
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Immunological detection of thymine dimers in indigenous genomic DNA from pre-disinfection drinking water as an ultraviolet disinfection dosimeter†
James
Blyth
 ,
Lucinda
Hazell
and
Michael R.
Templeton
*
,
Lucinda
Hazell
and
Michael R.
Templeton
*
Department of Civil and Environmental Engineering, South Kensington Campus, Imperial College London, London, UK. E-mail: m.templeton@imperial.ac.uk
First published on 7th September 2021
Abstract
Culture-based methods are the primary methods used for the routine detection and enumeration of bacteria and viruses in water samples. In the context of ultraviolet (UV) disinfection, they are also the basis for reactor validation in drinking water treatment systems. However, the majority of microorganisms in pre-disinfection drinking water are not culturable. In UV disinfection, the DNA of both the culturable and non-culturable microbial populations will form pyrimidine dimers in response to UV photon absorbance. In this research an enzyme-linked immuno-sorbent assay (ELISA) was used to detect thymine dimers in the extractable genomic DNA (gDNA) from the total microbial population in pre-disinfection drinking water as a UV disinfection dosimeter. The method was first optimised using “naked” (extracted prior to UV exposure) and in vivo (extracted post UV exposure) E. coli gDNA, and then tested using water samples from UK drinking water treatment plants. Samples were exposed to up to 120 mJ cm−2 of monochromatic (253.7 nm) UV light using a collimated beam device and an ELISA was applied to the gDNA. This approach, once optimised, resulted in linear relationships between the assay response and UV dose. This shows that ELISA-based enumeration of thymine dimers in total extractable gDNA from a mixed species population has the potential to provide a direct, relatively quick, sampling-based means of monitoring the UV disinfection dose being delivered by operating UV disinfection systems in drinking water treatment plants, without the need to spike a biodosimeter into the water nor take reactors out of service. Molecular techniques measuring dimer formation may also offer the UV disinfection industry a method of demonstrating dose delivery where the culturing of target organisms is problematic.
Water impactA molecular method has been used to measure the UV disinfection dose applied to water samples. The method uses indigenous DNA, meaning that nothing needs to be spiked into the water. This therefore represents a significant new water sample analysis method for routine monitoring of UV disinfection performance in water treatment works, to supplement existing online sensor-based methods. |
Introduction
The purpose of a drinking water UV disinfection system is to ensure that all water passing through it receives at least the minimum required dose of UV light in order to achieve the required levels of inactivation of target microorganisms found in the inflow stream. The intensity of UV light in a flow-through system decreases according to the square of the distance from the mercury arc lamp. Therefore, radially about the axis of a lamp there is a continually variable field of UV light intensity.1 In addition to a variable UV field, microorganisms behave as particles, and therefore traverse the system in a stochastic manner along discrete particle tracks.2 The combination of these two considerations gives rise to a population of microorganisms each of which has been exposed to a different UV dose. For this reason, flow-through UV disinfection systems deliver a distribution of UV doses to microorganisms as they traverse a reactor.3,4All municipal flow-through UV disinfection systems have at least one static UV sensor that measures the internal UV light intensity at a single point. However, because UV disinfection systems deliver a UV dose distribution, there is no direct correlation between the single-point UV intensity measured by the sensor and the dose delivered to microorganisms passing through the system. This, and the lack of a measurable chemical residual analogous to a chlorine residual, means all UV disinfection systems in water treatment works must be dose-validated using live biodosimeters or challenge organisms.3–7 Validation links the inactivation of challenge organisms achieved by a real system delivering a dose-distribution to a physically determined UV dose from a collimated beam procedure. The principle of the collimated beam is to irradiate microorganisms in an as near-ideal batch dose delivery system as possible such that average UV intensity can be determined using physical methods of measurement and calculation incorporating corrections for factors that influence UV intensity in the near-ideal system. The near-ideal system is important since in such systems all parts of the irradiated sample receive the calculated dose which is as close to the true arithmetic mean dose as possible. Calibration curves are then used to compare the inactivation achieved in the real system to the dose response from the collimated beam, and the reduced equivalent dose (RED) achieved in the reactor is determined. Protocols for UV flow-through system validation have been devised by several organisations, including: the Deutsche Vereinigung des Gas- und Wasserfaches (DGVW, Germany), United States Environmental Protection Agency (USEPA, USA), and Österreichisches Normungsinstitut (ÖNORM, Austria).
Since biodosimetry requires the injection of test microorganisms into the water flowing through the reactor, it cannot be used in operational UV dose verification,8,9i.e. it cannot be carried out in an “online” system. Therefore, UV disinfection guidance documents also describe how the data from validation should be used to develop system specific relationships between dose and operational conditions to allow in-service dose verification.10–13 These methods are required to demonstrate that the reactors are operating within the conditions used during dose validation and allow the recording of dose delivery in-service. The in-service dose is calculated based on the dose monitoring algorithm implemented in the control system, the variable parameters of which were determined during the dose validation procedure beforehand. Such telemetry-based verification of dose delivery in operational systems provides a robust method of linking operational conditions to delivered UV doses. However, the procedures rely on the condition of the reactor, particularly the state of the sensors, in order to be accurate. All guidance for UV disinfection is explicit in ensuring the telemetry of the system is maintained appropriately.11–13 There is currently no formal guidance for methods able to determine UV dose of operational UV systems that are independent of the condition of the reactor as measured by sensors.
The identification of an indigenous organism that is present in high numbers at the point of UV disinfection might form the basis of a simple sample measurement parameter for direct UV reactor performance verification, without the need for injecting a test organism.9 Samples taken upstream of the UV reactor could be subjected to a collimated beam test to determine dose–response and then compared with samples taken downstream of the reactor. However, the total number of individual heterotrophic bacterial species are unlikely to be sufficient to use in routine dose verification and the culturable population in drinking water is likely a minority in the context of the total microbial population.14–16 This suggests culture-based methods, while the basis of validation, are of limited use for routine verification.14–16
The biological mechanism of UV disinfection is based on the absorbance of UV light by nucleic acids, although a contribution for UV absorbance by proteins is increasingly becoming apparent.17,18 DNA, RNA and proteins absorb UV light over a broad range of wavelengths, but UV-C light (220–300 nm) is often referred to as the germicidal range. In this range the UV absorbance of water is at a minimum and there is a peak in the UV absorbance of nucleic acids at approximately 260–265 nm.19 All nucleotide bases absorb UV light, but the absorption by two adjacent pyrimidine bases is most critical for UV disinfection as this can form permanent covalent bonds between the adjacent bases known as a dimer. The most easily formed dimer pair in DNA is the thymine–thymine (TT) dimer (uracil in RNA), but cytosine–thymine (CT or TC) and cytosine–cytosine (CC) also occur.20 The resultant cyclobutane pyrimidine dimer, if not repaired, causes DNA replication to stall and prevents genome duplication which is required for cell replication.21 Dimer formation has been shown to be proportional to both UV dose and DNA adenine and thymine (AT) content.22–25
The molecular changes that occur in DNA upon exposure to UV light suggest that detection of dimers themselves could form the basis of a technique to determine dose delivery to pre-disinfection water samples. The dimers formed in DNA through absorption of UV photons can be detected directly by several molecular methods. These methods broadly fall into four principal types; those based on enzymatic degradation, DNA replication, chromatographic separation, and immunological detection. Table 1 summarises the advantages and disadvantages of these methods for the detection and measurement of dimer formation in DNA. It is important to note that in their simplest form molecular methods can only provide a relative response in a flow-through UV reactor and must therefore be used alongside collimated beam tests to relate the reactor to a reduced equivalent in-service dose.
| Method | Advantages | Disadvantages |
|---|---|---|
| Endonuclease digestion28 | • Relatively quick (hours) | • Not scalable |
| • Inexpensive (£1000) | • Not sensitive | |
| • Quantitation relies on densitometry | ||
| • A large quantity of sample is required compared to other techniques | ||
| Polymerase chain reaction (qPCR)26,27,29,30 | • Quantitative determination possible in the quantitative set-up that can read florescence from the PCR tubes directly | • Based on a decaying signal that saturates to zero |
| • Scalable | • If gels are used to visualise and quantify DNA replicated the process of band densitometry required to quantify the DNA introduces subjective quasi-quantitative element to the process | |
| • Sensitive | • Based on a small sequence of DNA often about 800–1000 bases | |
| • Relatively quick (hours) | • Relative measure | |
• Relatively inexpensive (£10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) 000) |
||
| High-pressure liquid chromatography-tandem mass spectroscopy (HPLC-MSMS)22,23,31,32 | • Able to detect numerous UV induced photoproducts | • Very expensive (over £100000) |
| • Quantitative | • Digestion of DNA introduces a processing step that could reduce sample amounts | |
| • Absolute measure | ||
| • Based on a positive signal | ||
| • Scalable | ||
| • Sensitive | ||
| • Relatively quick (hours) | ||
| Immuno-blotting8,22 | • Highly specific for the target of the primary antibody | • Not scalable |
| • Based on a positive signal | • Use of radioactive reporters | |
| • Relatively quick (hours) | • Quantitation relies on densitometry, introduces a subjective semi quantitative element to the process | |
| • Relatively inexpensive (£10000) |
• Requires determination of DNA amount deposited in the dot | |
| • Relative measure | ||
| Enzyme linked immunosorbent assay (ELISA)24 | • Quantitation not dependant on densitometry | • Requires determination of DNA amount deposited in the dot |
| • Highly scalable | • Relative measure | |
| • Highly specific for the target of the primary antibody | ||
| • Based on a positive signal | ||
| • Relatively quick (hours) | ||
| • Relatively inexpensive (over £10000) |
||
PCR based technology has previously been used by several authors to detect UV induced dimerisation in E. coli and P. aeruginosa, and in groundwater samples,26,27 however PCR detection of dimers relies on a negative response to increasing UV dose and this may influence bias, and limit application to high UV doses. Therefore, a signal based on an increase in response to increasing UV dose may increase the range of doses over which useful information can be obtained and have benefit in addressing bias. HPLC-MSMS is highly sensitive and discriminatory, but relies on very costly equipment. Immunological methods of binding antibodies to DNA extracted from water samples (i.e. immuno-blotting and ELISA) offer the potential to develop an economic sampling methodology that provides a positive signal in response to increasing UV dose. Of the two immunological methods considered, ELISA has advantages in not requiring densitometry in order to quantify the assay signal and in being scalable.
This study therefore explores the use of a monoclonal antibody to thymine-dimers in an ELISA to establish if UV dose response curves can be generated from the extractable gDNA of a mixed indigenous bacterial population in pre-disinfection water samples. Such a method may find application in independent verification of in-service reactors or evaluation of reactors for targeting non-culturable pathogens. An ELISA assay24 was tested and optimised to detect thymine dimers in gDNA from UV-treated E. coli cell suspensions at UV doses relevant to UV disinfection of drinking water (0–80 mJ cm−2) and to determine a UV dose response. The ELISA was also tested up to 120 mJ cm−2 on total gDNA from the mixed population of bacteria present at typical UV system installation locations in operating water treatment works.
2 Materials and methods
2.1 Naked E. coli gDNA preparation
All optimisation work for antibody dilutions and substrate concentrations was carried out using UV treated or untreated “naked” E. coli gDNA that was extracted prior to UV exposure. A single colony of E. coli cells (strain NCIMB 9481) maintained on a tryptone soy agar plate stored at 4 °C was transferred using a sterile bacteriological loop to 10 mL sterile tryptone soy broth media and incubated unshaken at 37 °C for 36 hours. E. coli gDNA was obtained by extraction of DNA directly from the 36 hour liquid culture by filtering the cell suspension using 22 μm filters. DNA extraction proceeded according to the methodology supplied with the PowerWater® DNA isolation kit (MoBio). The DNA was quantified using a nano-spectrophotometer (NanoDrop 1000, Thermo Scientific) and the concentration was adjusted to 1 μg mL−1 in phosphate buffered water. 5 mL samples of the stock solution were exposed to UV light in 5 cm Petri dishes, as described in section 2.4.2.2 In vivo UV treated E. coli gDNA preparation
Following initial optimisation, the ELISA was tested using E. coli gDNA exposed to a range of UV doses in vivo. A single colony of E. coli cells was transferred to tryptone soy broth media as described above and incubated unshaken at 37 °C for 18 hours. Two 1.5 mL samples were transferred to two 1.5 mL sterile Eppendorf tubes and cultures were pelleted in a benchtop centrifuge (Eppendorf) for 1 minute at 13000 rpm. The supernatant was decanted, and the cell pellets were resuspended in 200 mL of sterile PBS. 20 mL samples of the suspension were exposed to a range of UV doses in 90 mm Petri dishes (section 2.4). Then the E. coli gDNA was extracted into Buffer A (6 mM KH2PO4 4 mM K2HPO4) from Roza et al. (1988) and quantified as described before. The concentration of the in vivo UV treated E. coli gDNA was adjusted to a stock concentration of 1 ng μL−1 in Buffer A.
2.3 Water treatment plant samples
Water samples were obtained from two water treatment plants in the UK. The plants treat water from separate, predominantly rural, catchments with low population density. The main land use within the catchments is intensive agriculture with some light industry and point source discharges from industrial plants and sewage treatment works. The treatment scheme for both plants is illustrated in Fig. S.1 in the ESI.† During sampling, turbidity levels were below 1 NTU prior to disinfection. Water samples were collected post rapid gravity filters (PRGF) and post granular activated carbon (PGAC) in 2 L autoclaved borosilicate bottles and transported the same day in chilled cool bags to the laboratory. Upon arrival in the laboratory, samples were stored at 4 °C until processed, with a maximum time to processing of 48 hours. The 2 L bottles were mixed by shaking and 40 mL samples were aliquoted into 90 mm Petri dishes for the collimated beam tests (section 2.4).Following UV exposure, DNA from the water treatment plant samples was extracted, as described previously, into sterile water. The DNA concentration in these samples was below the detection limit of the nano-spectrophotometer, so a highly sensitive PicoGreen® (Inviltrogen) assay was used.33 Half of the DNA samples were used for DNA quantification and half for thymine dimer response determination by ELISA. The samples were adjusted to 1× concentration in Buffer A or PicoGreen development buffer such that 50 μL triplicates could be used for each of the ELISA and PicoGreen DNA quantifications. The fluorescence response was determined using a BMG Labtech POLARstar Omega plate reader fitted with a 485 nm 10 nm bandpass excitation filter and 520 nm and 10 nm bandpass emission filter. Mixing was by orbital shaking in the plate reader at 500 rpm for 1 minute prior to the first measurement. Determinations were done in triplicate for each sample. The DNA standard used was an E. coli gDNA preparation, prepared and quantified as described in section 2.1. Required DNA standard concentrations were made by serial dilutions in Tris-EDTA diluent.
2.4 UV exposure
The collimated UV beam experiments followed the protocol presented in Bolton and Linden (2003). Briefly, a 230 V low-pressure high output (LPHO) mercury lamp (Trojan Technologies) powered by a ballast from a UVMax C reactor was collimated using a dark coloured 140 mm internal diameter PVC tube to generate a quasi-parallel monochromatic UV beam. Prior to performing collimated beam experiments the UV lamp was turned on and allowed to stabilise for a minimum of 20 minutes. A magnetic stirrer was centred within the UV beam and the UV irradiance across two horizontal orthogonal axes of the beam was measured at the level of the sample surface at 5 mm intervals using an IL1700 radiometer with an SED240 sensor (International Light Technologies). The distance between the UV lamp and the sample surface was 64 cm and the average irradiance in the centre of the sample surface determined at the beginning of each experiment. At the end of the experiment the central irradiance value was checked to ensure it had not changed significantly during the time of the treatments. The absorbance of the samples was measured at 254 nm using a UV-vis spectrophotometer (Perkin Elmer Lambda 3) and the time required to deliver the indicated UV doses was calculated for each experiment using the samples UVT, its depth and volume, and accounted for the reflection from the matrix surface, divergence of the collimated beam, and variation of irradiance over the surface of the matrix, as per Bolton and Linden.34Treatments were carried out in polystyrene Petri dishes appropriate to the volume being treated. All samples were stirred with a sterile PTFE-coated 13 mm × 3 mm stir bar, the water depth varied depending on the sample volume. The stirring speed was set to avoid the formation of a vortex and the sample was exposed for the required time to achieve the desired UV dose. To ensure maximum DNA response in the optimisation experiments, naked gDNA samples (5 mL) were exposed to 0 and 500 mJ cm−2. E. coli suspensions (20 mL) and water treatment plant samples (40 mL) were exposed to 0, 10, 20, 40, 80 and 120 mJ cm−2 as indicated.
2.5 ELISA
Antibody dilutions and substrate concentrations were first optimised using naked E. coli gDNA. 50 ng of denatured naked E. coli gDNA, UV treated or untreated, was bound in triplicate to wells on a 96-well plate, a no DNA control was always included. Dilutions of primary and secondary antibodies are as indicated in Fig. S.3 to S.5 in the ESI.†The ELISA technique was similar to that previously described by Roza et al..24 A 96-well polystyrene plate was coated with poly-L-lysine (PLL) for 30 minutes at room temperature. Coated wells were washed 3 times with Tris-Buffered-Saline (TBS) and 50 μL of extracted and denatured DNA was incubated in coated wells for 16–20 hours at 37 °C. DNA was aspirated and wells washed 3 times with 0.05% TWEEN 20 in phosphate buffered saline (PBST). As a primary antibody, a mouse anti-thymine dimer monoclonal antibody (H3 clone, Sigma) was diluted to the optimal dilution of 1:1000 in 0.05% PBST containing 1% heat-inactivated foetal bovine serum (PBST-FCS). 100 μL of primary antibody (H3Ab) dilution was incubated in wells for 45 minutes at 37 °C. Wells were washed three times with PBST-FCS and incubated for 1 hour at room temperature with 100 μL of a 1:1000 optimal dilution of a polyclonal goat anti-mouse secondary antibody conjugated to horseradish peroxidase (HRP, Sigma) in PBST-FCS. Following aspiration of the secondary antibody, wells were washed 3 times with PBST-FCS (this wash step was included after initial optimisation of the primary antibody concentration suggested a high non-specific background binding occurred) and then 3 times with citrate buffer pH 5.0. After the third wash 25 μl of citrate buffer was added to each well. A two times stock of development buffer was made in citrate buffer pH 5.0 and contained 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) at 4.8 mg mL−1 and hydrogen peroxide at 180 μg mL−1. 25 μL of development buffer was added to each well and optical density was measured using a multi-well plate reader set at 405 nm every 2 minutes until one of the wells exceeded the measurement range of the instrument. Mixing was by orbital shaking in the plate reader at 500 rpm for 1 minute prior to the first measurement. Each sample was measured in triplicate.
2.6 Data analysis
Both endpoint and initial reaction rates were used to analyse dose response curves; a schematic representation of these two analysis types is shown in Fig. S.2 of the ESI.† For the endpoint analysis, the absorbance at 405 nm of the coloured ABTS end product was measured at the time point immediately prior to instrument saturation. Data was normalised to a unit mass of DNA. Data is reported as either the optical density (absorbance) at 405 nm (OD 405 nm) per unit mass of DNA or a ratio of OD 405 nm at specific doses per unit mass of DNA to OD 405 nm for the positive control per unit mass of DNA. Where replicates were used, data from the replicates of each dose on each plate were averaged.For the initial rate analysis, the rate of ABTS conversion at the start of the reaction was determined for each well by fitting a 2nd order polynomial regression to the timecourse data. The derivative of the resulting polynomial was evaluated at time = 0 minutes to give the initial rate of increase of OD 405 nm in each well and this was expressed as change in OD 405 nm per unit time. The initial rate from three wells for each dose was averaged to provide a mean initial rate value for each dose. Data was normalised to a unit mass of DNA loaded per well (expressed as change in OD 405 nm per unit time per unit mass of DNA). Data was reported as either initial rate of OD 405 nm increase per unit mass of DNA at specific doses or as a ratio of initial rate of increase of OD 405 nm at specific doses per unit mass of DNA to the initial rate of increase of OD 405 nm of the positive control per unit mass of DNA.
2.7 Statistical tests
Linear regression analysis was undertaken in Microsoft Excel with the significance level for p-values set at 95%. 95% confidence internals of the regression were determined from the Excel regression analysis. Significance testing of count data was by t-tests at the 95% significance level. Coefficient of variance was calculated as the standard deviation/mean. SlopesTest, implemented in the RealStats add-in to Microsoft Excel was used to perform a t-test based significance test of regression slopes.3 Results and discussion
3.1 Optimisation of antibody working concentrations
ELISA type protocols are highly sensitive, specific, scalable and commonly used in biomedical applications for detection of measurands in clinical samples.35,36 Roza et al. raised and characterised a mouse monoclonal antibody specific for thymine dimers in single-stranded DNA and developed an ELISA and immunofluorescence protocol for its use in human cells.24 Optimisation of the mouse monoclonal anti-thymine dimer H3 clone primary antibody (H3Ab) for detection of dimers in naked E. coli gDNA irradiated with 500 mJ cm−2 is shown in Fig. S.3 and S.4 in the ESI.† All signals appeared to be specific to the enzyme-linked reporter on the secondary antibody, as very low signal levels were seen in controls. Furthermore, in the absence of the primary antibody the same signal levels were seen for UV and non-UV irradiated DNA, indicating the H3Ab was responsible for the UV-specific signal. The signal strength of UV irradiated DNA was higher than unirradiated samples at all dilutions, and signal-to-noise ratio increases with increasing H3Ab concentration up to a 1:1000 dilution. There was a relatively high signal from the non-specific binding of the secondary antibody control which was a concern; therefore, an additional wash step was included in all further experiments. Further optimisation of H3Ab concentration between 1:100 and 1:1000 did not improve the signal to noise ratio (Fig. S.4†), therefore in agreement with Rosa et al.,24 optimum H3Ab concentration for 50 ng dsDNA was 1:1000. Reducing the concentration of H3Ab to 1:1000 for the no DNA control also reduced non-specific binding to background levels.
The ELISA protocol utilised a goat anti-mouse secondary antibody with a horseradish peroxidase (HRP) conjugated reporter enzyme. Fig. S.5† shows the results of the optimisation of the working concentration of this secondary antibody, confirming the optimal secondary antibody concentration was 1:1000. Controls for no primary, no secondary, and no DNA all show background levels. All further ELISA experiments used the goat anti-mouse HRP conjugated secondary antibody at 1:1000.
3.2 UV dose response profile of in vivo E. coli gDNA
Following optimisation with naked DNA, the ELISA assay was tested to determine if it could detect a UV dose response from in vivo gDNA. Fig. 1 shows data for E. coli gDNA extracted from filtered cell suspensions exposed to increasing UV dose. These data suggest the assay can detect linear UV dose responses between 0 and 20 mJ cm−2, however the response tailed above 20 mJ cm−2. While this may have reflected saturation of dimer sites in E. coli gDNA, it was also possible the assay conditions were causing substrate limitation in the enzymatic detection reaction.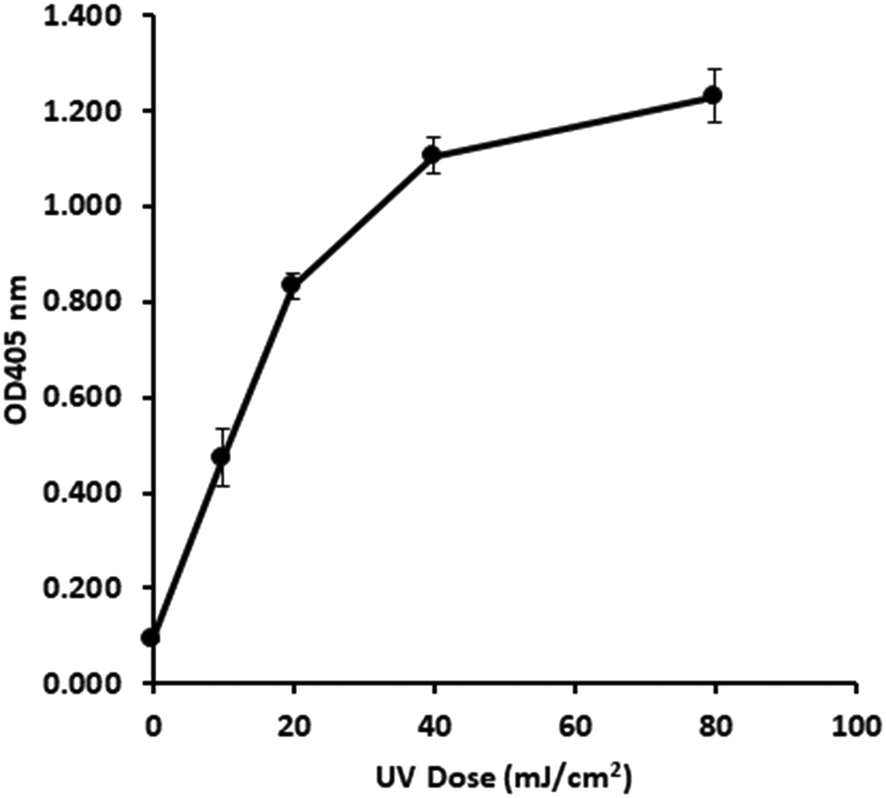 | ||
| Fig. 1
In vivo UV dose response of E. coli gDNA. ELISA development time t = 50 min. Error bars are 1.96 × standard error. Primary Ab 1:1000, secondary Ab 1:1000, [ABTS] 0.4 mg mL−1 and [H2O2] 90 μg mL−1. | ||
To investigate the possibility of the assay being limited by substrate, rather than dimer saturation, a series of assays with 500 mJ cm−2 irradiated naked E. coli gDNA were designed and followed over a timecourse after development buffer addition. Fig. 2A shows the timecourse of colorimetric product development for varying ABTS concentration in the assay development buffer. The thick line (0.4 mg mL−1) indicates the conditions used during the antibody optimisation experiments (Fig. S.3 to S.5†) and in the generation of the dose response curve in Fig. 1. Development time for these previous experiments was 50 minutes, at which point the product development had reached a plateau, as shown in Fig. 2A. Increasing ABTS concentration from 0.4 mg mL−1 to 2.4 mg mL−1 meant the reaction rate of DNA with high dimer content was linear over the first 20–30 minutes of the assay, after this the colour development exceeded the maximum range of the plate reader. Initial conditions for hydrogen peroxide appeared to be optimal (Fig. 2B). Increased hydrogen peroxide concentration decreased signal while decreased concentrations slightly decrease signal over the 0–25 minute range identified as optimal for coloured product development in Fig. 2A. Therefore, the tailing at higher doses in the dose response of Fig. 1 was likely due to substrate limitation with respect to ABTS concentration and not saturation of the dimer sites. To confirm this, dose response experiments were conducted with in vivo E. coli gDNA, using the optimised substrate concentrations (Fig. 3). Good linearity in response is obtained by endpoint OD determinations of the development reaction; as determined by the coefficient of determination R2 values of 0.998, 0.941, 0.985 for each of the three replicate experiments (Fig. S.6†). P-values indicate the slope is significant at or beyond the 98.5% level in all three determinations and the intercept does not differ significantly from zero in two out of the three data sets. The data sets that show an intercept indistinguishable from zero at the 95% confidence level are the two data sets with the larger upper and lower 95% confidence intervals of the regression. In the endpoint data, a non-zero intercept represents background activity in the development reaction. Coefficient of variation (CoV) range from 1.63% to 65.6%, but typically a CoV of 10% is achieved (Fig. S.6†). Pairwise statistical testing of the linear regressions was undertaken and indicated two of the three regression slopes were significantly different at the 95% confidence level, (p-values of 0.422, 0.002, 0.049) (Table S.1†). In order to address this issue, the endpoint data was normalised to the high dose positive control (500 mJ cm−2 treated naked E. coli gDNA) which improved the analysis, by reducing the number of pairs with significantly different slopes to one (p-values 0.391,0.003,0.098). We sought to improve the assay readout, and a second method of analysing the data was also undertaken in which the timecourse of ABTS development that was captured during data collection was used to determine the initial rate of increase in OD405 at time 0, (see section 2.6 for the method of calculating initial rates and Fig. S.2†). Since the calculation of initial rates uses all the data points captured within the timecourse, initial rates offer a method of improving sensitivity of assays by deconvoluting variability that may be introduced by using a single endpoint measure. These data are shown in Fig. S.7† for each of the replicate experiments, CoV values for the replicates ranged from 0–244.5% with a mean value of 38.8%. Regressions based on initial rate data all showed linear responses (R2 values of 0.977, 0.854, 0.951) with statistically significant gradients (p-values 0.0015, 0.0249, 0.0047), and intercepts not significantly different from zero (p-values 0.1467, 0.1893, 0.2381). Pairwise statistical testing of the slopes between replicates indicated there is no significant difference between the slopes for each replicate experiment when based on either initial rates (p-values 0.910, 0.187, 0.430) or the ratio of initial rates to those of the high dose control (p-values 0.086, 0.899, 0.138). Furthermore, in general the normalisation of rate data to the high dose control did not provide any benefit to the assay response. The means of the aggregated data for initial rates and the initial rate ratios were plotted and linear regression undertaken. The unnormalized (Fig. 3) versus normalised (Fig. S.8†) regressions of these data indicated R2 values of 0.936 vs. 0.945, p-values of gradient 0.0068 vs. 0.0056, and p-values of the intercept 0.191 vs. 0.188, respectively. Taken together these data indicate, that despite localised high coefficient of variation in some data, the assay can generate statistically significant linear responses between UV dose and the initial rate of the development reaction that can be compared between tests with little benefit, but no detriment, to normalising to an inter-test high-dose standard.
 | ||
| Fig. 2 Optimisation of substrate concentration in development buffer. A) ABTS concentration (legend values are mg ml−1). B) Hydrogen peroxide concentration (legend values are μg ml−1). Thick lines indicate the conditions used during the optimisation experiments. Primary Ab 1:1000, secondary Ab 1:1000, [ABTS] 0.4 mg mL−1 or as indicated and [H2O2] 90 μg mL−1 or as indicated. | ||
 | ||
| Fig. 3
E. coli gDNA optimised UV dose response based on initial rate data for three replicate experiments. OD 405 nm endpoint data. (○) are data points from three replicate experiments, dash (—) is the mean value. Light dotted lines represent the 95% confidence interval of the linear regression. Primary Ab 1:1000, secondary Ab 1:1000, [ABTS] 2.4 mg mL−1 and [H2O2] 90 μg mL−1. | ||
3.3 UV dose response profile of water treatment samples indigenous DNA
As a test of dimer detection on more complex samples, samples from water treatment processes were exposed to UV light at up to 120 mJ cm−2 using a collimated beam. Samples from two PGAC and one PRGF sampling locations were treated and gDNA was recovered from the treated samples. DNA concentration was below the detection limit of the nano-spectrophotometer used to measure the concentration in the E. coli gDNA preparations. Therefore, a more sensitive PicoGreen assay was used. Table 2 and Fig. S.9† shows DNA concentration as determined by PicoGreen in the UV treated PRGF and PGAC samples from plant 1. While CFU counts for this sample were not determined for these samples, PGAC samples usually gave fewer CFU mL−1 counts than PRGF samples but appear to have 3.05 times as much DNA on average.16 It is noted that high UV doses gave the lowest DNA recovery in both cases at plant 1, but this was not the case at Plant 2. UV dose did not seem to affect DNA recovery in a predictable fashion.| Sample point | UV dose (mJ cm−2) | pg μL−1 sample |
|---|---|---|
| PLANT 1 – PRGF | 0 | 13.51 |
| 10 | 8.73 | |
| 20 | 11.76 | |
| 40 | 9.96 | |
| 80 | 3.42 | |
| PLANT 1 – PGAC | 0 | 36.06 |
| 10 | 50.12 | |
| 20 | 21.69 | |
| 40 | 23.83 | |
| 80 | 13.31 | |
| PLANT 2 – PGAC | 0 | 9.25 |
| 10 | 4.13 | |
| 20 | 1.65 | |
| 40 | 5.93 | |
| 80 | 1.04 | |
| 120 | 2.91 |
The dimer detection ELISA for these samples was conducted and dimer levels corrected for the quantity of DNA loaded into each well. Some variability in the initial OD 405 nm at the start of the test meant the previously indicated initial rates data analysis was able to provide a dose dependant response. Fig. 4A and B shows the dimer dosimetry UV dose response curve for PGAC and PRGF samples from one water treatment works, and Fig. 4C the dimer dosimetry response from a PGAC sample at another treatment works. The dose range was increased to 120 mJ cm−2 for this sample. Fig. 4 indicates linear UV dose responses can be obtained using the optimised ELISA assay with an initial rate-based dependant variable, (rather than OD 405 nm endpoints) on DNA extracted from samples from real treatment processes. R2 values for the linear regressions are A: 0.914, B: 0.967, C: 0.946, gradient p-values are A: 0.011, B: 0.003, C: 0.001, and of the intercept are A: 0.130, B: 0.045, C: 0.586. These indicate good linearity, and significance in the link between dose and response in these samples. In the three samples, one indicated a non-zero intercept at the 95% significance level, but not at the 94.5% level. It is noted that the absolute response in the PGAC sample from plant 2 is significantly higher than those from plant 1. The reason for this is unclear, but highlights the relative nature of simple molecular methods such as these and potential for considerable variability in responses in real water samples. It also speaks to the critical and central importance of the collimated beam procedure for its ability to control for multiple variables by linking the dimer response in a real UV system to a dimer response in a calibrated near-ideal batch treatment system.
 | ||
| Fig. 4 Dose response curve for DNA indigenous to water from the water treatment works. (A) Plant 1 PRGF sample. (B) Plant 1 PGAC sample. (C) Plant 2 PGAC sample. Dotted lines are 95% confidence interval of the regression. Primary Ab 1:1000, secondary Ab 1:1000, [ABTS] 2.4 mg mL−1 and [H2O2] 90 μg mL−1. | ||
While acknowledging these data are from similar treatment processes, and limited in number, they are the first demonstration that measuring thymine dimers in indigenous multi-species DNA from a water treatment process could form the basis of a dose verification method for operational UV reactors that is independent of reactor conditions and does not rely on spiking dosimeters into a treatment process.
The initial idea driving the development of this proof of concept was the realisation that the viable but not culturable population of bacteria was potentially large,14,15 and carried with it an internal UV responsive element; its genomic DNA. Sequence characteristics differ between species of bacteria and therefore, each species gDNA reacts differently, in a sequence dependant manner, to UV irradiation. The primary sequence determinant is the number of pyrimidine-pyrimidine doublets that occurs in the sequence.23,37 Each doublet represents a potential dimer location following UV irradiation. As long as a collimated beam procedure is used to create a standard UV dose response profile, the total pool of DNA in a mixed population of bacteria can be considered a single UV dosimeter capable of responding to UV light in a linear dose-dependent manner.
Conclusions
Due to the lack of a measurable residual, verification of dose delivery by UV disinfection systems in operation relies on indirect methods. These methods ultimately relate readings taken at UV sensors, UVT meters and flow meters, via algorithmic determinations, to a reduction equivalent dose determined during the validation testing procedures. No direct sampling-based method for online dose estimation that is independent of the system currently exists. This study has demonstrated that the pool of genomic DNA in the whole indigenous bacterial population of a pre-disinfection partially treated drinking water treatment sample can function as a molecular measure of UV dose delivery. This method is based on quantifying dimerization of thymine nucleotide bases in DNA, and would not be expected to detect UV light dependant changes to other macromolecules including proteins. However, direct immunodetection of thymine dimers in genomic DNA of indigenous microorganisms exposed to UV light provides a dosimeter that is independent of UV disinfection system conditions (e.g. sensor and lamp conditions) and does not require the introduction of biodosimeters or reagents into a treatment process.Practically, ELISAs, and other molecular methods can be developed and optimised in high-throughput formats that will be essential for them to become routine analysis options. In order that such a sampling method could be used UV systems would require sampling taps upstream and immediately downstream of the UV reactor in order to draw off samples for testing. Bulk samples from each port taken at the sampling time would be required with samples stored in cold, dark conditions until processed. The upstream sample would be subjected to a bench scale collimated beam test in order to develop a dose–response curve for the bulk water sample. DNA would be extracted from collimated beam samples and samples downstream of the reactor and all samples analysed using the ELISA procedure described here. The collimated beam procedure is the key standardisation process in the procedure and its use allows a dose–response curve to be developed that is specific to the characteristics of the sample (UVT and DNA content). The response from the samples taken downstream of the UV reactor could then be compared to the collimated beam dose–response, to verify the dose delivered by the reactor. The feasibility of a similar procedure has been demonstrated using PCR to verify the performance of a full scale flow through UV reactor treating groundwater.26 Whilst the limited number of samples from real treatment works considered during this study are unable to unequivocally demonstrate the robustness of the ELISA, they demonstrate the potential of this approach. The following further work would extend the findings of this work:
1. Further testing of the ELISA assay with treatment process samples from treatment works with different types of source water, such as groundwater-dominated, pristine catchments, and different types of treatment process (e.g. post slow sand filter).
2. The testing of the ELISA assay with samples from a treatment works with an operational UV disinfection system. This work would enable the dose delivery reported by the telemetry of the UV reactor to be compared with the dose reported by the ELISA assay.
3. Other molecular methods have the potential to report UV dose–response including quantitative PCR. Such methods should be compared directly with the ELISA method demonstrated here.
4. The influence of clumping of organisms should be investigated, such clumping is common in pre-UV waters in wastewater treatment. The performance of this method in water with a higher concentration of organisms in clumped and particle-associated states would shed light on the methods applicability to UV treatment of wastewater.
5. Define the parameters that influence the magnitude of response in the response variable.
6. Define limits for practical sample processing considering systems are remote from laboratories.
Author contributions
James Blyth: conceptualisation, methodology, formal analysis, investigation, data curation, visualisation, project administration, writing – review and editing. Lucinda Hazell: writing – original draft and editing, Michael R. Templeton: writing – review and editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was funded by the UK Engineering and Physical Sciences Research Council via grant EP/G037094/1 and Trojan Technologies. The authors thank Bill Cairns at Trojan Technologies for helpful discussions throughout the period of this work.References
- H. B. Wright and S. P. Reddy, Factors impacting the precision of CFD-based modeling of UV reactor performance. In: Proceedings of the American Water Works Association Annual Conference & Exposition. Anaheim, CA: American Water Works Association; 2003.
- K. Chui, D. A. Lyn, P. Savoye and E. R. Blatchley III, Integrated UV Disinfection Model Based on Particle Tracking, J. Environ. Eng., 1999, 125(1), 7–16 CrossRef.
- H. B. Wright and Y. A. Lawrynshyn, An assessment of the bioassay concept for UV reactor validation, In: Disinfection 2000, Disinfection of Wastes in the New Millennium, New Orleans, LA, Water Environment Federation, 2000.
- A. Cabaj, R. Sommer and D. Schoenen, Biodosimetry: Model calculations for u.v. water disinfection devices with regard to dose distributions, Water Res., 1996, 30(4), 1003–1009 CrossRef CAS.
- R. Sommer, A. Cabaj, T. Sandu and M. Lhotsky, Measurement of UV radiation using suspensions of microorganisms, J. Photochem. Photobiol., B, 1999, 53(1–3), 1–6 CrossRef CAS.
- R. G. Qualls and J. D. Johnson, Bioassay and dose measurement in UV disinfection, Appl. Environ. Microbiol., 1983, 45(3), 872–877 CrossRef CAS PubMed.
- Y. A. Lawryshyn and B. Cairns, UV disinfection of water: the need for UV reactor validation, Water Sci. Technol.: Water Supply, 2003, 3(4), 293–300 CAS.
- P. A. Rochelle, E. R. Blatchley III, P. S. Chan, O. K. Scheible and C. Shen, Challenge organisms for inactivation of viruses by ultraviolet treatment. Denver, CO: Water Research Foundation; 2010. Report No.: Project #3105.
- H. Mamane-Gravetz and K. G. Linden, UV disinfection of indigenous aerobic spores: implications for UV reactor validation in unfiltered waters, Water Res., 2004, 38(12), 2898–2906 CrossRef CAS PubMed.
- R. Sommer, A. Cabaj, G. Hirschmann and T. Haider, Disinfection of drinking water by UV irradiation: Basic principles-specific requirements-international implementations, Ozone: Sci. Eng., 2008, 30(1), 43–48 CrossRef CAS.
- ÖNORM, Plants for disinfection of water using ultraviolet radiation: Requirements and testing. Part 1: Low pressure mercury lamp plants. ÖNORM M5873–1 E. Vienna, Austria, Osterreichisches Normungsinstitut, 2001.
- USEPA. Ultraviolet disinfection guidance manual for the final long term 2 enhanced surface water treatment rule: EPA 815-R-06-007. Washington DC, USA: United States Environmental Protection Agency, Office of Water (4601); 2006.
- DVGW. UV Disinfection Devices for Drinking Water Supply - Requirements and Testing. Bonn, Germany: German Gas and Water Mangement Union; 2006.
- J. T. Staley and A. Konopka, Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats, Annu. Rev. Microbiol., 1985, 39(1), 321–346 CrossRef CAS PubMed.
- R. I. Amann, W. Ludwig and K.-H. Schleifer, Phylogenetic identification and in situ detection of individual microbial cells without cultivation, Microbiol. Rev., 1995, 59(1), 143–169 CrossRef CAS PubMed.
- J. Blyth, M. R. Templeton, S.-J. Court, C. Luce, W. Cairns and L. Hazell, Assessment of indigenous surrogate microorganisms for UV disinfection dose verification, Water Environ. J., 2021, 00, 1–9, DOI:10.1111/wej.12722.
- A. C. Eischeid and K. G. Linden, Molecular Indications of Protein Damage in Adenoviruses after UV Disinfection, Appl. Environ. Microbiol., 2011, 77(3), 1145–1147 CrossRef CAS PubMed.
- Y. Gerchman, H. Mamane, N. Friedman and M. Mandelboim, UV-LED disinfection of Coronavirus: Wavelength effect, J. Photochem. Photobiol., B, 2020, 212, 112044 CrossRef CAS PubMed.
- J. R. Bolton and C. A. Cotton, The Ultraviolet Disinfection Handbook, American Water Works Association, Denver, CO, 2008 Search PubMed.
- R. B. Setlow and W. L. Carrier, Pyrimidine dimers in ultraviolet-irradiated DNA's, J. Mol. Biol., 1966, 17(1), 237–254 CrossRef CAS PubMed.
- R. B. Setlow, P. A. Swenson and W. L. Carrier, Thymine dimers and inhibition of DNA synthesis by ultraviolet irradiation of cells, Science, 1963, 142(3598), 1464–1466 CrossRef CAS PubMed.
- A. P. Schuch, G. R. da Silva, K. M. de Lima-Bessa, N. J. Schuch and C. F. M. Menck, Development of a DNA-dosimeter system for monitoring the effects of solar-ultraviolet radiation, Photochem. Photobiol. Sci., 2009, 8(1), 111–120 CrossRef CAS PubMed.
- R. Moeller, T. Douki, P. Rettberg, G. Reitz, J. Cadet and W. L. Nicholson, et al. Genomic bipyrimidine nucleotide frequency and microbial reactions to germicidal UV radiation, Arch. Microbiol., 2010, 192(7), 521–529 CrossRef CAS PubMed.
- L. Roza, K. J. van der Wulp, S. J. MacFarlane, P. H. M. Lohman and R. A. Baan, Detection of cyclobutane thymine dimers in DNA of human cells with monoclonal antibodies raised against a thymine dimer-containing tetranucleotide, Photochem. Photobiol., 1988, 48(5), 627–633 CrossRef CAS PubMed.
- T. Douki and J. Cadet, Individual determination of the yield of the main UV-induced dimeric pyrimidine photoproducts in DNA suggests a high mutagenicity of CC photolesions, Biochemistry, 2001, 40(8), 2495–2501 CrossRef CAS PubMed.
- L. Nizri, D. Vaizel-Ohayon, H. Ben-Amram, Y. Sharaby, M. Halpern and H. Mamane, Development of a molecular method for testing the effectiveness of UV systems on-site, Water Res., 2017, 127, 162–171 CrossRef CAS PubMed.
- M. Cristiani, M. J. Flores, R. J. Brandi, F. A. Tedeschi, F. E. Zalazar and M. D. Labas, ERIC-PCR technique applied to monitoring and quantification of DNA damage during water disinfection process, J. Photochem. Photobiol., B, 2020, 202, 111699 CrossRef CAS PubMed.
- B. M. Sutherland and A. G. Shih, Quantitation of pyrimidine dimer contents of nonradioactive deoxyribonucleic acid by electrophoresis in alkaline agarose gels, Biochemistry, 1983, 22(4), 745–749 CrossRef CAS PubMed.
- R. B. Setlow, P. A. Swenson and W. L. Carrier, Thymine dimers and inhibition of DNA synthesis by ultraviolet irradiation of cells, Science, 1963, 142(3598), 1464–1466 CrossRef CAS PubMed.
- A. C. Eischeid, J. N. Meyer and K. G. Linden, UV Disinfection of Adenoviruses: Molecular Indications of DNA Damage Efficiency, Appl. Environ. Microbiol., 2009, 75(1), 23–28 CrossRef CAS PubMed.
- B. H. Al-Adhami, R. A. B. Nichols, J. R. Kusel, J. O'Grady and H. V. Smith, Detection of UV-Induced Thymine Dimers in Individual Cryptosporidium parvum and Cryptosporidium hominis Oocysts by Immunofluorescence Microscopy, Appl. Environ. Microbiol., 2007, 73(3), 947–955 CrossRef CAS PubMed.
- T. Douki and J. Cadet, Individual determination of the yield of the main UV-induced dimeric pyrimidine photoproducts in DNA suggests a high mutagenicity of CC photolesions, Biochemistry, 2001, 40(8), 2495–2501 CrossRef CAS PubMed.
- V. L. Singer, L. J. Jones, S. T. Yue and R. P. Haugland, Characterization of PicoGreen reagent and development of a fluorescence-based solution assay for double-stranded DNA quantitation, Anal. Biochem., 1997, 249(2), 228–238 CrossRef CAS PubMed.
- J. R. Bolton and K. G. Linden, Standardization of methods for fluence (UV dose) determination in bench-scale UV experiments, J. Environ. Eng., 2003, 129(3), 209–215 CrossRef CAS.
- A. Voller, A. Bartlett and D. E. Bidwell, Enzyme immunoassays with special reference to ELISA techniques, J. Clin. Pathol., 1978, 31(6), 507–520 CrossRef CAS PubMed.
- R. M. Lequin, Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA), Clin. Chem., 2005, 51(12), 2415–2418 CrossRef CAS PubMed.
- W. J. Kowalski, Ultraviolet genomic modeling: Current research and applications. In: IOA & IUVA Joint World Congress & Exhibition, Ozone and UV Leading-Edge Science and Technologies, Paris, France, 2011, pp. 23–27.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ew00939c |
| This journal is © The Royal Society of Chemistry 2021 |