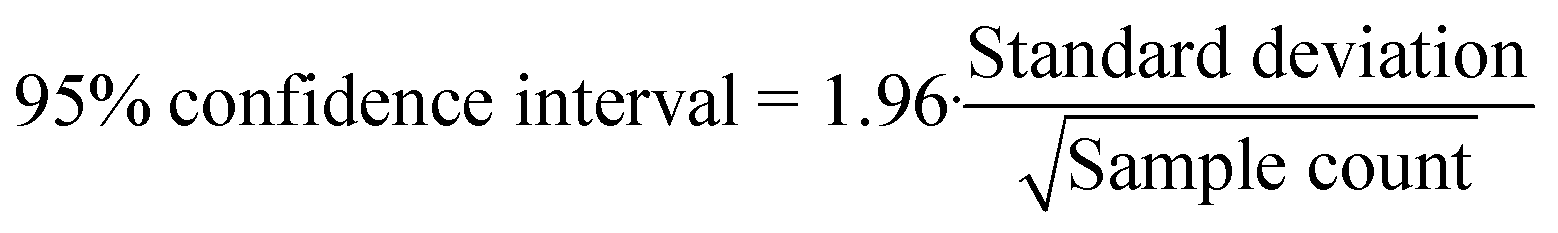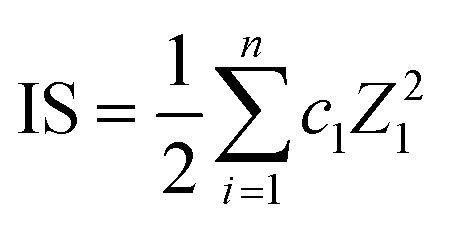Electrochemical system for selective oxidation of organics over ammonia in urine†
Received
1st December 2020
, Accepted 10th March 2021
First published on 18th March 2021
Abstract
Source-separated urine can enable efficient nutrient recycling, but the removal of the organic fraction that is required to ensure a safe nutrient product typically also removes the nitrogen in urine (present as total ammonium nitrogen, TAN). In this study, a reagent-free pH control method was used with a two-chamber electrochemical cell with a boron doped diamond (BDD) anode to oxidize synthetic and real urine at different anodic pH values. Without pH adjustment (pH 8.4), all TAN in urine was oxidized in synthetic urine, but at pH ≤3, 79% ± 5% of TAN was retained. Simultaneously, ≥90% of organic content was removed at all pH values with a pH-independent rate. Two different TAN oxidation regimes with corresponding zero-order (i.e. current limited) TAN oxidation rates of −0.02 h−1 and −0.002 h−1 were identified, separated by a chloride to TAN concentration ratio of approximately 0.2 M/M. The higher TAN oxidation rate is linked to a breakpoint chlorination-type oxidation pathway, whereas the lower rate is linked to a direct oxidation pathway on the BDD surface. In addition to TAN, potassium and phosphorus were conserved at 101% ± 6% and 89% ± 4%, respectively. The proposed technology allows for the selective oxidation of the organic fraction in urine while retaining a high proportion of all other key nutrients for potential reuse as a fertilizer.
Water impact
Urine source-separation offers a technological shift from WWTPs as centralized waste disposal/treatment infrastructure with high energy demand to a series of localized treatments with higher efficiency and concomitant nutrient capture for reuse. This article demonstrates how reagent-free anodic pH-control allows for the advanced oxidation of organics in source-separated urine and conserves nitrogen, potassium and phosphorous, representing a potential pathway for decentralized nutrient recovery and reuse.
|
1. Introduction
Source-separated human urine contains the majority of main nutrients consumed by adult humans (app. 86–65–76% N–P–K), while comprising less than 1% of produced wastewater.1–3 Nitrogen in urine comprises approximately 80% of total nitrogen in municipal wastewater, the removal of which forms a major portion of the total energy consumption of wastewater treatment.2,3 Even more energy is used for fixing the same amount of nitrogen from atmospheric N2 for fertilizer use using the Haber–Bosch-process, which accounts for 1.4% of global CO2 emissions.4 Technologies treating urine as a feed for nutrient recovery could reduce energy requirements from fixing and removal of nitrogen, redirect forms of used energy to electricity from fossil fuels, reduce nutrient runoff to the environment, reduce mining of phosphorus and potassium and thus form a component of the circular economy of nutrients.
While source-separated urine contains nutrients in ratios suitable for fertilizer use,3,5 direct reuse is not favoured in modern societies due to transportation costs, salinity and health and safety issues.3,6 Accordingly, the preferred route for urine-derived nutrient reuse is via separation and/or extraction, primarily of nitrogen and phosphorus, through a variety of technologies, particularly struvite precipitation7–10 and ammonia stripping.11,12 However, the sustainability of these approaches has been questioned by their extensive reagent requirement and the involved transport, storage, logistical and energy burdens.3 One potential alternative for nutrient removal/recovery from urine with little or no additional reagents required is electrochemical technology, which includes electrodialysis, electro-concentration, microbial electrochemical technologies and combined membrane stripping technologies.13–26
A category of electrochemical technologies called electrochemical advanced oxidation processes (EAOPs) involves water treatment processes using electricity to produce oxidative radicals on electrodes27,28 and can be well suited for source-separated urine due to urine's high conductivity.29 EAOPs can be operated without a supply of chemicals or other feeds, allowing for a simplified decentralized implementation, and can be viable even in locations with intermittent power supply from e.g. renewable sources.30 Development of small, flexible EAOPs for source-separated urine treatment could allow for a more widespread implementation of decentralized urine collection and nutrient recovery, provided that the latter can be stably preserved. EAOPs have been widely studied for organics oxidation,27,28,31 and specifically for the treatment of source-separated urine32–35 and latrine water.36–41 However, these studies have primarily focused on the complete oxidation of organic matter and TAN into CO2 and N2 as a means of localized wastewater treatment for safe release into the environment or for water reuse. The most widely utilized anodic electrode materials for EAOPs are dimensionally stable anodes (DSAs, e.g. Ti/IrO2 and Ti/RuO2 mixed metal alloys) and boron doped diamond (BDD). DSAs are termed active anodes and form a chemical-type sorption with the formed OH˙-radical during water electrolysis, while BDD electrodes are termed passive anodes and form a weaker, physical-type sorption with the OH˙-radical.27,28
In EAOPs, the oxidation pathways for organic materials (TOC, total organic carbon) and ammonium-nitrogen (total ammonium nitrogen, TAN = NH3 + NH4+) are complex and can differ based on multiple parameters including the electrode material, electrolyte composition, applied potential, mixing and temperature.27,42 Three distinct main oxidation pathways are identified: (i) direct oxidation through direct charge transfer on the electrode, (ii) indirect oxidation through reactive oxygen species, most prominently the electrode-bound hydroxide radical (M(OH˙)), and (iii) indirect oxidation through radical species such as oxocarbonates, oxosulfates, oxophosphates and especially chloride radicals (reactive chlorine species, RCS).27,28,31,43–45 Electrode oxidation chemistry can be separated into current controlled and mass transport controlled regimes, depending on the applied current density, mixing and concentration, and the chemistry can differ significantly between different regimes.27,46
BDD electrodes favour the formation of weakly adsorbed hydroxyl radicals BDD(OH˙) (1) that are very strong oxidizing agents (E0 = 2.8 V) and readily scavenged by organic molecules and inorganic anions to form oxidized products or radicals, respectively.27,28 In urine, chloride is present at significant concentrations and is readily oxidized by BDD(OH˙) to form chlorine which can subsequently dissolve to form hypochlorite (2) and (3), or react directly and form further oxidized species with the hydroxyl radical (see eqn (4)–(7)).28
| | | BDD + H2O → BDD(˙OH) + OH− + e− | (1) |
| | | Cl2(aq) + 2H2O → HClO + H3O+ + Cl− | (3) |
| | | Cl− + ˙OH → ClO− + H+ + e− | (4) |
| | | ClO− + ˙OH → ClO2− + H+ + e− | (5) |
| | | ClO2− + ˙OH → ClO3− + H+ + e− | (6) |
| | | ClO3− + ˙OH → ClO4− + H+ + e− | (7) |
While the hydroxyl radical is known to be a primary oxidant for most organic molecules, it is also reported to be a poor oxidant for certain inorganic molecules
e.g. ammonium and complex organics such as fulvic and humic acids.
28,34 These separate reaction pathways and oxidation affinities for organic materials and TAN remain a challenge for the practical implementation of EAOPs.
34 The relationship and situations where TAN is not oxidized in urine electro-oxidation have not been systematically studied and it remains unclear which parameters control the TOC and TAN oxidation capacity and rates during treatment. A better understanding of specific electro-oxidation pathways and rates under different parameters can allow for a better-informed design of electro-oxidation systems that perform as intended and could also enable selective organics electro-oxidation without TAN oxidation, allowing for selective nitrogen recovery from urine.
In this study, the electro-oxidation of synthetic and real ureolysed source-separated urine was investigated using a BDD anode under varying anodic pH conditions in a two-chamber electrochemical cell. The specific aims were to study the effects of pH on the (i) organics oxidation rate, (ii) TAN oxidation rate, and (iii) chloride oxidation rate and by-product formation. The outcomes provide a novel viewpoint into electro-chemical treatment of source-separated urine and expand the possibilities for selective nutrient recovery with concomitant organics removal.
2. Materials and methods
2.1 Medium composition
A synthetic urine solution representing human source-separated ureolysed urine was designed based on (i) previous investigations at the Swiss Federal Institute of Aquatic Science and Technology (EAWAG),47,48 (ii) a literature review of source-separated urine samples,5,49–57 (iii) the human urine metabolome,58 and (iv) real source-separated urine collected for research at the Advanced Water Management Centre at the University of Queensland, Australia.18 The four most prevalent organic compounds identified in the human urine metabolome database were included in the recipe: creatinine, hippuric acid, citric acid and glycine, while the rest of the organic loading was simulated by addition of acetic acid, comprising 15% of total theoretical chemical oxygen demand in the sample. The recipe consisted of (in g L−1): NH4HCO3 (22.14), NH4Cl (0.48), NaCl (2.69), KCl (4.10), NaH2PO4 (2.40), Na2SO4 (2.41), creatinine C4H7N3O (2.3), hippuric acid C9H9NO3 (0.88), citric acid C6H8O7 (0.88), glycine C2H5NO2 (0.17), ammonium acetate NH4CH3CO2 (2.06) and NH4OH (25% liquid, 13.8 mL L−1). This formulation simulates urine after ureolysis, which results in complete removal of Mg and Ca through precipitation with phosphate.47 This solution was employed as a feed, used as prepared and its composition was monitored through sampling at the start of each experiment. Synthetic urine was chosen to enable consistency when systematically varying the experimental factors.
Furthermore, human source-separated urine was collected from Hiedanranta Kuivaamo-cultural venue in Tampere (Finland), where Digi Toilet Systems Oy operates source-separating dry toilets. Urine was sampled from the male urinals' collection tank, filled during spring and summer 2020. This urine was stored as collected in a closed 1 m3 tank at room temperature until sampling. After sampling, the urine was stored in closed 20 L canisters at +4 °C in the dark until used. The main properties of this real source-separated urine are presented in Table 1. The sampled urine was significantly more dilute and had different TAN/Cl/TOC-ratios than the synthetic urine (Table 1). This can be due to the recreational quality of the collection venue, associated with evening and night-time use and consumption of large amounts of beverages. While not representative of an average 24/7 human urine sample, using this real urine is still beneficial for comparison with the synthetic formulation, as many real source-separation sites comprise similar venues. Due to long term storage, this urine was considered to be fully ureolysed.
Table 1 Measured real and synthetic urine properties. All values in mmol L−1, except pH (unitless) and conductivity (mS cm−1)
| Component |
Synthetic urine |
Real source-separated urine |
|
n.d. = not detected. IS = ionic strength.
|
| TAN |
522 ± 40 |
197 ± 3 |
| NO3− |
n.d. |
n.d. |
| NO2− |
n.d. |
n.d. |
| TOC |
286 ± 46 |
114 ± 17 |
| Cl− |
92 ± 5 |
24.7 ± 0.8 |
| ClO3− |
n.d. |
n.d. |
| ClO4− |
n.d. |
n.d. |
| K+ |
70 ± 6 |
10.4 ± 0.3 |
| Na+ |
117 ± 8 |
27.6 ± 1.0 |
| PO43− |
18 ± 2 |
5.1 ± 0.9 |
| SO42− |
15 ± 2 |
3.7 ± 0.3 |
| Cl/TAN |
0.23 ± 0.02 |
0.13 ± 0.01 |
| IS |
1.23 ± 0.05 |
0.393 ± 0.009 |
| pH |
8.35 ± 0.09 |
8.5 ± 0.3 |
| Conductivity |
37.7 ± 0.8 |
17.0 ± 0.2 |
2.2 Reactor and equipment
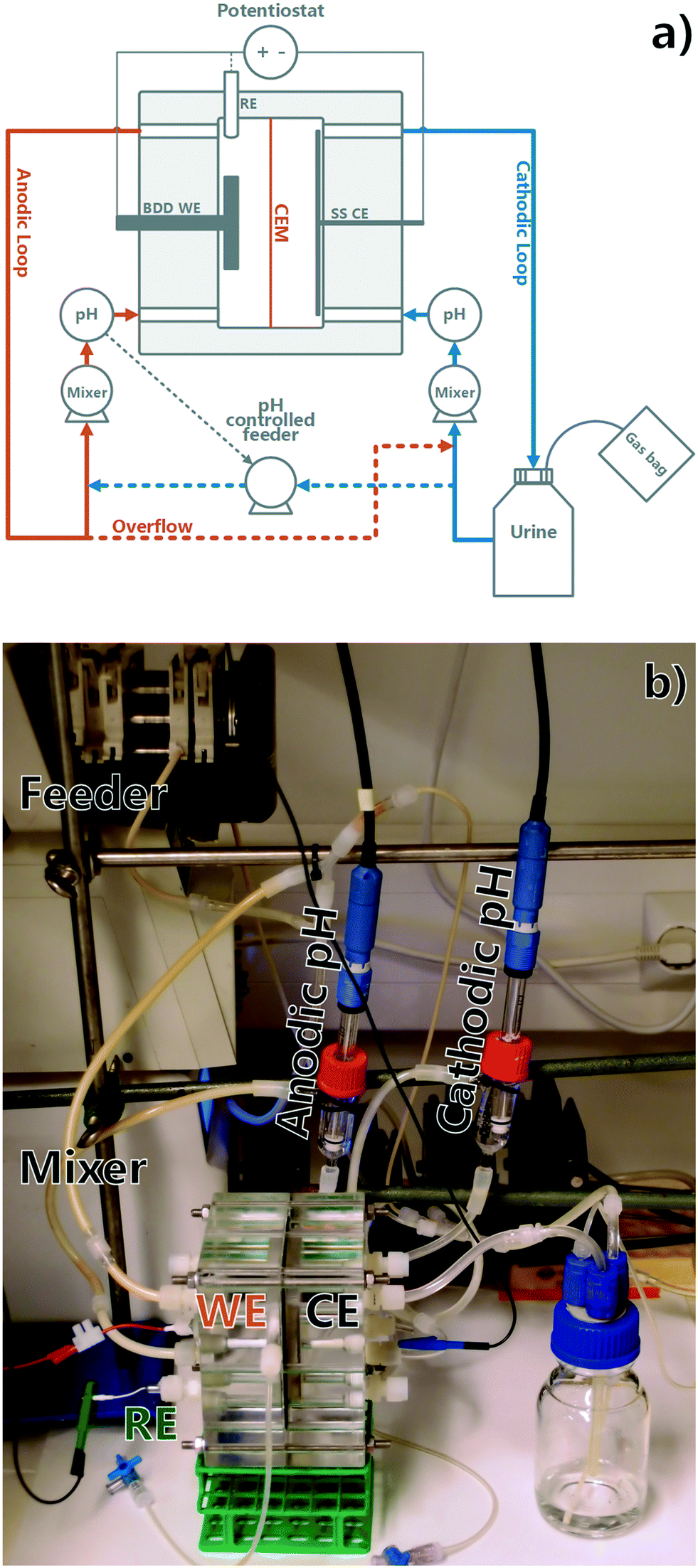
Custom reactors were used consisting of acrylic plates forming two parallel compartments of 70 mm × 50 mm × 10 mm each (see Fig. 1), sealed with triple rubber O-rings. Two identical reactor setups were utilized. The anodic compartment was fitted with a Condias Dischem 40 × 40 × 2 mm Nb-BDD electrode, with a plastic encased copper wire current collector. The current collector was attached using a conductive epoxy (Epo-Tek EJ2189-LV) and silicone sealant. An Ag/AgCl 3 M NaCl reference electrode (BASi, USA) was placed in the anodic chamber to monitor the anodic potential. The cathodic compartment contained a 70 mm × 50 mm stainless steel mesh as a cathode and was separated from the anodic compartment by a cation exchange membrane (CEM) (Membranes International Inc. CMI-7000). The projected surface area of the membrane was 35 cm2 and the volume of each chamber was 35 mL, rendering the total reactor working volume as 70 mL. The anodic loop volume containing the anodic reactor chamber and the mixing loop was 57 mL and remained constant throughout all the experiments, whereas the cathodic loop volume was variable due to reactor volume changes (from 150 mL to 89 mL). The anodic and cathodic compartments were connected to online pH meters (Endress+Hauser Liquiline CM448) and both compartments were mixed by circulation pumps, with a chamber mix time of app. 10 seconds (Cole Parmer Masterflex 7523-70). The cathodic loop was circulating from a 150 mL recirculation bottle, the content of which was considered as part of the cathodic loop volume and was added in the beginning of each batch experiment. The anodic loop was fed with a pH-controlled feeding pump (Watson-Marlow 205U) from the cathodic loop and overflowed to the cathodic chamber through gas tight connections. The anodic feeding pump was pH-relay controlled to keep the anodic loop pH constant. A potentiostat (Bio-Logic VMP-3) was used as a power source, recording the voltage applied and electrical energy used. An example of potentiostatic data is included in the ESI† (S7). A constant current of 350 mA, corresponding to a current density of 100 A m−2 based on the membrane surface area, was applied in all experiments. The anode surface area after current collector connections was estimated at 30 cm2, corresponding to an average current density of 117 A m−2 based on the anode surface area. Finally, the reactors' outlet gas composition was monitored using 100 mL 1 M HCl gas wash bottles or gas bags connected to the recirculation bottle. At the end of an experimental run, a 900 mL sample of gas bag content was transferred onto 1 L glass bottles using a displacement method, washed overnight in 1 M HCl, and analysed in the same way as the gas wash bottle samples. The runs where gas analysis was used and the method used are described in ESI† S1.
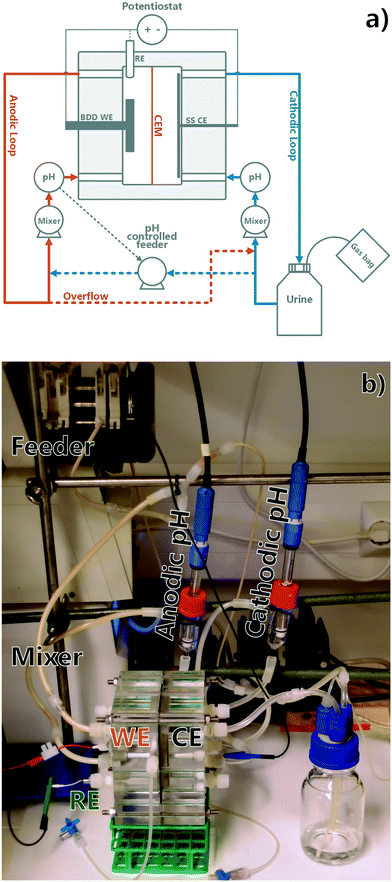 |
| | Fig. 1 a) Experiment flowchart (WE = working electrode, CE = counter electrode, RE = reference electrode, BDD = boron doped diamond, SS = stainless steel) and b) picture of the reactor setup. | |
2.3 Operation
Batch experiments, referred to as runs in this study, were started with 150 mL of feed in the reactor (anodic loop + cathodic loop). A 1 mL starting sample was taken from the feed outside the reactor. 1 mL samples were taken from the anodic and cathodic compartments twice a day during the run. Runs were continued for 4 or 5 days. After a run, all reactor content was collected, measured by volume by chamber, and sampled for chemical analyses. A control experiment, named “neutral”, was performed by connecting the anodic and cathodic loops into a single mixing loop, forming a continuously stirred-tank reactor. Other experiments are named based on anodic pH levels. A total of 19 runs under neutral conditions and for pH values of 5, 3, 2 and 1.5 were undertaken. A detailed list of all runs is presented in ESI† section S1.
2.4 Chemical analyses
Samples were analysed for cations and anions using a Thermo Scientific ICS-1600 ion chromatograph with a Dionex IonPac AS22 column (Cl−, ClO3−, ClO4−, NO2−, NO3−, SO42+, PO43+) and a Dionex IC-120 with an IonPac CS12A column (Na+, NH4+, K+, Mg2+, Ca2+) (Dionex, CA, USA), and total carbon species using a Shimadzu TOC-5000 total organic carbon analyser (Kyoto, Japan). Gas bags were analysed for N2 content using a Shimadzu GC-2014 GC-TCD with a Supelco Carboxen 1000 60/80 column. For analysis, liquid samples were filtered using 0.45 μm syringe filters into 1.5 ml microcentrifuge tubes, stored after sampling for a maximum of 4 days in a fridge and diluted 50× for TOC analysis and 500× for IC analysis using a two-step dilution method with 15 ml tubes.
2.5 Calculations
Fittings were made using the MatLab R2020a curve fitting tool. Graphs were drawn using Veusz 3.1 distributed under GNU public license. Data analysis was undertaken with Microsoft Excel 2019 Analysis Toolpak. Chemical equilibrium, speciation and precipitation were modelled using Visual MINTEQ 3.1 modelling software (see ESI† S6 for model parameters). Reactor concentrations were calculated from anodic and cathodic loop concentrations by multiplying the value measured by their respective loop volumes and normalizing by the total reactor volume. Due to the reactor configuration, the anodic loop volume remained constant through each run, while the cathodic loop volume was reduced by sampling from both sides and through electrolysis and evaporation (and potential gas leakage). When calculating mass balances, the proportion lost through sampling was accounted for and subtracted from the initial amount. Errors discussed in this article are 95% confidence intervals calculated from standard deviation and sample count using eqn (8), under the assumption that the data were normally distributed.| | 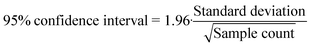 | (8) |
Results are presented in relation to the initial concentration C/C0 [mmol L−1/mmol L−1] and initial amount of substance mol/mol0 [mmol/mmol], simplifying the comparison between different operational parameters. TOC results were fitted with an exponential function, simulating a pseudo 1st-order decay, supplemented with a non-degradable residual TOC fraction as shown in eqn (9). TAN results were fitted with a linear function, simulating a zero-order decay as shown in eqn (10).59 Chloride and perchloride concentrations and masses were fitted with the experimental sigmoidal functions in eqn (11) and (12), which can be used to model combinations of reactions,60 enabling the comparison of fits between datasets.| | | TOC(t) = TOC0·e−k·x + TOCresidual | (9) |
| | | ClO4(t) = A/(B + e−Cx) | (12) |
Rate constants k with 95% confidence interval for both TOC and TAN decay were obtained through the MatLab R2020a curve fitting tool by fitting data into eqn (9) and (10), respectively. No similar rate constant was utilized for the experimental sigmoidal functions (eqn (11) and (12)).
Ionic strength (IS) to compare salinities of samples was approximated using eqn (13).
| | 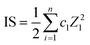 | (13) |
where
ci is molarity (mol L
−1) and
Zi charge for each measured ion (Cl
−, ClO
3−, ClO
4−, NO
2−, NO
3−, SO
42+, PO
43+, Na
+, NH
4+, K
+, Mg
2+, Ca
2+). Only measured ions were considered, and no estimation on the effect of organic fraction was made nor corrections for activities.
3. Results and discussion
3.1 Reagent-free pH control
The anodic pH was controlled using a two-chamber cell, where the cathodic eluent is pumped to the anodic chamber loop by a relay pump controlled by the anodic loop pH (see Fig. 1). pH-Graphs can be found in Fig. S1 in the ESI.† For all experiments, an anodic pH-equilibration period (approx. 1–2 h) was observed at the beginning of the relay-pump controlled experiments, as the pH in the anodic compartment dropped to the desired value before the relay-controlled circulation pump actuates for the first time. This period can influence the results in the first measuring interval as approx. 30% of the reactor volume is in the anodic loop during this time period and the pH is initially slightly alkaline (pH 8.4) and heavily buffered. After this initial equilibration period, the pH remained stable at the target value, demonstrating the effectiveness of the reagent-free pH control system (see Fig. S1 in the ESI†).
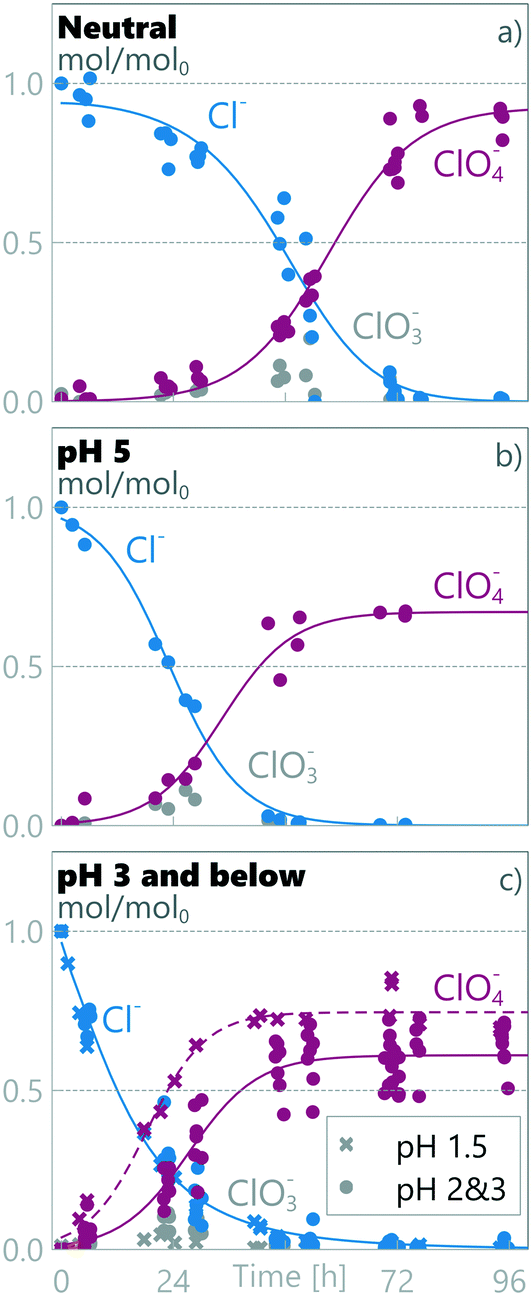
3.2 TAN decay rate is linked to anodic chloride concentration
3.2.1 Measured decay rates at different pH regions.
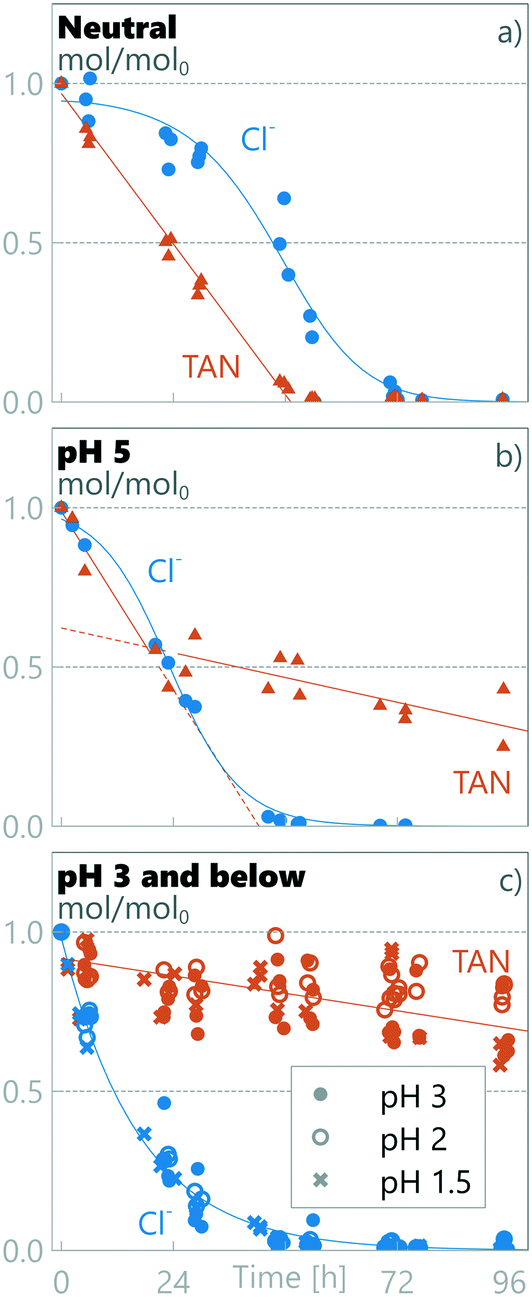
Two separate regimes were observed for TAN decay as different regimes were observed for different anodic pH values (see Fig. 2). The results obtained at anodic pH values of 3, 2 and 1.5 are grouped together (Fig. 2c), as no statistically-significant differences between them were observed in measured chloride or TAN. At neutral pH, the TAN decay fitted a single zero order decay rate. This implies that TAN removal under these conditions is not limited by TAN mass transfer, but by current density.27,46 Mass balance analysis (see later in this section) shows that TAN is oxidized to N2, but as we did not measure any specific chemical pathways, we refer to the disappearance of TAN as decay, encompassing destruction through all potential pathways.
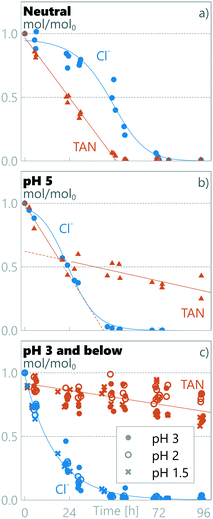 |
| | Fig. 2 Measured relative amount of TAN and chloride in synthetic urine in a) neutral pH, b) pH 5 and c) pH 3 and below. | |
At pH 5 (Fig. 2b), TAN decay showed two different linear decay regions, with a regime change at ≈24 h. If the initial decay in pH 5 is assumed to continue until 24 h, a similar initial rate of zero-order decay is observed to the neutral-pH runs (Fig. 2a) (kneutral = −0.020 ± 0.002 h−1, k1ph=5 = −0.023 ± 0.006 h−1), conforming with the same 95% confidence interval. Interestingly, the decay rate observed after the transition in pH 5 is an order of magnitude lower (k2ph=5 = −0.003 ± 0.002 h−1). This same order of magnitude lower decay rate was also measured in pH values ≤3 from the beginning of the experiments (kph≤3 = −0.002 ± 0.001 h−1), the rates being similar within a 95% confidence interval.
Clear differences in removal behaviour were also observed for chloride, with different rates of change for the three pH regions: neutral, pH 5 and pH ≤3. In neutral runs (Fig. 2a), the chloride concentration initially remained high (0.83 ± 0.04 Cl/Cl0 at ≈24 h), but after a marked drop between 30 and 48 h, it reached 0.02 ± 0.03 Cl/Cl0 at ≈72 h. In contrast at pH 5, the chloride concentration started dropping immediately at the start of the experiment and reached near-zero values (0.03 ± 0.02 Cl/Cl0) within 48 h. Finally, for the experimental runs at anodic pH values ≤3, the chloride concentration dropped rapidly, reaching 0.03 ± 0.02 Cl/Cl0 within the first 48 h. For pH values ≤3, the chloride decay followed pseudo first-order kinetics, implying limitation by mass transfer to the electrode surface and not limitation by current, whereas in higher pH values a first order model did not produce a good fit, and an experimental fit was used (see section 2.5). The chloride decay was more rapid at lower pH: at ≈24 h the Cl/Cl0 was 0.83 ± 0.04, 0.51 ± 0.09 and 0.21 ± 0.05, for neutral, pH 5 and pH ≤3, respectively.
3.2.2 Cl/TAN ratio defines two separate TAN decay regimes.
According to the published literature, TAN oxidation on the BDD anode in urine is expected to be linked to the availability of chloride, whereas oxidation of organics can readily proceed through hydroxyl radical oxidation.28,34 Breakpoint chlorination is a widely studied chemical phenomenon of organics and TAN oxidation via active chlorine (Cl2 + HOCl + OCl−), a type of reactive chlorine species (RCS), in bulk water near neutral pH,61,62 and is often cited as a mechanism that also takes place during electro-oxidation.37–39,41,63 Electro-oxidation chemistry can however be dominated by boundary layer phenomena, and a more complicated system of reaction pathways exists, the details of which are yet to be fully elucidated.27,28,64 The basis of TAN oxidation via RCS is that TAN readily reacts with hypochlorite to form chloramines, including monochloramine, dichloramine and trichloramine, which can further react to form N2, oxidize to nitrate or reduce back to ammonium at the cathode. The conventional breakpoint chlorination mechanism's sequential reaction pathways, and equations for overall oxidation are presented in eqn (14)–(19).| | | NH4+ + HOCl → NH2Cl + H2O + H+ | (14) |
| | | NH2Cl + HOCl → NHCl2 + H2O | (15) |
| | | NHCl2 + HOCl → NCl3 + H2O | (16) |
| | | HOCl + 2/3NH3 → 1/3N2 + H2O + H+ + Cl− | (17) |
| | | HOCl + 2/3NH4+ → 1/3N2 + H2O + 5/3H+ + Cl− | (18) |
| | | HOCl + 1/4NH4+ → 1/4NO3− + 1/4H2O + 3/2H+ + Cl− | (19) |
Gendel and Lahav (2012)64 have argued for an alternative TAN chlorination reaction pathway suggesting direct formation of trichloramine with chlorine as per eqn (20), followed by complex reactions pathways, two sum iterations of which are presented in eqn (21) and (22).| | | 3Cl2 + NH4+ → NCl3 + 3Cl− + 4H+ | (20) |
| | | 2NCl3 + 60H− → N2 + 3OCl− + 3Cl− + 3H2O | (21) |
| | | 2NCl3 + 3NH3 + 3OH− → 3NH2Cl + N2 + 3OCl− + 3Cl− + 3H2O | (22) |
For breakpoint chlorination to proceed, a limiting ratio of RCS to TAN needs to be exceeded for the oxidation to proceed. This value is typically described as a molar ratio near 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 active chlorine to nitrogen (i.e. Cl2 to NH3–N), depending on water characteristics. Below this threshold, active chlorine can be bound as chloramines, and no TAN oxidation via breakpoint chlorination mechanisms is observed.65 In the absence of chloride, TAN is reported to be oxidized on the electrode surface via the direct pathway (see eqn (23)), not through hydroxyl radical oxidation.66
:1 active chlorine to nitrogen (i.e. Cl2 to NH3–N), depending on water characteristics. Below this threshold, active chlorine can be bound as chloramines, and no TAN oxidation via breakpoint chlorination mechanisms is observed.65 In the absence of chloride, TAN is reported to be oxidized on the electrode surface via the direct pathway (see eqn (23)), not through hydroxyl radical oxidation.66| | | 2NH3 → N2 + 6H+ + 6e− | (23) |
While the exact reaction pathways and rates between RCS and TAN on the electrode surface and bulk remain unclear, they are tightly linked to the chloride reaction chemistry at the electrode boundary layer. In our experiments, the rapid TAN decay observed under neutral conditions is expected to proceed through indirect oxidation via the hypochlorite radical.27,28,64 The observed rapid oxidation under neutral and initial pH 5 conditions in this study is observed to follow zero-order kinetics and is likely limited by RCS production on the electrode (or RCS diffusion from the surface to the bulk). An order of magnitude lower TAN decay is present at pH ≤3 and after transition in pH 5, most likely representing the direct oxidation rate (eqn (23)). To understand this difference, more parameters were examined.
To study the defining parameters for the transition, chloride concentrations (see Fig. 2 and S3 in the ESI†) and chloride to TAN concentration ratios (Cl/TAN) were analysed. The Cl/TAN ratio of the synthetic urine is 0.23 ± 0.02 M/M, and in neutral runs it rises from this initial value as TAN decays more rapidly than Cl. In pH 5, Cl/TAN remains constant for the first ≈24 h (averaging 0.27 ± 0.02 for the first 24 h), after which it drops sharply to zero. This drop coincides with a clear TAN decay transition. In pH ≤3, Cl/TAN starts dropping instantly, averaging 0.18 ± 0.02 at ≈6 h, after which it continues to drop reaching zero by 24 h. Under the same conditions (pH ≤3), no rapid TAN decay is observed. While all Cl/TAN values are lower than active chloride to TAN of 3 (or Cl2/TAN of 1.5, in M/M), commonly described as necessary for breakpoint chlorination, the observation that Cl/TAN is a defining parameter in changing the TAN decay kinetics supports the hypothesis that breakpoint chlorination-type pathways are operational for TAN decay when a high enough Cl/TAN ratio is present. When Cl/TAN is below a threshold, the RCS produced can be absorbed by the chloramines formed from TAN and later reduced back to TAN and chloride.61 From the results in this study, an experimental threshold Cl/TAN-ratio of ≈0.2 M/M can be approximated for this system. Above this ratio, a higher TAN decay rate of −0.02 h−1 is measured, while below this ratio an order-of-magnitude lower TAN decay rate of −0.002 h−1 occurs. These observations about different reaction rates based on the presence of chloride are supported by a previous investigation by Zöllig et al.34
3.2.3 TAN mass balances.
Depending on the anodic pH, 0.01 ± 0.01, 0.44 ± 0.15 and 0.79 ± 0.05 of TAN mol/mol0 were retained as TAN in the system in neutral pH, pH 5 and pH ≤3, respectively (Fig. 5). TAN oxidation pathways and potential stripping were examined through nitrogen mass balance. The nitrogen mass balances in this study were established by measuring TAN from the reactor gas outlet through gas bags or gas washing bottles, as well as measuring TAN, nitrite and nitrate in solution in both chambers of the reactor. N2 measurements from gas bags confirmed formation of N2 but were qualitative in nature and were not used for mass balance calculations. TAN oxidation via RCS produces nitrogen gas (N2), or nitrate (NO3−) (eqn (17)–(22)). Nitrate was formed initially at the anode in all runs (average detected maximum in all runs 0.03 ± 0.01 CNO3/CNtot0), with lower concentrations detected uniformly at the cathodic compartment. Nitrite was detected almost only at the cathodic compartment (average detected maximum 0.010 ± 0.002 CNO2/CNtot0). No NOx species were measurable by the end of the run, and the nitrate and nitrite concentration patterns suggest that they were lost through nitrate reduction at the cathode, with nitrite as an intermediate reduction product (see ESI† section S4 for details).
TAN was detected in the effluent gas, but regardless of operating parameters, it amounted to ≤1% of the initial TAN mass (mol0), being below the level of error for TAN analyses in the reactor. Nitrogen stripping as NH3 therefore had no significant role in the TAN balances and conservation in our setup. While the cathodic loop pH was above 9 (reaching 9.5 in pH = 1.5 runs), the cathodic hydrogen generation and mixing was not strong enough to induce significant NH3 stripping. This is encouraging in the context of nitrogen removal and recovery with EAOPs, as loss of TAN through stripping would require nitrogen re-capture with an acid (e.g. H2SO4), creating the need for unwanted chemical dosing and added complexity and risk of ammonia gas management. With stripping discarded as a significant N-removal mechanism in all experiments, the most probable reaction pathway for the observed rapid TAN decay was a combination of oxidation reactions with RCS to form N2 (eqn (14)–(22)) with some oxidation to NO3− (eqn (19)) with subsequent reduction via NO2− at the cathode to N2 or TAN (see eqn (24)–(26)).64,67–70 The most probable reaction pathway for the observed slow TAN decay is through direct oxidation to form N2 through adsorption on the BDD surface (see eqn (23)).66 The mass balance calculations show that no other statistically significant species for TAN loss from the system were identified than loss as N2 gas.
| | | NO3− + 2H+ + 2e− → NO2− + H2O | (24) |
| | | 2NO3− + 12H+ + 10e− → N2 + 6H2O | (25) |
| | | 2NO3− + 9H+ + 8e− → NH3 + 3H2O | (26) |
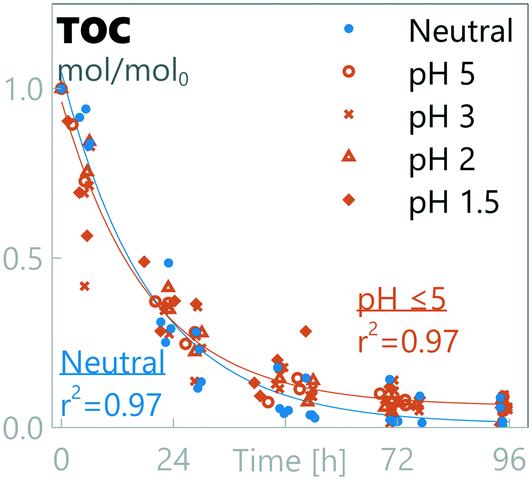
3.3 TOC decay rate is pH independent
The oxidation of organic matter was monitored through TOC-measurements (Fig. 3). TOC decay followed pseudo 1st-order kinetics in all experiments, irrespective of the anodic pH level. A residual TOC fraction was observed when the anodic pH was at pH ≤5, indicating that not all TOC could be removed during the timeframe of the experiment (results at pH ≤5 were grouped together as they were not statistically significantly different). The resulting TOC concentration decay rates show no statistically-significant difference between all pH regions (kpH=neutral = 0.052 ± 0.010 h−1, kpH≤5 = 0.054 ± 0.007 h−1), although a difference in the residual TOC concentration was observed between neutral conditions (0.01 ± 0.04 TOCresidual/TOC0) and pH ≤5 results (0.06 ± 0.03 TOCresidual/TOC0).
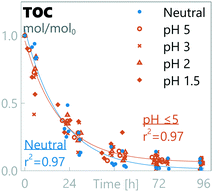 |
| | Fig. 3 Relative amount of total organic carbon in synthetic urine at different pH values. | |
To understand the decay kinetics of TOC, known decay mechanisms are examined: organic molecules in synthetic urine are considered prone to electrochemical oxidation through the complex interplay of both BDD(OH˙)-radicals and indirect oxidation through hypochlorite radicals (or other RCS).34,42,71 Zöllig et al.34 were able to detect distinct decay rates for chemical oxygen demand (COD) in urine before and after complete oxidation of chloride, showing a higher decay rate in the presence of chloride. In this study, such a distinction in the organics decay rate was not observed. However, it's worth noting that this study was not designed to test the effect of chloride on the TOC decay rate, and all experiments did contain chloride in the beginning of the experiment, a factor which can inhibit detection of change in the decay rate. Regardless of this, the observed pseudo 1st-order decay kinetics match previously-reported observations and support mass-transfer-limited reaction kinetics, considered characteristic of high applied current densities as done in this study.27
The residual TOC at pH ≤5 is most likely linked to the presence of RCS. At pH ≤5, chloride is depleted before TOC and none or little RCS is expected to be formed, whereas under neutral conditions TOC is oxidized before chloride is depleted and RCS is expected to be the main chloride oxidation product. RCS is described as a weaker oxidant than BDD(OH˙) with measured E0vs. SHE potentials of 1.36 V for Cl2(aq), 1.49 V for HClO, and 0.89 V for ClO−,72versus 1.65 V for BDD(OH˙).73 These species have a different reach in the system as BDD(OH˙) has a very short lifespan of ≈10−9 s (ref. 28) compared to potentially days or weeks for RCS in clean water,61 and BDD(OH˙) most likely acts as an oxidant only close to the boundary layer.42 BDD(OH˙) has been reported to react poorly with some substances, including ammonium and some organic acids.28 Our data suggest that TOC contains recalcitrant organic components that remain in the solution when RCS is not available. In synthetic urine, the organic constituents are known, and recalcitrant components are most likely degradation products, but in real urine source components can also form a component of the recalcitrant fraction. Another potentially-linked source of residual TOC can be the formation of recalcitrant chlorinated by-products, as the initial presence of RCS can induce the formation of organochlorinated by-products that can be recalcitrant to oxidation by hydroxyl radicals and other subsequent reactive oxygen species (ROS).42,74
3.4 Chloride by-products
The chloride oxidation was examined through chloride (Cl−), chlorate (ClO3−) and perchlorate (ClO4−) concentrations and through a calculated mass balance. This does not account for active chlorine, chloramines, organochlorinated by-products or chlorine lost as Cl2. The mass balances under different pH conditions are presented in Fig. 4. The results show that, independent of pH values, low levels of chlorate were measured during the runs, corresponding to a middle product in a sequential oxidation from chloride to perchlorate (see eqn (5)–(7)). The average relative maximum chlorate concentration was 0.08 ± 0.03 CClO3/CCltot0 (8% of initial chloride was present as chlorate). For perchlorate measurements, the pH 1.5 data is statistically different from the combined pH 3 & pH 2 dataset and is processed separately. There were clear differences in perchlorate formation between different pH values and final relative perchlorate mass balances of 0.93, 0.67, 0.61 and 0.75 ClClO4−/Cltot0 were obtained in neutral pH, pH 5, combined pH 3 & pH 2, and pH 1.5, respectively, using eqn (12). These results imply a change in the chloride oxidation pathway as the pH is changed. Less perchlorate is detected at lower pH, which is most likely due to increased Cl2 gas production but can also be partly due to increased formation of organochlorinated by-products or other chloramines.42 While retaining the same total contact times, lower pH corresponds to a lower exchange rate between anodic and cathodic compartments, enabling a faster decrease in chloride, and thus a lower chloride concentration at the anode. This is confirmed by comparing anodic loop samples, which show lower anodic chloride concentration in pH ≤3 compared to pH 5 and neutral runs (see Fig. S3†). The final perchloride rises slightly at pH 1.5 compared to pH 3 & 2, with a clearly different form of reaction pathway, which is not visible in the Cl− concentration decay rate. This implies a faster or more direct oxidation pathway at pH 1.5 for chloride, with rate changes in the steps after the initial chloride oxidation.
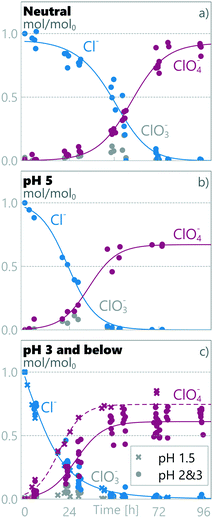 |
| | Fig. 4 Relative amount of chloride species in synthetic urine in a) neutral pH, b) pH 5 and c) pH 3 and below. | |
While the previously presented results on preservation of TAN at acidic pH values are encouraging, it should be noted that chlorate and perchlorate are toxic and persistent substances that are a serious impediment to the implementation of BDD based electro-oxidation technologies in the field,75,76 such as the one described in this paper. More research is underway to potentially overcome this challenge using current BDD or DSA materials in novel ways (e.g. combination with electroreduction) or by developing novel electrode materials that allow for safe electro-oxidation in chlorine-rich media.43,77–79 There are numerous electrochemical treatment and nutrient recovery technologies tested for source-separated urine (see section 1), each with their own advantages and caveats. Most of these can offer ample opportunities for the development of practical solutions that do not produce toxicity and are able to operate intermittently without addition of reagents. In this development, EAOPs can have a strong role to play as they differ from pure electro-concentration and stripping in their rapid organics oxidation and biological inactivation capabilities.27
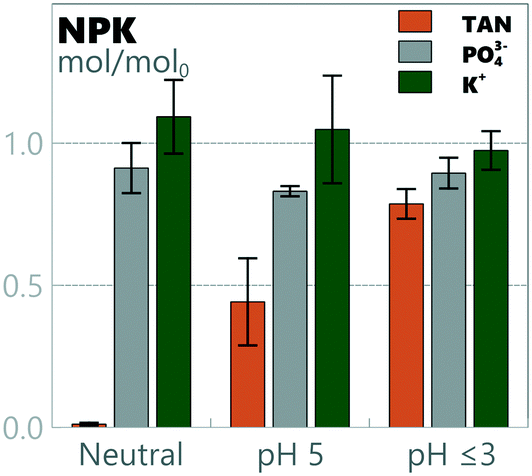
3.5 Nutrient balances and potential for recovery
Preservation of the main nutrients in urine during electro-oxidation and removal of organic materials is potentially a benefit for subsequent processing for utilization as a liquid fertilizer. TAN, phosphate and potassium mass balances for all runs are presented in Fig. 5. TAN retainment varied significantly depending on the pH: 0.01 ± 0.01, 0.44 ± 0.15 and 0.79 ± 0.05 of TAN mol/mol0 were retained in neutral pH, pH 5 and pH ≤3, respectively. Conservation of K+ and PO43− was 1.01 ± 0.06 and 0.89 ± 0.04 mol/mol0 over all pH values, with no statistically significant differences observed between different pH levels. While no loss of K+ was detected in the system, a systemic loss of PO43− of 11 ± 4% was detected, most likely due to precipitation. While precipitates were not visually detected, speciation was simulated using Visual MINTEQ 3.1.80 According to the model used by MINTEQ, even a small addition of calcium (5–10 mmol L−1) would have induced precipitation of hydroxyapatite (Ca5(PO4)3(OH)) in the cathodic chamber at the pH values it was operated at (8.5–9.5). While calcium was not added to the medium, it was detected in the ≈10 mmol L−1 range in the samples, making precipitation a likely source of phosphorus loss. The final concentrations of TAN in neutral pH, pH 5 and pH ≤3 were 0.008 ± 0.005 mmol L−1, 0.24 ± 0.09 mmol L−1, and 0.46 ± 0.04 mmol L−1, respectively. The final concentrations of K+ and PO43− over all pH values were 0.083 ± 0.008 mmol L−1 and 0.021 ± 0.002 mmol L−1, respectively.
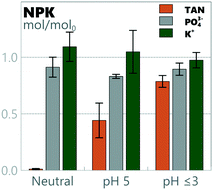 |
| | Fig. 5 Nitrogen, phosphorus and potassium (NPK) mass balances for full experiments with synthetic urine at different pH values. | |
Na+ and SO42 remained in the solution during treatment (average mass balances of 1.07 ± 0.08 and 0.95 ± 0.05 were measured, respectively). Conversely, total inorganic carbon (TIC = CO2 + HCO3− + CO32−) conservation depended on the anodic pH: 0.00 ± 0.00 mol/mol0 in neutral pH, 0.16 ± 0.06 mol/mol0 in pH 5, 0.46 ± 0.13 mol/mol0 in combined pH 2 & 3 and 0.83 ± 0.20 mol/mol0 in pH 1.5 TIC retainment was measured. While TIC is expected to be stripped by low pH, the circulation speed could be a key parameter for the inverse behaviour observed, as lower pH runs have lower volumetric flow of CO2(g) returning to the cathodic loop for redissolution. Chloride was removed completely, replaced by oxidized chlorates and partly removed as chlorine gas (see section 3.4.). These results show that while further work is required for chloride by-product removal, the current method has the capacity to selectively retain the valuable macro-nutrients N, P, K and S, while removing organic carbon from urine using a reagent-free pH-control system.
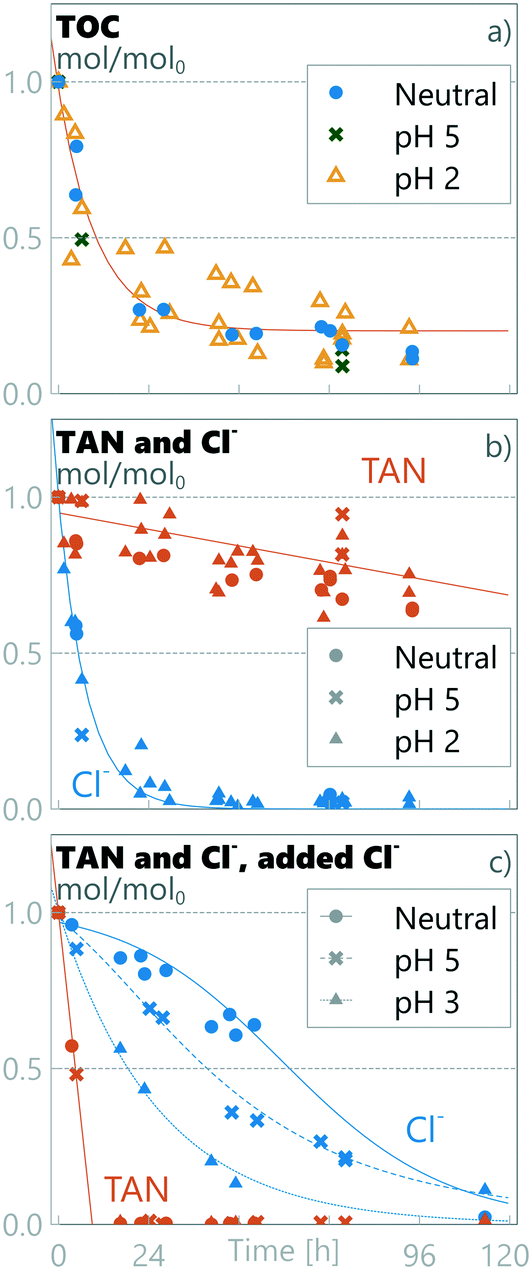
3.6 Real source-separated urine results
The real source-separated human urine collected at a social venue in Tampere, Finland, had different properties to the synthetic urine (see Table 1). The collected urine had only 0.32 ISreal/ISsynthetic, 0.38 TANreal/TANsynthetic and 0.22 Clreal/Clsynthetic compared to the synthetic urine used. Using these real urine samples, the TOC was oxidized at all pH levels following a 1st order reaction rate (eqn (9); kurine = 0.10 ± 0.04 h−1) significantly higher than that measured for synthetic urine (ksynthetic = 0.05 h−1) (see Fig. 6). The remaining TOC relative to TOC0 (0.20 ± 0.04 TOCresidual,urine/TOC0) was however higher than in the synthetic urine (0.06 ± 0.03 TOCresidual,pH=5/TOC0), but still in a similar absolute measured TOC level (0.020 ± 0.005 mmol L−1 in real urine vs. 0.013 ± 0.009 mmol L−1 in synthetic urine on average). TAN was oxidized following a zero-order constant decay rate of kurine = −0.002 ± 0.001 h−1, the same as the slow decay rate measured for synthetic urine at pH ≤3 within a 95% confidence interval (see section 3.2). As TAN was following a similar decay pattern to that observed for synthetic urine at pH ≤3, so was chloride, following pseudo 1st-order kinetics and rapidly oxidized within the first 24 h for all pH values to form perchlorate (mass balance of 1.07 ± 0.04 ClO4−/Cl0), while similar nitrate and nitrite results were recorded to the synthetic urine (see ESI† section S5).
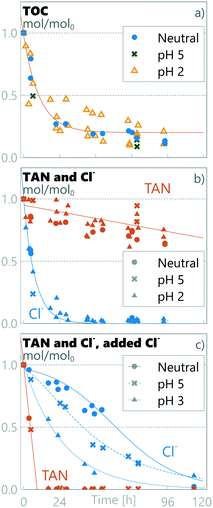 |
| | Fig. 6 Relative amount of substances in real urine at different pH values. a) TOC and b) TAN and Cl− in real urine, and c) TAN and Cl− in real urine with surplus chloride. Notice that the initial absolute chloride concentration in real urine with surplus chlorine c) is much higher than in real urine b) (540 vs. 197 mmol L−1). | |
As discussed in section 3.2, the experiments with synthetic urine indicated that the Cl/TAN ratio has a significant role in TAN oxidation chemistry. When real urine was employed, no rapid TAN oxidation in any pH settings was observed (see Fig. 6b). Real urine had a Cl/TAN-ratio of 0.13 ± 0.01, lower than experimentally required (≈0.2 Cl/TAN) for the rapid oxidation of TAN observed for the synthetic urine. To confirm the effect of the chloride ratio on the complexity of real urine, 150 mL of the collected source-separated urine was supplemented with 28 mL of 3 M KCl, resulting in a chloride concentration of 540 ± 60 mmol L−1, 20× and 5× higher than that in real and synthetic urine samples, respectively, corresponding to seawater salinity and a measured Cl/TAN ratio of 4.1 ± 0.4. In the presence of this surplus chloride, TAN was rapidly oxidized under all pH conditions following zero-order kinetics with an observed decay rate of ksurplusCl = −0.11 ± 0.01 h−1 (see Fig. 6c), significantly higher than that observed in neutral pH experiments for synthetic urine (kneutral = −0.020 ± 0.002 h−1). TAN retainment in real urine for all pH values was 0.82 ± 0.07 TAN mol/mol0 (Fig. 6b); when surplus chloride was added, no TAN was retained (Fig. 6c). The P mass balance in real urine was 1.05 ± 0.2 mol/mol0 over all runs and the K mass balance in runs without KCl addition was 1.9 ± 0.4 mol/mol0. The reason for the measured K increase is unknown and was not investigated but could be related to nutrient release from organic content. Chloride mass balance did not close in the surplus run, and the ClOx measured could not account for the chloride lost (see ESI† Fig. S5). The discrepancy could be due to oxidation to chlorine gas, but more likely due to precipitation of a perchlorate salt.59
While the pH had no impact on TOC or TAN decay in real urine with surplus chloride, the pH did have a significant effect on the chloride decay rate, with a similar relation to pH as observed for synthetic urine (see section 3.2.1 and eqn (11)). These results suggest that, in our setup, the determining characteristic for TAN preservation was the Cl/TAN ratio, with the anodic pH playing a role in mitigating the removal of chloride to Cl2 gas and perchlorate, lowering hypochlorite diffusion to the bulk and therefore limiting breakpoint chlorination chemistry for TAN oxidation. In our experiments, the effect of pH was visible only in a specific range of chloride concentrations, where the Cl/TAN-ratio passes through ≈0.2, when a transition from a fast to slow TAN oxidation rate was observed. For synthetic urine, Cl/TAN was at 0.23 ± 0.02 and the anodic pH defines the chloride oxidation pathway and rate, subsequently defining the rate for TAN oxidation. In the real urine, Cl/TAN was naturally lower (0.13 ± 0.01), and no rapid TAN oxidation rate was observed. However, with surplus chloride, Cl/TAN remains above 0.2 (starting at 4.1 ± 0.3) until all TAN is oxidized and no transition was observed regardless of the anodic pH. Based on literature sources (see section 2.1), source-separated urine Cl/TAN is expected to be in the proximity of ≈0.2, and improper storage can further increase the ratio if TAN is lost. Accordingly, pH control could be essential for selective TAN oxidation in most real urine situations.
3.7 TAN decay and boundary layer chloride chemistry
Based on the measurements done in this study, as well as the published literature, the mechanisms for selective TAN preservation are most probably related to the chloride oxidation pathways at the boundary layer on the electrode surface. In our experiments, the chloride to TAN molar ratio, at a level of ≈0.2, was identified as the defining characteristic for the TAN oxidation rate change. While a more systematic study and detailed measurements are required to confirm a precise limit, a hypothesis for the basis of this limit can be formulated based on the literature.
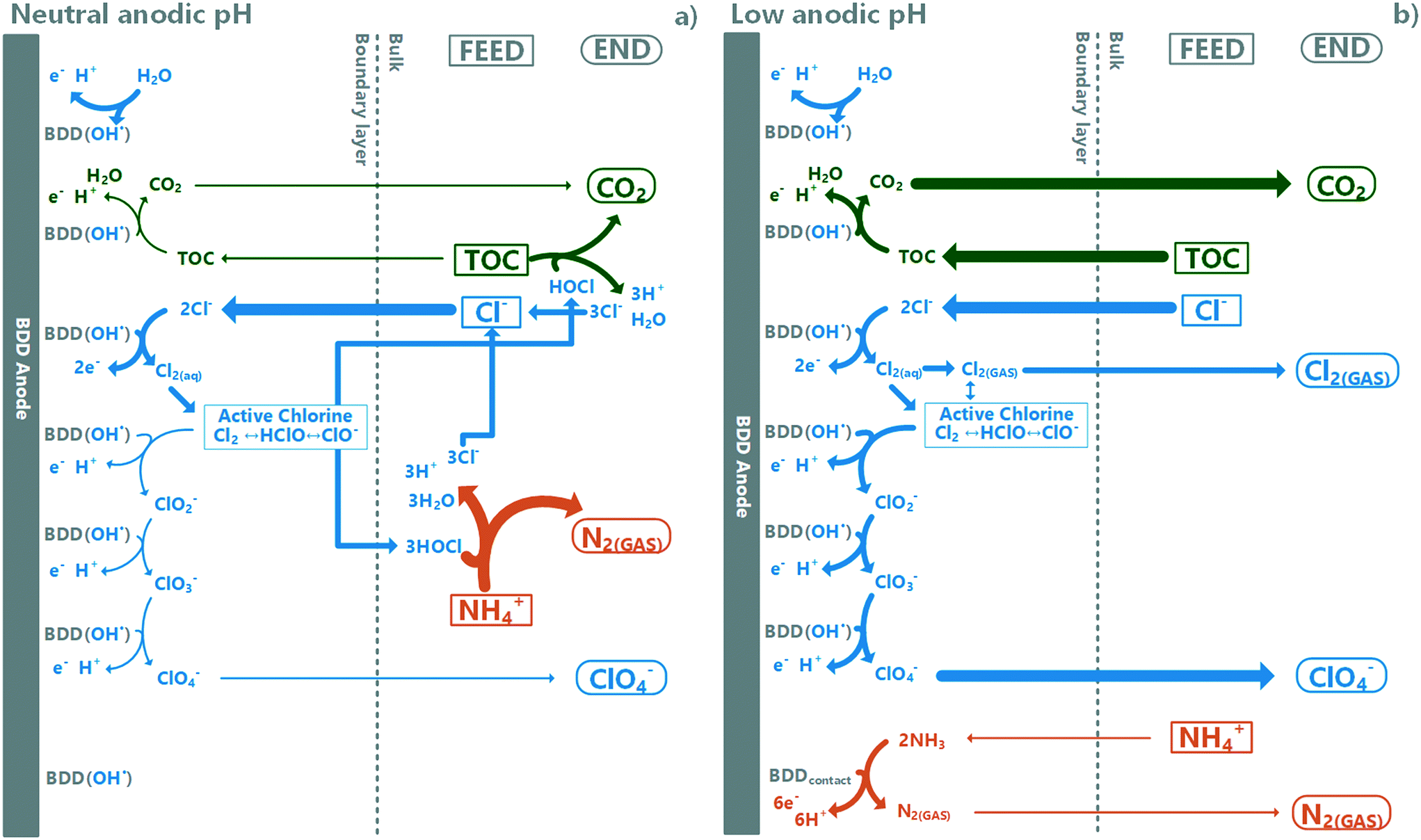
The electrode surface BDD(OH˙)-radical readily reacts with chloride to form RCS (eqn (2) and (3)). It can be hypothesised that in neutral pH, there are enough reaction pathways available to scavenge surface BDD(OH˙)-radicals to a low level, allowing the formed RCS to diffuse off the boundary layer, and further oxidize TAN (and TOC) in the bulk through breakpoint chemistry in regions where the active chloride/TAN ratio is high enough. The dominating RCS in the bulk, hypochlorite, forms chloride after reacting with TAN (it can react also with TOC). This circulation allows for the retainment of chloride in the system and continuation of this process until TAN depletion. In low pH however, the rate for further oxidation reactions from RCS to chlorite dominates over RCS diffusion to the bulk electrolyte, causing a rapid drop in chloride concentration, subsequentially preventing further TAN oxidation through the breakpoint chlorination mechanism as the hypochlorite concentration does not reach high enough concentrations.27,33,81,82 In the absence of chloride, the mechanism for slow TAN decay is most likely ammonia oxidation to N2via direct oxidation on the BDD surface, while TOC oxidation proceeds through BDD(OH˙)-radicals.66 These hypotheses are summarized in Fig. 7.
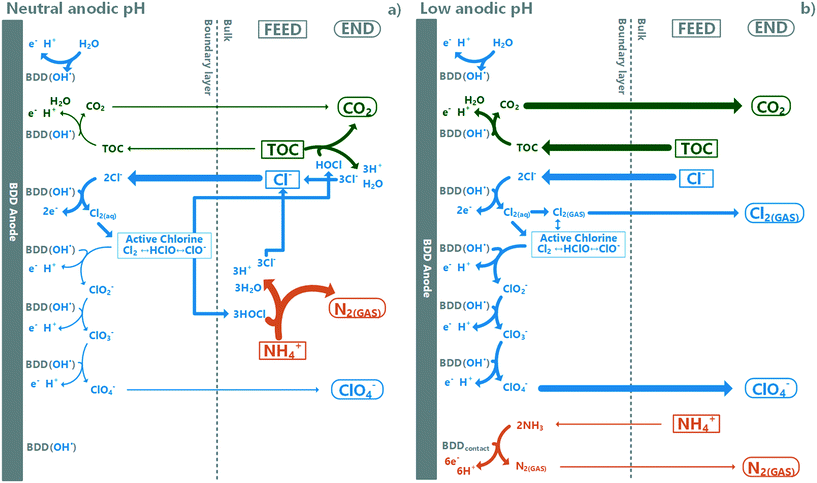 |
| | Fig. 7 Hypothesised pathways (simplified) dominating TOC and TAN oxidation under a) neutral and b) low anodic pH-conditions. Boxes indicate feed substances, bubbles end products and arrow widths relative scale of reaction rates. Under neutral conditions, active chlorine diffuses to the bulk and is responsible for most of the TOC and TAN decay measured. At low pH, formed chloride radicals (RCS) have reaction pathways with competitive reaction rates including further oxidation and removal as gaseous chlorine, and diffused active chlorine to the bulk is low and cannot induce breakpoint-chlorination-type TAN decay. | |
While this study did not focus specifically on chloride chemistry or speciation, it demonstrates that for future research, such emphasis is required if a method for separating TOC and TAN oxidation without perchlorate accumulation is to be realised for the decentralized EAOP treatment of source-separated urine with concomitant nutrient recovery. Based on these results, the key to TAN recovery with selective TOC oxidation relies on controlling the chloride chemistry defining the TAN oxidation regime. It can also require monitoring the composition of the source-separated urine feed, which can be highly variable depending on the source. Also, to make this technology meet regulatory limits, an alternate RCS quenching mechanism or end-pathway will be required to prevent chloride oxidation to perchlorate.
4. Conclusions
• Electrochemical oxidation of urine in low pH allows for the selective oxidation of organics with retainment of all main nutrients N, P and K, enabling subsequent product utilization as a liquid fertilizer through e.g. electro-concentration.
• Lowering pH to 3 or below at the anodic chamber using a reagent-free control system stops electrochemical oxidation of TAN in synthetic urine. A retainment of 79 ± 5% of TAN was measured compared to 1 ± 1% in neutral pH.
• Over 90% oxidation of organic material TOC was measured in all pH levels. This can enable selective electrochemical oxidation of the urine organics fraction without oxidation of TAN.
• In real urine samples, TAN was retained under all pH levels. When surplus chloride was added, TAN was lost under all conditions. These results confirm the central role of the chloride to ammonium nitrogen ratio as the main parameter defining TAN retainment in the reactor, with a molar ratio of approximately 0.2 Cl/TAN found defining in this system.
• In all experiments, chloride was mainly oxidized to perchlorate, a persistent toxic by-product. To make the proposed technology feasible in the field, an alternate active chloride quenching mechanism or end-pathway for chlorine still needs to be developed.
Ethical statement
The human urine used was collected from a toilet block in the cultural venue “Hiedanlahti-Kuivaamo” in Tampere, operated by a private company, Digi-Toilet Systems Oy. All experiments were performed in accordance with the guidelines of ‘Responsible Conduct of Research and Procedures for Handling Allegations of Misconduct in Finland’. The human participants were clearly informed of the research use of the collected source-separated urine.
Funding
Johannes Jermakka was supported financially by Maj and Tor Nessling foundation, Walter Ahlström Foundation and The Finnish Cultural Foundation. Pablo Ledezma acknowledges a Development Fellowship from The University of Queensland and the Australian Research Council project LP 150100402 in partnership with Queensland Urban Utilities (QUU) and ABR Process Development.
Author contributions
Johannes Jermakka: conceptualization, methodology, formal analysis, investigation, writing – original draft, visualization, funding acquisition. Marika Kokko: validation, resources, writing – review & editing, supervision, project administration. Stefano Freguia: conceptualization, methodology, writing – review & editing. Pablo Ledezma: methodology, validation, writing – review & editing, supervision.
Conflicts of interest
None.
References
-
E. Friedler, D. Butler and Y. Alfiya, in Source Separation and Decentralization for Wastewater Management, ed. T. A. Larsen, K. M. Udert and J. Lienert, IWA Publishing, Oxford, 2013, pp. 241–258 Search PubMed.
- D. G. Randall and V. Naidoo, Urine: The liquid gold of wastewater, J. Environ. Chem. Eng., 2018, 6, 2627–2635 CrossRef CAS.
- P. Ledezma, P. Kuntke, C. J. N. Buisman, J. Keller and S. Freguia, Source-separated urine opens golden opportunities for microbial electrochemical technologies, Trends Biotechnol., 2015, 33, 214–220 CrossRef CAS PubMed.
- M. Capdevila-Cortada, Electrifying the Haber-Bosch, Nat. Catal., 2019, 2, 1055 CrossRef.
- H. Jönsson, T. A. Stenström, J. Svensson and A. Sundin, Source separated urine-nutrient and heavy metal content, water saving and faecal contamination, Water Sci. Technol., 1997, 35, 145–152 CrossRef.
-
T. A. Larsen, K. M. Udert and J. Lienert, Source separation and decentralization for wastewater management, IWA, London, 2013 Search PubMed.
- S. Antonini, S. Paris, T. Eichert and J. Clemens, Nitrogen and Phosphorus Recovery from Human Urine by Struvite Precipitation and Air Stripping in Vietnam, Clean: Soil, Air, Water, 2011, 39, 1099–1104 CAS.
- B. Etter, E. Tilley, R. Khadka and K. M. Udert, Low-cost struvite production using source-separated urine in Nepal, Water Res., 2011, 45, 852–862 CrossRef CAS PubMed.
- A. Hug and K. M. Udert, Struvite precipitation from urine with electrochemical magnesium dosage, Water Res., 2013, 47, 289–299 CrossRef CAS PubMed.
- J. A. Wilsenach, C. A. Schuurbiers and M. C. van Loosdrecht, Phosphate and potassium recovery from source separated urine through struvite precipitation, Water Res., 2007, 41, 458–466 CrossRef CAS PubMed.
- T. Duong, Z. L. Xie, D. Ng and M. Hoang, Ammonia removal from aqueous solution by membrane distillation, Water Environ. J., 2013, 27, 425–434 CAS.
- A. Zarebska, K. V. Christensen and B. Norddahl, The Application of Membrane Contactors for Ammonia Recovery from Pig Slurry, Procedia Eng., 2012, 44, 1642–1645 CrossRef.
- M. Ronteltap, M. Maurer and W. Gujer, Struvite precipitation thermodynamics in source-separated urine, Water Res., 2007, 41, 977–984 CrossRef CAS PubMed.
- M. Rodríguez Arredondo, P. Kuntke, A. ter Heijne, H. V. M. Hamelers and C. J. N. Buisman, Load ratio determines the ammonia recovery and energy input of an electrochemical system, Water Res., 2017, 111, 330–337 CrossRef PubMed.
- R. C. Tice and Y. Kim, Energy efficient reconcentration of diluted human urine using ion exchange membranes in bioelectrochemical systems, Water Res., 2014, 64, 61–72 CrossRef CAS PubMed.
- J. Jermakka, E. Thompson Brewster, P. Ledezma and S. Freguia, Electro-concentration for chemical-free nitrogen capture as solid ammonium bicarbonate, Sep. Purif. Technol., 2018, 203, 48–55 CrossRef CAS.
- P. Ledezma, J. Jermakka, J. Keller and S. Freguia, Recovering Nitrogen as a Solid without Chemical Dosing: Bio-Electroconcentration for Recovery of Nutrients from Urine, Environ. Sci. Technol. Lett., 2017, 4, 119–124 CrossRef CAS.
- S. Freguia, M. E. Logrieco, J. Monetti, P. Ledezma, B. Virdis and S. Tsujimura, Self-powered bioelectrochemical nutrient recovery for fertilizer generation from human urine, Sustainability, 2019, 11, 1–10 CrossRef.
- P. Kuntke, M. Rodríguez Arredondo, L. Widyakristi, A. Ter Heijne, T. H. J. A. Sleutels, H. V. M. Hamelers and C. J. N. Buisman, Hydrogen Gas Recycling for Energy Efficient Ammonia Recovery in Electrochemical Systems, Environ. Sci. Technol., 2017, 51, 3110–3116 CrossRef CAS PubMed.
- A. K. Luther, J. Desloover, D. E. Fennell and K. Rabaey, Electrochemically driven extraction and recovery of ammonia from human urine, Water Res., 2015, 87, 367–377 CrossRef CAS PubMed.
- J. Desloover, A. Abate Woldeyohannis, W. Verstraete, N. Boon and K. Rabaey, Electrochemical resource recovery from digestate to prevent ammonia toxicity during anaerobic digestion, Environ. Sci. Technol., 2012, 46, 12209–12216 CrossRef CAS PubMed.
- J. Desloover, J. De Vrieze, M. Van De Vijver, J. Mortelmans, R. Rozendal and K. Rabaey, Electrochemical nutrient recovery enables ammonia toxicity control and biogas desulfurization in anaerobic digestion, Environ. Sci. Technol., 2015, 49, 948–955 CrossRef CAS PubMed.
-
P. Kuntke, Nutrient and energy recovery from urine, PhD Thesis, Wageningen University, 2013 Search PubMed.
- M. Rodríguez Arredondo, P. Kuntke, A. W. Jeremiasse, T. H. J. A. Sleutels, C. J. N. Buisman and A. ter Heijne, Bioelectrochemical systems for nitrogen removal and recovery from wastewater, Environ. Sci.: Water Res. Technol., 2015, 1, 22–33 RSC.
- S. Gildemyn, A. K. Luther, S. J. Andersen, J. Desloover and K. Rabaey, Electrochemically and bioelectrochemically induced ammonium recovery, J. Visualized Exp., 2015, 95, e52405 Search PubMed.
- P. Kuntke, P. Zamora, M. Saakes, C. J. N. Buisman and H. V. M. V. Hamelers, Gas-permeable hydrophobic tubular membranes for ammonia recovery in bio-electrochemical systems, Environ. Sci.: Water Res. Technol., 2016, 2, 261–265 RSC.
- C. A. Martínez-Huitle, M. A. Rodrigo, I. Sirés and O. Scialdone, Chem. Rev., 2015, 115, 13362–13407 CrossRef PubMed.
- S. O. Ganiyu and C. A. Martínez-Huitle, ChemElectroChem, 2019, 6, 2379–2392 CrossRef CAS.
-
K. Udert, S. Brown-Malker and J. Keller, in Source Separation and Decentralization for Wastewater Management, ed. T. A. Larsen, K. M. Udert and J. Lienert, IWA Publishing, Oxford, 2013, pp. 321–336 Search PubMed.
- P. Ma, H. Ma, S. Sabatino, A. Galia and O. Scialdone, Electrochemical treatment of real wastewater. Part 1: Effluents with low conductivity, Chem. Eng. J., 2018, 336, 133–140 CrossRef CAS.
- M. Panizza and G. Cerisola, Direct And Mediated Anodic Oxidation of Organic Pollutants, Chem. Rev., 2009, 109, 6541–6569 CrossRef CAS PubMed.
- H. Zöllig, C. Fritzsche, E. Morgenroth, K. M. Udert, H. Zollig, C. Fritzsche, E. Morgenroth and K. M. Udert, Direct electrochemical oxidation of ammonia on graphite as a treatment option for stored source-separated urine, Water Res., 2015, 69, 284–294 CrossRef PubMed.
- H. Zöllig, A. Remmele, C. Fritzsche, E. Morgenroth and K. M. Udert, Formation of Chlorination Byproducts and Their Emission Pathways in Chlorine Mediated Electro-Oxidation of Urine on Active and Nonactive Type Anodes, Environ. Sci. Technol., 2015, 49, 11062–11069 CrossRef PubMed.
- H. Zöllig, A. Remmele, E. Morgenroth and K. M. Udert, Removal rates and energy demand of the electrochemical oxidation of ammonia and organic substances in real stored urine, Environ. Sci.: Water Res. Technol., 2017, 3, 480–491 RSC.
- K. Cho and M. R. Hoffmann, Urea Degradation by Electrochemically Generated Reactive Chlorine Species: Products and Reaction Pathways, Environ. Sci. Technol., 2014, 48, 11504–11511 CrossRef CAS PubMed.
- J. T. Jasper, O. S. Shafaat and M. R. Hoffmann, Electrochemical Transformation of Trace Organic Contaminants in Latrine Wastewater, Environ. Sci. Technol., 2016, 50, 10198–10208 CrossRef CAS PubMed.
- J. T. Jasper, Y. Yang and M. R. Hoffmann, Toxic Byproduct Formation during Electrochemical Treatment of Latrine Wastewater, Environ. Sci. Technol., 2017, 51, 7111–7119 CrossRef CAS PubMed.
- K. Cho, D. Kwon and M. R. Hoffmann, Electrochemical treatment of human waste coupled with molecular hydrogen production, RSC Adv., 2014, 4, 4596–4608 RSC.
- Y. Yang, L. Lin, L. K. Tse, H. Dong, S. Yu and M. R. Hoffmann, Membrane-separated electrochemical latrine wastewater treatment, Environ. Sci.: Water Res. Technol., 2019, 5, 51–59 RSC.
- X. Huang, Y. Qu, C. A. E. Cid, C. Finke, M. R. Hoffmann, K. Lim and S. C. Jiang, Electrochemical disinfection of toilet wastewater using wastewater electrolysis cell, Water Res., 2016, 92, 162–172 CrossRef PubMed.
- C. M. Chung, S. W. Hong, K. Cho and M. R. Hoffmann, Degradation of organic compounds in wastewater matrix by electrochemically generated reactive chlorine species: Kinetics and selectivity, Catal. Today, 2018, 313, 189–195 CrossRef CAS.
- S. O. Ganiyu and C. A. Martínez-Huitle, Nature, Mechanisms and Reactivity of Electrogenerated Reactive Species at Thin-Film Boron-Doped Diamond (BDD) Electrodes During Electrochemical Wastewater Treatment, ChemElectroChem, 2019, 6, 2379–2392 CrossRef CAS.
- S. Garcia-Segura, J. D. Ocon and M. N. Chong, Process Saf. Environ. Prot., 2018, 113, 48–67 CrossRef CAS.
- S. T. McBeath, D. P. Wilkinson and N. J. D. Graham, Environ. Sci.: Water Res. Technol., 2019, 5, 2090–2107 RSC.
- E. Brillas and C. A. Martínez-Huitle, Decontamination of wastewaters containing synthetic organic dyes by electrochemical methods. An updated review, Appl. Catal., B, 2015, 166–167, 603–643 CrossRef CAS.
- C. Comninellis, A. Kapalka, S. Malato, S. A. Parsons, I. Poulios and D. Mantzavinos, Advanced oxidation processes for water treatment: Advances and trends for R&D, J. Chem. Technol. Biotechnol., 2008, 83, 769–776 CrossRef CAS.
- K. M. Udert, T. A. Larsen, M. Biebow and W. Gujer, Urea hydrolysis and precipitation dynamics in a urine-collecting system, Water Res., 2003, 37, 2571–2582 CrossRef CAS PubMed.
- K. M. Udert, T. A. Larsen and W. Gujer, Fate of major compounds in source-separated urine, Water Sci. Technol., 2006, 54, 413–420 CrossRef CAS PubMed.
-
C. Lentner, Geigy scientific tables, Basel, Switzerland, 8th edn, 1977 Search PubMed.
- S. T. Jaatinen, M. R. T. Palmroth, J. A. Rintala and T. A. Tuhkanen, The effect of urine storage on antiviral and antibiotic compounds in the liquid phase of source-separated urine, Environ. Technol., 2016, 37, 2189–2198 CrossRef CAS PubMed.
- H. Kirchmann and S. Pettersson, Human urine - Chemical composition and fertilizer use efficiency, Fert. Res., 1994, 40, 149–154 CrossRef.
- P. Kuntke, K. M. Śmiech, H. Bruning, G. Zeeman, M. Saakes, T. H. J. A. Sleutels, H. V. M. Hamelers and C. J. N. Buisman, Ammonium recovery and energy production from urine by a microbial fuel cell, Water Res., 2012, 46, 2627–2636 CrossRef CAS PubMed.
- M. Maurer, W. Pronk and T. A. Larsen, Treatment processes for source-separated urine, Water Res., 2006, 40, 3151–3166 CrossRef CAS PubMed.
-
F. Tettenborn, J. Behrendt and R. Otterpohl, Final report for task 7 of the demonstration project “Sanitation Concepts for Separate Treatment of Urine, Faeces and Greywater” (SCST) Resource recovery and removal of pharmaceutical residues Treatment of separate collected urine, 2007 Search PubMed.
- P. Zamora, T. Georgieva, I. Salcedo, N. Elzinga, P. Kuntke and C. J. N. Buisman, Long-term operation of a pilot-scale reactor for phosphorus recovery as struvite from source-separated urine, J. Chem. Technol. Biotechnol., 2017, 92, 1035–1045 CrossRef CAS.
- K. M. Udert, M. Wachter and M. Wächter, Complete nutrient recovery from source-separated urine by nitrification and distillation, Water Res., 2012, 46, 453–464 CrossRef CAS PubMed.
-
B. Etter, A. Hug and K. M. Udert, Total Nutrient Recovery from Urine – Operation of a Pilot-Scale Nitrification Reactor, WEF/IWA Int. Conf. Nutr. Remov. Recover, 2013, pp. 1–4 Search PubMed.
- S. Bouatra, F. Aziat, R. Mandal, A. C. Guo, M. R. Wilson, C. Knox, T. C. Bjorndahl, R. Krishnamurthy, F. Saleem, P. Liu, Z. T. Dame, J. Poelzer, J. Huynh, F. S. Yallou, N. Psychogios, E. Dong, R. Bogumil, C. Roehring and D. S. Wishart, The Human Urine Metabolome, PLoS One, 2013, 8(9), e73076 CrossRef CAS PubMed.
-
S. Zumdahl and D. DeCoste, Chemical Principles, 2012 Search PubMed.
- N. Kyurkchiev and S. Markov, Sigmoidal Functions: Some Computational and Modelling Aspects, Biomath Commun., 2014, 1(2) DOI:10.11145/j.bmc.2015.03.081.
-
S. J. Randtke, in White's Handbook of Chlorination and Alternative Disinfectants, John Wiley and Sons, 5th edn, 2010, pp. 68–173 Search PubMed.
-
E. A. Kobylinski and A. Bhandari, in White's Handbook of Chlorination and Alternative Disinfectants, John Wiley and Sons, 5th edn, 2010, pp. 326–362 Search PubMed.
- A. Romano, A. M. Urtiaga and I. Ortiz, Optimized energy consumption in electrochemical-based regeneration of RAS water, Sep. Purif. Technol., 2020, 240, 116638 CrossRef CAS.
- Y. Gendel and O. Lahav, Revealing the mechanism of indirect ammonia electrooxidation, Electrochim. Acta, 2012, 63, 209–219 CrossRef CAS.
-
H. Shorney-Darby and L. L. Harms, in White's Handbook of Chlorination and Alternative Disinfectants, John Wiley and Sons, 5th edn, 2010, pp. 230–325 Search PubMed.
- N. L. Michels, A. Kapałka, A. A. Abd-El-Latif, H. Baltruschat and C. Comninellis, Enhanced ammonia oxidation on BDD induced by inhibition of oxygen evolution reaction, Electrochem. Commun., 2010, 12, 1199–1202 CrossRef CAS.
- M. J. Martin De Vidales, M. Millán, C. Sáez, P. Cañizares and M. A. Rodrigo, What happens to inorganic nitrogen species during conductive diamond electrochemical oxidation of real wastewater?, Electrochem. Commun., 2016, 67, 65–68 CrossRef CAS.
- S. Garcia-Segura, E. Mostafa and H. Baltruschat, Electrogeneration of inorganic chloramines on boron-doped diamond anodes during electrochemical oxidation of ammonium chloride, urea and synthetic urine matrix, Water Res., 2019, 160, 107–117 CrossRef CAS PubMed.
- Y. J. Shih, Y. H. Huang and C. P. Huang, Oxidation of ammonia in dilute aqueous solutions over graphite-supported α- and β-lead dioxide electrodes (PbO2@G), Electrochim. Acta, 2017, 257, 444–454 CrossRef CAS.
- Y. Zeng, C. Priest, G. Wang and G. Wu, Restoring the Nitrogen Cycle by Electrochemical Reduction of Nitrate: Progress and Prospects, Small Methods, 2020, 4, 2000672 CrossRef CAS.
- J. Boudreau, D. Bejan and N. J. Bunce, Competition between electrochemical advanced oxidation and electrochemical hypochlorination of acetaminophen at boron-doped diamond and ruthenium dioxide based anodes, Can. J. Chem., 2010, 88, 418–425 CrossRef CAS.
- C. A. Martínez-Huitle and E. Brillas, Decontamination of wastewaters containing synthetic organic dyes by electrochemical methods: A general review, Appl. Catal., B, 2009, 87, 105–145 CrossRef.
- E. Brillas, I. Sirés and M. A. Oturan, Electro-fenton process and related electrochemical technologies based on fenton's reaction chemistry, Chem. Rev., 2009, 109, 6570–6631 CrossRef CAS PubMed.
- B. P. Chaplin, G. Schrader and J. Farrell, Electrochemical destruction of N -Nitrosodimethylamine in reverse osmosis concentrates using boron-doped diamond film electrodes, Environ. Sci. Technol., 2010, 44, 4264–4269 CrossRef CAS PubMed.
- J. Radjenovic and D. L. Sedlak, Challenges and Opportunities for Electrochemical Processes as Next-Generation Technologies for the Treatment of Contaminated Water, Environ. Sci. Technol., 2015, 49, 11292–11302 CrossRef CAS PubMed.
- S. Garcia-Segura, J. Keller, E. Brillas and J. Radjenovic, Removal of organic contaminants from secondary effluent by anodic oxidation with a boron-doped diamond anode as tertiary treatment, J. Hazard. Mater., 2015, 283, 551–557 CrossRef CAS PubMed.
- M. Herraiz-Carboné, S. Cotillas, E. Lacasa, Á. Moratalla, P. Cañizares, M. A. Rodrigo and C. Sáez, Improving the biodegradability of hospital urines polluted with chloramphenicol by the application of electrochemical oxidation, Sci. Total Environ., 2020, 725, 138430 CrossRef PubMed.
- S. Cotillas, E. Lacasa, M. Herraiz, C. Sáez, P. Cañizares and M. A. Rodrigo, The Role of the Anode Material in Selective Penicillin G Oxidation in Urine, ChemElectroChem, 2019, 6, 1376–1384 CrossRef.
- G. O. S. Santos, K. I. B. Eguiluz, G. R. Salazar-Banda, C. Sáez and M. A. Rodrigo, Understanding the electrolytic generation of sulfate and chlorine oxidative species with different boron-doped diamond anodes, J. Electroanal. Chem., 2020, 857, 113756 CrossRef CAS.
-
J. P. Gustafsson, Visual MINTEQ 3.1 user guide, Dep. L. Water Resour. Eng. R. Inst. Technol., Stokholm, Sweden, 2014, pp. 1–73 Search PubMed.
- L. R. Czarnetzki and L. J. J. Janssen, Formation of hypochlorite, chlorate and oxygen during NaCl electrolysis from alkaline solutions at an RuO2/TiO2 anode, J. Appl. Electrochem., 1992, 22, 315–324 CrossRef CAS.
- C. D. N. Brito, D. M. De Araújo, C. A. Martínez-Huitle and M. A. Rodrigo, Understanding active chlorine species production using boron doped diamond films with lower and higher sp3/sp2 ratio, Electrochem. Commun., 2015, 55, 34–38 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ew01057j |
| ‡ Permanent address: Tampere University, Faculty of Engineering and Natural Sciences, PO Box 541, 33014 Tampere, Finland. E-mail: johannes.jermakka@tuni.fi |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Stefano
Freguia
c,
Marika
Kokko‡
a and
Pablo
Ledezma
*ab,
Stefano
Freguia
c,
Marika
Kokko‡
a and
Pablo
Ledezma