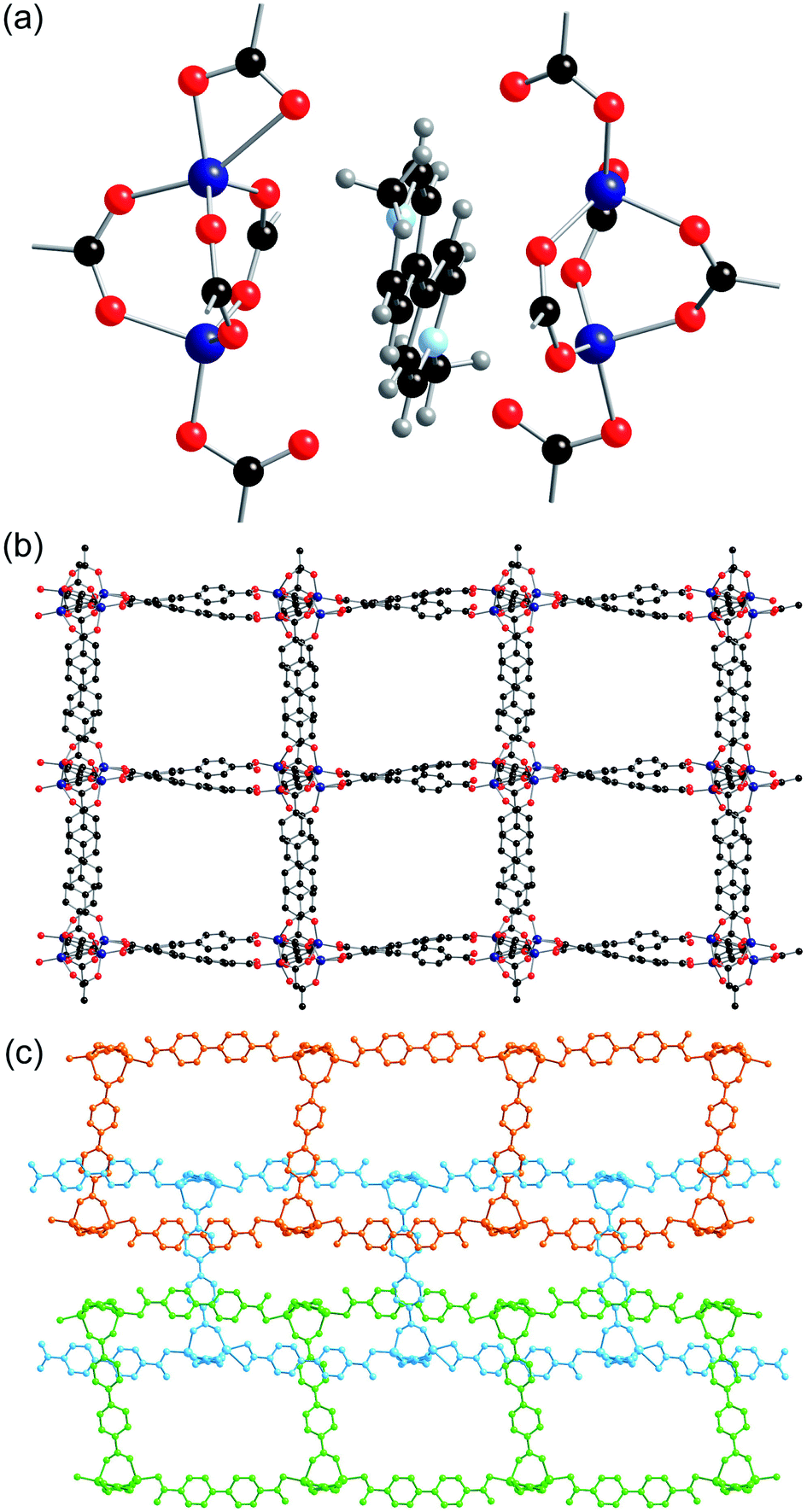 | ||
| Fig. 6 The structure of 5-ht, showing (a) independent Zn2(CO2)5 SBUs sandwiching the MV˙+ cation, (b) the two-dimensional sheets viewed down the c-axis, and (c) the interpenetrated three-dimensional structure viewed down the b-axis. | ||
The SBUs are linked together by the bpdc linkers to form a two-dimensional network (Fig. 6b). The networks are interpenetrated, and the interpenetration generates a three-dimensional structure (Fig. 6c).
The gross structure contains one-dimensional channels along the b-axis which are occupied by DMF solvent molecules and methyl viologen cations. The MV˙+ cations exhibit close to planar geometry and alternate with DMF molecules down the channels. As was observed in both 2-ht and 3-ht, uncoordinated oxygen atoms from the dicarboxylate ligands are directed towards the methyl viologen cations, with N⋯O distances of 3.084 and 3.097 Å. Charge transfer between the framework and the methyl viologen is therefore likely to occur during the synthesis, forming the MV˙+ radical cation species and accounting for the observed blue colour of the product. The N⋯O interactions are supported by C–H⋯O contacts within the hydrogen bonding range [C(163)–H(163)⋯O(7): C⋯O 3.295, H⋯O 2.40 Å, C–H⋯O 156°; C(148)–H(148)⋯O(38): C⋯O 3.198, H⋯O 2.30 Å, C–H⋯O 159°].
Relative stability to air of the methyl viologen containing MOFs
In the bdc-containing MOFs, 2-ht and 3-ht, the MV˙+ cations oxidise in air to MV2+ within a few minutes. However, for both [MV][Zn3(mbdc)4], 1-ht, and [MV]0.5[Zn(btc)]·DMF, 4-ht, the methyl viologen radical persists in air for several weeks. Analysis of the crystal structures enables this difference in behaviour to be rationalised. The frameworks of 1-ht and 4-ht both contain one-dimensional channels that are occupied by MV˙+ cations and/or solvent molecules. Scrutiny of the voids within these structures calculated based on the contact surface shows that in 1-ht only small, isolated pockets remain unoccupied, forming 1.6% of the unit cell volume. This is supported by the reported thermogravimetric analysis of this compound, in which no mass loss corresponding to solvent within the pores of the structure is observed. In the case of 4-ht, no unoccupied accessible pores are available. This means that the MV˙+ radical cations remain isolated from the external environment of the MOF, allowing them to persist even when the MOF is exposed to air.The structure of 2-ht also contains one-dimensional channels occupied by MV˙+ cations and solvent molecules. Void calculations show that in addition to these channels, two further pore types are present, making up a total of 11.8% of the unit cell volume. Some of these regions contain DMF and water solvent molecules that were not resolved in the crystal structure but were found to be present through thermogravimetric analysis. The larger of the two pore types link together along the b-axis of the structure and form channels through which oxygen can conceivably migrate. This offers an explanation as to why the MV˙+ radical cations within the pores of the 2-ht/3-ht framework are so rapidly oxidised. Despite these differences in structure and behaviour, none of the networks showed significant nitrogen adsorption after attempted activation by solvent exchange and heating.
Ethyl viologen containing MOFs
Following the successful inclusion of MV˙+ cations into anionic zinc-carboxylate MOFs, the investigation was extended to include ethyl viologen cations into similar frameworks. This aimed to determine if any differences in the product structure, radical formation or stability occurred with variation of the viologen substituents. Two systems were investigated with ethyl viologen as the cation, those based on the [Zn3(mbdc)4]2− and [Zn3(bdc)4]2− frameworks. These frameworks were selected as they displayed differences to one another in their ability to stabilise the radical form of the MV˙+ species.The reaction between Zn(NO3)2·6H2O, H2mbdc and [EV]Br2 was carried out in DMF at 120 °C for 48 h, and yielded blue crystals of 6. The powder X-ray diffraction pattern for 6 showed Bragg peaks with similar positions and relative intensities as that for 1-ht (Fig. S21†), suggesting both frameworks have similar topologies. The blue colour of 6 suggests that ethyl viologen is present in the radical cation state (EV˙+) within the pores of the MOF, and in samples exposed to air the colour is maintained for at least four weeks, suggesting similar stability to the methyl viologen analogue 1-ht.
Digestion of 6 in DCl/D2O/DMSO-d6 occurred with a colour change to orange, consistent with the conversion of EV˙+ to EV2+. The 1H NMR spectrum (Fig. S22†) revealed the presence of both D2mbdc (δ/ppm 7.64t, 8.15d and 8.45s) and EV2+ (δ/ppm 1.59t, 4.75q, 8.83d, 9.46d). Analysis of the integrals revealed a mbdc:EV ratio of 4:1, and thermogravimetric analysis (Fig. S23†) showed no mass loss associated with included solvent in the structure. Taken together, this gives a formula for 6 as [EV][Zn3(mbdc)4], which is directly analogous to the product formed with methyl viologen.
Zn(NO3)2·6H2O was also reacted with H2bdc and [EV]Br2 in DMF at 120 °C for 48 h. The reaction yielded blue crystals of 7 that rapidly turned orange (7′) upon exposure to air. Analysis of the PXRD pattern (Fig. S24†) showed that 7 was isostructural to 2-ht. The 1H NMR spectrum of 7 following digestion in DCl/D2O/DMSO-d6 (Fig. S25†) showed the presence of D2bdc (δ/ppm 8.02s), EV2+ (δ/ppm 1.59t, 4.72q, 8.82d and 9.44d) and a third species with similar symmetry to EV2+ (δ/ppm 1.50t, 4.60q, 8.01(sh) and 9.07d). By comparison with 2-ht, this species was identified as H2EV2+, and it is likely that this was formed during the synthesis as a result of acidification of EV2+.
A crystal from this reaction was suitable for single crystal X-ray diffraction analysis. The crystal was orange in colour (i.e.7′) when the data collection was carried out, indicating the ethyl viologen cations within the crystal are not present as EV˙+ ions. The asymmetric unit of 7′ contains two crystallographically independent zinc atoms, one present at full occupancy and the other at 50% occupancy and proximate to an inversion centre. One full bdc ligand is present, along with two half bdc ligands. Two molecules of DMF and one H2O molecule, each with partial site occupancy, are also present, along with half an EV2+ dication with attributed site occupancy of 40%. The SBUs and the gross structure of 7′ are similar to those observed for 2-ht.
A total solvent content of 1.4 DMF and 0.7 H2O molecules per three zinc centres was refined crystallographically. Treatment with PLATON SQUEEZE revealed additional electron density in the pores of the framework, giving a total solvent content of 1.4 DMF and 1.3 H2O molecules. The composition of 7′ in the crystal structure was therefore determined to be [EV]0.54[H2EV]0.26[NMe2H2]0.4[Zn3(bdc)4]·1.4DMF·1.3H2O.
The viologen moiety shows positional disorder, which required atomic displacement, planarity and distance restraints to achieve a chemically sensible convergence. The EV2+ and H2EV2+ moieties could not be resolved in the crystal structure due to smearing of the electron density and the relatively low proportion of H2EV2+.
The EV2+/H2EV2+ moiety displays twisting of the two rings relative to one another, with a plane to plane angle of 52°, which is slightly less than the equivalent angle present in 2-ht. The position of the viologen cations in the pores of 7′ is comparable to that of 2-ht, and the O⋯N distance between the viologen and non-coordinating oxygen of the bdc ligand (3.400 Å) is within the expected range for charge transfer to occur.
Conclusions
The formation of four zinc MOFs with MV˙+ cations has been successfully achieved through direct synthesis with [MV]I2. In three of the systems investigated, 1-ht, 2-ht and 4-ht, the MOFs were found to have a similar framework topology to the structures in which NMe2H2+ was the counterion. In contrast, inclusion of [MV]I2 in the synthesis with Zn(NO3)2·6H2O and H2bpdc yielded a new interpenetrated framework structure with the formula [MV][Zn4(bpdc)5]·8DMF·10H2O.In the case of the [Zn3(bdc)4]2− framework, direct synthesis with [MV]I2 resulted in the formation of the protonated species H2MV2+, which was included into the framework along with MV˙+ or MV2+ cations. H2MV2+ is believed to form under the reaction conditions employed in the synthesis of the MOF. The generation of this protonated viologen species was avoided by using an alternative synthesis in which the methyl viologen was formed in situ from viologen diacetic acid.
All methyl and ethyl viologen-containing MOFs were formed as blue crystalline products when synthesised at 120 °C. This colour is consistent with the formation of MV˙+ and the presence of this radical cation was confirmed through EPR spectroscopy. Single crystal X-ray analysis of the products showed that, in all cases, the methyl viologen species are in close proximity to non-coordinated oxygen atoms of the framework (less than 3.7 Å apart), allowing the frameworks to act as electron donors in the reduction of viologens from their dicationic to radical cationic states during the reaction procedure.
For the bdc-containing products, 2-ht and 7, the radical was rapidly oxidised to its dicationic form on exposure to air, mimicking the behaviour of methyl viologen in the solution phase. Such materials may have applications in areas such as oxygen detection. In contrast, for [MV][Zn3(mbdc)4], 1-ht, [MV]0.5[Zn(btc)]·DMF, 4-ht, and [EV][Zn3(mbdc)4], 6, the blue colour of the crystals persisted when exposed to air for multiple weeks. This demonstrates that these frameworks shield the radical cations from oxidation in air, providing a route to long-lived radical species. This has potential applications in the field of molecular electronics, in which nanoscale materials can behave as electronic components.19
Current work is focused on exploring the generality of the observations in this paper, principally by investigating the incorporation of viologen cations into anionic MOFs based on other metals, with a view to controlling the lifetime of the radical cation-containing species and exploring the use of these materials as oxygen sensors.
Experimental
Synthetic details and characterisation data are given in the ESI.† Compounds 2-ht, 4-ht, 5-ht and 7′ were structurally characterised using single-crystal X-ray diffraction techniques. While full data have been provided in the ESI,† key data are summarised below:2-ht
C44.2H35.32N2.6O16.6Zn3 (M = 1064.58 g mol−1): monoclinic, space group C2/c (no. 15), a = 32.7998(10) Å, b = 9.7204(4) Å, c = 18.3042(6) Å, β = 96.453(3)°, V = 5798.9(3) Å3, Z = 4, T = 150.00(10) K, μ(CuKα) = 1.956 mm−1, Dcalc = 1.219 g cm−3, 13781 reflections measured (5.42° ≤ 2θ ≤ 145.7°), 5632 unique (Rint = 0.0366, Rsigma = 0.0656) which were used in all calculations. The final R1 was 0.0655 (I ≥ 2σ(I)) and wR2 was 0.2023 (all data).
4-ht
C36H34N4O14Zn2 (M = 877.41 g mol−1): monoclinic, space group P21/n (no. 14), a = 9.6981(2) Å, b = 16.1078(5) Å, c = 11.5458(4) Å, β = 90.923(3)°, V = 1803.41(9) Å3, Z = 2, T = 150.01(10) K, μ(CuKα) = 2.305 mm−1, Dcalc = 1.616 g cm−3, 7613 reflections measured (9.426° ≤ 2θ ≤ 146.132°), 3527 unique (Rint = 0.0382, Rsigma = 0.0515) which were used in all calculations. The final R1 was 0.0430 (I ≥ 2σ(I)) and wR2 was 0.1157 (all data).5-ht
C212H260N20O76Zn8 (M = 4827.34 g mol−1): orthorhombic, space group Pca21 (no. 29), a = 36.4926(13) Å, b = 14.6058(4) Å, c = 45.4204(13) Å, V = 24209.2(13) Å3, Z = 4, T = 150.00(10) K, μ(CuKα) = 1.588 mm−1, Dcalc = 1.324 g cm−3, 52150 reflections measured (6.052° ≤ 2θ ≤ 136.502°), 32875 unique (Rint = 0.0508, Rsigma = 0.0940) which were used in all calculations. The final R1 was 0.0788 (I ≥ 2σ(I)) and wR2 was 0.2252 (all data).
7′
C48.2H46.52N3.4O18.7Zn3 (M = 1168.72 g mol−1): monoclinic, space group C2/c (no. 15), a = 32.6406(6) Å, b = 9.63384(16) Å, c = 18.4404(4) Å, β = 102.534(2)°, V = 5660.48(19) Å3, Z = 4, T = 150.01(10) K, μ(CuKα) = 2.085 mm−1, Dcalc = 1.371 g cm−3, 26374 reflections measured (9.592° ≤ 2θ ≤ 145.74°), 26374 unique (Rint = 0.0437, Rsigma = 0.0523) which were used in all calculations. The final R1 was 0.0568 (I ≥ 2σ(I)) and wR2 was 0.1597 (all data).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the EPSRC for a PhD studentship to LKC and support for the National Service for EPR Spectroscopy and the Material and Chemical Characterisation Facility (MC2) at University of Bath (https://doi.org/10.15125/mx6j-3r54) for technical support and assistance in this work.References
- B. Li, H.-M. Wen, Y. Cui, W. Zhou, G. Qian and B. Chen, Adv. Mater., 2016, 28, 8819 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- S. M. Cohen, Chem. Rev., 2012, 112, 970 CrossRef CAS.
- A. D. Burrows, Post-synthetic Modification of MOFs in Metal Organic Frameworks as Heterogeneous Catalysts, ed. F. X. Llabrés i Xamena and J. Gascon, RSC Publishing, 2014, p. 31 Search PubMed.
- M. D. Allendorf, M. E. Foster, F. Léonard, V. Stavila, P. L. Feng, F. P. Doty, K. Leong, E. Y. Ma, S. R. Johnston and A. A. Talin, J. Phys. Chem. Lett., 2015, 6, 1182 CrossRef CAS.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782 CrossRef CAS.
- Y. Lin, C. Kong, Q. Zhang and L. Chen, Adv. Energy Mater., 2017, 7, 1601296 CrossRef.
- H. K. Chae, D. Y. Siberio-Pérez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O’Keeffe and O. M. Yaghi, Nature, 2004, 427, 523 CrossRef CAS.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232 CrossRef CAS.
- Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2011, 3, 349 CrossRef CAS.
- J. V. Knichal, H. J. Shepherd, C. C. Wilson, P. R. Raithby, W. J. Gee and A. D. Burrows, Angew. Chem., Int. Ed., 2016, 55, 5943 CrossRef CAS.
- M. Müller, A. Devaux, C.-H. Yang, L. De Cola and R. A. Fischer, Photochem. Photobiol. Sci., 2010, 9, 846 RSC.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Leonard and M. D. Allendorf, Science, 2014, 343, 66 CrossRef CAS.
- C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49 RSC.
- K. Madasamy, D. Velayutham, V. Suryanarayanan, M. Kathiresan and K.-C. Ho, J. Mater. Chem. C, 2019, 7, 4622 RSC.
- M. Mohammad, J. Org. Chem., 1987, 52, 2779 CrossRef CAS.
- Y.-N. Gong and T.-B. Lu, Chem. Commun., 2013, 49, 7711 RSC.
- A. S. Hopkins, A. Ledwith and M. F. Stam, J. Chem. Soc. D, 1970, 494 RSC.
- J.-K. Sun and J. Zhang, Dalton Trans., 2015, 44, 19041 RSC.
- Y. Tan, Z. Fu, Y. Zeng, H. Chen, S. Liao, J. Zhang and J. Dai, J. Mater. Chem., 2012, 22, 17452 RSC.
- Y. Zeng, Z. Fu, H. Chen, C. Liu, S. Liao and J. Dai, Chem. Commun., 2012, 48, 8114 RSC.
- A. D. Burrows, K. Cassar, R. M. W. Friend, M. F. Mahon, S. P. Rigby and J. E. Warren, CrystEngComm, 2005, 7, 548 RSC.
- M. Venturi, Q. G. Mulazzani and M. Z. Hoffman, Radiat. Phys. Chem., 1984, 23, 229 CrossRef CAS.
- L. Xie, S. Liu, B. Gao, C. Zhang, C. Sun, D. Li and Z. Su, Chem. Commun., 2005, 2402 RSC.
- S. A. Sapchenko, D. N. Dybtsev, D. G. Samsonenko and V. P. Fedin, New J. Chem., 2010, 34, 2445 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses and characterisation of products. X-ray crystal structures. CCDC 1970065–1970068 for 2-ht, 4-ht, 5-ht and 7′, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9fd00137a |
| This journal is © The Royal Society of Chemistry 2021 |