
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceToxicity and chemical transformation of silver nanoparticles in A549 lung cells: dose-rate-dependent genotoxic impact†
Laure
Bobyk
a,
Adeline
Tarantini
a,
David
Beal
a,
Giulia
Veronesi
 b,
Isabelle
Kieffer
c,
Sylvie
Motellier
d,
Eugenia
Valsami-Jones
e,
Iseult
Lynch
e,
Pierre-Henri
Jouneau
f,
Karin
Pernet-Gallay
g,
Catherine
Aude-Garcia
b,
Sylvie
Sauvaigo
h,
Thierry
Douki
a,
Thierry
Rabilloud
b and
Marie
Carriere
*a
b,
Isabelle
Kieffer
c,
Sylvie
Motellier
d,
Eugenia
Valsami-Jones
e,
Iseult
Lynch
e,
Pierre-Henri
Jouneau
f,
Karin
Pernet-Gallay
g,
Catherine
Aude-Garcia
b,
Sylvie
Sauvaigo
h,
Thierry
Douki
a,
Thierry
Rabilloud
b and
Marie
Carriere
*a
aUniv. Grenoble Alpes, CEA, CNRS, IRIG, SyMMES, Chimie Interface Biologie pour l'Environnement, la Santé et la Toxicologie (CIBEST), F-38000 Grenoble, France. E-mail: marie.carriere@cea.fr; Tel: +33438780328
bChemistry and Biology of Metals, Univ. Grenoble Alpes, CNRS UMR5249, CEA, IRIG/DIESE/CBM, F-38054 Grenoble, France
cBM30B/FAME beamline, ESRF, Observatoire des Sciences de l'Univers de Grenoble, 38000 38043, Grenoble, France
dUniv. Grenoble Alpes, CEA/DRT/LITEN, Laboratoire Mesures, Sécurisation et Environnement, Grenoble, France
eSchool of Geography Earth and Environmental Sciences, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK
fUniv. Grenoble Alpes, CEA, CNRS, IRIG, MEM, F-38000 Grenoble, France
gUniv. Grenoble Alpes, Inserm, U1216, CHU Grenoble Alpes, CEA, Grenoble Institut Neurosciences, Grenoble, France
hLXRepair, BIOPOLIS, 5 avenue du grand sablon, 38700 La Tronche, France
First published on 26th February 2021
Abstract
Silver nanoparticles (Ag-NPs) are widely used as biocides, leading to contamination of the environment and possible adverse effects on humans. Recent studies revealed that the cellular response to acute exposure to Ag-NPs differs from the response to chronic exposure, although we currently lack systematic studies comparing responses to different dosing regimens. In this study, A549 lung epithelial cells were exposed to 15 nm NM300K or 59 nm PVP-coated Ag-NPs under two different conditions. Under these two conditions, the cells received the same total sub-lethal concentration of Ag-NPs, but the dose was either administered over a 24 hour period (acute exposure) or split over four successive days (repeated exposure). These two types of Ag-NPs were chosen as PVP-coated particles were hypothesized to dissolve more slowly than NM300K particles. EXAFS measurements confirmed this hypothesis, showing more rapid oxidation of Ag0-NPs to AgI in cells exposed to NM300K. The intracellular Ag content was higher in cells exposed to NM300K, and higher in cells following acute exposure than cells exposed to repeated doses. Whatever the exposure scenario, AgI bound to thiol-containing intracellular proteins. Both exposure regimens altered cellular metabolism, caused intracellular ROS accumulation and blocked cell cycle progression. DNA damage was only observed following acute exposure, as strand breaks in cells exposed to NM300K and oxidized DNA bases in cells exposed to Ag-PVP. This damage was concomitant with decreased DNA repair activities. Together, these results show that acute exposure of A549 cells to Ag-NPs induces stronger effects on DNA integrity than repeated exposure. Nevertheless, repeated exposure to a low concentration of Ag-NPs profoundly altered the cell's metabolism and blocked cell cycle progression, confirming that both exposure regimens have detrimental effects.
Environmental significanceSilver nanoparticles (Ag-NPs) are widely used for their biocidal properties in everyday products and healthcare units, which leads to environmental contamination and human exposure. The toxicity of Ag-NPs derives from their oxidative degradation to Ag(I) species. While most previously published toxicity studies report their toxicological impact upon acute exposure, we provide evidence that the exposure dose rate is a key determinant of their toxicity by comparing the impact of acute vs. repeated exposure scenarios. We show that it influences NP dissolution kinetics, impact on cell metabolism, ROS accumulation, DNA damage and cell cycle. Moreover, the size and surface coating of Ag-NPs influence their intracellular fate and distribution, possibly explaining their different toxicological impact. |
Introduction
Silver nanoparticles (Ag-NPs) are widely used for their antibacterial properties in everyday products, including textiles, electronics, paints, sanitization products, food packaging and personal care products.1,2 This widespread use has led to extensive release of these particles into the environment. Consequently, humans may be exposed to Ag-NPs via inhalation both in their everyday life and in occupational settings. For example, Ag-NPs can be released into the ambient air when handling textiles into which Ag-NPs have been integrated, or Ag-NPs can be inhaled directly when breathing through face masks if they are impregnated with Ag-NPs as a biocidal agent. The toxic potential of Ag-NPs has been assessed in a wide range of in vitro models.3–7 Acute exposure to Ag-NPs leads to apoptotic and/or necrotic cell death, characterized by morphological changes, cell membrane permeabilization and cell growth arrest.6 The mechanism behind these effects is mainly linked to the accumulation of reactive oxygen species (ROS),4 which first triggers a protective response from the cell, eventually leading to lipid peroxidation, oxidatively generated DNA lesions, gene mutations, mitochondrial swelling, lysosomal leakage, and blocking of the autophagic flux.6,8–13 Chronic exposure to Ag-NPs, i.e. exposure to a low concentration over long periods of time, triggers cellular responses that differ from those observed with acute exposure.14,15 In primary murine macrophages, chronic exposure to Ag-NPs triggers inflammatory and thiol responses, causes mitochondrial damage, reduces phagocytic capacity, and increases nitric oxide production upon lipopolysaccharide stimulation. Similar, but less intense, responses are observed in cells exposed repeatedly to NPs.14 In HaCat keratinocytes, chronic exposure to Ag-NPs leads to an amplified, sustained stress reaction compared to that caused by acute exposure.15 Moreover, BEAS-2B bronchial epithelial cells chronically exposed to Ag-NPs for 6 weeks or 4 months acquire tumorigenic cellular characteristics, with anti-apoptotic responses, anoikis resistance, and the capacity to migrate and invade.16,17The chemical and physicochemical transformation of Ag-NPs, including dissolution, agglomeration and surface modification, is known to play a key role in their toxicity. The hypothesis of intracellular release of Ag ions (AgI) from Ag-NPs and of a Trojan horse-type toxicity mechanism has been widely discussed.7,18 In mammalian cells, following internalization, oxidation transforms Ag0-NPs into AgI, which interacts with cellular ligands containing thiol functional groups.13,18–20 Due to their abundance in the cell, the most likely ligands are glutathione (GSH), cysteine-containing and thiolated proteins including metallothioneins and Zn-finger proteins.21 Depending on the cell type, AgI binds either to glutathione, e.g. in macrophages,22 or to both glutathione and thiolated proteins, e.g. in Hep-G2 hepatocytes.19 This binding coincides with the upregulation of genes encoding metallothioneins.13 These molecules maintain redox homeostasis, and their complexation may reduce the availability of their free thiols, therefore hampering their functions. In THP-1 monocytes, Wang et al.13 described the gradual chemical transformation of Ag0-NPs, first into AgI-O and subsequently into AgI-S species. In microglial cells and astrocytes, the Hsiao team18 concluded that intracellular dissolution of uncoated Ag0-NPs was promoted by H2O2 and subsequent formation of AgI–cysteine complexes.
The aim of the present study was to compare the physicochemical transformation and impact of Ag-NPs following either acute or repeated exposure. In both scenarios the total exposure concentration was the same. Particular attention was paid to DNA damage incurred. For these experiments, A549 cells were exposed to two distinct Ag-NPs, varying in both their size and surface coating. The selected NPs were a PVP-coated Ag-NP that was previously used in macrophage studies from our team14,22 and NM300K Ag-NP provided by the EU Joint Research Center (JRC, Ispra, Italy). As PVP-coated Ag-NPs are known to dissolve more slowly than other Ag-NPs,19,23,24 we hypothesized that the two types of Ag-NPs would undergo chemical transformation at different rates and with distinct kinetics within cells. Acute exposure consisted of application of a high concentration of Ag-NPs in a single dose (25 μg mL−1) for 24 h or 48 h. Repeated exposure entailed application of one-fourth of this high concentration of NPs every day for four days (6.25 μg mL−1 of Ag-NPs per day; cumulative dose: 25 μg mL−1). Intracellular transformation of Ag0 was monitored by X-ray absorption spectroscopy (XAS), and transformation products were identified. Combined with ICP-MS quantification, these analyses allowed us to determine the Ag intracellular content and intracellular proportion of Ag0-NP and AgI transformation products over time. Finally, the impact of Ag-NPs on cells, including cytotoxicity, oxidative stress, genotoxicity, and effects on the cell cycle and DNA repair systems were assessed.
Materials and methods
Chemicals and reagents
Unless otherwise indicated, chemicals and reagents were >98% pure and were from Sigma-Aldrich. Ag-NPs were obtained either from the JRC NM repository (NM300K) or from Sigma-Aldrich. NM300K (<20 nm) is provided as a liquid suspension (100 mg mL−1) in 4% polyoxyethylene glycerol trioleate, 4% sorbitan monolaurate (Tween) and 7% ammonium nitrate. Ag-PVP (Sigma-Aldrich, reference 758329, <100 nm) is provided as a liquid suspension (50 mg mL−1) stabilized at 5 wt% in ethylene glycol. Both NPs were aliquoted and stored in the dark under argon at 4 °C until use.Cell culture and exposure
A549 human epithelial lung alveolar carcinoma cells (CCL-185) were purchased from ATCC. This cell line was selected because the alveolar area is the most vulnerable region of the lungs to NP exposure, particularly epithelial alveolar type I cells that have been shown to accumulate NPs at intense levels.25 A549 is the only commercially available cell line representative of this region of the lung. These cells derive from alveolar type II cells, but nevertheless have specific characteristics of alveolar type I cells.26,27 Although their use in toxicity studies has been criticized, particularly because they constitutively express active NRF-2,28 these cells are widely used in nanoparticle toxicology studies. They possess wild-type active p53, and we previously showed that they display similar behavior to BEAS-2B cells in response to TiO2-NP, in terms of genotoxicity and oxidative stress.29 Cells were maintained at 37 °C in a 5% CO2 and 100% humidity incubator and grown in 4.5% glucose DMEM-GlutaMAX supplemented with 1% (V/V) non-essential amino acids, penicillin/streptomycin, and 10% (V/V) fetal bovine serum. Cells were sub-cultured twice a week following trypsin–EDTA treatment.For acute exposure assays, cells were seeded in multi-well plates. Plate type was determined by the assay to be performed: 96 well transparent (WST-1 assay), black (H2-DCF-DA assay) or black plates with a transparent bottom (53BP1 immunostaining and micronucleus assay); 12 well plates were used for the comet assay and ICP-MS and 6-well plates for RT-qPCR and DNA repair assays. 55 cm2 Petri dishes were used for 8-oxo-dGuo measurement and XAS. Cells were seeded at 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per cm2 (WST-1, H2-DCF-DA, 8-oxo-dGuo and comet assay), 15000 cells per cm2 (53BP1 immunostaining) or 9500 cells per cm2 (micronucleus assay) the day before exposure. Cells were exposed to the indicated concentrations of NPs for 24 h (and 48 h for the XAS experiment): 10, 25, 50, 100 and 200 μg mL−1 in cytotoxicity assays, 6.25, 12.5 and 25 μg mL−1 in H2-DCF-DA assays and 25 μg mL−1 for all other assays.
000 cells per cm2 (WST-1, H2-DCF-DA, 8-oxo-dGuo and comet assay), 15000 cells per cm2 (53BP1 immunostaining) or 9500 cells per cm2 (micronucleus assay) the day before exposure. Cells were exposed to the indicated concentrations of NPs for 24 h (and 48 h for the XAS experiment): 10, 25, 50, 100 and 200 μg mL−1 in cytotoxicity assays, 6.25, 12.5 and 25 μg mL−1 in H2-DCF-DA assays and 25 μg mL−1 for all other assays.
In the case of repeated exposure, cells were seeded at one-fourth the concentration to allow for cell division and population doubling approximately every 24 hours. Cells were exposed to repeated doses of NPs; the total exposure concentration was fractionated into four concentrations (X/4 μg mL−1), and NPs were applied to cells once a day for four successive days without removing the exposure medium from the previous day. The corresponding concentrations in μg cm−2 are reported in Table S1.† For example, when the acute exposure concentration was chosen to be 25 μg mL−1 Ag-NPs (and 10 μg mL−1 Ag-lactate), in the repeated exposure scheme cells were exposed to 6.25 μg mL−1 Ag-NPs (or 2.5 μg mL−1 Ag-lactate) on the first day, then 12.5 μg mL−1 Ag-NPs (or 5 μg mL−1 Ag-lactate) on the second day, 18.75 μg mL−1 Ag-NPs (or 7.5 μg mL−1 Ag-lactate) on the third day and 25 μg mL−1 Ag-NPs (or 10 μg mL−1 Ag-lactate) on the fourth day. As a consequence of this dose rate, in the repeated exposure regimen, on the last day, the exposure medium contained the same total quantity of Ag-NPs as the acute exposure medium, but the NPs had been applied progressively, i.e., at a lower dose rate. This regimen mimics daily, cumulative exposure to a low concentration of Ag-NPs. Cells were sampled after 24 h of exposure (acute exposure) or 24 h after the last exposure (repeated exposure), i.e., on day 5 (Fig. S1†).
Cytotoxicity assessment
To assess cytotoxicity, because of its low interference from NPs,30 the WST-1 assay (Roche) was used according to the supplier's protocol. Cells were seeded in 96 well plates (n = 5) and exposed to freshly prepared NP suspensions for 24 h. The exposure medium was then removed and replaced by 100 μL WST-1 solution diluted in phosphate-buffered saline (PBS) (1/10, V/V). Absorbance was measured at 450 nm and 650 nm to correct for background absorbance. Polystyrene-amine NPs (PS-NH2, 200 μg mL−1) were used as positive control;31 they completely inhibited cell metabolic activity and thus were considered to correspond to 100% cell mortality. Since the exposure medium containing NPs was removed before measuring absorbance, potential optical interference of NPs with the assay should be minimal. Interference was assessed using two 96 well plates seeded with A549 cells. The first one was exposed to 200 μg mL−1 PS-NH2 for 24 h, leading to 100% cell death (100% decrease in metabolic activity). The second one was not exposed to PS-NH2; therefore the metabolic activity of cells in this plate was maximal. In these conditions, after incubation of cells with WST-1, the absorbance at 450 nm was approximately 4 for unexposed cells (maximal metabolic activity), and 0.25 in cells exposed to 200 μg mL−1 PS-NH2 (minimal metabolic activity). These plates were then exposed to 3.125, 6.25, 12.5 and 25 μg mL−1 Ag-NPs for 24 h. The exposure medium was removed and replaced by PBS, therefore only the Ag-NPs which had settled down and attached onto cell membranes remained at the bottom of the wells. Absorbance at 450 nm was measured once again. In the plate with living cells (not exposed to PS-NH2), absorbance at 450 nm did not increase significantly compared to pre-NP-exposure (up to ∼4 absorbance units), indicating minimal optical interference from Ag-NPs. In the plate with dead cells (exposed to PS-NH2), absorbance at 450 nm increased by up to 0.67 units (Table S2†). This degree of optical interference of Ag-NPs with the WST-1 assay was considered negligible.Quantification of reactive oxygen species with H2-DCF-DA
Reactive oxygen species (ROS) were quantified using dichlorodihydrofluorescein diacetate (H2-DCF-DA) (Thermo Fisher Scientific). H2-DCF-DA is a cell permeant, non-fluorescent dye, which becomes fluorescent when its acetate groups are cleaved by intracellular esterases and oxidized by reaction with ROS. It mainly detects H2O2, ONOO−, and ROO˙. The cell culture medium was removed and 100 μL of 25 μM H2-DCF-DA prepared in PBS was added to each well. After 40 min at 37 °C under 5% CO2, the H2-DCF-DA was replaced by NP suspensions prepared in complete cell culture medium. Fluorescence (λexc/λem 480/530 nm) was recorded immediately after the addition of NPs and then 2 h, 4 h and 24 h later. tert-Butyl-hydroperoxide (1 mM, not shown) and KBrO3 (2 mM) were used as positive controls. Interference of Ag-NPs with this assay was assessed by measuring the fluorescence emitted by the two Ag-NPs at 6.25, 12.5 and 25 μg mL−1. Both NM300K and Ag-PVP partly quenched the fluorescence emitted by the cell culture medium (Table S2†), resulting in a reduction of 182000 and 167000 fluorescence units (f.u.) for NM300K and Ag-PVP, respectively. In positive controls, fluorescence measurement was 912000 f.u. at t0; it reached 6.68 × 106 f.u. after 24 h of incubation. Therefore the optical interference of Ag-NPs with the H2-DCF-DA assay was negligible.
Inductively coupled plasma mass spectrometry (ICP-MS)
Intracellular Ag content was quantified by ICP-MS. After exposure, cells were rinsed four times with PBS, and then harvested and suspended in 200 μL PBS. Protein concentration was measured using the Bradford assay. Samples for ICP-MS were dissolved in 32.5% ultrapure HNO3 overnight at room temperature under constant stirring. Samples were then diluted in ultrapure grade HNO3 (1% vol/vol) and analyzed on a 7700 Series ICP-MS (Agilent 7700) instrument operated in standard mode. Calibration curves were obtained from a certified ionic Ag solution (silver standard for ICP, Fluka). The concentration of 107Ag was determined.X-ray absorption spectroscopy (XAS)
After exposure to NPs, cells were rinsed three times with PBS before harvesting by trypsinization. Cell pellets were suspended in Ag-free culture medium containing 20% glycerol. Samples were deposited in the XAS sample holder and flash-frozen by immersion in liquid nitrogen. Measurements were performed at 15 K in a He cryostat, on the BM30B (FAME) beamline at ESRF (Grenoble, France).32 XAS spectra for samples and reference compounds were recorded at the Ag K-edge by scanning in the energy range 25.200–26.260 keV with a nitrogen-cooled Si(220) double-crystal monochromator.33 The incoming photon energy was calibrated against Ag metallic foil by defining the first inflection point of the spectrum at 25.514 keV. Spectra were recorded in fluorescence mode using a 30-element solid-state Ge detector (Canberra). Data were analyzed using the Athena and Artemis graphic packages running in the Ifeffit suite34 according to the previously described strategy.22 Briefly, as a first step the near-edge spectrum region was fitted as a linear combination of reference compounds to account for any Ag species encountered in cells. Then, for the conditions in which significant dissolution of Ag-NPs and formation of Ag–thiolate complexes was observed, the EXAFS oscillations were extracted and fitted as a linear combination of reference compounds representing the possible Ag–S binding geometries (digonal vs. trigonal). Finally, the EXAFS spectra were fitted using an ab initio method based on the two-component model developed for our previous study.22 The goodness of fit was indicated by the R factor, defined as ∑(xexp − xfit)2/∑(xexp)2, where xexp corresponds to the experimental data points and xfit is the corresponding point on the best-fitting curve.Transmission electron microscopy (TEM) and energy dispersive spectroscopy (EDS)
For TEM imaging, after exposure to NPs, cells were rinsed with PBS, fixed in 2% glutaraldehyde prepared in cacodylate buffer followed by 1% osmium tetroxide. Cells were then dehydrated in graded ethanol baths before embedding in Epon resin. Ultra-thin sections were cut and stained with 1% uranyl acetate. Sections were observed on a JEOL 1200EX TEM operating at 80 kV (Grenoble Institut des Neurosciences, Grenoble, France).For EDS analysis, samples were carbon-coated, then imaged by scanning-transmission electron microscopy (STEM); EDS was recorded on zones of interest using an FEI/Tecnai OSIRIS microscope operating at 200 kV.
Comet assay
NP-induced DNA strand breaks and alkali-labile sites were assessed using the alkaline version of the comet assay. Fpg-sensitive sites, such as 8-oxo-dGuo oxidized guanine were quantified using the bacterial DNA repair enzyme formamidopyrimidine-DNA glycosylase (Fpg). Following exposure to NPs, cells were harvested and stored at −80 °C in sucrose (85.5 g L−1) and DMSO (50 mL L−1) prepared in citrate buffer (11.8 g L−1), pH 7.6. Ten thousand cells were mixed with 0.6% low melting point agarose (LMPA) and deposited on an agarose-coated slide (n = 6). The cell/LMPA mix was allowed to solidify on ice for 10 min, then immersed in cold lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, 10% DMSO, 1% Triton X-100, pH 10) and incubated overnight at 4 °C. Slides were then rinsed in 0.4 M Tris pH 7.4. Slides were incubated for 45 min at 37 °C, three with 5 U Fpg (Invitrogen, prepared in 100 μL Fpg buffer) and three in Fpg buffer. DNA was allowed to unwind for 30 min in electrophoresis buffer (300 mM NaOH, 1 mM EDTA, pH >13) and electrophoresis was performed at 0.7 V cm−1 and 300 mA for 30 min. Slides were then neutralized in 0.4 M Tris pH 7.4 and stained with ethidium bromide (20 mg mL−1; Life Technologies). As a positive control, 50 μM H2O2 was deposited onto the agarose layer containing the cells and incubated for 5 min on ice. As a positive control for comet-Fpg, A549 cells were exposed to 1 μM riboflavin for 20 min at 37 °C, followed by UVA irradiation (10 J cm−2). Fifty comets were analyzed per slide using Comet IV software (Perceptive Instruments, Suffolk, UK).8-Oxo-dGuo measurement by HPLC/MS-MS
To quantify 8-oxo-dGuo and other oxidized bases by high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS),35 DNA was extracted and digested as described by Ravanat et al.36 Briefly, cell pellets were lysed with Triton X-100. Nuclei were collected by centrifugation and further lysed in 10% sodium dodecyl sulfate. The sample was incubated with a mixture of RNAse A and RNAse T1, followed by proteinase K. DNA was precipitated in isopropanol and concentrated sodium iodide. Deferoxamine was added to all buffers to prevent spurious oxidation. DNA was then digested into a mixture of nucleosides first by incubation with nuclease P1, DNAse II and phosphodiesterase II at pH 6 (2 h), followed by alkaline phosphatase and phosphodiesterase I (2 h, pH 8). The solution was neutralized with 0.1 μM HCl and centrifuged. The supernatant was collected and analyzed by HPLC-MS/MS. An API 3000 mass spectrometer (SCIEX) was used in the multiple reaction monitoring mode with positive electrospray ionization. 8-Oxo-dGuo fragmentation (m/z 284 [M + H]+ → m/z 168 [M + H − 116]+) was monitored. Chromatographic separation was achieved on a C18 reversed phase Uptisphere ODB column (Interchim, Montluçon, France). Fragments were eluted by a gradient of methanol in 2 mM ammonium formate at a flow rate of 0.2 mL min−1. The retention time for the fragment of interest was around 29 min. In addition to MS analysis, the HPLC eluent was analyzed in a UV detector set at 270 nm to quantify unmodified nucleosides. Levels of 8-oxo-dGuo were expressed as number of lesions per million bases.Reverse-transcription quantitative polymerase chain reaction (RT-qPCR)
Gene expression was quantified by RT-qPCR. RNA was extracted and reverse-transcribed using the GenElute™ mammalian total RNA miniprep kit with the optional DNAse treatment step and the SuperScript II Reverse Transcriptase kit (Life Technologies) according to the manufacturers' protocols. RNA concentration and purity were assessed using a Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific) based on A260/A280 and A260/A230 absorbance ratios. Duplicate cDNA aliquots from each of the three biological replicates for each exposure condition were loaded on a 96 well plate. qPCR was performed on an MX3005P Multiplex Quantitative PCR thermocycler (Stratagene) using SYBR Green (Eurogentec) and the following thermal cycling steps: 95 °C for 5 min, then 95 °C for 15 s, 55 °C for 20 s and 72 °C for 40 s 40 times and finally 95 °C for 1 min, 55 °C for 30 s and 95 °C for 30 s for the dissociation curve. Cq was determined using Mx-Pro 3.20 software with default settings. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 18S ribosomal 1 (S18) and cyclophilin B (CycloB) were chosen as reference genes for normalization and validated using BestKeeper.37 All primer sequences are indicated in Table S3.† mRNA expression analysis, normalization and statistical analysis were performed with REST 2009 software38 using the ΔΔCq method and a pairwise fixed reallocation randomization test. PCR efficiencies were experimentally verified for compliance using a mix of all samples, with a quality criterion of 2 ± 0.3.Multiplex array for DNA repair activity
The base excision repair (BER) and nucleotide excision repair (NER) abilities of A549 cells exposed to Ag-NPs were determined using a multiplexed excision/synthesis repair assay (LXRepair, Grenoble, France) composed of damaged plasmids spotted on a slide, as described previously.29,39 Damaged plasmids contained either photoproducts (cyclobutane pyrimidine dimers and 6–4 photoproducts (CPD-64)), 8-oxo-dGuo (8oxoG), abasic sites (Abasic), etheno bases (etheno or pyrimidine glycols (Glycols)). 8oxoG, Abasic, Etheno and Glycols are classically repaired by BER, whereas CPD-64 are classically repaired by NER. Nuclear extracts from exposed cells were prepared as described previously40 and applied to the microarray. Excision/synthesis reactions were run for 3 h at 30 °C using 0.3 mg mL−1 of each nuclear extract, 1 mM adenosine triphosphate and 0.25 μM dCTP-Cy3 (GE Healthcare). For each lesion, the total fluorescence incorporated into the damaged plasmid was quantified using the Innoscan 710AL scanner (Innopsys, Toulouse, France) and normalized relative to the fluorescence incorporated into an undamaged plasmid. Each extract was tested twice and the experiment was repeated three times.Cell cycle analysis
After exposure (106 cells per condition), cells were harvested and rinsed in PBS containing 2 mM EDTA (PBS-EDTA), then fixed in ice-cold 70% ethanol for 30 min and washed with PBS-EDTA. Cells were suspended in PBS-EDTA containing 25 μg mL−1 propidium iodide (Life Technologies) and 25 μg mL−1 RNAse A (Sigma-Aldrich). A minimum of 20000 events per condition was measured by flow cytometry (FACSCalibur, BD Biosciences, Franklin Lanes, NJ, USA) equipped with CXP software (Beckman Coulter Inc., Pasadena, CA, USA). Cell cycle data were fitted using Flowing Software 2.5.1 (http://www.flowingsoftware.com/). The experiment was reproduced three times, with three technical replicates per experiment.
Statistical analyses
All experiments were performed at least three times. Statistical tests were run using the Statistica 7.1 software (Statsoft, Chicago, IL, USA). As normality assumptions for valid parametric analyses were not satisfied, non-parametric Kruskal–Wallis one-way analysis of variance was used to determine statistical significance. When significance was demonstrated, paired comparisons were performed using Mann–Whitney U tests. Results were considered statistically significant (*) when the P value was <0.05.Results
NP characterization
The physicochemical characteristics of the NPs used in this study have been studied and are reported elsewhere;14,41 they are summarized in Table S4.† TEM images of NM300K and Ag-PVP are shown in Fig. 1A and B, respectively. NM300K consisted of spherical NPs with a bimodal size distribution, i.e., primary diameters of 5 and 15 nm, as indicated by the supplier. Ag-PVP was composed of spherical, polydisperse particles with a mean primary diameter of 59 nm. Their size distributions were characterized in MilliQ water (Fig. 1C) and in exposure medium (Fig. 1D). Hydrodynamic diameters in water were 33.6 nm and 103.3 nm for NM300K and Ag-PVP, respectively; they were 40.3 nm and 109.8 nm in exposure medium. Their zeta potentials, measured in MilliQ water, were −4 mV (NM300K) and −23 mV (Ag-PVP) (Table S4†). | ||
| Fig. 1 Characterization of Ag-NPs sizes. Transmission electron microscopy images of NM300K (A, scale bar: 50 nm) and Ag-PVP (B, scale bar: 500 nm); size distribution measured by dynamic light scattering, on NM300K and Ag-PVP suspensions at 10 μg mL−1 prepared either in water (C) or in exposure medium, i.e., DMEM containing 10% FBS (D). | ||
Cytotoxicity
The cytotoxicity of NM300K and Ag-PVP NPs was assessed based on the metabolic activity of cells post-exposure. In the acute exposure scheme (Fig. 2A), cell metabolic activity was maintained with up to 200 μg mL−1 Ag-PVP. Conversely, with just 100 μg mL−1 NM300K for 24 h, the metabolic activity of A549 cells was reduced to 4% of the value measured in unexposed cells. The impact of Ag-lactate was even stronger, with 85% residual metabolic activity in cells exposed to 10 μg mL−1, but only 25% in cells exposed to 20 μg mL−1. In the repeated exposure scheme, cell viability measured on the day after the 4 day exposure period (i.e., on day 5), was more affected than in the acute exposure regimen (Fig. 2B). Thus, metabolic activity was reduced by 50% and 25% in cells exposed to 12.5 μg mL−1 per day (i.e., a cumulative concentration of 50 μg mL−1) of NM300K or Ag-PVP, respectively. This concentration of both types of NPs only had a minor impact in the acute exposure regimen. As with acute exposure, metabolic activity decreased further with increasing Ag-NP cumulative concentration. Once again, Ag-lactate was more cytotoxic than Ag-PVP and NM300K in the repeated exposure condition. In subsequent experiments, cells were exposed to 25 μg mL−1 for NM300K or Ag-PVP or to 10 μg mL−1 for AgI-lactate (acute); and 6.25 μg mL−1 Ag-NPs or 2.5 μg mL−1 AgI-lactate per day for four days, which correspond to cumulative concentrations of 25 μg mL−1 of Ag-NPs or 10 μg mL−1 of Ag-lactate (repeated). At these concentrations, less than 25% cell death was observed in the acute exposure condition, and all cells received an equivalent overall concentration, with only the dose rate differing between the acute and the repeated regimens. | ||
| Fig. 2 Cytotoxicity of Ag-NPs and Ag-lactate. The cytotoxicity of Ag-NPs was assessed based on measurement of cellular metabolic activity, using the WST-1 assay. Cells were exposed to 10, 25, 50, 100 or 200 μg mL−1 Ag-NPs or Ag-lactate, either as an acute dose for 24 h (A) or to repeated doses of 1/4 of these concentrations per day for four successive days (B). Three independent experiments with five replicates per experiment were performed. | ||
Intracellular physicochemical transformation of Ag-NPs
The toxicity of Ag-NPs has been linked to their oxidative dissolution, with dissolved AgI causing the damage rather than Ag0 (Ag-NPs). This effect was the reason behind comparing NM300K and Ag-PVP for this study, as we hypothesized that they would undergo chemical transformation at different rates and with distinct kinetics within cells, with PVP-coated Ag-NPs dissolving more slowly than other Ag-NPs.19,23,24 To test this hypothesis, the chemical transformation of Ag was monitored in exposed cells using XAS, which is an element-selective method for speciation analysis. XAS can be applied without the need for extraction or centrifugation during sample preparation, which could modify the native chemical species. First, the near-edge region of XAS spectra (XANES) were analyzed by linear combination fitting (LCF), using spectra from Ag0-NP, Ag-GSH, AgCl, AgNO3 and AgO as references. These spectra represent all possible silver species encountered in cells (Fig. 3A). The AgI-GSH spectrum was used as a proxy for AgI combined with thiol-bearing ligands (Ag-SR).20 Incubation of NM300K for 48 h in culture medium, at 37 °C, led to the transformation of 12 ± 3% of Ag0 from NPs to AgI-thiol. In contrast, Ag-PVP remained as Ag0 (Fig. 3B). In cells exposed to Ag-NPs, Ag0 was progressively observed to transform into AgI. Upon acute exposure to NM300K, 54% of Ag0 transformed to AgI-thiol within 24 h of exposure, and 80% within 48 h of exposure. In the repeated exposure scheme, 78% of Ag0 from NM300K was transformed to AgI-thiol 24 h after the last of the four daily exposures (Fig. 3B). Ag-PVP was slower to transform, and transformed to a lesser degree, with 13% or 14% transformation in cells exposed for 24 h or 48 h, respectively. In the repeated exposure scheme, 41% of Ag-PVP was transformed to AgI (Fig. 3B). LCF with spectra for Ag0-NP and AgI-GSH were sufficient to obtain good quality fits. Thus, Ag0-NPs transformed in such a way that AgI only recombined with thiol-containing molecules, not forming AgO or AgCl, as reported in other exposure conditions.13 | ||
| Fig. 3 Ag K-edge XANES analysis of cells exposed to NM300K or PVP-coated Ag-NPs (A). Ag K-edge experimental XANES spectra (black dots) of A549 cells after 4 days of repeated exposure to PVP-coated and NM300K Ag-NPs. The corresponding best-fitting curves (solid red lines) based on the linear combination of reference compounds (dashed black lines) are reported on each experimental spectrum. Analysis by linear combination fitting of XANES spectra, using reference spectra for Ag-NPs (Ag–Ag) and Ag-GSH (for details, see ref. 20), used as a proxy for Ag-thiolate sites (B). The standard deviation error relative to the last digit is reported in parenthesis. NM300K and Ag-PVP were also analyzed by LCF under acellular conditions, i.e., NP suspensions diluted in cell culture medium and incubated for 96 h at 37 °C under a 5% CO2 atmosphere (“Ag-PVP, acellular” and “NM300K, acellular” in B). | ||
The intracellular transformation product that formed was identified thanks to ab initio fitting of EXAFS spectra.22 The average Ag–S bond length was 2.45–2.47 Å under all exposure conditions (Table 1). This bond distance is consistent with Ag atoms occupying trigonal AgS3 coordination sites, such as in AgI–cysteine or AgI–metallothioneins (2.47–2.48 Å), rather than a digonal AgS2 coordination such as in AgI-GSH complexes (2.36–2.38 Å).21 Moreover, the quality of the fit was enhanced when an Ag–Ag contribution was introduced. The bond distance for this contribution was 2.87 Å, consistent with the distances reported for Ag–Ag in such complexes.21 This result suggests the presence of S2Ag–(S)–AgS2 interactions within oligomeric species with trigonal AgS3 coordination, as previously described in AgI–cysteine or AgI–metallothionein complexes.21 The predominance of AgS3 sites was also confirmed by LCF analysis of EXAFS spectra (Table S5 and Fig. S2†), using model compounds representing distinct coordination geometries (discussed in the ESI†). Therefore, in A549 cells exposed to NM300K or Ag-PVP NPs, oxidized Ag recombined with cysteine or thiol-containing proteins present in cells, such as metallothionein, rather than with glutathione.
| n (%) | R Ag–Ag (Å) | σ 2 Ag–Ag (10−3 Å2) | R Ag–S (Å) | σ 2 Ag–S (10−3 Å2) | ΔE0 (eV) | R fit (%) | |
|---|---|---|---|---|---|---|---|
| a EXAFS spectra for A549 cells exposed to Ag-PVP repeatedly (“rep.”) for four days (6.25 μg mL−1 per day), or to NM300K acutely (25 μg mL−1 for 24 h, or 48 h), or repeatedly for four days (6.25 μg mL−1 per day) were fitted using the described strategy.22 Free parameters of the fit were the fraction of Ag atoms in undissolved NPs (n); interatomic distances (R) and Debye–Waller factors (σ2) relative to the first-shell photoelectron paths (Ag–Ag and Ag–S). An energy shift common to all photoelectron paths (ΔE0) was also included in the fit. Rfit (%) is used to evaluate the goodness of fit (see Materials and methods). This fitting procedure was possible only on EXAFS spectra with a good signal-to-noise ratio; therefore, it could not be applied to A549 cells exposed to Ag-PVP in the acute regimen. The standard deviation error relative to the last digit is reported in brackets. | |||||||
| Ag-PVP, rep. | 50 (2) | 2.872 (2) | 2.4 (3) | 2.450 (20) | 8 (3) | 2.1 (4) | 1.3 |
| NM300K, 24 h | 39 (2) | 2.872 (3) | 3.0 (3) | 2.463 (13) | 8 (2) | 2.4 (4) | 1.5 |
| NM300K, 48 h | 18.6 (14) | 2.874 (3) | 2.8 (4) | 2.473 (7) | 6.2 (7) | 2.0 (5) | 1.6 |
| NM300K, rep. | 19.5 (16) | 2.875 (4) | 3.2 (5) | 2.464 (8) | 7.8 (9) | 2.3 (6) | 2.2 |
Intracellular Ag content
Intracellular accumulation of Ag was measured by ICP-MS. The values obtained, combined with the proportions of Ag0-NPs and AgI determined by XAS analysis, are presented in Fig. 4. When using the repeated exposure scheme, Ag content in cells was 6 to 7 times lower than with acute exposure, suggesting either lower intracellular accumulation or progressive release of Ag from the cells in the repeated exposure scheme. In the acute exposure scheme (Fig. 4A), the overall intracellular Ag content was three times higher in cells exposed to NM300K compared to cells exposed to Ag-PVP. Interestingly, the content of Ag as Ag0-NP was similar in cells exposed to Ag-PVP and NM300K; whereas the amount of Ag present as AgI was 12-fold higher in cells exposed to NM300K than in cells exposed to Ag-PVP. In the repeated exposure scenario (Fig. 4B), the trend was similar to that for the acute exposure scenario, with comparable levels of Ag0-NPs in cells exposed to both NPs, but higher Ag content and higher levels of AgI in cells treated with NM300K than in cells exposed to Ag-PVP.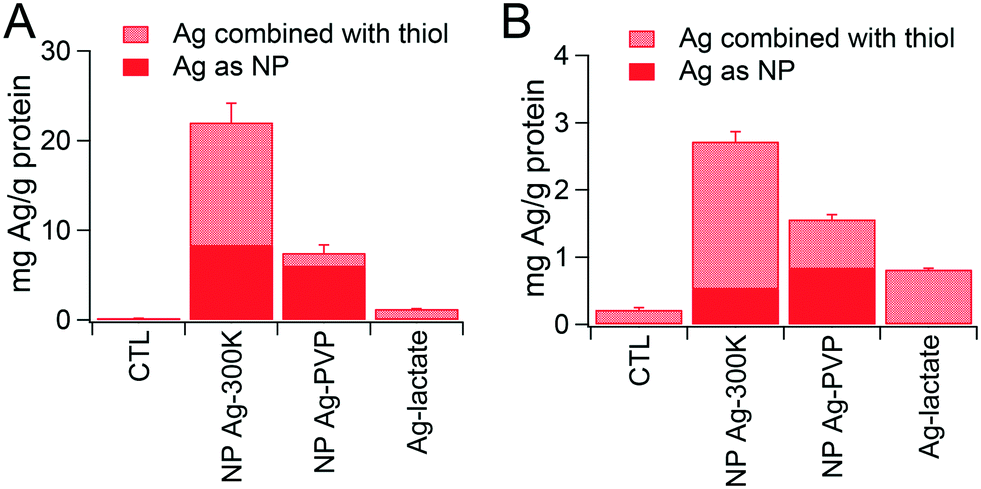 | ||
| Fig. 4 Intracellular content of Ag and proportion of Ag-NPs and their transformation product. Intracellular Ag content was measured by ICP-MS, then the % of each species as determined by EXAFS LCF fitting was applied to these values to determine the proportion of Ag as Ag-NPs and Ag combined with thiolated ligands. Ag content was measured in three independent replicates of cells exposed to 25 μg mL−1 Ag-NPs or 10 μg mL−1 Ag-lactate for 24 h (A) or 6.25 μg mL−1 Ag-NPs or 2.5 μg mL−1 Ag-lactate per day for four consecutive days (B). | ||
Intracellular distribution of Ag-NPs and their transformation products
To further explore the intracellular fate of Ag, cells exposed to Ag-NPs were observed by TEM. Large vesicles filled with electron-dense spherical or needle-like structures were observed in the cytoplasm of cells exposed to NM300K (Fig. 5A and B). Further analysis of these structures by EDS showed that they contained both Ag and S (Fig. 5C–E). The S/Ag ratio was higher in the sheet-like structures than in the spherical structures (in particular, see the agglomerate of spherical structures in Fig. 5D and E (arrow), in which S was undetectable). These sheet-like structures could be formed either outside cells before being internalized or inside cells after accumulation of spherical Ag-NPs. However, TEM observation of the exposure medium after 24 h of incubation at 37 °C under 5% CO2 revealed no obvious morphological modification of Ag-NPs (Fig. S3†). This finding suggests that NM300K particles are accumulated in cells as spherical particles and then transformed inside cells into needle-like structures, which could be sheets that have become sectioned during TEM preparation. These sheet-like structures could result from deposition of released AgI on sheets of cellular protein, or from aggregation of proteins together with AgI as sheets. | ||
| Fig. 5 TEM images of cells exposed to Ag-NPs. TEM images (A, B, F and G), STEM images using the HAADF detector (C and H), and EDS images (D, E, I, J, D and I: distribution of Ag; E and J: distribution of S) of A549 cells exposed to 25 μg mL−1 of NM300K (A–E) or Ag-PVP (F and G). | ||
In cells exposed to Ag-PVP, only spherical structures composed of Ag and S were observed, and these structures were contained within cytoplasmic vesicles (Fig. 5F and G). The diameter of these structures was ∼60 nm (Fig. 5G), which is close to the average primary diameter of the Ag-PVP NPs. The presence of S in these structures (Fig. 5J) suggests that the surface of Ag-PVP has undergone some chemical transformation. Around these deposits, dot-like structures were observed, also composed of Ag and S (Fig. 5G, arrows), which could be intracellularly transformed NPs.
DNA damage, reactive oxygen species, and impact on the cell cycle
Acute exposure to Ag-NPs significantly altered DNA integrity. Exposure to NM300K resulted in a significantly increased incidence of DNA strand breaks and/or alkali-labile sites, as revealed by the alkaline comet assay, but not of Fpg-sensitive sites such as 8-oxo-dGuo (Fig. 6A). An opposing scenario was observed in cells acutely exposed to Ag-PVP, i.e., few DNA strand breaks detected in the alkaline comet assay, but a significant extent of damage detected in the comet-Fpg assay (Fig. 6A). In contrast, in the repeated exposure scenario, no DNA damage was observed with Ag-NPs (Fig. 6B). Since interference of Ag-NPs with Fpg has been documented (for a review, see ref. 42), this result was confirmed by direct quantification of 8-oxo-dGuo by HPLC/MS-MS. This analysis confirmed increased 8-oxo-dGuo content in cells following acute exposure to Ag-PVP, but not to NM300K (Fig. S4†). This oxidative damage to DNA was associated with a significant increase in intracellular ROS levels in all conditions tested (Fig. 6C and D). As only acute exposure significantly altered DNA integrity, the genotoxicity of Ag-NPs was further explored in this condition. Neither NM300K nor Ag-PVP had a significant impact on results of the micronucleus assay (aneugenic and clastogenic events) or 53BP1 foci count (double-strand breaks), whereas the corresponding positive controls (etoposide and mitomycin c, respectively) did induce significant increases (Table S6†). | ||
| Fig. 6 Impact of Ag-NP on DNA integrity, level of reactive oxygen species and cell cycle progression. In all these experiments, cells were exposed to an acute dose of 25 μg mL−1 Ag-NPs or 10 μg mL−1 Ag-lactate for 24 h (A, C and E), or to repeated doses of 6.25 μg mL−1 Ag-NPs or 2.5 μg mL−1 Ag-lactate per day for four successive days (B, D and F). Damage to DNA, assessed based on the comet assay, either alkaline (positive control: 50 μM H2O2) or Fpg-modified (positive control: riboflavin + UVA) (A, acute exposure; B, repeated exposure). Intracellular levels of reactive oxygen species assessed using the H2-DCF-DA assay, positive control: KBrO3 2 mM (C, acute exposure; D, repeated exposure). Analysis of cell cycle and distribution of cells in the G2, S, G0/G1 and sub-G1 phases (E, acute exposure; F, repeated exposure). Graphs represent mean ± standard deviation for three independent experiments (DCFH-DA and cell cycle analysis) with five (H2-DCF-DA) or three (cell cycle) replicates per experiment. For comet assays, five independent experiments were performed with three comet slides per condition (one gel per slide) and per experiment. Statistical significance: *p < 0.05, exposed vs. control. | ||
The DNA damage observed in the comet assay was associated with a modification of the cell cycle distribution, characterized by a higher proportion of cells in S and G2/M following acute exposure to Ag-NPs or AgI-lactate, and a lower proportion of cells in G1, indicating cell cycle arrest in G2/M (Fig. 6E). This cell cycle modification was even more intense in cells following repeated exposure to Ag-NPs and Ag-lactate. In these conditions, cells were distributed equally between G1 and G2/M, and the S phase was no longer visible, demonstrating that the cell cycle was totally arrested (Fig. 6F). A marginal but statistically significant increase of the proportion of cells in the sub-G1 phase was observed in cells exposed repeatedly to NM300K, which suggests the onset of apoptosis (Fig. 6F).
Impact of acute exposure to Ag-NPs on DNA repair processes
The impact of Ag-NPs on the capacity of A549 cells to repair damaged DNA was then assessed using a multiplexed array, as previously for TiO2-NPs.29,39 This array measures the repair efficiency for five DNA lesions, repaired either by BER (8oxoG, AbaS, Etheno and Glycols) or by NER (CPD-64). Data were normalized for the total fluorescence detected on the arrays in order to reveal whether a specific repair pathway played a more extensive role when compared to the overall cellular DNA repair activity. Results are presented as a heat map, with blue boxes representing reduced DNA repair activity and red boxes corresponding to increased activity. Following acute exposure to both Ag-NPs, the overall fluorescence intensity decreased, indicating that the cells' capacity to repair DNA lesions by both BER and NER pathways was decreased (Fig. 7A). Conversely, in cells exposed to AgI-lactate, the repair of AbaS and 8oxoG increased while the repair of other lesions decreased (Fig. 7A), suggesting that the repair of oxidized bases by the BER pathway was activated. This activation was associated with significant and intense downregulation of PARP1 and APE1 mRNA expression, both of which are involved in DNA repair via the BER pathway (Fig. 7B). Significant downregulation of OGG1 was also observed in cells exposed to NM300K and Ag-lactate in the acute regimen, of XRCC1 in cells following acute exposure to NM300K, of PCNA in cells repeatedly exposed to Ag-PVP, and of POLβ in all conditions tested except cells with acute exposure to Ag-PVP (Fig. 7B). Since oxidative damage to DNA and elevated levels of intracellular ROS were detected, the mRNA expression of antioxidant defense enzymes was measured (Fig. 7B). Downregulation of superoxide dismutase (SOD1 and SOD2), catalase (CAT) and glutathione reductase (GSR) was observed in cells exposed to both Ag-NPs and to Ag-lactate in both the acute and the repeated exposure schemes. The glutamate–cysteine ligase modifier subunit (GCLM), which is essential to the glutathione synthesis pathway, and nuclear factor (erythroid-derived 2)-like 2 (NRF-2), which controls the expression of a number of genes encoding antioxidant enzymes, were also downregulated. This overall downregulation was similar in the acute and the repeated exposure schemes (Fig. 7B). Moreover, expression levels for metallothioneins are known to be induced by a number of metallic stresses, and more generally in conditions causing oxidative stress.43 A strong induction of the expression of both metallothionein 1 (MT1) and 2 (MT2) was observed in cells exposed to NM300K and Ag-PVP, with 250- to 300-fold increases measured in the acute exposure regimen (Fig. 7B). A less intense regulation of MT gene expression was observed in the repeated exposure scheme, with 10-fold changes in expression levels measured in exposed cells following the four exposures compared to control cells (Fig. 7B). Induction was consistently more extensive in cells exposed to Ag-NPs than in cells exposed to Ag-lactate. | ||
| Fig. 7 DNA repair capacities of cells exposed to Ag-NPs. Heat map representation of DNA excision/synthesis repair activities in A549 cells exposed to 25 μg mL−1 Ag-NPs or 10 μg mL−1 Ag-lactate for 24 h (A). Color scale ranges between blue (lower value) and red (higher value). Each value represents the relative contribution of a specific repair pathway to total DNA repair activity in the cell after normalization of data. For instance, when considering DNA repair activity in a chosen condition (Ag-lactate, Ag-PVP or NM300K), the pathway with the most red is the most active, while the pathway with the most blue is the least active. The experiment was performed once with three independent replicates. Fold-change in mRNA expression of genes encoding proteins involved in DNA repair, oxidative stress response, and metallothioneins (B). Results are also expressed as a heat map, with color scale ranging between blue (lower value) and red (higher value). Fold-changes are indicated only in conditions where statistically significant (p < 0.05) changes to gene expression were observed. | ||
Discussion
With this study, we examined the accumulation, physicochemical transformation and toxicity of two Ag-NPs in A549 cells. Ag-NPs are used as biocides in consumer products and are therefore continuously released into the environment, especially in ambient air. This study was conducted to model human exposure via inhalation to Ag-NPs released from consumer products, e.g. when handling textiles impregnated with NP-Ag or when wearing a silver nanoparticle-containing respiratory protection mask. Two types of Ag-NPs were chosen as models, one of them being a highly soluble Ag-NP (NM300K) and the other dissolving to a lesser extent (Ag-PVP). Cells were exposed using either an acute or a repeated exposure regimen, with 25 μg mL−1 Ag-NP applied directly for 24 h or split across four days (i.e., 6.25 μg mL−1 per day for a total concentration of 25 μg mL−1), respectively. This high exposure concentration was justified by the cytotoxicity profile of these Ag-NPs. This concentration corresponded to the lethal dose 25 (LD25) in the repeated exposure scenario or the LD10–20 in the acute exposure scenario, which is high enough to observe some response in cells, but sufficiently low to ensure the absence of false-positive outcomes as a result of DNA fragmentation in dying cells in genotoxicity assessments. As discussed by Gliga et al.,23 10 μg mL−1 would approximately correspond to the load deposited on alveolar cells after 74 days of exposure to ambient air, 8 hours per day and 5 days per week in an Ag-NP manufacturing facility where the maximum level is 289 μg m−3. The concentration used here, 25 μg mL−1, is 2.5-fold higher than this potential occupational exposure level; this type of exposure load could be encountered following accidental exposure or after repeated exposure of workers by inhalation over the course of a year.Both the acute and the repeated exposures regimens affected cellular metabolism and caused accumulation of ROS in cells. However, while acute exposure led to significant oxidative damage to DNA, no DNA damage was observed in cells following repeated exposure, suggesting that either DNA was not damaged or damaged DNA has been repaired. The cell cycle was totally blocked at the end of the repeated exposure scheme. Cell cycle arrest is known to be a crucial stage in the DNA damage response, leaving cells enough time to repair damaged DNA before the onset of mitosis, hence limiting the risk of propagating DNA damage in daughter cells.44 Consequently, the strong cell cycle blockade observed with repeated exposure of cells to Ag-NPs would be a protective response allowing cells to repair damaged DNA; acute exposure does not appear to induce this type of response. Moreover, intracellular accumulation of Ag after repeated exposure to Ag-NPs was approximately ten-fold lower than following acute exposure. Lower intracellular accumulation would lead to a less toxic outcome, particularly a reduced impact on DNA. Overall, both reduced intracellular accumulation of Ag, leading to a reduced impact on DNA, and cell cycle blockade, facilitating DNA repair, might explain the absence of DNA lesions in cells exposed to Ag-NPs at a low dose rate.
Since the total Ag to which cells were exposed was the same in the acute and repeated exposure regimens, intracellular accumulation should be the same. The reduced intracellular accumulation of Ag observed following repeated exposure could be explained by cell division and subsequent splitting of the Ag load at each cell division. However, as the cell cycle slowed down and then was completely arrested in this exposure regime other factors must limit intracellular Ag accumulation. A possibility is that cell division resulted in more confluent cultures, i.e., each cell had more neighbor cells. This would reduce the cell surface interacting with the exposure medium and thus limit Ag intracellular accumulation. Other possible explanations could be progressive release of Ag from cells, or adaptation of cells, reducing their capacity to accumulate Ag-NPs. Indeed, cellular adaptation to chronic exposure to Ag-NPs has been reported when cells are exposed for much longer periods of time, either 3 months,15 4 months (ref. 16) or 6 weeks.17 In the study by Comfort et al.,15 chronic exposure resulted in an amplified, sustained stress reaction, while the studies by Gliga et al.17 and Choo et al.16 suggested that chronic exposure to Ag-NPs could enhance malignant transformation in non-tumorigenic cells. For the present study, the duration of exposure was limited to 4 days; therefore the response observed could correspond to the first stages of this adaptive response, characterized by reduced intracellular Ag accumulation, cell cycle blockade, decreased overall metabolism and profound modification of cellular activities.
Whatever the exposure condition and NP, physicochemical transformation of Ag0-NPs occurred within cells, characterized by oxidation of Ag0 to AgI and combination of AgI with thiolated ligands identified as thiol-containing proteins. The most abundant thiol-containing proteins in A549 cells are metallothioneins and zinc-finger proteins, it is therefore probable that AgI combines with these proteins. Therefore, the transformation product of Ag-NPs that forms in A549 cells differs from those reported previously in other cell types.19,22 This transformation product is AgI-GSH in mouse primary macrophages22 and a mixture of AgI-GSH and AgI-thiol-containing proteins, thought to be metallothioneins, in Hep-G2 hepatocytes.19 This difference could be due to a higher content of thiol-containing proteins and/or a lower GSH content in A549 cells compared to hepatocytes and mouse primary macrophages. The total glutathione content is assumed to be higher in hepatocytes, which export GSH, than in other cell types, with concentrations of approximately 10 mM versus 1–2 mM, respectively.45 Indeed, the basal level of total glutathione content was shown to be more than three times higher in Hep-G2 (225 nmol mg−1 protein) than in A549 cells (66 nmol mg−1 protein), whereas the metallothionein content was approximately 1.5 times higher in Hep-G2 than in A549 cells.46 The glutathione content in macrophage cell lines is reported to be around 4 nmol mg−1 protein,47,48 10 nmol mg−1 proteins,49 or 16 nmol mg−1 proteins,50 all of which are much lower than the values presented for A549 and Hep-G2 cells. Our previous work was performed in primary murine macrophages,22 for which the glutathione content may not be directly comparable to the glutathione content in cell lines. Interestingly, the genotoxicity of NM300K – a representative Ag nanomaterial distributed by the European Commission Joint Research Center (JRC, Ispra, Italy) – has been reported for A549 cells, hepatocytes and macrophages. In A549 cells, it produces strand breaks but no Fpg-sensitive sites,51 consistent with the results presented here. In J774A mouse macrophages, it is reported to increase the level of 8-OHdG.52 In 3D liver microtissues, NM300K causes the formation of both strand breaks and Fpg-sensitive sites.53 This result suggests that when Ag ions released from Ag-NPs are complexed by GSH, the main DNA damage would be oxidized DNA bases, whereas it would be strand breaks when the released Ag ions are trapped by thiol-containing proteins. Consequently, several different mechanisms probably converge to explain Ag-NP genotoxicity and, depending on the ligand that complexes Ag ions, one mechanism becomes predominant over the others. The first mechanism would be impaired removal of ROS by GSH. The cellular metabolism naturally produces some reactive species, which are scavenged by GSH and other antioxidant systems.45 If GSH is engaged in complexing Ag ions released from Ag-NPs, it is no longer available to scavenge reactive species. These species can consequently migrate to the cell nucleus and oxidize DNA bases. This process would be particularly problematic in phagocytes, where the mechanisms employed to kill microorganisms involve the production of ROS, the excess of which are subsequently scavenged by GSH.45 Another mechanism would be through alteration of the main function of metallothioneins, which are antioxidants and are responsible for the detoxification of heavy metals and the maintenance of the homeostasis of essential metal ions in cells.54 The proteins that complex Ag ions released from Ag-NPs can also be other thiol-containing proteins, such as any zinc-finger protein. As discussed previously,55 Ag could displace Zn in the zinc-binding sites of metallothioneins or of zinc-finger proteins, therefore distorting their active site and leading to their partial or total inactivation. This would lead to the release of Zn into cells, which could affect a wide range of cellular functions.56 The amplified expression of mRNA for metallothionein-encoding genes suggests that the cell responds either to the presence of Ag ions or to an increased level of free Zn2+ ions in cytoplasm following their release from the active sites of metallothioneins or other proteins using Zn as a cofactor. Moreover, several zinc-finger proteins are involved in DNA repair processes, particularly PARP1, XPA and p53. Cu ions, CuO-NPs and micron-sized particles can all inactivate PARP1.57,58 Given the similar coordination properties of Cu and Ag,20 Ag could also inactivate PARP1. This enzyme plays a central role in DNA strand-break repair;59 consequently, its inactivation would affect this repair process. Indeed, our data confirm that the DNA damage induced by Ag-NPs correlates with a decreased ability to repair DNA via the BER and NER pathways, as previously suggested by others.60–63 This inhibition of DNA repair is observed in cells treated with both NM300K and PVP-coated Ag-NPs, even though the two NP forms induce different types of DNA lesions. Inhibited repair is therefore not a consequence of the DNA lesion per se, but probably results from the depletion of thiol-containing proteins due to their complexation with Ag ions.
Finally, the extent of physicochemical transformation of Ag-NPs depends on the type of Ag-NP to which the cells are exposed, with more intense transformation of NM300K compared to Ag-PVP. The toxicity of Ag-NPs depends on their size and surface coating, which are related to their dissolution potential. Smaller NPs dissolve more extensively and lead to a greater cytotoxic impact than larger NPs.23,24 Moreover, PVP-coated Ag-NPs have been shown to be less toxic than citrate-coated Ag-NPs,23,24 possibly because PVP protects the Ag-NP surface from degradation and/or because PVP binds silver ions that are released when the particles dissolve. This is consistent with our results showing more extensive dissolution of NM300K (15 nm), which is smaller and not coated with PVP, compared to the Ag-PVP NPs used here (59 nm). This more extensive dissolution of NM300K as well as the higher intracellular accumulation is certainly the reason for its higher toxicity.
Conclusions
Ag-NPs used as biocides in a plethora of daily products are continuously released into the environment; they could potentially be released as airborne particles, although this would have to be demonstrated. This release scenario raises questions as to their impact on humans, especially following inhalation exposure, which is the most problematic route of human exposure because the lungs are very difficult to protect from nanoparticles. In the present study, we show that two Ag-NPs with distinct dissolution potentials lead to the same transformation product in A549 cells: Ag complexed with thiol-containing proteins. This transformation product differs from the one observed in hepatocytes and macrophages in previous studies, indicating that it is cell-type-dependent. In addition, different rates of Ag-NP exposure resulted in non-identical intracellular accumulation and toxicity outcomes, with repeated exposure causing a more intense reduction in cell metabolism and a stronger cell cycle blockade, whereas DNA damage was observed only in response to acute exposure. These distinctions were all the more striking in that the cells received the same total quantity of Ag-NPs over 24 h or over 4 days. Intracellular accumulation of Ag was six to seven times higher after acute exposure than after repeated exposure. PVP-coated Ag-NPs, which show low dissolution and lower intracellular accumulation than NM300K, were less cytotoxic than NM300K. These two Ag-NPs induced distinct types of DNA lesions: oxidized DNA bases for Ag-PVP, and DNA strand breaks for NM300K. Taken together, these results show that the dose rate rather than the concentration explains Ag-NP toxicity, with lower concentrations over longer periods allowing cells to adapt.Author contributions
LB performed most experiments and prepared all samples. DB performed qPCR experiments. AT performed 53BP1 experiments and reproduced some comet assays. IK was responsible for the EXAFS experiment and GV analyzed the EXAFS data. SM and IL performed ICP-MS measurements. KPG prepared samples for TEM and recorded TEM images; PHJ performed STEM-EDS analyses on these samples. SS was responsible for DNA repair activity assays. MC, the principal investigator of this study, conceptualized the whole study with the help of TR, CAG and EVJ. MC wrote the manuscript, which was edited and approved by all authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work leading to these results received funding from the European Union's Seventh Framework Programme for research, technology development and demonstration under grant agreement no. 310451 (NanoMILE) and from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 760928 (BIORIMA). The study was performed in the framework of the hCOMET COST action (CA15132) and of the Labex Serenade (no. ANR-11-LABX-0064) funded by the Investissements d'Avenir French Government program of the French National Research Agency (ANR) through the A*MIDEX project (no. ANR-11-IDEX-0001-02). The TECNAI OSIRIS, operated by P. H. Jouneau, and the ICP-MS equipment are part of the NanoID platform supported by the French Investissements d'Avenir (ANR-10-EQPX-39). The authors acknowledge the ESRF for providing access to FAME-BM30B beamline (experiment LS-2331). The FP7 research infrastructure project QualityNano (grant agreement no. 262163) provided funding for Transnational Access to ICP-MS facilities and expertise at the University of Birmingham. We further acknowledge the GRAL-supported platforms, funded through the University Grenoble-Alpes Graduate School (Ecoles Universitaires de Recherche). The authors would like to thank Anne Bertrand for help in preparing TEM samples, Véronique Collin-Faure for flow cytometry experiments, Pascale Delangle for fruitful discussions related to the chemistry of Ag-NPs and metal–ligand interactions and M. Gallagher (TWS Editing) for English proofreading of the manuscript.References
- D. M. Mitrano, S. Motellier, S. Clavaguera and B. Nowack, Review of nanomaterial aging and transformations through the life cycle of nano-enhanced products, Environ. Int., 2015, 77, 132–147 CrossRef CAS.
- B. Nowack, H. F. Krug and M. Height, 120 years of nanosilver history: implications for policy makers, Environ. Sci. Technol., 2011, 45, 1177–1183 CrossRef CAS.
- P. Dubey, I. Matai, S. U. Kumar, A. Sachdev, B. Bhushan and P. Gopinath, Perturbation of cellular mechanistic system by silver nanoparticle toxicity: Cytotoxic, genotoxic and epigenetic potentials, Adv. Colloid Interface Sci., 2015, 221, 4–21 CrossRef CAS.
- H. J. Johnston, G. Hutchison, F. M. Christensen, S. Peters, S. Hankin and V. Stone, A review of the in vivo and in vitro toxicity of silver and gold particulates: particle attributes and biological mechanisms responsible for the observed toxicity, Crit. Rev. Toxicol., 2010, 40, 328–346 CrossRef CAS.
- D. McShan, P. C. Ray and H. Yu, Molecular toxicity mechanism of nanosilver, J. Food Drug Anal., 2014, 22, 116–127 CrossRef CAS.
- Z. Wang, T. Xia and S. Liu, Mechanisms of nanosilver-induced toxicological effects: more attention should be paid to its sublethal effects, Nanoscale, 2015, 7, 7470–7481 RSC.
- S. W. Wijnhoven, W. Peijnenburg, C. A. Herberts, W. I. Hagens, A. G. Oomen, E. H. W. Heugens, B. Roszek, J. Bisschops, I. Gosens, D. Van De Meent, S. Dekkers, W. H. De Jong, M. van Zijverden, A. J. A. M. Slips and R. E. Geertsma, Nano-silver - a review of available data and knowledge gaps in human and environmental risk assessment, Nanotoxicology, 2009, 3, 109–138 CrossRef CAS.
- A. Hudecova, B. Kusznierewicz, E. Runden-Pran, Z. Magdolenova, K. Hasplova, A. Rinna, L. M. Fjellsbo, M. Kruszewski, A. Lankoff, W. J. Sandberg, M. Refsnes, T. Skuland, P. Schwarze, G. Brunborg, M. Bjoras, A. Collins, E. Miadokova, E. Galova and M. Dusinska, Silver nanoparticles induce premutagenic DNA oxidation that can be prevented by phytochemicals from Gentiana asclepiadea, Mutagenesis, 2012, 27, 759–769 CrossRef CAS.
- A. Huk, E. Izak-Nau, N. El Yamani, H. Uggerud, M. Vadset, B. Zasonska, A. Duschl and M. Dusinska, Impact of nanosilver on various DNA lesions and HPRT gene mutations - effects of charge and surface coating, Part. Fibre Toxicol., 2015, 12, 25 CrossRef.
- Z. Magdolenova, A. Collins, A. Kumar, A. Dhawan, V. Stone and M. Dusinska, Mechanisms of genotoxicity. A review of in vitro and in vivo studies with engineered nanoparticles, Nanotoxicology, 2014, 8, 233–278 CrossRef CAS.
- P. Nymark, J. Catalan, S. Suhonen, H. Jarventaus, R. Birkedal, P. A. Clausen, K. A. Jensen, M. Vippola, K. Savolainen and H. Norppa, Genotoxicity of polyvinylpyrrolidone-coated silver nanoparticles in BEAS 2B cells, Toxicology, 2013, 313, 38–48 CrossRef CAS.
- A. Rinna, Z. Magdolenova, A. Hudecova, M. Kruszewski, M. Refsnes and M. Dusinska, Effect of silver nanoparticles on mitogen-activated protein kinases activation: role of reactive oxygen species and implication in DNA damage, Mutagenesis, 2015, 30, 59–66 CrossRef CAS.
- L. Wang, T. Zhang, P. Li, W. Huang, J. Tang, P. Wang, J. Liu, Q. Yuan, R. Bai, B. Li, K. Zhang, Y. Zhao and C. Chen, Use of Synchrotron Radiation-Analytical Techniques To Reveal Chemical Origin of Silver-Nanoparticle Cytotoxicity, ACS Nano, 2015, 9, 6532–6547 CrossRef CAS.
- C. Aude-Garcia, F. Villiers, V. Collin-Faure, K. Pernet-Gallay, P. H. Jouneau, S. Sorieul, G. Mure, A. Gerdil, N. Herlin-Boime, M. Carriere and T. Rabilloud, Different in vitro exposure regimens of murine primary macrophages to silver nanoparticles induce different fates of nanoparticles and different toxicological and functional consequences, Nanotoxicology, 2016, 10, 586–596 CrossRef CAS.
- K. K. Comfort, L. K. Braydich-Stolle, E. I. Maurer and S. M. Hussain, Less is more: long-term in vitro exposure to low levels of silver nanoparticles provides new insights for nanomaterial evaluation, ACS Nano, 2014, 8, 3260–3271 CrossRef CAS.
- W. H. Choo, C. H. Park, S. E. Jung, B. Moon, H. Ahn, J. S. Ryu, K. S. Kim, Y. H. Lee, I. J. Yu and S. M. Oh, Long-term exposures to low s of silver nanoparticles enhanced in vitro malignant cell transformation in non-tumorigenic BEAS-2B cells, Toxicol. In Vitro, 2016, 37, 41–49 CrossRef CAS.
- A. R. Gliga, S. Di Bucchianico, J. Lindvall, B. Fadeel and H. L. Karlsson, RNA-sequencing reveals long-term effects of silver nanoparticles on human lung cells, Sci. Rep., 2018, 8, 6668 CrossRef.
- I. L. Hsiao, Y. K. Hsieh, C. F. Wang, I. C. Chen and Y. J. Huang, Trojan-horse mechanism in the cellular uptake of silver nanoparticles verified by direct intra- and extracellular silver speciation analysis, Environ. Sci. Technol., 2015, 49, 3813–3821 CrossRef CAS.
- G. Veronesi, A. Deniaud, T. Gallon, P. H. Jouneau, J. Villanova, P. Delangle, M. Carriere, I. Kieffer, P. Charbonnier, E. Mintz and I. Michaud-Soret, Visualization, quantification and coordination of Ag+ ions released from silver nanoparticles in hepatocytes, Nanoscale, 2016, 8, 17012–17021 RSC.
- G. Veronesi, T. Gallon, A. Deniaud, B. Boff, C. Gateau, C. Lebrun, C. Vidaud, F. Rollin-Genetet, M. Carriere, I. Kieffer, E. Mintz, P. Delangle and I. Michaud-Soret, XAS Investigation of Silver(I) Coordination in Copper(I) Biological Binding Sites, Inorg. Chem., 2015, 54, 11688–11696 CrossRef CAS.
- B. O. Leung, F. Jalilehvand, V. Mah, M. Parvez and Q. Wu, Silver(I) complex formation with cysteine, penicillamine, and glutathione, Inorg. Chem., 2013, 52, 4593–4602 CrossRef CAS.
- G. Veronesi, C. Aude-Garcia, I. Kieffer, T. Gallon, P. Delangle, N. Herlin-Boime, T. Rabilloud and M. Carriere, Exposure-dependent Ag+ release from silver nanoparticles and its complexation in AgS2 sites in primary murine macrophages, Nanoscale, 2015, 7, 7323–7330 RSC.
- A. R. Gliga, S. Skoglund, I. O. Wallinder, B. Fadeel and H. L. Karlsson, Size-dependent cytotoxicity of silver nanoparticles in human lung cells: the role of cellular uptake, agglomeration and Ag release, Part. Fibre Toxicol., 2014, 11, 11 CrossRef.
- R. F. Hamilton, S. Buckingham and A. Holian, The effect of size on Ag nanosphere toxicity in macrophage cell models and lung epithelial cell lines is dependent on particle dissolution, Int. J. Mol. Sci., 2014, 15, 6815–6830 CrossRef.
- S. J. Kemp, A. J. Thorley, J. Gorelik, M. J. Seckl, M. J. O'Hare, A. Arcaro, Y. Korchev, P. Goldstraw and T. D. Tetley, Immortalization of human alveolar epithelial cells to investigate nanoparticle uptake, Am. J. Respir. Cell Mol. Biol., 2008, 39, 591–597 CrossRef CAS.
- W. H. O. (WHO), principles and methods to assess the risk of immunotoxicity associated with exposure to nanomaterials (Environmental criteria: n. 244), Geneva, 2019.
- R. J. Swain, S. J. Kemp, P. Goldstraw, T. D. Tetley and M. M. Stevens, Assessment of cell line models of primary human cells by Raman spectral phenotyping, Biophys. J., 2010, 98, 1703–1711 CrossRef CAS.
- M. H. Kweon, V. M. Adhami, J. S. Lee and H. Mukhtar, Constitutive overexpression of Nrf2-dependent heme oxygenase-1 in A549 cells contributes to resistance to apoptosis induced by epigallocatechin 3-gallate, J. Biol. Chem., 2006, 281, 33761–33772 CrossRef CAS.
- M. Biola-Clier, D. Beal, S. Caillat, S. Libert, L. Armand, N. Herlin-Boime, S. Sauvaigo, T. Douki and M. Carriere, Comparison of the DNA damage response of BEAS-2B and A549 cells exposed to titanium dioxide nanoparticles, Mutagenesis, 2017, 31(1), 161–172 CrossRef.
- R. Guadagnini, B. Halamoda Kenzaoui, L. Walker, G. Pojana, Z. Magdolenova, D. Bilanicova, M. Saunders, L. Juillerat-Jeanneret, A. Marcomini, A. Huk, M. Dusinska, L. M. Fjellsbo, F. Marano and S. Boland, Toxicity screenings of nanomaterials: challenges due to interference with assay processes and components of classic in vitro tests, Nanotoxicology, 2015, 9(Suppl 1), 13–24 CrossRef CAS.
- I. Hansjosten, J. Rapp, L. Reiner, R. Vatter, S. Fritsch-Decker, R. Peravali, T. Palosaari, E. Joossens, K. Gerloff, P. Macko, M. Whelan, D. Gilliland, I. Ojea-Jimenez, M. P. Monopoli, L. Rocks, D. Garry, K. Dawson, P. J. F. Rottgermann, A. Murschhauser, J. O. Radler, S. V. Y. Tang, P. Gooden, M. A. Belinga-Desaunay, A. O. Khan, S. Briffa, E. Guggenheim, A. Papadiamantis, I. Lynch, E. Valsami-Jones, S. Diabate and C. Weiss, Microscopy-based high-throughput assays enable multi-parametric analysis to assess adverse effects of nanomaterials in various cell lines, Arch. Toxicol., 2018, 92, 633–649 CrossRef CAS.
- O. Proux, X. Biquard, E. Lahera, J. J. Menthonnex, A. Prat, O. Ulrich, Y. Soldo, P. Trevisson, G. Kapoujyan, G. Perroux, P. Taunier, D. Grand, P. Jeantet, M. Deleglise, J. P. Roux and J. L. Hazemann, FAME: A new beamline for X-ray absorption investigations of very-diluted systems of environmental, material and biological interests, Phys. Scr., 2005, 115, 970–973 CrossRef.
- O. Proux, V. Nassif, A. Prat, O. Ulrich, E. Lahera, X. Biquard, J. J. Menthonnex and J. L. Hazemann, Feedback system of a liquid-nitrogen-cooled double-crystal monochromator: design and performances, J. Synchrotron Radiat., 2006, 13, 59–68 CrossRef CAS.
- B. Ravel and M. Newville, Athena, artemis, hephaestus: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- S. Frelon, T. Douki, J. L. Ravanat, J. P. Pouget, C. Tornabene and J. Cadet, High-performance liquid chromatography--tandem mass spectrometry measurement of radiation-induced base damage to isolated and cellular DNA, Chem. Res. Toxicol., 2000, 13, 1002–1010 Search PubMed.
- J. L. Ravanat, T. Douki, P. Duez, E. Gremaud, K. Herbert, T. Hofer, L. Lasserre, C. Saint-Pierre, A. Favier and J. Cadet, Cellular background level of 8-oxo-7,8-dihydro-2′-deoxyguanosine: an isotope based method to evaluate artefactual oxidation of DNA during its extraction and subsequent work-up, Carcinogenesis, 2002, 23, 1911–1918 CrossRef CAS.
- M. W. Pfaffl, A. Tichopad, C. Prgomet and T. P. Neuvians, Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations, Biotechnol. Lett., 2004, 26, 509–515 CrossRef CAS.
- M. W. Pfaffl, A new mathematical model for relative quantification in real-time RT-PCR, Nucleic Acids Res., 2001, 29, e45 CrossRef CAS.
- M. L. Jugan, S. Barillet, A. Simon-Deckers, N. Herlin-Boime, S. Sauvaigo, T. Douki and M. Carriere, Titanium dioxide nanoparticles exhibit genotoxicity and impair DNA repair activity in A549 cells, Nanotoxicology, 2012, 6, 501–513 CrossRef CAS.
- J. F. Millau, A. L. Raffin, S. Caillat, C. Claudet, G. Arras, N. Ugolin, T. Douki, J. L. Ravanat, J. Breton, T. Oddos, C. Dumontet, A. Sarasin, S. Chevillard, A. Favier and S. Sauvaigo, A microarray to measure repair of damaged plasmids by cell lysates, Lab Chip, 2008, 8, 1713–1722 RSC.
- C. L. Klein, S. Comero, B. Stahlmecke, J. Romazanov, T. A. J. Kuhlbusch, E. Van Doren, P.-J. De Temmerman, J. Mast, P. Wick, H. Krug, G. Locoro, K. Hund-Rinke, W. Kördel, S. Friedrichs, G. Maier, J. Werner, T. Linsinger and B. M. Gawlik, NM-300 Silver. Characterisation, Stability, Homogeneity, http://publications.jrc.ec.europa.eu/repository/handle/JRC60709.
- P. Moller, D. M. Jensen, D. V. Christophersen, A. Kermanizadeh, N. R. Jacobsen, J. G. Hemmingsen, P. H. Danielsen, D. G. Karottki, M. Roursgaard, Y. Cao, K. Jantzen, H. Klingberg, L. G. Hersoug and S. Loft, Measurement of oxidative damage to DNA in nanomaterial exposed cells and animals, Environ. Mol. Mutagen., 2015, 56, 97–110 CrossRef CAS.
- A. Hartwig, Metal interaction with redox regulation: an integrating concept in metal carcinogenesis?, Free Radical Biol. Med., 2013, 55, 63–72 CrossRef CAS.
- J. W. Harper and S. J. Elledge, The DNA damage response: ten years after, Mol. Cell, 2007, 28, 739–745 CrossRef CAS.
- H. J. Forman, H. Zhang and A. Rinna, Glutathione: overview of its protective roles, measurement, and biosynthesis, Mol. Aspects Med., 2009, 30, 1–12 CrossRef CAS.
- N. Arnal, M. J. de Alaniz and C. A. Marra, Effect of copper overload on the survival of HepG2 and A-549 human-derived cells, Hum. Exp. Toxicol., 2013, 32, 299–315 CrossRef CAS.
- M. Diotallevi, P. Checconi, A. T. Palamara, I. Celestino, L. Coppo, A. Holmgren, K. Abbas, F. Peyrot, M. Mengozzi and P. Ghezzi, Glutathione Fine-Tunes the Innate Immune Response toward Antiviral Pathways in a Macrophage Cell Line Independently of Its Antioxidant Properties, Front. Immunol., 2017, 8, 1239 CrossRef.
- V. Venketaraman, Y. K. Dayaram, A. G. Amin, R. Ngo, R. M. Green, M. T. Talaue, J. Mann and N. D. Connell, Role of glutathione in macrophage control of mycobacteria, Infect. Immun., 2003, 71, 1864–1871 CrossRef CAS.
- S. R. Yang, A. S. Chida, M. R. Bauter, N. Shafiq, K. Seweryniak, S. B. Maggirwar, I. Kilty and I. Rahman, Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages, Am. J. Physiol., 2006, 291, L46–L57 CrossRef CAS.
- B. Dalzon, J. Bons, H. Diemer, V. Collin-Faure, C. Marie-Desvergne, M. Dubosson, S. Cianferani, C. Carapito and T. Rabilloud, A Proteomic View of Cellular Responses to Anticancer Quinoline-Copper Complexes, Proteomes, 2019, 7(2), 26 CrossRef CAS.
- N. El Yamani, A. R. Collins, E. Runden-Pran, L. M. Fjellsbo, S. Shaposhnikov, S. Zienolddiny and M. Dusinska, In vitro genotoxicity testing of four reference metal nanomaterials, titanium dioxide, zinc oxide, cerium oxide and silver: towards reliable hazard assessment, Mutagenesis, 2017, 32, 117–126 CrossRef CAS.
- K. C. Nguyen, L. Richards, A. Massarsky, T. W. Moon and A. F. Tayabali, Toxicological evaluation of representative silver nanoparticles in macrophages and epithelial cells, Toxicol. In Vitro, 2016, 33, 163–173 CrossRef CAS.
- A. Kermanizadeh, M. Lohr, M. Roursgaard, S. Messner, P. Gunness, J. M. Kelm, P. Moller, V. Stone and S. Loft, Hepatic toxicology following single and multiple exposure of engineered nanomaterials utilising a novel primary human 3D liver microtissue model, Part. Fibre Toxicol., 2014, 11, 56 CrossRef.
- B. Ruttkay-Nedecky, L. Nejdl, J. Gumulec, O. Zitka, M. Masarik, T. Eckschlager, M. Stiborova, V. Adam and R. Kizek, The role of metallothionein in oxidative stress, Int. J. Mol. Sci., 2013, 14, 6044–6066 CrossRef CAS.
- M. Carriere, S. Sauvaigo, T. Douki and J. L. Ravanat, Impact of nanoparticles on DNA repair processes: current knowledge and working hypotheses, Mutagenesis, 2017, 32, 203–213 CrossRef CAS.
- D. Beyersmann and H. Haase, Functions of zinc in signaling, proliferation and differentiation of mammalian cells, BioMetals, 2001, 14, 331–341 CrossRef CAS.
- T. Schwerdtle, I. Hamann, G. Jahnke, I. Walter, C. Richter, J. L. Parsons, G. L. Dianov and A. Hartwig, Impact of copper on the induction and repair of oxidative DNA damage, poly(ADP-ribosyl)ation and PARP-1 activity, Mol. Nutr. Food Res., 2007, 51, 201–210 CrossRef CAS.
- A. Semisch, J. Ohle, B. Witt and A. Hartwig, Cytotoxicity and genotoxicity of nano - and microparticulate copper oxide: role of solubility and intracellular bioavailability, Part. Fibre Toxicol., 2014, 11, 10 CrossRef.
- R. Abbotts and D. M. Wilson, 3rd, Coordination of DNA single strand break repair, Free Radical Biol. Med., 2017, 107, 228–244 CrossRef CAS.
- H. J. Eom, N. Chatterjee, J. Lee and J. Choi, Integrated mRNA and micro RNA profiling reveals epigenetic mechanism of differential sensitivity of Jurkat T cells to AgNPs and Ag ions, Toxicol. Lett., 2014, 229, 311–318 CrossRef CAS.
- P. Kovvuru, P. E. Mancilla, A. B. Shirode, T. M. Murray, T. J. Begley and R. Reliene, Oral ingestion of silver nanoparticles induces genomic instability and DNA damage in multiple tissues, Nanotoxicology, 2015, 9, 162–171 CrossRef CAS.
- H. K. Lim, P. V. Asharani and M. P. Hande, Enhanced genotoxicity of silver nanoparticles in DNA repair deficient Mammalian cells, Front. Genet., 2012, 3, 104 Search PubMed.
- M. Wojewodzka, A. Lankoff, M. Dusinska, G. Brunborg, J. Czerwinska, T. Iwanenko, T. Stepkowski, I. Szumiel and M. Kruszewski, Treatment with silver nanoparticles delays repair of X-ray induced DNA damage in HepG2 cells, Nukleonika, 2011, 56, 29–33 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0en00533a |
| This journal is © The Royal Society of Chemistry 2021 |