
Elemental iron: reduction of pertechnetate in the presence of silica and periodicity of precipitated nano-structures†
Daria
Boglaienko
 a,
Odeta
Qafoku
a,
Ravi K.
Kukkadapu
a,
Libor
Kovarik
a,
Yelena P.
Katsenovich
b,
Denis E.
Cherkasov
a,
Hilary P.
Emerson
a and
Tatiana G.
Levitskaia
*a
a,
Odeta
Qafoku
a,
Ravi K.
Kukkadapu
a,
Libor
Kovarik
a,
Yelena P.
Katsenovich
b,
Denis E.
Cherkasov
a,
Hilary P.
Emerson
a and
Tatiana G.
Levitskaia
*a
aPacific Northwest National Laboratory, USA. E-mail: Tatiana.Levitskaia@pnnl.gov
bFlorida International University, USA
First published on 10th November 2020
Abstract
Nano-structural transformation of iron minerals in the natural environment is altered and often retarded in the presence of silica (e.g., impeded transformation of ferrihydrite) resulting in a modulated interaction with constituents or contaminants present in groundwater. This phenomenon can significantly affect molecular mechanisms of reduction, precipitation, and sequestration of pertechnetate (TcO4−), the most prevalent chemical form of radioactive contaminant technetium-99 in the environment, by elemental iron Fe0 often referred to as zero valent iron (ZVI). Understanding the role of silica in moderating the reactivity of Fe0 toward reduction of TcO4− to Tc4+ and its interaction with in situ formed iron minerals (ferrihydrite, magnetite) is crucial for successful design of a practical separation system and can be related to similar environmental systems. This study was designed to evaluate silica-modified ZVI systems with two commercially available iron materials. The results revealed that the efficiency of TcO4− reduction by Fe0 increased in the presence of silica due to inhibited transformation of iron oxyhydroxide into non-stoichiometric magnetite. Moreover, microscopic evaluation of the newly formed iron mineral phases, both in the presence and absence of silica, revealed unique morphologies related to geological phenomena, such as orbicular rocks and Liesegang rings, suggesting that iron dissolution/re-precipitation is a rhythmical reaction–diffusion process, which occurs in both micro-scaled and macro-geological environments resulting in layered structures of iron oxidation products.
Environmental significanceRadioactive contaminants pose risks upon their spread in the environment. Pertechnetate, studied here, is a highly soluble anion of technetium-99, found in subsurface environments near nuclear waste storage and reprocessing sites. Application of metallic iron for reductive removal of pertechnetate is a promising method due to its availability, efficiency, low toxicity, and low cost; however, its performance and oxidative transformations are altered in the presence of silica species, common in natural environments. Our results demonstrate the enhanced effect of silica on the pertechnetate reduction with in situ formed iron minerals. Additionally, evaluation of iron minerals revealed rhythmical formations common in geological structures, relating phenomena such as orbicular rocks and Liesegang rings to both macro- and micro-scaled systems. |
Introduction
The presence of silica in the natural environment affects the formation of mineral phases, e.g. transformation of iron minerals in aqueous media. For example, silica-rich groundwaters in Finland and New Zealand contain precipitated ferrihydrite with silica adsorbed to its surface.1 Ferrihydrite is a poorly crystalline iron oxyhydroxide, whose crystallization and transformation into well-ordered iron minerals (e.g., lepidocrocite, goethite, magnetite, and green rust) is stimulated by the electron donor Fe2+ and can be hindered by sorbed species, i.e. silica, sustaining ferrihydrite's disordered structure and linking its nanoparticles into an immobile network.2,3 In pedogenic environments, it is commonly detected together with lepidocrocite and goethite; however, elevated concentrations of silica impede the formation of crystalline lepidocrocite in favor of that of ferrihydrite.4 Ferrihydrite has a high surface area and reactivity, which makes it an effective sorbent in water treatment.5–7 For instance, reductive removal of the radioactive contaminant technetium-99 (Tc) predominantly found in subsurface plumes in the form of pertechnetate (TcO4−) is a heterogeneous process and requires a solid phase for the efficient exchange of electrons, e.g. in situ precipitating ferrihydrite.8Zero-valent iron (ZVI) is a common reductant, effectively used for decontamination of aqueous waste streams from inorganic and organic compounds.9–13 It has been investigated as a commercially available non-modified material14,15 and as an engineered material with enhanced efficiency via a silica support or cover.16–21 Supported with silica gel or porous silica, ZVI is less prone to agglomeration,16,22 and covered with a silica shell, ZVI nanoparticles are characterized by a larger surface area, higher mobility, and reduced oxidation.19 However, the enhanced effect of silica on reduction, sorption, and co-precipitation processes through alteration of iron mineral transformation during Fe0 oxidation has not been evaluated, and there are no known studies conducted for such systems in the presence of the pertechnetate anion (TcO4−).
Technetium-99 (Tc) is a long-lived radionuclide (2.1 × 105 years),23 produced during operation of nuclear reactors and accumulated in the liquid phase of radioactive waste contained in underground tanks of decommissioned complexes, on sites of nuclear fuel reprocessing plants, and in underground plumes. The pertechnetate anion, a dominant form of Tc in systems with oxygen, is highly mobile in subsurface environments and poses a significant risk during radioactive waste processing and disposal. Removal of Tc7+via its reduction to insoluble Tc4+ by Fe0 (ZVI) is one of the treatment options that can be easily implemented due to the availability of ZVI materials and their low cost. Our previous studies compared a wide variety of commercially available iron materials14 and the feasibility of ZVI for TcO4− reduction at high Tc to Fe loading (1 to 53 mol ratio) and concomitant fractional incorporation into in situ spontaneously formed magnetite.15 Evidence of Tc-incorporated iron minerals (magnetite, hematite, goethite) was also reported in other studies,24–28 relating this process to the advantages of more environmentally stable forms of mononuclear Tc4+ sequestered in the lattice of iron minerals.
Among previously tested iron products of high purity (97–99.9%, metal basis), the highest efficiency for reduction of pertechnetate in aqueous solutions was demonstrated with a ZVI material of 75 μm particle size, manufactured by an electrolytic method.14 However, our further investigations revealed that this material was not as efficient as ZVI containing relatively high levels of impurities, mainly silica. Hence, the objective of this study was to investigate the mechanism of this enhanced reductive removal of TcO4− by ZVI in the presence of silica in relation to the altered iron mineral transformation. The experimental design of this work is based on evaluation of two commercially available ZVI materials, and is organized as a comparative analysis of series of experiments conducted in different modes of silica addition to ZVI and silica co-precipitation with in situ formed iron oxy/hydroxides. The widespread co-existence of silica and iron minerals relates this work to subsurface environments with natural processes of silica dissolution and precipitation and iron mineral transformation.
Materials and methods
Caution! 99Tc is a β− emitter with an energy of 0.29 MeV.23 The experimental work was conducted at a research nuclear facility by trained personnel.Materials
Zero valent iron powder, 75 μm (200 mesh), denoted in this work as ZVI-A, was obtained from Alfa Aesar. This iron material was manufactured by an electrolytic method using steel anodes and a ferrous sulfate solution and was of high purity, 99+% (metals basis). Ferox PRB reactive iron powder, 297 μm (50 mesh), was provided by Hepure Technologies Inc. This material, designated as ZVI-B, was produced from cast iron utilizing grinding and pulverizing methods and was composed of 95+% iron, carbon (∼2.0%), oxygen (<1%), silicon (1–1.5%) and trace amounts of phosphorus (<0.1%) and sulfur (∼0.1%) according to technical specifications from the manufacturer.Sodium metasilicate nonahydrate (crystalline certified from Fisher Scientific, Na2SiO3 ≥ 95%) was used to prepare an amorphous silica gel by dissolution in deionized water followed by direct precipitation with an acid. Hydrochloric acid and sodium hydroxide (Fisher Scientific) were used to adjust the pH, starting from 12.7 to 5.4. The silica gel was shaken overnight, centrifuged, washed two times with deionized water, and dried in a vacuum oven at 50 °C. The theoretical yield of amorphous silica from 1 g of Na2SiO3·9H2O is 0.2420 g of SiO2. The weight of SiO2 was monitored daily and drying was stopped as the weight of amorphous silica solids stabilized after 12 days, giving an amount of 0.2795 g. The difference between the theoretical and experimental yields is due to the presence of hydration water which is challenging to completely remove.
Solutions were prepared using an NH4TcO4 stock (prepared at the Radiochemical Processing Laboratory within the Pacific Northwest National Laboratory) and NaCl salt (ACS reagent grade, Fisher Chemical). Deionized water (>18 MΩ cm) was used to prepare all solutions and reagents.
Study design
The schematic of the experimental design is presented in Fig. 1. The small grey boxes on top denote labels of the experimental series (E0–E6). Amorphous silica prepared by direct precipitation with an acid was used without iron material as a control (E0). ZVI-A (99+% Fe0) was used as received (E1). Another series was composed of a physical mixture of this material with amorphous silica to investigate the effect of the presence of silica not yet reacted with iron (E2). For this purpose, amorphous silica was manually ground into powder and added to ZVI-A as a mixture of 95 wt% ZVI-A and 5 wt% SiO2. Additionally, this material (ZVI-A) was surface treated via co-precipitation with silica, following the same acid precipitation protocol described earlier, but in the presence of ZVI-A together with Na2SiO3·9H2O in deionized water (E3); adjustment of pH from the initial value of 12.3 to 5.6 was done using 1 M and 0.1 M HCl. In order to see the effect of the acid treatment on iron, a series of ZVI-A without silica, but acid-treated, mimicking the silica precipitation protocol, was prepared as a control for surface treatment of ZVI-A (E4). Finally, a series of samples using ZVI-B (95+% Fe0 and 1–1.5% of Si) as received (E5) was added, and a series of ZVI-B surface treated with an acid (E6) was prepared as a comparison to the series E4 of the surface treated ZVI-A.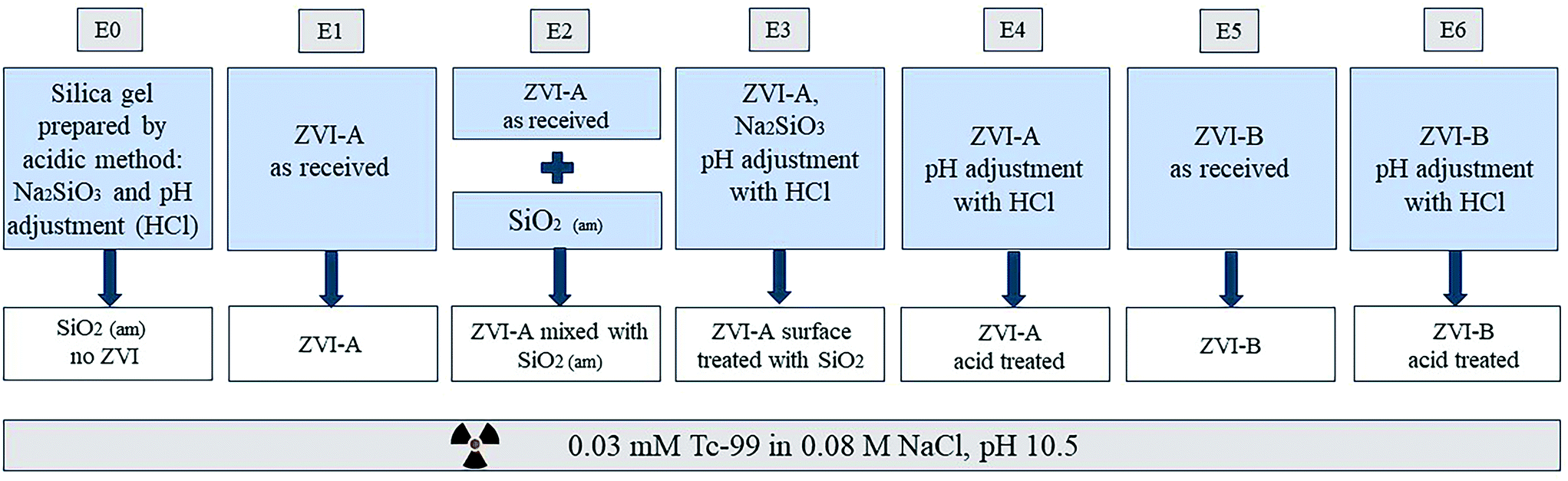 | ||
| Fig. 1 Schematic of the experimental design. ZVI-A is electrolytically manufactured iron (99+% purity, 75 μm; Alfa Aesar); ZVI-B is iron powder produced from cast iron (95+% purity; 297 μm; Hepure Technologies Inc). The grey boxes on top represent labels of the series (E0–E6) used in the figures, and the white boxes represent names of the series referred to in the main text. The large blue boxes contain short descriptions of the series, which are fully explained in the study design. The box on the bottom describes the stock solution all the series of experiments were conducted in. | ||
Batch contacts
The samples were prepared in duplicate with 150 mg of ZVI in 40 mL of a solution (phase ratio 0.27 L g−1, or concentration 3.75 g L−1) containing nominally 0.03 mM TcO4− in 0.08 M NaCl, pH 10.50 ± 0.04 (adjusted with 0.1 M NaOH and HCl). The samples were mixed using a shaking table for 20 hours, centrifuged, decanted and filtered to separate the supernatant for analysis. The concentration of TcO4− was analyzed using a liquid scintillation counter (Tri-Carb 2910 TR, PerkinElmer), with the minimum detectable activity corresponding to 3 × 10−8 M Tc, with a high sample load scintillation cocktail (Optiphase Hisafe 3, PerkinElmer).The dissolution kinetics of ZVI-A was studied previously.14 The kinetics of iron and silicon dissolution for the pristine ZVI-B (150 mg) in 40 mL of 0.08 M NaCl (initial pH 10.50 ± 0.04) together with pH measurements were studied in triplicate at time points of 10 min, 20 min, 30 min, 1 h, 2 h, 3 h, 5 h, 8 h and 24 h. Control samples (no ZVI; only a solution of 0.08 M NaCl, pH 10.50 ± 0.04) were analyzed after reaction for 10 min, 3 h, 8 h, and 24 h. The pH was chosen to achieve relatively slow oxidation of Fe0 and, thus, reduction of Tc7+, which would allow comparison of the iron materials' reactivities. The samples were centrifuged, and the supernatants (acidified with 0.3 M HNO3) were analyzed for dissolved iron and silicon with a commercial ICP-OES instrument (Avio 500, PerkinElmer, USA) with the following detection limits for iron and silicon of 0.3 μg L−1 and 30.2 μg L−1, respectively. Data were processed accounting for background iron and silicon (subtraction based on blanks). ICP-OES instrumental parameters: RF power 1300 W; viewing distance 15 mm; integration window 0.02–5 s; sample uptake rate 1.0 mL min−1; plasma flow 15 L min−1; aux. flow 0.2 L min−1; nebulizer flow 0.45 L min−1.
All series of batch experiments were conducted at ambient temperature (22 ± 2 °C) and pressure in an aerobic environment.
Solid characterization
Chemical composition data and mappings were collected using an Oxford X-Max 80 mm2 solid state EDX detector. The EDX analyses were performed using an acceleration voltage of 30 keV and a current of 4 nA. For all the elemental analysis, the Kα positions were considered. The EDX point analyses were performed using an acquisition time of 60 seconds and elemental mappings were collected with an acquisition time of 300 to 600 seconds.
The areas that showed morphological changes and unique patterns were selected and prepared for analysis with transmission electron microscopy (TEM). These areas were extracted from the bulk sample using FIB Ga liquid metal ion source milling and the lift-out technique was employed to prepare thin lamellae for TEM analysis. Prior to ion milling, the areas of interest were protected by deposition of a 1–2 μm Pt layer using a Quanta GIS (gas injection system). The specimens were thinned to ∼80–100 nm by using lower beam currents to below 100 pA.
STEM analysis was carried out with an aberration-corrected JEOL-ARM200F microscope operated at 200 kV. The instrument is equipped with a CEOS GmbH double-hexapole aberration corrector (CESCOR) for the probe-forming lens. The images were acquired with a high angle annular dark field detector (HAADF) in scanning transmission electron imaging mode. Compositional analysis was performed with a JEOL Centurio high-collection angle silicon drift detector (100 mm2).
Mössbauer spectra were collected at room temperature for all samples. The 50 mCi 57Co/Rh source and velocity transducer MVT-1000 (WissEL) were operated in constant acceleration mode (23 Hz, ±12 mm s−1). The signal was transmitted through a holder where radiation was detected by an Ar–Kr proportional counter. The counts were stored in a multichannel scalar as a function of energy, utilizing a 1024-channel analyzer. Data were folded to 512 channels to give a flat background and a zero-velocity position corresponding to the center shift (CS or δ) of metal Fe foil at room temperature. A 25 μm thick Fe foil piece (Amersham, England) was placed in the same position as the samples to obtain calibration spectra. The Mössbauer data were modeled using the Recoil software (University of Ottawa, Canada) and a Voigt-based structural fitting routine.
Results and discussion
Characterization of the pre-contact iron materials
Solid characterization of the ZVI-A and ZVI-B materials via scanning electron microscopy (SEM) before being in contact with aqueous media (Fig. SI-1) and via dispersive X-ray analysis (EDX), which provides visual evidence of silica particles among the particles of iron in ZVI-B as received (Fig. SI-2†), can be found in the ESI.† Room temperature comparative Mössbauer analysis between ZVI-A and ZVI-B (Fig. SI-3†) supported the evidence of additional sites in ZVI-B, which were previously studied as structural silicon in an Fe–Si alloy.29 Thus, both structural silicon and particulate silica were identified in the ZVI-B material.The dissolution kinetics of ZVI-A were studied before14 and revealed the highest initial (within 10 min sampling time) concentrations of dissolved Fe2+, which declined rapidly with time. The dissolution kinetics of ZVI-B (Fig. 2) showed similar behavior in which the concentration of dissolved Fe increased significantly within the first 20 min, followed by a sharp decrease at up to one hour with the possible formation of precipitated iron oxyhydroxides, and reaching steady-state for Fe2+ dissolution (eqn (1)) and precipitation (eqn (2)). The changes in pH (Fig. 2) with an overall decrease to 9.93 ± 0.09 towards a time period of 24 hours are indicative of the dominant processes of iron oxyhydroxide and oxide formation and precipitation during that time. The differences between concentrations of dissolved iron from ZVI-A as received (up to 12 μM after 3 hours for an initial concentration of 0.5 g L−1)14 and ZVI-B as received (predominantly below 35 μM after 3 hours for an initial concentration of 3.75 g L−1) can be related to the effect of the presence of silica/silicon in ZVI-B.
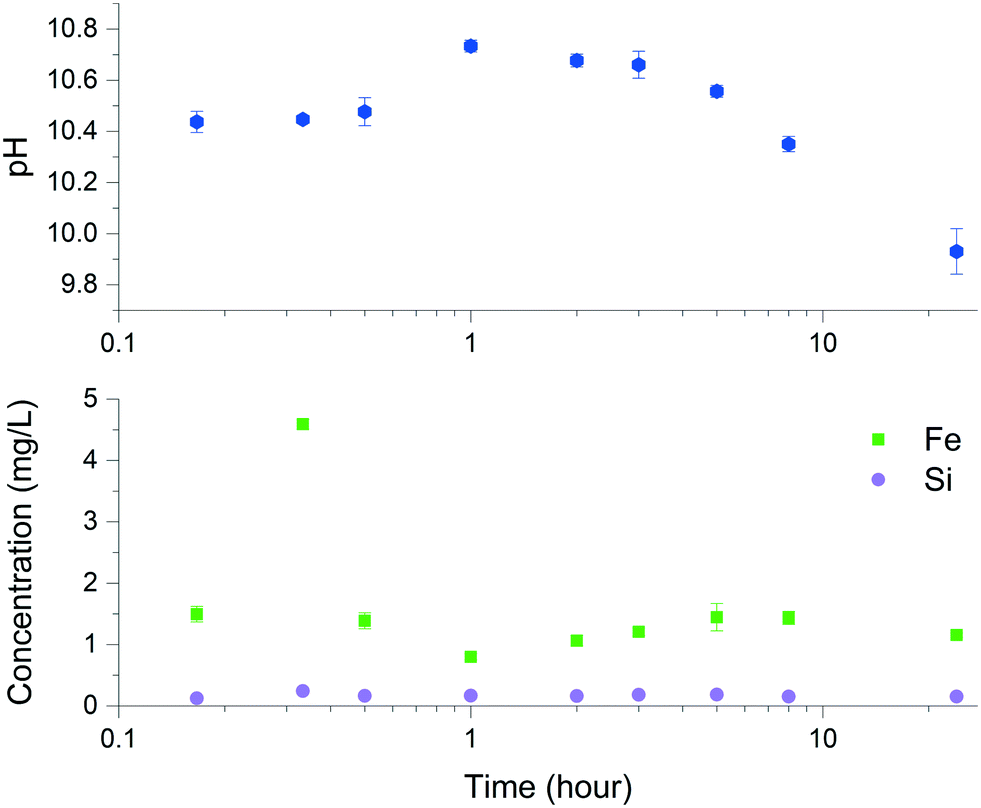 | ||
| Fig. 2 Iron and silicon dissolution using ZVI-B (as received) in 0.08 M NaCl and pH measurements upon dissolution of this material. | ||
The general process of metallic iron dissolution in the presence of silica can be presented as follows. Oxidation of Fe0 by water under aerated conditions:30
| 2Fe0 + O2 + 2H2O → 2Fe2+ + 4OH− | (1) |
Further oxidation of Fe2+ in the presence of oxygen can be written as:
| 2Fe(OH)2(aq) + O2 → 2FeOOH + 2OH− | (2) |
The effect of 0.08 M NaCl, which composes the solution matrix, is not considered to be significant here, as the FeCl+ concentrations decline far below 1 μM at pH >9 (Geochemist's workbench version 12.0 modelling for a similar system with metallic iron dissolved in 0.08 M NaCl).14
Iron oxyhydroxide, which can include goethite (α-FeOOH), lepidocrocite (γ-FeOOH), or ferrihydrite (several chemical formulas exist, 5Fe2O3·9H2O, Fe5HO8·4H2O, or FeOOH·0.4H2O),31 is an intermediate product in alkaline solutions, where magnetite (Fe3O4) forms via adsorption of Fe2+ on FeOOH, eqn (3), and via transformation of iron oxyhydroxide, eqn (4), as was investigated for γ-FeOOH under alkaline conditions by Tamaura et al.:32
| 2FeOOH + Fe2+ + H2O → [FeOOH]2FeOH+ + H+ | (3) |
| [FeOOH]2FeOH+ → Fe3O4 + H2O + H+ | (4) |
The effect of silica on iron transformation is known in the literature31,33,34 and is expected to impact TcO4− reduction by metallic iron. Thus, experimental series E2 and E3 were designed to amend ZVI-A (99+% pure Fe0) with silica as a physical mixture (E2) and surface treated iron (E3) and compare its reductive efficiency to ZVI-B. Silica gel was synthesized according to eqn (5):
| Na2SiO3 + H2O + 2HCl → Si(OH)4 + 2NaCl | (5) |
The dissolution kinetics of particulate silica and silicon incorporated into the ZVI-B material during its manufacturing reveals steadily low concentrations of silicon in the supernatant amounting to less than 0.5% of the total Si in the system, which is estimated to be approximately 35.7 mg L−1 in 3.75 g L−1 ZVI-B, if a more conservative number of 1 wt% Si impurities is considered according to the manufacturer specification. Undissolved Si particles are visible on the EDX map obtained after ZVI-B is brought into contact with 0.03 mM TcO4− in 0.08 M NaCl solution (Fig. SI-4†).
The amount of dissolved Si is significantly less than that reported previously in the range from 100 to 150 mg L−1 over a wide pH range at room temperature in the absence of iron.36 Low dissolution rates of silicon can be explained by complexation of silicic acid and dissolved Fe2+ species, as well as sorption to the surface of iron hydroxides and oxyhydroxides. The existence of monomeric and dimeric silica surface complexes on iron sites has been examined in previous studies.35,37,38 Monomeric species FeSiO2+(OH)2+ and FeSiO(OH)32+ are formed during the contact of iron hydroxides with silicic acid, whereas soluble dimeric silica can be formed according to the reaction37 with liberation of H+ ions:
| 2Si(OH)4 → Si2O2(OH)5− + H+ + H2O | (6) |
Reduction of TcO4−
Oxidation of Fe2+ to Fe3+ allows reduction of Tc7+ to Tc4+ which proceeds accordingly:| TcO4− + 3Fe2+ + (n + 7)H2O → TcO2·nH2O(s) + 3Fe(OH)3(s) + 5H+ | (7) |
The results obtained within a time period of 20 hours showed complete reduction of TcO4− in all systems, except the control composed of silica without ZVI (Fig. 3, series E0). The differences in reduction can be discerned for the 3 hour period of time, where the aqueous non-reduced fraction of TcO4− was below 0.3 in all the series of metallic iron containing silica/silicon, i.e., ZVI-A amended with silica (series E2 and E3), and ZVI-B (series E5 and E6) which initially had particulate silica and structural silicon. The series with ZVI-A as received (E1) showed the lowest TcO4− removal efficiency (Fig. 3).
 | ||
| Fig. 3 Reduction of Tc7+ (TcO4−) in the six series (E1–E6) of two ZVI materials (ZVI-A and ZVI-B), pristine and modified with silica and acid treatment (see Fig. 1); E0 is the control series with amorphous silica and no ZVI. | ||
Treatment of iron (ZVI-A) with sodium silicate and an acid did not enhance its performance in comparison to ZVI-B as received and ZVI-B treated in the same manner with an acid (E3 vs. E5 and E6). However, the presence of sodium silicate enhanced iron reactivity as compared to the sample without silica but similarly treated with HCl to reduce pH (E3 vs. E4). Overall, acid treatment slightly improved reduction efficiency, as evident from ZVI-A treated (E4) and ZVI-A non-treated (E1); acid treated samples of ZVI-B (E6) showed almost complete reductive removal of Tc from the aqueous fraction, as well as non-treated samples of ZVI-B (E5). Addition of amorphous silica to the ZVI-A samples (E2, no acid treatment) resulted in the most drastic improvement of the Tc reduction compared to the ZVI-A material as received. This series, E2, was intended to simulate the ZVI-B series of samples, E5, and succeeded in the goal of TcO4− reduction efficiency improvement, hence, revealing the role of silica in the ZVI-B material, where the presence of silica “impurities” should be considered as an “enhancing agent”.
Characterization of the post-contact solid phase
Mössbauer analysis was carried out on all the series with ZVI-A and ZVI-B to compare the formation of different iron minerals after a 20 hour reaction time of iron materials with TcO4−. According to Fig. 4, all samples are dominated by Fe0 sextets, implying a significant amount of unreacted ZVI in the samples.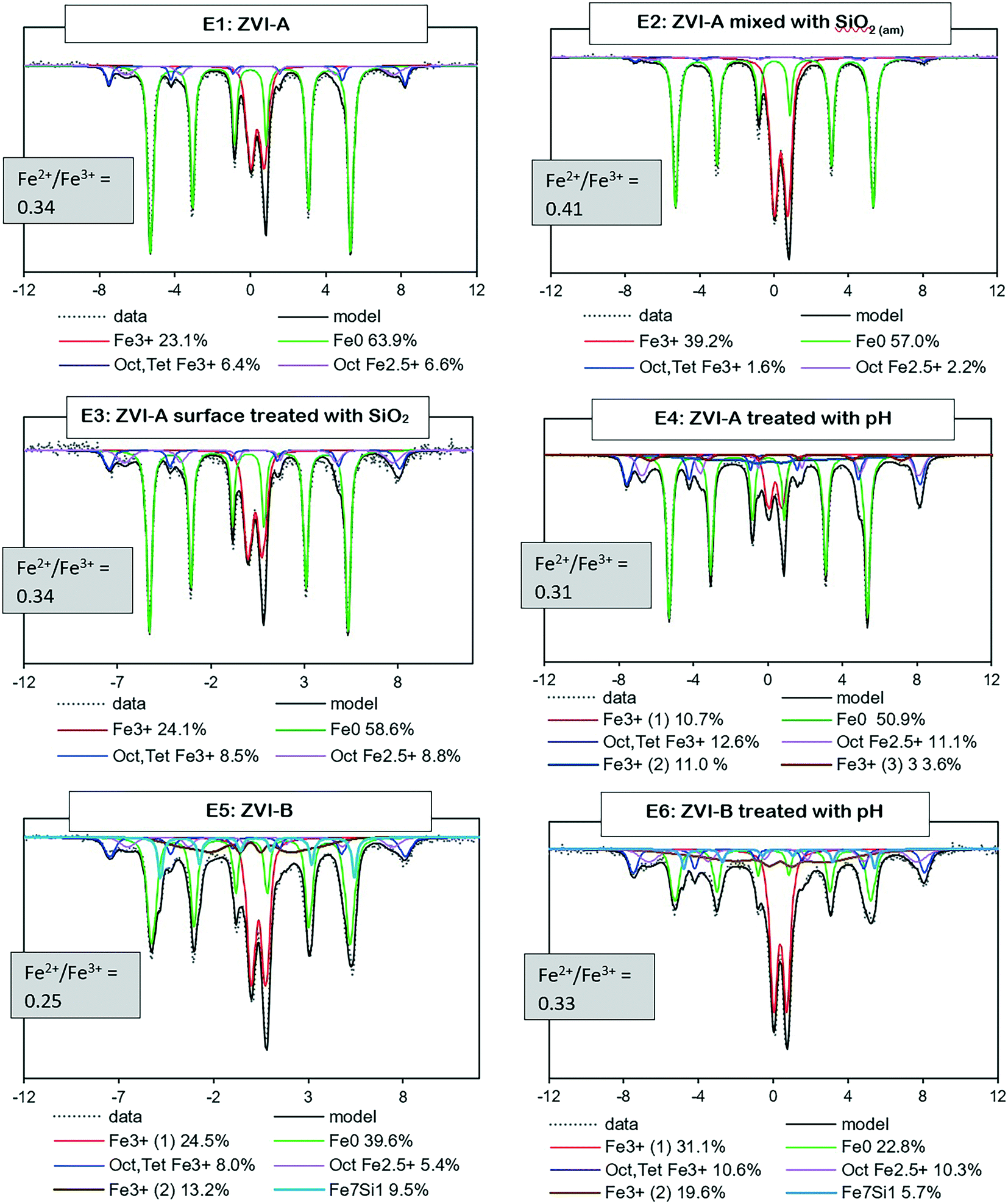 | ||
| Fig. 4 Room temperature Mössbauer spectra for the six series (E1–6) of two ZVI materials (ZVI-A and ZVI-B) and the Fe2+/Fe3+ ratio as evidence of partial oxidation of magnetite (<0.5). | ||
Two overlapping minor sextets of octahedral–tetrahedral Fe3+ and octahedral Fe2.5+, which belong to non-stoichiometric magnetite, and/or a magnetite and maghemite mixture, are present in all series as well. Magnetite, which is composed of both Fe3+ and Fe2+ atoms, would oxidize to maghemite in an aerobic environment.42 The Fe2+/Fe3+ ratio43 serves as evidence of magnetite partial oxidation, ranging from 0.5 for pure magnetite to 0 for pure maghemite. The ratio estimates are given for each sample in Fig. 4 and are indicative of partially oxidized magnetite in all series. Even though a magnetite and maghemite mixture can also be characterized by an Fe2+/Fe3+ ratio <0.5, it is impossible to differentiate a magnetite and maghemite mixture from partially oxidized (non-stoichiometric) magnetite via Mössbauer analysis.
Further, a doublet of Fe3+ with a quadrupole splitting of, averaged for six samples, ∼0.5 mm s−1 is distinguished in all the collected spectra. It can be a composite of multiple phases including ferrihydrite, lepidocrocite, and nano-sized goethite, magnetite or maghemite.8,44–47 Considering the initial conditions of pH 10.5 ± 0.04, an ambient atmosphere, 0.08 M NaCl, and a relatively short reaction time (20 hours), the iron transformation pathway would likely be directed towards ferrihydrite formation with further transformation to magnetite during adsorption of Fe2+ in alkaline solutions, eqn (3) and (4),32 and to goethite and lepidocrocite during dissolution–precipitation and crystallization of ferrihydrite or iron hydroxide. However, lepidocrocite formation is known to be suppressed in the presence of silica, but the ferrihydrite and goethite couple is often found in natural environments with a relatively high Si content in water, e.g., bog iron ores, lake ores, and placic horizons.1 Green rust is another theoretically possible product of ferrihydrite transformation, but it forms in weakly acidic or weakly alkaline solutions, and microscopy analysis (absence of hexagonal plates) and Mössbauer data (no evidence of Fe2+ signal) do not support its presence in these samples.
There are known effects of common natural environment impurities, Si and Al, on iron mineral transformation; however the mechanisms are different.2,4,31,33,48 Aluminum impedes transformation of ferrihydrite by decreasing Fe2+ retention on ferrihydrite.48 Binding of silica monomers or oligomers to the ferrihydrite crystal growth sites inhibits its further transformation by blocking crystal growth sites and stabilizing the disordered structure.2,31,33 However, the inhibition depends on the molar ratio of Si to Fe, and the transformation of ferrihydrite still proceeds at a ratio of approximately 3%, with almost complete inhibition at a ratio of 5.8% and higher.2 In the present study, at ratios of 2–4.7%, transformation of ferrihydrite is slowed down but not fully suppressed.
It is important to note that a clear Mössbauer signal related to Fe0 with structural silicon is identified in the ZVI-B samples (E5 and E6), Fig. 4. This signal is supported via analysis of the pristine ZVI-B materials (Fig. S3a†), where signals from the Fe environment containing from 1 to 3 Si atoms are identified. Such Fe–Si alloys were studied in detail by Overman et al.,29 whose results, in relation to our ZVI-B material, suggest approximately 5 wt% silicon in iron.
All the silica containing samples (E2, E3, E5, and E6) exhibited more efficient TcO4− reduction (Fig. 3), and most of them are characterized by their higher Fe3+ mineral content than the no-silica containing samples (predominantly, ferrihydrite, as discussed in the following section). This can be related to the effect of the high surface area of ferrihydrite (>200 m2 g−1,31 or 200–800 m2 g−1), and, more importantly, to higher concentrations of sorbed Fe2+, which can be more than three times that observed for ferrihydrite compared to that on goethite with a similar surface area.49 This can also be explained by the retardation effect of silica on ferrihydrite transformation, i.e. complexation on the surface of ferrihydrite and with dissolved Fe2+, as discussed earlier, which impedes its crystallization. Thus, ferrihydrite serves as an amorphous medium with a highly developed surface for more efficient heterogeneous Tc reduction, i.e., the Fe0–Tc7+ redox process that occurs not as extensively and rapidly in the aqueous phase as that on the surface of the in situ formed iron minerals.8
Microscopic evidence of the unique morphology
Scanning electron microscopy and transmission electron microscopy (SEM and TEM) together with energy dispersive X-ray analysis (EDX) were employed to reveal the morphological nature of the post-contact samples of series E1–E6 (Fig. 5 and SI-5a†). Surprisingly, orbicular formations with layered or concentric structures can be discerned in all the series, especially in the series E1, E2, and E5, which were not pre-treated with low pH solutions; hence, the iron oxidation steps differed. The layered structure was also observed in the series not yet exposed to the Tc solution (Fig. SI-5b†). | ||
| Fig. 5 SEM images of the experimental series (E1–E6) after being in contact with 0.03 mM TcO4− in 0.08 M NaCl solution. The red shape on the central image in series E5 shows the area selected for FIB extraction. | ||
Close-up micrographs of each post-contact series are represented by the right image in the triads, where rather amorphous and round formations suggest iron hydroxide and ferrihydrite in all the series, and needle-like crystals suggestive of goethite are noticeable in series E4 (acid treated ZVI-A without silica). The layered structure of oxidized iron is clearly seen in the right image of the series E1 (ZVI-A), and EDX analysis was carried out aiming to compare the composition of each of the layers (Table SI-1, Fig. SI-6†). As presented in Table SI-1,† the iron content in locations 2, 4, and 6 is slightly higher than that in locations 1, 3, 5, and 7. This provides evidence of different densities of iron oxide or oxyhydroxide in the layers, or periodicity in layering of different species of iron hydrolysis. Liesegang ring patterns formed during iron oxide growth on carbon steel coupons were also reported by Do et al.,50 coupling this phenomenon to Ostwald ripening of oxides (supersaturation theory), which is one of the theoretical approaches among others including adsorption, sol coagulation, diffusion wave, and phase separation theories.51,52 More studies are warranted to investigate this process.
TEM and EDX analyses were conducted on the series E5 sample (ZVI-B). The area selected for FIB extraction is shown on the center image in the E5 triad (Fig. 5). The lamella presents the layered structure previously observed with SEM. TEM analysis revealed that the layered structure was composed of metallic iron (Fig. 6a) along with 2-line ferrihydrite as a matrix embedding possibly goethite and/or lepidocrocite “wavy” structures, identified via selected area electron diffraction (SAED) patterns (Fig. 6b). EDX maps show concentrated silica along the edges of metallic iron, which might imply the bonding of silica polymers with the newly formed products of iron oxidation on the surface of the metal. Environmental levels of TcO4− used in this study (0.03 mM) did not allow for Tc identification via EDX analysis.
 | ||
| Fig. 6 TEM images and EDX analyses of the FIB extraction of the E5 sample (FIB area is shown in Fig. 6 on the central image belonging to E5). (a) Selected area electron diffraction (SAED) pattern showing iron BCC structure, and (b) ferrihydrite with lepidocrocite or goethite. (c) TEM image of a sample and EDX maps of Fe, Si, and O. | ||
Relation to geological phenomena
The layered morphology of iron oxyhydroxides, discovered via SEM analysis, is analogous to phenomena known in geology such as orbicular rocks and Liesegang rings. Orbicular rocks are formed due to rhythmical layering, resulting in concentric shells of different textures and compositions, and are common throughout the world without relation to a specific geological setting or chemical environment.53 They are often compared to Liesegang rings, which are formed in gelatinous or porous substrates of reaction–diffusion systems. There is no single theory agreed upon to explain the process governing all the varieties of such phenomena;51,54 however, an explanation given in the review by L'Heureux proposes an interaction between the processes of diffusion of dissolved species and kinetics of formed precipitates.55A mathematical model developed by H. Meinhardt56 successfully replicates the elementary steps of the process of biological pattern formation, and is based on autocatalytic activator–inhibitor or, in our case, activator–substrate interactions, expressed as:
 | (8) |
 | (9) |
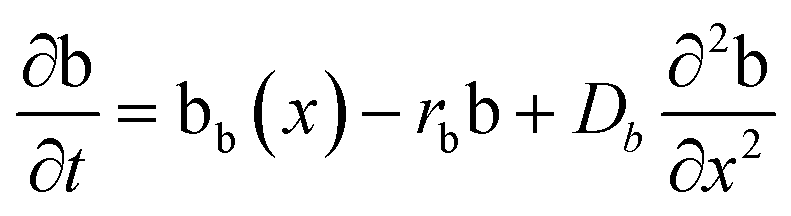 | (10) |
Formation of a layer of iron hydroxide/oxyhydroxide leads to depletion of dissolved iron and inhibition of the layer growth, which reoccurs once there is enough substrate, i.e. time-dependent supply of the dissolved iron. A code based on this model56 simulates a rich variety of patterns, found in nature, throughout different display modes and controllable parameters from eqn (8)–(10). Fig. 7 presents the modelled pattern and its comparison to the dominant morphologies observed in this study. Overall, our study reveals the phenomena of periodicity and rhythmical layering observed at the micro/nano-scale during iron dissolution and re-precipitation, similar to the geological structures occurring at the macro scale (orbicular rocks and Liesegang rings).
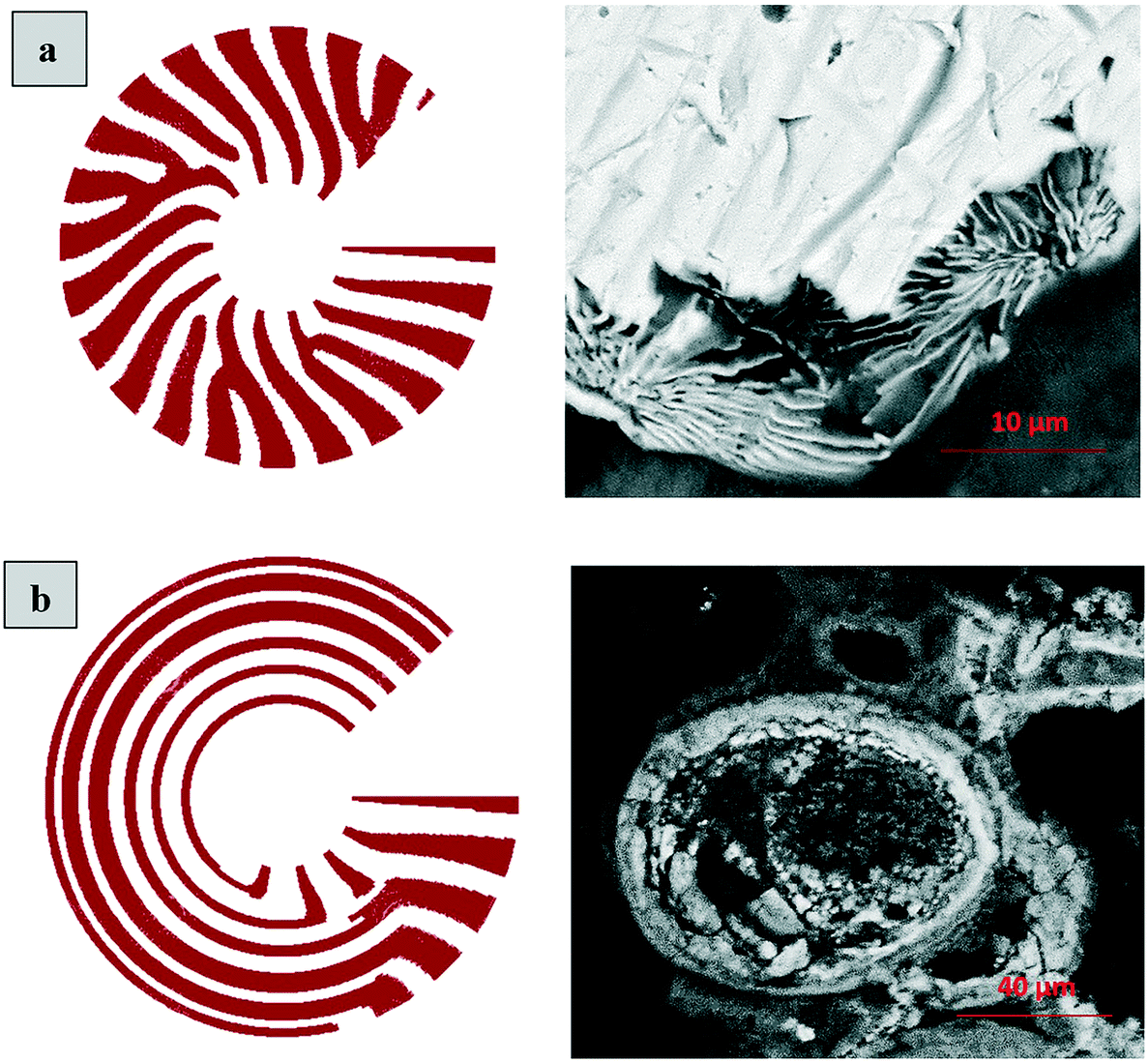 | ||
| Fig. 7 Iron hydroxide rings simulated and observed. (a) Simulation of the layered structure observed in sample E5; (b) simulation of concentric or orbicular structure observed in sample E2. Changes in the simulation model are due to different diffusion coefficients, (a): Da = 0.06 and Db = 0.15; (b): Da = 0.002 and Db = 0.35, adjusted empirically. | ||
Conclusions
Oxidation of metallic iron and concurrent in situ transformation of iron oxides and oxyhydroxides are complex processes that can be altered by the presence of other dissolved or incorporated species, i.e. silica monomers or polymers. The mechanisms of these processes have relevance to both natural environments, where concomitant dissolution of iron minerals and silica is common in aqueous systems, i.e. groundwater, and anthropogenically impacted environments with introduced contaminants, i.e. TcO4− which highly mobile in subsurfaces. As shown in this study, the effect of silica on TcO4− reduction is indirect, via impeded transformation of iron oxyhydroxides (mainly ferrihydrite) into non-stoichiometric magnetite, which in turn provides a highly developed surface of ferrihydrite for heterogeneous reduction of TcO4−. Here, heterogeneity implies that the redox reaction occurs on the surface of a solid phase, and the co-existence of both dissolved iron (reductant) and pertechnetate (oxidant) in the aqueous phase is not sufficient for the effective process of electron exchange. Furthermore, the complexity of iron dissolution and re-precipitation manifests itself through the rhythmical layering that is initiated and observable on a micro/nano-scale and is related to the natural phenomena of orbicular rocks and Liesegang rings commonly found among geological structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
It is our pleasure to acknowledge the help from T. H. Beasley (FIU FCAEM) and PNNL researchers C. T. Resch, N. L. D'Annunzio, S. Chatterjee, G. B. Hall, S. I. Sinkov, and C. H. Delegard. This research was supported by the U.S. Department of Energy's Office of Environmental Management and performed as part of the Technetium Management Hanford Site project at the Pacific Northwest National Laboratory (PNNL) operated by Battelle for the U.S. Department of Energy under Contract No. DE-AC05-76RL01830. This work was in part supported by the Department of Energy Minority Serving Institution Partnership Program (MSIPP) managed by the Savannah River National Laboratory under SRNS contract DE-AC09-08SR22470. Part of this research was performed at EMSL, a national scientific user facility at PNNL managed by the Department of Energy's Office of Biological and Environmental Research. Postdoctoral appointment of DB at PNNL is gratefully acknowledged.References
- U. Schwertmann, The effect of pedogenic environments on iron oxide minerals, Advances in Soil Science, Springer, New York, NY, 1985, vol. 1, pp. 171–200 Search PubMed.
- X. Wang, M. Zhu, S. Lan, M. Ginder-Vogel, F. Liu and X. Feng, Formation and secondary mineralization of ferrihydrite in the presence of silicate and Mn(II), Chem. Geol., 2015, 415, 37–46 CrossRef CAS.
- R. M. Cornell, R. Giovanoli and P. W. Schindler, Effect of silicate species on the transformation of ferrihydrite into goethite and hematite in alkaline media, Clays Clay Miner., 1987, 35, 21–28 CrossRef CAS.
- U. Schwertmann, Occurrence and formation of iron oxides in various pedoenvironments, in, Iron in Soils and Clay Minerals, NATO ASI Series (Series C: Mathematical and Physical Sciences), ed. J. W. Stucki, B. A. Goodman and U. Schwertmann, Springer, Dordrecht, 1988, vol. 217 Search PubMed.
- G. A. Waychunas, B. A. Rea, C. C. Fuller and J. A. Davis, Surface chemistry of ferrihydrite: Part 1. EXAFS studies of the geometry of coprecipitated and adsorbed arsenate, Geochim. Cosmochim. Acta, 1993, 57, 2251–2269 CrossRef CAS.
- A. C. Scheinost, S. Abend, K. I. Pandya and D. L. Sparks, Kinetic controls on Cu and Pb sorption by ferrihydrite, Environ. Sci. Technol., 2001, 35, 1090–1096 CrossRef CAS.
- L. Tian, Z. Shi, Y. Lu, A. C. Dohnalkova, Z. Lin and Z. Dang, Kinetics of cation and oxyanion adsorption and desorption on ferrihydrite: Roles of ferrihydrite binding sites and a unified model, Environ. Sci. Technol., 2017, 51, 10605–10614 CrossRef CAS.
- J. M. Zachara, S. M. Heald, B.-H. Jeon, R. K. Kukkadapu, C. Liu, J. P. McKinley, A. C. Dohnalkova and D. A. Moore, Reduction of pertechnetate [Tc(VII)] by aqueous Fe(II) and the nature of solid phase redox products, Geochim. Cosmochim. Acta, 2007, 71, 2137–2157 CrossRef CAS.
- Y. Zou, X. Wang, A. Khan, P. Wang, Y. Liu, A. Alsaedi, T. Hayat and X. Wang, Environmental remediation and application of nanoscale zero-valent iron and its composites for the removal of heavy metal ions: A review, Environ. Sci. Technol., 2016, 50, 7290–7304 CrossRef CAS.
- B. Gu, L. Liang, M. J. Dickey, X. Yin and S. Dai, Reductive precipitation of uranium(VI) by zero-valent iron, Environ. Sci. Technol., 1998, 32, 3366–3373 CrossRef CAS.
- F. Fu, D. D. Dionysiou and H. Liu, The use of zero-valent iron for groundwater remediation and wastewater treatment: A review, J. Hazard. Mater., 2014, 267, 194–205 CrossRef CAS.
- J. A. de Lima Perini and R. F. P. Nogueira, Effect of particle size, iron ligands and anions on ciprofloxacin degradation in zero-valent iron process: Application to sewage treatment plant effluent, J. Chem. Technol. Biotechnol., 2017, 92, 2300–2308 CrossRef.
- A. B. Cundy, L. Hopkinson and R. L. D. Whitby, Use of iron-based technologies in contaminated land and groundwater remediation: A review, Sci. Total Environ., 2008, 400, 42–51 CrossRef CAS.
- D. Boglaienko, H. P. Emerson, Y. Katsenovich and T. G. Levitskaia, Comparative analysis of ZVI materials for reductive separation of 99Tc(VII) from aqueous waste streams, J. Hazard. Mater., 2019, 380, 120836 CrossRef CAS.
- D. Boglaienko, J. A. Soltis, R. K. Kukkadapu, Y. Du, L. E. Sweet, V. E. Holfeltz, G. B. Hall, E. C. Buck, C. U. Segre, H. P. Emerson and Y. Katsenovich, Spontaneous redox continuum reveals sequestered technetium clusters and retarded mineral transformation of iron, Commun. Chem., 2020, 3(1), 1–11 CrossRef.
- M. Jang, B. Park, H. Lee, T.-Y. Kim and Y. Kim, Removal of hexavalent chromium ion from aqueous solution using nanoscale zero-valent iron particles immobilized on porous silica support prepared by polymer template method, Korean J. Chem. Eng., 2018, 35, 2015–2023 CrossRef CAS.
- D. Shi, G. Zhu, X. Zhang, M. Cheng, T. Wu, K. Zhang and J. Fan, Decorating of ultra small and recyclable nanoscale zero-valent iron on NH2-SiO2 for enhanced high-performance removal of water pollutants, J. Alloys Compd., 2019, 782, 183–192 CrossRef CAS.
- N. Shukla, V. Gupta, A. S. Rawat, V. K. Gahlot, S. Shrivastava and P. K. Rai, 2, 4-Dinitrotoluene (DNT) and 2, 4, 6-Trinitrotoluene (TNT) removal kinetics and degradation mechanism using zero valent iron-silica nanocomposite, J. Environ. Chem. Eng., 2018, 6, 5196–5203 CrossRef CAS.
- S. Chen, J. Bedia, H. Li, L. Y. Ren, F. Naluswata and C. Belver, Nanoscale zero-valent iron@mesoporous hydrated silica core-shell particles with enhanced dispersibility, transportability and degradation of chlorinated aliphatic hydrocarbons, Chem. Eng. J., 2018, 343, 619–628 CrossRef CAS.
- L. Honetschlägerová, P. Janouškovcová, M. Velimirovic, M. Kubal and L. Bastiæns, Using silica coated nanoscale zerovalent particles for the reduction of chlorinated ethylenes, Silicon, 2018, 10, 2593–2601 CrossRef.
- H. Lu, J. Dong, M. Zhang, W. Hu, C. Wen, C. Yang and Y. Wu, SiO2-coated zero-valent iron nanocomposites for aqueous nitrobenzene reduction in groundwater: Performance, reduction mechanism and the effects of hydrogeochemical constituents, Colloids Surf., A, 2018, 558, 271–279 CrossRef CAS.
- J. G. Darab, A. B. Amonette, D. S. D. Burke and R. D. Orr, Removal of pertechnetate from simulated nuclear waste streams using supported zerovalent iron, Chem. Mater., 2007, 19, 5703–5713 CrossRef CAS.
- K. Schwochau, Technetium, Wiley-VCH, 2000 Search PubMed.
- T. A. Marshall, K. Morris, G. T. W. Law, J. F. W. Mosselmans, P. Bots, S. A. Parry and S. Shaw, Incorporation and retention of 99-Tc(IV) in magnetite under high pH conditions, Environ. Sci. Technol., 2014, 48, 11853–11862 CrossRef CAS.
- S. A. Saslow, W. Um, C. I. Pearce, M. H. Engelhard, M. E. Bowden, W. Lukens, I. I. Leavy, B. J. Riley, D.-S. Kim, M. J. Schweiger and A. A. Kruger, Reduction and simultaneous removal of 99Tc and Cr by Fe(OH)2(s) mineral transformation, Environ. Sci. Technol., 2017, 51, 8635–8642 CrossRef CAS.
- E. Yalçintaş, A. C. Scheinost, X. Gaona and M. Altmaier, Systematic XAS study on the reduction and uptake of Tc by magnetite and mackinawite, Dalton Trans., 2016, 45, 17874–17885 RSC.
- W. W. Lukens and S. A. Saslow, Facile incorporation of technetium into magnetite, magnesioferrite, and hematite by formation of ferrous nitrate in situ: precursors to iron oxide nuclear waste forms, Dalton Trans., 2018, 47, 10229–10239 RSC.
- W. Um, H.-S. Chang, J. P. Icenhower, W. W. Lukens, R. J. Serne, N. P. Qafoku, J. H. Jr. Westsik, E. C. Buck and S. C. Smith, Immobilization of 99-Technetium (VII) by Fe(II)-Goethite and limited reoxidation, Environ. Sci. Technol., 2011, 45, 4904–4913 CrossRef CAS.
- N. R. Overman, X. Jiang, R. K. Kukkadapu, T. Clark, T. J. Roosendaal, G. Coffey, J. E. Shield and S. N. Mathaudhu, Physical and electrical properties of melt-spun Fe-Si (3–8 wt.%) soft magnetic ribbons, Mater. Charact., 2018, 136, 212–220 CrossRef CAS.
- X. Zhao, W. Liu, Z. Cai, B. Han, T. Qian and D. Zhao, An overview of preparation and applications of stabilized zero-valent iron nanoparticles for soil and groundwater remediation, Water Res., 2016, 100, 245–266 CrossRef CAS.
- J. Zhao, F. E. Huggins, Z. Feng and G. P. Huffman, Ferrihydrite: Surface structure and its effects on phase transformation, Clays Clay Miner., 1994, 42, 737–746 CrossRef CAS.
- Y. Tamaura, K. Ito and T. Katsura, Transformation of γ-FeO(OH) to Fe3O4 by adsorption of iron(II) ion on γ-FeO(OH), J. Chem. Soc., Dalton Trans., 1983, 189–194 RSC.
- P. J. Swedlund and J. G. Webster, Adsorption and polymerization of silicic acid on ferrihydrite, and its effect on arsenic adsorption, Water Res., 1999, 33, 3413–3422 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, The iron oxides: structure, reactions, occurrences and uses, Wiley-VCH, 2nd edn, 2003 Search PubMed.
- I. M. Ali, E. S. Zakaria, M. M. Ibrahim and I. M. El-Naggar, Synthesis, structure, dehydration transformations and ion exchange characteristics of iron-silicate with various Si and Fe contents as mixed oxides, Polyhedron, 2008, 27, 429–439 CrossRef CAS.
- R. K. Iler and R. Iler, The chemistry of silica: solubility, polymerization, colloid and surface properties, and biochemistry, J. Wiley & Sons, NY, 1979 Search PubMed.
- C. C. Davis, H.-W. Chen and M. Edwards, Modeling silica sorption to iron hydroxide, Environ. Sci. Technol., 2002, 36, 582–587 CrossRef CAS.
- X. Wang, J. D. Kubicki, J.-F. Boily, G. A. Waychunas, Y. Hu, X. Feng and M. Zhu, Binding geometries of silicate species on ferrihydrite surfaces, ACS Earth Space Chem., 2018, 2, 125–134 CrossRef CAS.
- A. C. Makrides, M. Turner and J. Slaughter, Condensation of silica from supersaturated silicic acid solutions, J. Colloid Interface Sci., 1980, 73, 345–367 CrossRef CAS.
- X. Gao, R. A. Root, J. Farrell, W. Ela and J. Chorover, Effect of silicic acid on arsenate and arsenite retention mechanisms on 6-L ferrihydrite: A spectroscopic and batch adsorption approach, Appl. Geochem., 2013, 38, 110–120 CrossRef CAS.
- X. Yang, P. Roonasi, R. Jolsterå and A. Holmgren, Kinetics of silicate sorption on magnetite and maghemite: An in situ ATR-FTIR study, Colloids Surf., A, 2009, 343(1–3), 24–29 CrossRef CAS.
- T. W. Swaddle and P. Oltmann, Kinetics of the magnetite–maghemite–hematite transformation, with special reference to hydrothermal systems, Can. J. Chem., 1980, 58(17), 1763–1772 CrossRef CAS.
- C. A. Gorski and M. M. Scherer, Determination of nanoparticulate magnetite stoichiometry by Mössbauer spectroscopy, acidic dissolution, and powder X-ray diffraction: A critical review, Am. Mineral., 2010, 95, 1017–1026 CrossRef CAS.
- P. Larese-Casanova, A. Kappler and S. B. Haderlein, Heterogeneous oxidation of Fe(II) on iron oxides in aqueous systems: Identification and controls of Fe(III) product formation, Geochim. Cosmochim. Acta, 2012, 91, 171–186 CrossRef CAS.
- E. Brok, C. Frandsen, D. E. Madsen, H. Jacobsen, J. O. Birk, K. Lefmann, J. Bendix, K. S. Pedersen, C. B. Boothroyd, A. A. Berhe, G. G. Simeoni and S. Mørup, Magnetic properties of ultra-small goethite nanoparticles, J. Phys. D: Appl. Phys., 2014, 47, 365003 CrossRef.
- G. F. Goya, T. S. Berquó and F. C. Fonseca, Static and dynamic magnetic properties of spherical magnetite nanoparticles, J. Appl. Phys., 2003, 94, 3520–3528 CrossRef CAS.
- G. M. da Costa, E. De Grave and R. E. Vandenberghe, Mössbauer studies of magnetite and Al-substituted maghemites, Hyperfine Interact., 1998, 117, 207–243 CrossRef CAS.
- Y. Masue-Slowey, R. H. Loeppert and S. Fendorf, Alteration of ferrihydrite reductive dissolution and transformation by adsorbed As and structural Al: Implications for As retention, Geochim. Cosmochim. Acta, 2011, 75(3), 870–886 CrossRef CAS.
- A. G. B. Williams and M. M. Scherer, Spectroscopic evidence for Fe(II)-Fe(III) electron transfer at the iron oxide-water interface, Environ. Sci. Technol., 2004, 38, 4782–4790 CrossRef CAS.
- T. V. Do, J. M. Joseph, G. Whitaker, B. Noye, V. Bostan, D. Jarron and J. C. Wren, Methodology for Volume Reduction of Radioactive Metallic Waste, WM 2019 Symposia, March 3–7, Phoenix, AZ, 2019.
- P. Hantz, Pattern formation in a new class of precipitation reactions, PhD diss., Université de Genève, 2006 Search PubMed.
- K. H. Stern, The Liesegang Phenomenon, Chem. Rev., 1953, 54(1), 79–99 CrossRef.
- D. J. Leveson, Orbicular rocks: A Review, Geol. Soc. Am. Bull., 1966, 77, 409–426 CrossRef CAS.
- J. George and G. Varghese, Intermediate colloidal formation and the varying width of periodic precipitation bands in reaction-diffusion systems, J. Colloid Interface Sci., 2005, 282, 397–402 CrossRef CAS.
- I. L'Heureux, Self-organized rhythmic patterns in geochemical systems, Philos. Trans. R. Soc., A, 2013, 371(2004), 20120356 CrossRef.
- H. Meinhardt, The algorithmic beauty of sea shells, Springer, 4th edn, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0en00897d |
| This journal is © The Royal Society of Chemistry 2021 |