
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRelease of gold (Au), silver (Ag) and cerium dioxide (CeO2) nanoparticles from sewage sludge incineration ash†
Jonas
Wielinski
 ab,
Alexander
Gogos
ac,
Andreas
Voegelin
a,
Christoph R.
Müller
d,
Eberhard
Morgenroth
ab and
Ralf
Kaegi
*a
ab,
Alexander
Gogos
ac,
Andreas
Voegelin
a,
Christoph R.
Müller
d,
Eberhard
Morgenroth
ab and
Ralf
Kaegi
*a
aEawag, Swiss Federal Institute of Aquatic Science and Technology, Überlandstrasse 133, 8600 Dübendorf, Switzerland. E-mail: ralf.kaegi@eawag.ch; Fax: +41587655802; Tel: +41587655273
bETH Zürich, Institute of Environmental Engineering, 8093 Zürich, Switzerland
cEMPA, Swiss Federal Laboratories for Materials Science and Technology, 9014 St. Gallen, Switzerland
dETH Zürich, Institute of Energy Technology, 8092 Zürich, Switzerland
First published on 11th October 2021
Abstract
Engineered nanoparticles (NPs) that are released into wastewater are retained by wastewater treatment plants (WWTPs) and accumulate in sewage sludge. Increasing shares of sludge are incinerated and landfilled, especially in industrialized countries. It is debated whether certain types of NPs can outlive the incineration process and subsequently be released from sewage sludge ash (SSA) landfills. To investigate the release of different types of NPs from SSA, we spiked gold (Au), silver (Ag) and cerium dioxide (CeO2) NPs to a pilot WWTP increasing the Au, Ag and Ce concentrations to 30, 43 and 389 mg kg−1 (dry matter basis) in the digested sludge. The spiked sludge was incinerated in a pilot fluidized bed reactor resulting in SSA with Au, Ag and Ce concentrations of 61, 103 and 854 mg kg−1. In addition, two sludge samples from a full-scale WWTP with Au concentrations of 5 and 16 mg kg−1 were incinerated, resulting in SSA with 9 mg kg−1 and 30 mg kg−1 Au. The spiked Au-NPs remain largely unaltered during the wastewater treatment and incineration process, whereas Ag-NPs and CeO2-NPs undergo transformation. During simulated landfill leaching in columns flushed with 400 to 500 pore volumes of artificial rainwater, Ag and Ce were retained in the ash, whereas about 17% of the spiked Au was released, mainly in particulate form. Lower fractions of mostly particulate Au were released from the ashes (3 and 9%) of unspiked SSA. In conclusion, unaltered Au-NPs significantly leach from landfilled SSA, whereas the incorporation of Ag-NPs and CeO2-NPs as transformed species into the SSA matrix limits the leaching of (nano)particulate and dissolved Ag and Ce compounds.
Environmental significanceStudies quantifying the release of engineered nanoparticles (ENPs) from landfill sites are scarce, although this information is required for drafting legislation regulating the end-of-life of ENPs. In this study, we distinguish between ‘transient’ ENPs that undergo transformation during wastewater treatment and sludge incineration and ‘conservative’ ENPs that remain unaltered. We show that conservative ENPs are released to a greater extent from sewage sludge ash compared to transient ENPs which are firmly embedded in the ash matrix. |
1 Introduction
Engineered nanoparticles (ENPs) are applied in industrial processes and consumer products due to their beneficial properties. After their use phase, major shares of the ENPs are released to wastewater streams. In wastewater treatment plants (WWTPs), more than 95% of the discharged ENPs are eliminated from the wastewater stream and accumulated in the sewage sludge.1 Thus, sewage sludge is considered as a major sink for ENPs.1,2 In industrialized high-income countries, sewage sludge is commonly digested anaerobically.3 Agricultural use of digested sewage sludge (in the following, only referred to as “sludge”) becomes increasingly restricted due to concerns about the negative impacts of organic micropollutants and heavy metals from the sludge on the environment and human health.4,5 Therefore, sludge incineration is a viable disposal alternative, additionally leading to a significant reduction in the handling volumes.6–8 In Switzerland, sewage sludge ash (SSA) resulting from mono-incineration is landfilled with the intent to recycle phosphorus from the ash in the near future.9Previous studies have shown that sewage sludge digestion and incineration lead to the same Cu and Zn speciation in ash independent of the form of Cu and Zn spiked into the sludge (copper oxide (CuO)-NPs, Cu(aq), zinc oxide (ZnO)-NPs, Zn(aq)).10,11 Cerium dioxide (CeO2)-NPs were partially transformed during anaerobic digestion, whereas titanium dioxide (TiO2)-NPs (anatase) remained largely unaffected by the anaerobic digestion, but both ENP types were sequestered into newly formed mineral phases during incineration.12,13 Silver (Ag)-NPs discharged to wastewater streams transform into silver sulfide (Ag2S)-NPs2,14–16 and through subsequent incineration, around 80% of these Ag2S-NPs transformed into (metallic) Ag-NPs with diameters of <5 nm.17 We classify these ENPs as “transient ENPs” that transform into other (nanoparticulate) chemical species in sewer systems and WWTPs, including anaerobic digestion, and during sludge incineration.2,10,12,13,17–19 “Conservative ENPs” such as gold (Au)-NPs or platinum (Pt)-NPs form another group of commercially available ENPs,18 which are more resistant to transformation in managed waste facilities. These ENPs are expected to undergo only minor transformations during wastewater treatment, sludge digestion and incineration. Knowledge on the potential release of these different types of ENPs or their transformation products during subsequent rainwater leaching of landfill sites is required to estimate the extent to which ENPs can be mobilized from landfill sites, and to evaluate whether nano-specific regulations are required. However, studies investigating the fate of conservative ENPs beyond anaerobic digestion are currently lacking. Therefore, we investigated the fate of Au-NPs representing conservative ENPs during sludge incineration and assessed their release from SSA landfill sites. The release of Au-NPs was compared to the release of transient ENPs and their transformation products, represented by Ag-NPs and CeO2-NPs.
To investigate the possible release of conservative ENPs, transient ENPs, and their transformation products from SSA, we spiked Au-NPs, Ag-NPs, and CeO2-NPs into a pilot WWTP including an activated sludge process followed by anaerobic digestion and collected the sludge for incineration in a pilot fluidized bed reactor. The resulting ash was packed into a glass column, artificial rainwater was passed through the column and the Au, Ag and Ce concentrations in the leachate were determined. As a comparison, additional leaching experiments were conducted with glass columns that were packed with SSA containing high Au concentrations originating from industrial sources.
2 Materials and methods
2.1 Engineered nanoparticle characterization
Three types of ENPs were used for the experiments: Au-NPs (PVP coated, nanoComposix, 50 nm nominal diameter), Ag-NPs (AppNano, 50 nm nominal diameter) and CeO2-NPs (Envirox, Energenics). Au-NPs and CeO2-NPs were characterized by transmission electron microscopy (TEM, Hitachi HT7700) and Ag-NPs were characterized by scanning TEM (STEM, Talos FEI). Further, Au-NPs and Ag-NPs were characterized by single particle inductively coupled plasma mass spectrometry (spICP-MS, see section 2.3).Au-NPs were mostly spherical, between 40 and 50 nm in diameter and either present as individual particles or as aggregates of 2–4 primary particles (Fig. S1a†). The spICP-MS results indicated a size range between 30 and 55 nm with a median diameter of 41 nm, and the diameter obtained through image analysis was around 50 nm (Fig. S1b–d†). The dissolved Au signal was below the detection limit (Fig. S1b†). The results from STEM measurements showed that Ag-NPs were slightly elongated and aggregated (Fig. S2a and b†) and the results from spICP-MS measurements indicated a median diameter of 49 nm (Fig. S2c and d†), in excellent agreement with the nominal diameter. CeO2-NPs had primary diameters between 5 and 7 nm, but were aggregated to clusters of around 20 nm (Fig. S3†).
2.2 Digested sewage sludge and ash samples
The ENPs (Ag-NPs, Au-NPs, and CeO2-NPs) were spiked into a pilot WWTP at Eawag (Duebendorf, Switzerland). The pilot WWTP consisted of conventional activated sludge treatment including denitrification (V = 92 L), nitrification (V = 147 L), a secondary clarifier (V = 120 L), a gravity thickener (V = 24 L) and an anaerobic digester (V = 300 L) (Fig. S4†). A return sludge stream pumped sludge from the secondary clarifier to the denitrification tank (47 L h−1). A nearby, larger, experimental WWTP (called “WWTP Eawag”, treatment capacity of 300 population equivalents) receiving wastewater from the municipality of Duebendorf operated a primary clarifier to separate primary sludge from the primary effluent. This primary effluent of WWTP Eawag was used as the influent to the pilot WWTP (15 L h−1, Fig. S4†). The thickener was filled with 50% excess activated sludge from the secondary clarifier of the pilot WWTP and 50% primary sludge from WWTP Eawag (12 L d−1 each). The mixture was stirred for 10 min and allowed to settle for 3 h. Once per day, 12 L of sludge were transferred from the bottom of the sludge thickener into the digester. The remaining sludge and supernatant were discarded. Thus, the hydraulic residence time in the anaerobic digestion unit was 25 days. The digested sludge was collected in a storage tank (12 L d−1).| Item/spiked ENPs | Au | Ag | CeO2a |
|---|---|---|---|
| a Particles were added as CeO2 but the concentrations and masses listed in Table 1 refer to elemental Ce. | |||
| Target concentration in the digested sludge (mg kg−1) | 100 | 100 | 500 |
| Measured concentration in the digested sludge (mg kg−1) | 30 | 43 | 389 |
| Recovery in the digested sludge | 30% | 43% | 78% |
| Mass added to the denitrification to reach steady-state concentrations (mg) | 57.2 | 57.2 | 285.4 |
| Mass added to the thickener to reach steady-state concentrations (mg) | 41.3 | 41.3 | 211.7 |
| Mass added to the denitrification during continuous spiking (mg) | 131.2 | 131.2 | 655.8 |
| Mass added to the thickener during continuous spiking (mg) | 28.8 | 28.8 | 152.6 |
| Elemental concentration of the dispersion spiked into the denitrification (mg L−1) | 2.9 | 2.9 | 14.2 |
| Elemental concentration of the dispersion spiked into the thickener (mg L−1) | 5.0 | 5.0 | 26.6 |
| Concentration in fly ash (mg kg−1) | 61 | 103 | 854 |
A detailed explanation of the methodology and mass balances covering the entire experiment can be found elsewhere.19 Briefly, the Au, Ag and Ce concentrations and total solid concentrations were determined continuously in the nitrification tank, the anaerobic digester and the secondary effluent and compared to the effective amount of Au, Ag and Ce added to the denitrification and the thickener. The mass recoveries (difference between the expected and measured concentrations) of Au, Ag and Ce spiked into the system in the digested sludge were 30, 43 and 78%, respectively (Table 1). We occasionally observed sludge flotation in the thickener, which most likely explains the lower than expected Au/Ag/Ce concentrations in the digested sludge.
Digested sludge was collected while spiking was ongoing (steady state operation). After the spiking ended, the content of the anaerobic digester (300 L) was mixed with the previously collected digested sludge (720 L) leading to a total of 1020 L of digested sludge. The sludge was dewatered using an industrial scale sludge decanter (Z-Series, Flottweg AG) and dried at 105 °C. The dried sludge was crushed in pieces of a few cm and 500 g were incinerated in a pilot scale bubbling bed type fluidized bed reactor (FBR) at 820–840 °C under conventional fluidization conditions over the course of 1–2 h (Table 2). The fly ash was collected using two electrostatic precipitators and stored at room temperature in the dark until further processing. The bottom ash was collected from the sand bed after termination of the incineration experiment. The incineration conditions and phase transformations occurring in the pilot FBR resemble those occurring in a full-scale incinerator.10,13 Details on the pilot FBR and its operation can be found elsewhere.10 The Au, Ag and Ce mass balances and enrichment factors (EFs) were calculated as indicated below and derived during earlier studies.10,13 In eqn (1), the recovery of ash content is determined using the mass of the collected fly ash (mfly), the mass of the collected bottom ash (mbottom), the mass of the incinerated sludge (msludge) and the ash content in the dried sludge (Xash).
 | (1) |
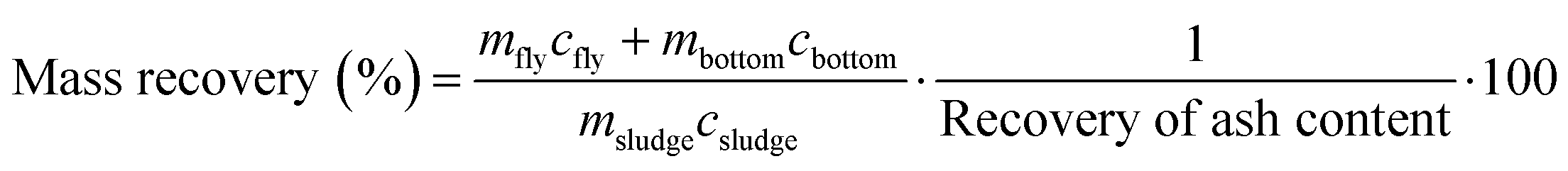 | (2) |
 | (3) |
| Item/experiment | A | B | C | ||
|---|---|---|---|---|---|
| Incinerated sludge (g) | 500.0 | 650.0 | 1000.0 | ||
| Fly ash (g) | 22.85 | 48.92 | 56.48 | ||
| Bottom ash (g) | 149.59 | 172.03 | 386.29 | ||
| X ash | 33% | 38% | 50% | ||
| Ash recovery | 106% | 90% | 89% | ||
| Element | Au | Ag | Ce | Au | Au |
| Concentration in sludge (mg kg−1) | 30 | 43 | 389 | 5 | 16 |
| Concentration in fly ash (mg kg−1) | 61 | 103 | 854 | 9 | 30 |
| Concentration in bottom ash (mg kg−1) | 99 | 105 | 1145 | 15 | 29 |
| X mass recovery | 103% | 88% | 93% | 97% | 92% |
| EF | 3.2 | 2.4 | 2.8 | 2.6 | 1.9 |
| Theoretical EF (1/Xash) | 3.1 (identical for all elements) | 2.6 | 2.0 | ||
In addition to experiment A, dewatered digested sludge was obtained from the WWTP at Chiasso (Canton of Ticino, Switzerland, “experiment B”) and La Chaux-de-Fonds (Canton of Neuchâtel, Switzerland, “experiment C”), dried at 105 °C and incinerated under similar conditions to those indicated above (details in Table 2). In contrast to that in experiment A, the air volume rate passed through the reactor had to be increased by 15% to sustain a continuous reaction at 820–840 °C. The WWTPs for experiments B and C were selected based on the observation of elevated Au concentrations resulting from industrial activities in the catchments of these WWTPs.20
2.3 Sample digestions and analytical techniques
For elemental analysis, sludge, fly ash and bottom ash aliquots were freeze-dried and homogenized in stainless steel containers using a ball mill (17 Hz, 4 min, MM 400, Retsch). Three aliquots of the homogenized sludge (≈20 mg each) were digested using 4 mL 30% H2O2 (Sigma-Aldrich), 3 mL 69% HNO3 (Supra-quality, Carl Roth) and 6 mL 35% HCl (Supra-quality, Carl Roth) and processed using a microwave-assisted acid-digestion system (MLS GmbH) for the subsequent determination of Au concentrations. In triplicate, total acid digestions of milled fly and bottom ash samples were performed. For this purpose, ≈20 mg of ash powder were mixed with 3 mL 69% HNO3, 6 mL 35% HCl (Supra-quality, Carl Roth) and 200 μL 40% HF (Suprapur, Merck) and also processed using the microwave digestion system for the subsequent determination of Au concentrations. For the preparation of digests for the determination of Ag, Ce and Ca, the HCl addition was omitted. Instead, 2.4 mL of 5% boric acid were added to the digests after the microwave treatment (primary digestion) and the primary digests were subsequently treated by microwave-assisted acid-digestion (secondary digestion) a second time to dissolve CeF3 and other potential precipitates such as CaF2 or AgF, especially where HF was used for the primary digestions. All sample types (sludge and ash for Au or other elements) were completely mineralized, resulting in clear digests that were diluted to 50 mL with DI water. The National Institute of Standardization and Technology (NIST) Standard Reference Materials (SRM) 2782 industrial sludge and NIST SRM 1633c coal fly ash were digested using the respective sludge and ash digestion protocols to determine the analytical recovery of Ce (both SRM samples) and Ag (SRM 2782 only). A comparable SRM including a certified Au concentration was unavailable, however the NIST SRM 2782 analysis certificate contains a non-certified information value for Au (2.2 mg kg−1) which we recovered to around 95%.Elemental concentrations in the primary (Au) and secondary digests (Ag, Ce, Ca), leachates and filtrates (see section 2.4) were determined by inductively coupled plasma mass spectrometry (ICP-MS) measurements (8900QQQ, Agilent) or using an ICP optical emission spectrometer (OES, Spectro Arcos). In ICP-MS, samples were introduced using an Integrated Sample Introduction System (ISIS 3, Agilent). Au and Ce were calibrated externally from 0.01 to 2 μg L−1 with eight calibration points using ICP-MS standard solutions (both from Sigma-Aldrich). Ag and Ca were calibrated externally from 0.01 to 10 μg L−1 using the multi-element ICP-MS standard solution “Merck IV” (Merck). Au and Ag were measured on-mass in the He gas mode, and Ce was measured in no-gas-mode. For Au measurements, Pt sampling and skimmer cones were used. Diluted HCl was used instead of diluted HNO3 to flush the tubes to prevent cross-contamination. The Au, Ag and Ce concentrations in the digestion blanks were below the detection limits (DL = 3σB × C/(S − B) of the ICP-MS measurements, where σB corresponds to the standard deviation of the counts of the calibration blank, C corresponds to the concentration of the calibration standard, S corresponds to the counts of the calibration standard, and B refers to the counts of the blank, which were around 0.002 μg L−1 for all analytes. Calcium was much higher in concentration and thus determined by ICP-OES. Here, the DL was around 1 μg L−1. For quality control, we periodically measured the diluted NIST SRM 1643f (“Trace elements in water”) and a diluted 100 μg mL−1 Au standard (Inorganic Ventures).
For single particle ICP-MS (spICP-MS) measurements, selected fractions collected from the column experiments (see section 2.4) were diluted in DI water to about or below 25 ng L−1 Au (lowest dilution was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) and thereafter directly introduced into the nebulization chamber via a peristaltic pump mounted to the ICP-MS. A 1.0 mm diameter torch (Agilent) and Pt sampling and skimmer cones were used. The nebulization efficiency (4.8%) was determined with the Agilent MassHunter single particle module software using Au-NPs (nominal diameter of 50 nm, NanoXact, nanoComposix) diluted to 25 ng L−1 Au. The signal integration time was 3 ms and the effective measurement time per sample was 180 s with a sample introduction flow rate of 0.346 mL min−1. The background equivalent diameter (BED, lower particulate size detection limit) was 9 nm for the 50 nm Au-NPs (NanoXact, nanoComposix) in DI water and 18 nm for Au-NPs in the diluted experimental electrolytes. Particle size distributions were extracted from raw data using an algorithm described in Pace et al.21
:10) and thereafter directly introduced into the nebulization chamber via a peristaltic pump mounted to the ICP-MS. A 1.0 mm diameter torch (Agilent) and Pt sampling and skimmer cones were used. The nebulization efficiency (4.8%) was determined with the Agilent MassHunter single particle module software using Au-NPs (nominal diameter of 50 nm, NanoXact, nanoComposix) diluted to 25 ng L−1 Au. The signal integration time was 3 ms and the effective measurement time per sample was 180 s with a sample introduction flow rate of 0.346 mL min−1. The background equivalent diameter (BED, lower particulate size detection limit) was 9 nm for the 50 nm Au-NPs (NanoXact, nanoComposix) in DI water and 18 nm for Au-NPs in the diluted experimental electrolytes. Particle size distributions were extracted from raw data using an algorithm described in Pace et al.21
BET-Surface area measurements of the fly ash samples were conducted on a NOVA 3000e (Quantachrome Instruments). The residual water and ash content (the non-combustibles, Xash) were determined by heating 1 g of dried sludge (105 °C) to 950 °C for 2 h and keeping the materials at this temperature for 1 h. The residual mass was used to determine Xash. The ash particle volume distribution between 0.1 and 2000 μm particle diameter was determined by multi-angle static light scattering using the universal liquid module of an LS 13 320 particle size analyzer (Beckman Coulter).
STEM measurements including high angular annular dark field (HAADF) imaging and energy dispersive X-ray spectroscopy (EDX) analyses were performed on an FEI Talos (F200X, Super-X EDS, 4 detector configuration, FEI) with an acceleration voltage of 200 kV. For STEM analyses, 200 mg of dried sludge or ash were dispersed in 0.2% NovaChem and centrifuged on poly-L-lysine (Sigma-Aldrich) functionalized Cu TEM grids (EM Resolutions) or dispersed in ethanol and deposited on a Cu TEM grid with a holey carbon film.
2.4 Column setup and leaching experiment
An artificial rainwater electrolyte was used for the leaching experiments. For its preparation, a concentrated electrolyte was prepared by dissolving selected salts in DI water (composition in Table 3). The concentrated electrolyte was diluted to 1:100 in DI water and the resulting solutions were equilibrated with the atmosphere for 72 h. Thereafter, the pH value, which was controlled by the carbonate equilibrium with the atmosphere, was measured at 5.60. The composition of the rainwater corresponds to the average rainwater composition in Switzerland.22 However, the reported ionic concentrations were not charge balanced; therefore, a system of linear equations was solved to determine the smallest deviation in ionic concentrations using the available salts (Table 3).
| Compound | mg L−1 |
|---|---|
| NaCl | 36.1 |
| KCl | 17.2 |
| NH4NO3 | 103.2 |
| (NH4)2SO4 | 205.7 |
| Mg(NO3)2 | 40.9 |
| Ca(NO3)2 | 106.6 |
A BioRad liquid chromatography (LC) glass column (length: L = 50 mm, diameter: d = 8 mm) was filled with ≈800 mg of fly ash (Table 4). Glass wool was used to confine the ash in the LC column extending 36 mm in the direction of flow. A piston pump (M6 Vici) was used to pass the artificial rainwater electrolyte through the ash column in upflow mode (Q = 1 mL min−1). For each experiment, 40 10 mL fractions were collected using a fraction collector (LKB Bromma 2212 Helirac). Immediately after the collection of each fraction, a 10 kDa filtrate (Vivaspin, Sartorius) was prepared and the pH was determined using a pH electrode (Van London). The column was fully saturated during the entire experiment. To assess the hydraulic parameters and evaluate the occurrence of preferential flow paths, a 1 mM KCl solution was injected into the column after each experiment using a Rheodyne injection valve with an injection loop. Conductivity was determined on-line using an 80 μL flow cell including a Pt-electrode (Microelectrodes Inc.) in the column effluent. The conductivity data were fitted to a 1-D solution of the advection–dispersion equation to obtain the effective diffusion coefficients and flow velocities (section S3, Fig. S5–S7†).23 The results indicated that preferential flow was insignificant in all columns. After termination of the experiment, the electrolyte-saturated ash was completely removed from the column and weighed before and after drying at 105 °C for 24 h to determine the effective pore volume.
| Item/experiment | A | B | C |
|---|---|---|---|
| Ash in column (mg) | 800.0 | 800.3 | 801.0 |
| Ash filled length (mm) | 36 | 36 | 36 |
| Ash volume (mL) | 1.8 | 1.8 | 1.8 |
| Pore volume (mL) | 0.9 | 1.0 | 0.8 |
| Effective porosity | 49% | 55% | 43% |
| Collected fractions (10 mL per fraction) | 40 | 40 | 40 |
| Collected pore volumes | 453 | 400 | 514 |
| Au fly ash (mg kg−1) | 60.50 | 8.78 | 30.12 |
| Au in column before leaching (μg) | 48.40 | 7.03 | 24.10 |
| Total Au released (μg) | 8.00 | 0.64 | 0.66 |
| Dissolved Au released (μg) | 2.38 | 0.06 | 0.06 |
| Particulate Au released (μg) | 5.62 | 0.58 | 0.60 |
| Fraction of Au released (%) | 17% | 9% | 3% |
| Share of dissolved Au (%) | 30% | 9% | 9% |
| Share of particulate Au (%) | 70% | 91% | 91% |
| Electrolyte velocity (u, mm s−1) | 0.35 | 0.36 | 0.46 |
| Effective diffusion coefficient (×10−7 m2 s−1) | 1.40 | 3.08 | 6.78 |
| BET surface area (m2 g−1) | 10.00 | 9.45 | 3.59 |
| Mode ash grain diameter (μm) | 48 | 650 | 540 |
2.5 Thermodynamic equilibrium calculations
Thermodynamic equilibrium calculations were performed in Phreeqc Interactive v3.6.2 (released January 28th 2020) using the Minteq v4 database to determine the saturation index (SI) of Ca(OH)2 (portlandite) in selected fractions. The pH and the concentration of Ca, Na, and Cl (assumed equivalent to Na) were fixed at the measured values.3 Results and discussion
3.1 Experiment A: characterization of ENP spiked samples
The Au, Ag and Ce concentrations in the spiked sludge were 30, 43, and 389 mg kg−1 and increased to 61, 103, and 854 mg kg−1 in the fly ash. In the bottom ash, 99 mg kg−1 Au were measured leading to an Au recovery of 103% in the incineration experiment (Table 2). The Au EF was 3.2 which was close to the theoretical EF of 3.1. The selective accumulation of ENPs in bottom ash has been previously observed for TiO2-NPs.13,24 The excellent recovery of Au in the fly and bottom ash and the agreement between the calculated and ideal EFs indicated that during the incineration experiment, all relevant Au mass flows were collected, i.e. Au volatilization was insignificant. The Ag and Ce recoveries were 88 and 93%, respectively. The Ag and Ce EFs were 2.4 and 2.8, respectively, and thus below the theoretical EF of 3.1. Given the complexity of the experiments, the recoveries and EFs are sufficient and indicate that the major flows of Ag and Ce were collected. For a detailed analysis of the behavior of Ag and Ce during sludge incineration, the reader is referred to the publications by Meier et al. (2016)17and Gogos et al. (2018).12The spatial distribution of Au, Ag and Ce in sludge and ash particles was assessed by HAADF imaging complemented with EDX analyses. Selected results of this characterization were reported previously.19 In the sludge, the size of Au-NPs was similar to the size in the stock dispersion (Fig. 1a–c and S1†). Ag was associated with S (Ag2S) in the sludge, which is in agreement with previous research (Fig. 1a, b and d).2,25,26 Ce was associated with sludge flocs and formed aggregates from <10 nm up to ≈100 nm (Fig. 1a, b and e). It was previously reported that during activated sludge treatment and anaerobic digestion, around 30% of Ce+IV originating from CeO2-NPs were reduced to Ce+III.12,27
 | ||
| Fig. 1 (a) HAADF image recorded on the sludge including ENPs in experiment A. (b) Corresponding elemental distribution maps for Au (yellow), Ag (blue), S (violet) and Ce (orange). Insets show the EDX spectra extracted from the areas marked with the white rectangles. (c)–(e) EDX line profiles along Au, Ag and Ce particles indicated by the grey bars in (b). The profile direction runs from left to right/top to bottom. Modified after Gogos et al.29 | ||
Through incineration, the distribution and morphology of the engineered (Au-NPs, CeO2-NPs) or incidental (Ag2S) NPs changed to a variable extent. The Au-NPs did not significantly transform during the incineration and were present as ≈50 nm particles in the ash (Fig. 2a–c). Ag was present as ≈5 nm incidental NPs and the Ag signal was not correlated with S (Fig. 2d–f). These findings are in agreement with a previous study reporting that around 80% of Ag2S-NPs transform into incidental Ag-NPs during incineration in a bench scale fluidized bed reactor.17 The CeO2-NPs changed in morphology (Fig. 2a, b and g–i) and it was reported that CeO2-NPs transformed into a different mineral structure during sewage sludge incineration.12
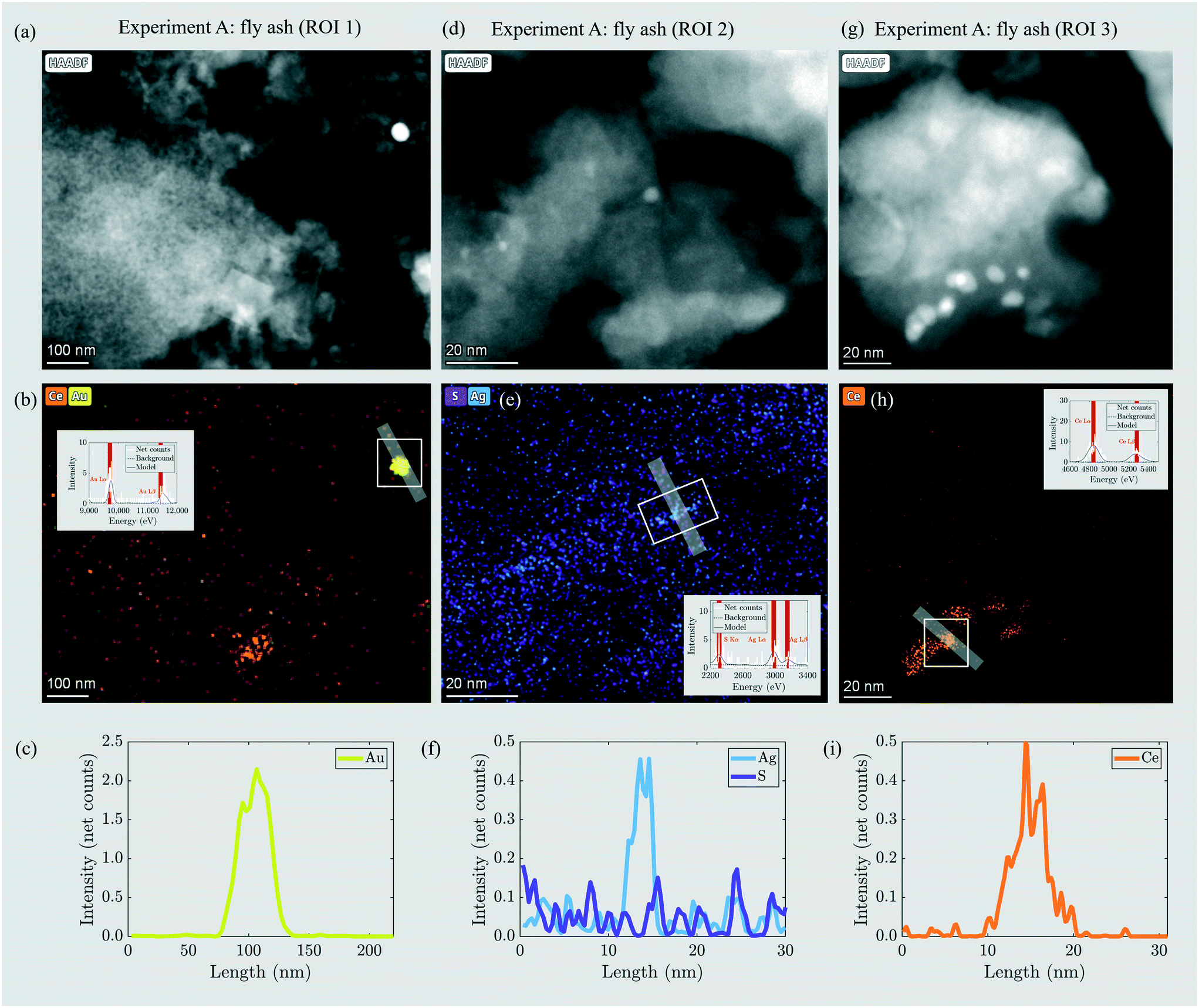 | ||
| Fig. 2 (a, d and g) HAADF images of three regions of interest (ROI) in the fly ash including ENPs in experiment A. Corresponding elemental distribution maps for Au (yellow, b), Ag and S (blue and violet, e; the EDX signal is slightly blurred due to sample drift during recording of the elemental distribution maps), and Ce (orange, b and h). Insets show the EDX spectra extracted from the areas marked with the white rectangles. (c, f and i) EDX line profiles through Au, Ag and Ce particles. Their direction is indicated by the grey bars in the corresponding EDX mappings. The profile direction runs from left to right/top to bottom. Modified after Gogos et al.29 | ||
The BET specific surface area of the fly ash was 10 m2 g−1, which is in agreement with previously published data for sewage sludge fly ash.28 The ash grains were between 1 and 300 μm in diameter and the mode size of the ash grains was 48 μm, also in agreement with published values (Fig. S8†).28
3.2 Experiment A: release of selected elements in column experiments
Artificial rainwater was passed through the column for 453 pore volumes (pv) and 40 fractions of 11.33 pv (10 mL) each were collected for elemental analysis and pH measurements. We used an aliquot of 800 μL from each fraction, which was centrifuged (3500 rpm, 5 min) to separate the particulate/particle bound Au from the dissolved fractions with a 10 kDa filter. The Au concentrations (total, dissolved, particulate), the total Ca concentration and the pH are displayed as a function of pv (Fig. 3a). Neither Ag nor Ce was released in measurable quantities during the entire experiment, i.e. the concentrations were below 0.02 μg L−1, accounting for a 1:10 dilution necessary for ICP-MS measurements. This shows that throughout the experiments, less than 0.008 μg of Ag or Ce were released which translate into 0.009% and 0.0009% of the total Ag and Ce in the ash, respectively.
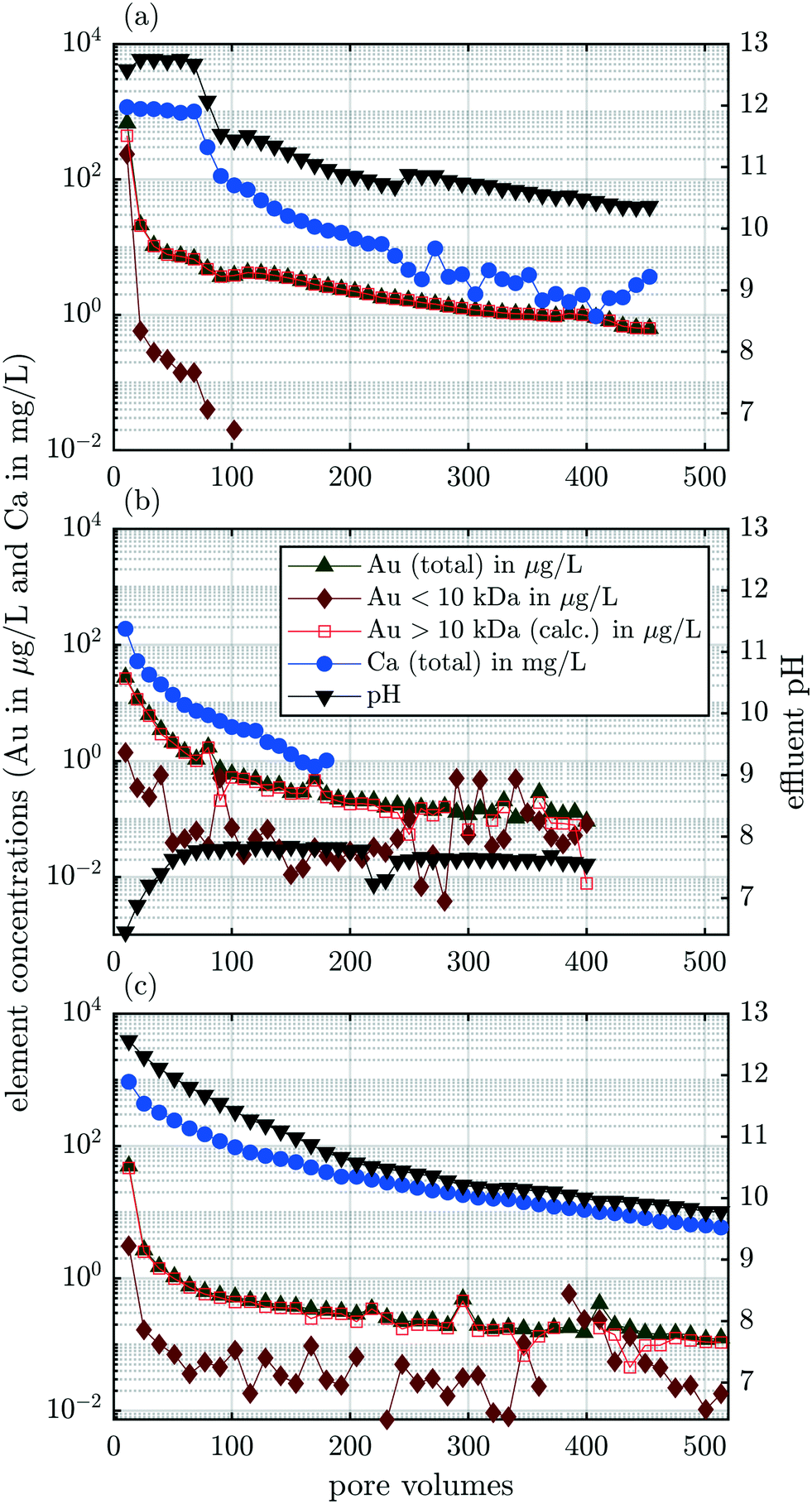 | ||
| Fig. 3 Results from experiments A–C in panels (a)–(c), respectively. In each panel, the concentrations of total Au (grey, upward triangles), dissolved Au (10 kDa filtrate, dark red diamonds), particulate Au (open, red squares), total Ca (blue dots) and the pH (right axis, black downward triangles) versus the volume of column effluent expressed as pore volumes are shown. | ||
The total Au concentration was almost 680 μg L−1 in the first fraction (11.33 pv); however, the Au concentration decreased to 20 μg L−1 in the second fraction and fell to below 10 μg L−1 in fraction #4. The concentration of dissolved Au (10 kDa ≅ 1.20 nm diameter of an Au particle, details in section S5†) was almost 240 μg L−1 in the first fraction, but decreased to 0.6 μg L−1 in the second fraction (22.7 pv) and fell below the DL after ≈100 pv. This indicated that a limited amount of soluble Au-species formed during waste processing. Soluble Au species, for example, formed through complexation with thiosulfate, during wastewater processing (activated sludge process), would be transferred to the effluent of the treatment plant and, thus, separated from the retained solids. The dissolution of Au using various leaching agents, such as cyanide, thiosulfate and thiourea, requires molecular oxygen (O2).30 During the anaerobic digestion, however, O2 is absent. We, thus, conclude, that the formation of soluble Au species most likely occurred during the incineration process and these water soluble compounds were released during the first few pore volumes. Integrated over the entire experimental run time, about 30% of the total released Au was present as dissolved species and 70% in particulate form (Table 4). Throughout the entire experiment, 8.0 μg Au were released which corresponds to 17% of the total 60.5 μg Au contained in the column at the beginning of the experiment (Table 4). In the dispersions of Au-NPs that were spiked into the pilot WWTP, the dissolved background of Au was below the detection limit (Fig. S1b and c†). Therefore, the concentrations of released Au show that at least 5.1% (0.3 Audissolved × 0.17 share of Aureleased = 5.1% Autransformed) of the spiked nanoparticulate Au in the sludge transformed to water-soluble Au species during the incineration process.
The pH was around 12.7 for the first 70 pv and thereafter dropped sharply to around 11.5 during 30 pv (Fig. 3a). From there, the pH continuously decreased with a brief increase after 260 pv and stabilized at 10.4 after 440 pv. The strongly alkaline leachate is in agreement with previous studies.31
The mobilization of colloids depends on the ionic strength of the carrier solution and on the charge of the cations.32 High ionic strength and high concentrations of divalent cations destabilize colloids in solution, favor their attachment to other surfaces (deposition) and thus, inhibit their release from and transport through porous media. The most important cation in our system is Ca2+ and we, therefore, focus on the release of Ca from the experimental columns. The Ca concentration was well correlated to the pH, suggesting that the Ca content in the leachate was controlled by the dissolution of Ca(OH)2. The formation of Ca(OH)2 in the ash column occurred through the initial contact of the electrolyte with CaO which forms during sewage sludge incineration and potentially induces pozzolanic activity.31,33–36 The Ca concentration in the fly ash was 24000 mg kg−1. The dissolution of Ca(OH)2 released 2 mol of OH− per mol of Ca and thus buffered the pH at the measured value of 12.7 during the first 70 pv. Thermodynamic equilibrium calculations, however, suggested that the electrolyte was supersaturated (saturation index (SI) = 0.30) with respect to Ca(OH)2 (portlandite). We, thus, hypothesize that a fraction of the total Ca in the aqueous phase was complexed by compounds that were not considered in the thermodynamic equilibrium calculations, which explains the discrepancy between the observations (dissolution) and the thermodynamic calculations (supersaturation). This is in agreement with results from a previous study reporting Ca(OH)2 supersaturated pore water extracted from a high-volume fly ash-Portland cement binder mixture (SI = 0.10) and pure Portland cement binder (SI = 0.87) after 2 h of curing.37 The authors attributed the apparent early stage supersaturation to the formation of complex ions in solution, e.g., Ca-sulphonates that reduced the activity of Ca2+ but were not accounted for by thermodynamic calculations.37 In our experiment, the slope of the pH curve flattened after 90 pv when the solution became undersaturated (SI < 0) with respect to Ca(OH)2. Despite the previously reported influence of the Ca concentration in the carrier solution on colloid mobilization,32 the release of particulate Au (>10 kDa) was not correlated to the Ca concentration.
3.3 Experiments B and C: characterization of fly ash samples
The sludge samples of experiments B and C contained 5 and 16 mg kg−1 Au resulting from industrial sources located in the catchment of the two WWTPs (Table 2). In the fly and bottom ash, the Au concentrations were 9 and 15 mg kg−1 (experiment B) and 30 and 29 mg kg−1 (experiment C). Thus, in B, Au enriched selectively in the bottom ash, whereas in C, the Au was evenly distributed between the fly ash and bottom ash. The Au mass recoveries were 97% and 92%, thus indicating the excellent recovery of Au during the incineration experiment. The BET surface areas were 9.45 m2 g−1 and 3.59 m2 g−1 for the fly ashes B and C, respectively (Table 4). The fly ash grains of experiments B and C were mostly between 100 and 1000 μm in diameter and have their modes at 650 and 540 μm, respectively, i.e. one order of magnitude larger compared to the mode of the grain size distribution in experiment A (Fig. S8†). This increase in diameter resulted from the 15% increased air volume rate in experiments B and C compared to that in experiment A, which was necessary to sustain the incineration at 820–840 °C (section 2.2). The enhanced air volume rate led to the pneumatic transport of larger ash grains out of the fluidized bed reactor.Nanoscale Au particles appeared as bright spots in HAADF images (Fig. 4a–c) and were identified by EDX (Fig. 4d–i). In the fly ash of experiment B, Au-NPs were observed in two regions of interest (ROI) (Fig. 4a and b). Elemental distribution maps (Fig. 4d and e) and line profiles across the particles (Fig. 4g and h) indicated the occurrence of an individual Au-NP with a diameter of 20 nm (ROI 1) and two individual or agglomerated Au-NPs stacked in the Z-direction and extending around 50 and 20 nm in the XY-direction, respectively (ROI 2). In the fly ash of experiment C, an individual Au-NP of around 40 nm in diameter was identified (Fig. 4c, f and i).
 | ||
| Fig. 4 STEM data including HAADF images (panels a–c), Au distribution maps in yellow (panels d–f) and Au line profiles (panels g–i) recorded on fly ash produced in experiments B and C. Insets in panels d–f show EDX spectra between 9000 and 12000 eV extracted from the respective white squares. The insets contain the net instrument counts (white), the background signal (dotted, black line) and a model fit including all identified elements (black line). The vertical, red bars indicate the energy positions of the Au Lα and Au Lβ fluorescence lines, which confirmed the presence of Au. The transparent, grey bars indicate the orientation of the line profiles. The profile orientation is from left to right in all Au distribution maps. | ||
3.4 Experiments B and C: elemental release
000 mg kg−1 Ca in sludge and fly ash, respectively) can be traced back to the hard water (French degree hardness (fH) = 22–25) in the catchment of the WWTP in La Chaux-de-Fonds originating from the limestone rich lithology of the Jurassic mountain range in the western part of Switzerland. As observed in experiment A, the release of particulate Au was independent of the Ca concentration.
3.5 Au particle sizes and inter-experiment data comparison
spICP-MS data were collected on 197Au to determine the diameter of the particulate Au present in the collected fractions #1, 2, 5, 10, 20, 30, and 40 of experiments A–C. The results indicated that the particle diameter was independent of the time at which the fractions were collected over the experiment. Therefore, only the mean and standard deviation of the cumulated mass versus the particle diameter for all three experiments are given for each experiment (Fig. 5). In addition, spICP-MS data on the spiking material are included in Fig. 5. A comparison of the concentrations determined by spICP-MS and bulk ICP-MS revealed that spICP-MS in general estimated higher total Au concentrations but lower particulate Au concentrations (Table S1†). The latter can be explained by a BED of around 20 nm in spICP-MS measurements versus a 1.2 nm “BED” determined with the 10 kDa filter.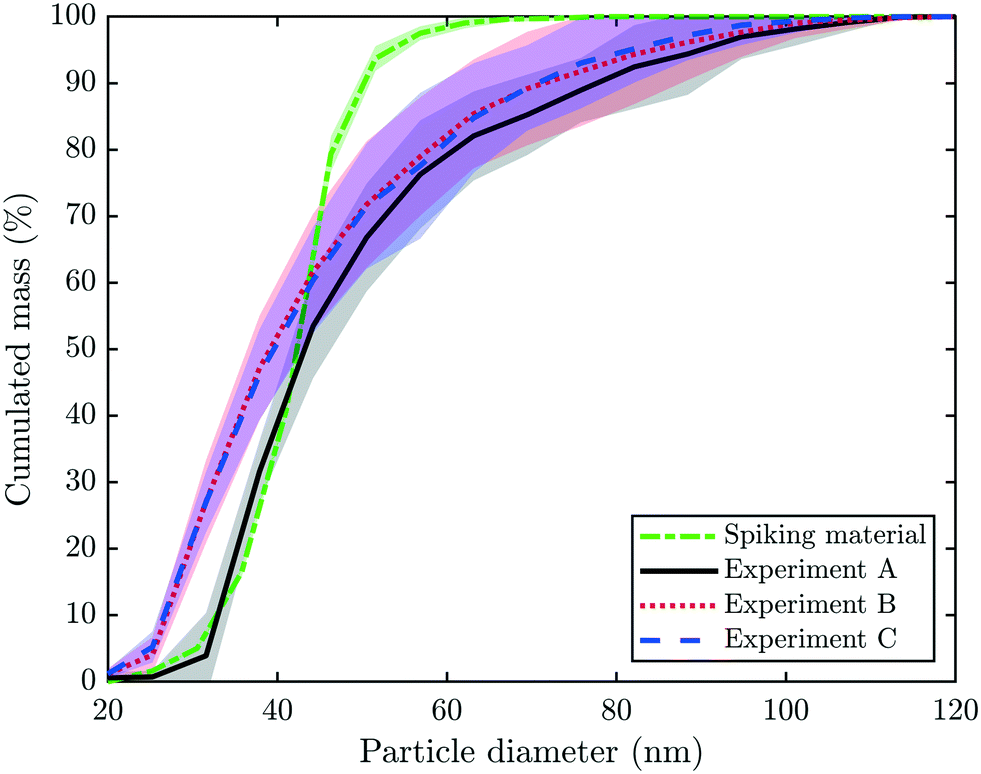 | ||
| Fig. 5 Single particle (sp)-ICP-MS data collected on 197Au. Displayed is the cumulated mass (%) versus the particle diameter (nm). The dashed, green line indicate the mean and standard deviation of three measurements of the spiking material (Au-NPs) diluted to 25 ng L−1 Au (compare also Fig. S1b and c†). The bold, black line and grey shaded area indicate the mean signal and two standard deviations collected on fractions #1, 2, 5, 10, 20, 30 and 40 of experiment A. The corresponding information is given for experiments B (dotted, red line) and C (dashed, blue line). | ||
In experiment A, approximately 45% of the mass results from particles ≤41 nm and the remaining 55% is from particles between 41 and 100 nm. Since the median diameter of the spiked particles determined by spICP-MS was 41 nm (section 2.1, Fig. 5), the larger particles most likely resulted from Au-NP homo-aggregates already formed in the stock solution during spiking or immediately after spiking into the pilot WWTP.38 However, aggregates may also have formed during downstream processes, although we consider this as rather unlikely. The size distributions measured for the (diluted) Au stock suspensions and the experimental ash leachates were in excellent agreement for particles below the median primary particle size of the Au-NPs (41 nm). This suggests that the incineration process only marginally affected the size of the primary Au-NPs. Based on our results from filtering the experimental leachates, we have estimated that 5.1 wt% Au-NPs transformed into a water soluble species. Assuming a primary Au-NP diameter of 41 nm, a transformation of 5% would correspond to a reduction of the primary particle diameter of the Au-NPs of 0.7 nm, which is beyond the precision of the spICP-MS measurements. The deviation between the Au-NP size distributions of the (diluted) stock suspension and the experimental ash leachates for particles larger than the primary size of the Au-NPs most likely reflects the formation of Au-NP (homo)aggregates, as outlined above.
The cumulative mass distribution of Au-NPs in the samples from experiments B and C follows a similar trend; however, the data suggest that a significant share of mass (≈25%) results from small particles ≤30 nm. The mean Au particle size distributions of B and C are almost indistinguishable and consistently indicate higher shares of mass resulting from smaller particles compared to the mean Au-NP size distribution from experiment A. Thus, we conclude that Au discharged by industrial companies (B, C) is present as smaller particles with a broader particle size distribution in the fly ash compared to the Au-NPs obtained from nominally 50 nm particles. These results were in good agreement with the HAADF images and the corresponding Au-distribution maps from experiments B and C, where Au-NPs with diameters between 20 and 50 nm were identified (Fig. 4).
In experiments A–C, the fractions of dissolved Au in the column effluents were 30, 9 and 9%, respectively, meaning that 5.1, 0.81 and 0.27 wt% of the total Au in the ash were present as water soluble species. The dissolution and complexation of Au-NPs by CN− through freshwater macrophytes under thermodynamically unfavorable conditions have been previously observed in a freshwater mesocosm.39 In our experiments, the concentrations of dissolved Au were high in the beginning of each experiment, but towards the end of the leaching experiments, they were mostly at least an order of magnitude lower compared to the concentration of particulate Au. The mode diameter of the ash grains was 48, 650 and 540 μm in experiments A–C and the larger extent of the formation of water soluble Au in experiment A may thus be related to the smaller ash grains. During the sludge incineration, hydrogen cyanide (HCN) and hydrogen chloride (HCl) are produced in the flue gas and for the latter compound, mole fractions of up to ≈2 × 10−4 were estimated.40,41 The bases of both acids efficiently complex Au+ ions in the aqueous phase and (crystalline) solid phases (AuCl and AuCN) can precipitate during drying. We hypothesize that the surface of individual Au-NPs is oxidized during the incineration process, where elevated temperatures up to 1000 °C in the fluidized bed and freeboard can be reached8,13 enabling the formation of AuCl and AuCN precipitates. This mechanism requires the exposure of the surface of Au-NPs to the flue gas. Besides the formation of soluble Au-phases, the formation of metallic Au clusters with diameters ≈1 nm may also contribute to the dissolved Au fraction observed in the fly ash leachates.42 Assuming an even distribution of the Au-NPs within the ash particles, the probability of an Au-NP being at the surface of the ash particles instead of being well embedded within the ash particles increases with decreasing diameter of the ash particles. Therefore, we propose that the Au-NPs present in the ash from experiment A were transformed to a higher degree compared to the Au-NPs present in the other ashes which exhibited a considerably larger (ash) particle diameter. However, an effect (e.g., reduced stability) caused by different properties of the spiked Au-NPs compared to the Au-NPs already present in the sludge (experiments B and C) may also have contributed to the higher extent of soluble Au species formed in experiment A.
Conclusion
In this study, we compared the fate of three types of ENPs, namely Au-NPs, Ag-NPs and CeO2-NPs in managed waste facilities including WWTPs, fluidized bed incinerators and SSA landfill sites. We found that Au was released from sewage sludge fly ash in significant quantities (17% of the total Au contained in the ash), whereas Ag and Ce were retained (less than 0.01% of Ag and 0.001% of Ce were released). Au originating from industrial discharges was released in lower shares (3 and 9%) from sewage sludge fly ash and the released Au was dominantly (91%, n = 2) present as nanoparticles (d = 20–100 nm). In contrast, leachates from the sewage sludge fly ash containing spiked Au-NPs contained a larger fraction of soluble Au-species (30%) originating from transformation reactions occurring during sludge incineration. Most likely, the reduced ash grain size ≈50 μm versus 540–650 μm led to a higher fraction of Au-NPs exposed to flue gas at the surface of the ash grains resulting in a higher degree of Au-NP transformation.Au-NPs represent a class of conservative ENPs that are unaffected by anaerobic digestion and only weakly affected by sludge incineration. Based on data obtained in an experiment where Au-NPs were spiked to a pilot WWTP, we saw that 30% (by mass) of released Au was present as species <10 kDa, indicating that at least 5.1% of the Au in the fly ash occurred as water soluble compounds. Thus, Au-NPs transformed to a limited degree during sludge incineration, most likely through reactions with Cl− or CN−, which led to the formation of water soluble AuCl or AuCN precipitates attached to the ash particles. In contrast, Ag-NPs and CeO2-NPs represent transient ENPs that are entirely transformed into novel phases during anaerobic digestion and/or sludge incineration. Through their reaction during digestion and incineration, Ce and Ag became tightly embedded into the ash matrix resulting in the negligible release of the respective transformation products from sewage sludge fly ash. In conclusion, our data show that conservative (or chemically stable) ENPs like Au-NPs are released to a greater extent from sewage sludge fly ash compared to the transformation products of transient ENPs like Ag-NPs, CeO2-NPs, TiO2-NPs,13 CuO-NPs or ZnO-NPs.10 A classification according to the “reactivity” or the degree of transformation of ENPs in managed waste facilities may therefore be an appropriate approach to assess the release potential of ENPs including their (nano)particulate transformation products from landfill sites. However, the parameters determining the quantity of the release of conservative ENPs remain to be elucidated.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Brian Sinnet, Mathias Phillip, Nadine Fritschi and Numa Pfenninger for their support in the laboratory and Marco Kipf for his support during the incineration experiments. We acknowledge funding for this research, which was provided through the Swiss National Science Foundation (grant no. 165911), and the European Union‘s Horizon 2020 research and innovation program (grant agreement no. 646002). We thank Stefano Airaghi (wastewater treatment plant of Chiasso) and Jacques Vidal (wastewater treatment plant of La Chaux-de-Fonds) for providing us dewatered sludge samples. The authors acknowledge the ETH Zürich Microscopy Center (ScopeM) for providing access to their electron microscopes.References
- P. Westerhoff, G. Song, K. Hristovski and M. A. Kiser, Occurrence and removal of titanium at full scale wastewater treatment plants: implications for TiO2 nanomaterials, J. Environ. Monit., 2011, 13(5), 1195–1203 RSC.
- R. Kaegi, A. Voegelin, B. Sinnet, S. Zuleeg, H. Hagendorfer, M. Burkhardt and H. Siegrist, Behavior of Metallic Silver Nanoparticles in a Pilot Wastewater Treatment Plant, Environ. Sci. Technol., 2011, 45(9), 3902–3908 CrossRef CAS PubMed.
- G. Tchobanoglous, H. D. Stensel, R. Tsuchihashi and F. L. Burton, Wastewater engineering: treatment and resource recovery, McGraw-Hill Education, New York, NY, USA, 2013 Search PubMed.
- P. Mantovi, G. Baldoni and G. Toderi, Reuse of liquid, dewatered, and composted sewage sludge on agricultural land: effects of long-term application on soil and crop, Water Res., 2005, 39(2), 289–296 CrossRef CAS PubMed.
- F. Laturnus, K. von Arnold and C. Grøn, Organic Contaminants from Sewage Sludge Applied to Agricultural Soils. False Alarm Regarding Possible Problems for Food Safety? (8 pp), Environ. Sci. Pollut. Res., 2007, 14(1), 53–60 CrossRef CAS PubMed.
- A. Laube and A. Vonplon, BUWAL, Klärschlammentsorgung in der Schweiz, Mengen- und Kapazitätserhebung, 2004 Search PubMed.
- B. Wiechmann, D. Claudia, K. Christian, B. Simone, V. Ines and R. Andrea, Sewage Sludge Management in Germany, Umweltbundesamt Deutschland, 2015 Search PubMed.
- J. Werther and T. Ogada, Sewage sludge combustion, Prog. Energy Combust. Sci., 1999, 25(1), 55–116 CrossRef CAS.
- L. Morf, S. Schlumberger, F. Adam and G. Díaz Nogueira, Urban Phosphorus Mining in the Canton of Zurich: Phosphoric Acid from Sewage Sludge Ash, in Phosphorus Recovery and Recycling, ed. H. Ohtake and S. Tsuneda, Springer Singapore, Singapore, 2019, pp. 157–177 Search PubMed.
- J. Wielinski, A. Gogos, A. Voegelin, C. Müller, E. Morgenroth and R. Kaegi, Transformation of Nanoscale and Ionic Cu and Zn during the Incineration of Digested Sewage Sludge (Biosolids), Environ. Sci. Technol., 2019, 53(20), 11704–11713 CrossRef CAS PubMed.
- J. Wielinski, F. F. Marafatto, A. Gogos, A. Scheidegger, A. Voegelin, C. R. Müller, E. Morgenroth and R. Kaegi, Synchrotron hard X-ray chemical imaging of trace element speciation in heterogeneous samples: development of criteria for uncertainty analysis, J. Anal. At. Spectrom., 2020, 35(3), 567–579 RSC.
- A. Gogos, J. Wielinski, A. Voegelin, H. Emerich and R. Kaegi, Transformation of cerium dioxide nanoparticles during sewage sludge incineration, Environ. Sci.: Nano, 2019, 6(6), 1765–1776 RSC.
- J. Wielinski, A. Voegelin, B. Grobéty, C. R. Müller, E. Morgenroth and R. Kaegi, Transformation of TiO2 (nano)particles during sewage sludge incineration, J. Hazard. Mater., 2021, 411, 124932 CrossRef CAS PubMed.
- B. Thalmann, A. Voegelin, E. Morgenroth and R. Kaegi, Effect of humic acid on the kinetics of silver nanoparticle sulfidation, Environ. Sci.: Nano, 2016, 3(1), 203–212 RSC.
- B. Thalmann, A. Voegelin, B. Sinnet, E. Morgenroth and R. Kaegi, Sulfidation Kinetics of Silver Nanoparticles Reacted with Metal Sulfides, Environ. Sci. Technol., 2014, 48(9), 4885–4892 CrossRef CAS PubMed.
- R. Kaegi, B. Sinnet, S. Zuleeg, H. Hagendorfer, E. Mueller, R. Vonbank, M. Boller and M. Burkhardt, Release of silver nanoparticles from outdoor facades, Environ. Pollut., 2010, 158(9), 2900–2905 CrossRef CAS PubMed.
- C. Meier and A. Voegelin, Pradas del Real, A.; Sarret, G.; Mueller, C. R.; Kaegi, R., Transformation of Silver Nanoparticles in Sewage Sludge during Incineration, Environ. Sci. Technol., 2016, 50(7), 3503–3510 CrossRef CAS PubMed.
- A. Lazareva and A. A. Keller, Estimating Potential Life Cycle Releases of Engineered Nanomaterials from Wastewater Treatment Plants, ACS Sustainable Chem. Eng., 2014, 2(7), 1656–1665 CrossRef CAS.
- A. Gogos, R. Kaegi, J. Wielinski, M. Matzke, B. Stahlmecke, H. Kaminski, C. Asbach and G. Cornelis, Report the Behavior of ENM in Reactors; H2020 NanoFASE Project, 2019 Search PubMed.
- B. Vriens, A. Voegelin, S. J. Hug, R. Kaegi, L. H. E. Winkel, A. M. Buser and M. Berg, Quantification of Element Fluxes in Wastewaters: A Nationwide Survey in Switzerland, Environ. Sci. Technol., 2017, 51(19), 10943–10953 CrossRef CAS PubMed.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Determining Transport Efficiency for the Purpose of Counting and Sizing Nanoparticles via Single Particle Inductively Coupled Plasma Mass Spectrometry, Anal. Chem., 2011, 83(24), 9361–9369 CrossRef CAS PubMed.
- W. Shotyk and P. Steinmann, Pore-water indicators of rainwater-dominated versus groundwater-dominated peat bog profiles (Jura Mountains, Switzerland), Chem. Geol., 1994, 116(1), 137–146 CrossRef CAS.
- J. Crank, The mathematics of diffusion, Oxford University Press, 1979 Search PubMed.
- J. Oischinger, M. Meiller, R. Daschner, A. Hornung and R. Warnecke, Fate of nano titanium dioxide during combustion of engineered nanomaterial-containing waste in a municipal solid waste incineration plant, Waste Manage. Res., 2019, 37(10), 1033–1042 CrossRef CAS PubMed.
- C. Levard, E. M. Hotze, G. V. Lowry and G. E. Brown, Environmental Transformations of Silver Nanoparticles: Impact on Stability and Toxicity, Environ. Sci. Technol., 2012, 46(13), 6900–6914 CrossRef CAS PubMed.
- R. Kaegi, A. Voegelin, C. Ort, B. Sinnet, B. Thalmann, J. Krismer, H. Hagendorfer, M. Elumelu and E. Mueller, Fate and transformation of silver nanoparticles in urban wastewater systems, Water Res., 2013, 47(12), 3866–3877 CrossRef CAS PubMed.
- L. E. Barton, M. Auffan, M. Bertrand, M. Barakat, C. Santaella, A. Masion, D. Borschneck, L. Olivi, N. Roche, M. R. Wiesner and J.-Y. Bottero, Transformation of Pristine and Citrate-Functionalized CeO2 Nanoparticles in a Laboratory-Scale Activated Sludge Reactor, Environ. Sci. Technol., 2014, 48(13), 7289–7296 CrossRef CAS PubMed.
- K.-S. Wang, I.-J. Chiou, C.-H. Chen and D. Wang, Lightweight properties and pore structure of foamed material made from sewage sludge ash, Constr. Build. Mater., 2005, 19(8), 627–633 CrossRef.
- A. Gogos, M. Matzke and R. Kaegi, Report on completion of Task 5.1& Task 5.6.; H2020 NanoFASE Project, 2019 Search PubMed.
- A. Birich, S. Stopic and B. Friedrich, Kinetic Investigation and Dissolution Behavior of Cyanide Alternative Gold Leaching Reagents, Sci. Rep., 2019, 9(1), 7191 CrossRef PubMed.
- T. L. Theis and L. E. Padgett, Factors Affecting the Release of Trace Metals from Municipal Sludge Ashes, J. - Water Pollut. Control Fed., 1983, 55(10), 1271–1279 CAS.
- D. Grolimund, M. Elimelech, M. Borkovec, K. Barmettler, R. Kretzschmar and H. Sticher, Transport of in Situ Mobilized Colloidal Particles in Packed Soil Columns, Environ. Sci. Technol., 1998, 32(22), 3562–3569 CrossRef CAS.
- J. Wielinski, C. Müller, A. Voegelin, E. Morgenroth and R. Kaegi, Combustion of Sewage Sludge: Kinetics and Speciation of the Combustible, Energy Fuels, 2018, 32(10), 10656–10667 CrossRef CAS.
- D. Nakić, D. Vouk, S. Donatello and A. Anić Vučinić, Environmental impact of sewage sludge ash assessed through leaching, Eng. Rev., 2017, 37(2), 222–234 Search PubMed.
- S. Donatello, A. Freeman-Pask, M. Tyrer and C. R. Cheeseman, Effect of milling and acid washing on the pozzolanic activity of incinerator sewage sludge ash, Cem. Concr. Compos., 2010, 32(1), 54–61 CrossRef CAS.
- M. Willems, B. Pedersen and S. S. Jørgensen, Composition and Reactivity of Ash from Sewage Sludge, Ambio, 1976, 5(1), 32–35 CAS.
- E. E. Berry, R. T. Hemmings and B. J. Cornelius, Mechanisms of hydration reactions in high volume fly ash pastes and mortars, Cem. Concr. Compos., 1990, 12(4), 253–261 CrossRef CAS.
- M. C. Surette, J. A. Nason and R. Kaegi, The influence of surface coating functionality on the aging of nanoparticles in wastewater, Environ. Sci.: Nano, 2019, 6(8), 2470–2483 RSC.
- A. Avellan, M. Simonin, E. McGivney, N. Bossa, E. Spielman-Sun, J. D. Rocca, E. S. Bernhardt, N. K. Geitner, J. M. Unrine, M. R. Wiesner and G. V. Lowry, Gold nanoparticle biodissolution by a freshwater macrophyte and its associated microbiome, Nat. Nanotechnol., 2018, 13(11), 1072–1077 CrossRef CAS PubMed.
- G. T. Daigger and T. E. Sadick, Evaluation of methods to detect and control nitrification inhibition with specific application to incinerator flue-gas scrubber water, Water Environ. Res., 1998, 70(7), 1248–1257 CrossRef CAS.
- M. Hartman, K. Svoboda, M. Pohořelý and O. Trnka, Combustion of Dried Sewage Sludge in a Fluidized-Bed Reactor, Ind. Eng. Chem. Res., 2005, 44(10), 3432–3441 CrossRef CAS.
- K. Michaelian, N. Rendón and I. L. Garzón, Structure and energetics of Ni, Ag, and Au nanoclusters, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60(3), 2000–2010 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1en00497b |
| This journal is © The Royal Society of Chemistry 2021 |