
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface-water/groundwater boundaries affect seasonal PFAS concentrations and PFAA precursor transformations†
Andrea K.
Tokranov
 ab,
Denis R.
LeBlanc
b,
Heidi M.
Pickard
a,
Bridger J.
Ruyle
a,
Larry B.
Barber
c,
Robert B.
Hull
b,
Elsie M.
Sunderland
ad and
Chad D.
Vecitis
*a
ab,
Denis R.
LeBlanc
b,
Heidi M.
Pickard
a,
Bridger J.
Ruyle
a,
Larry B.
Barber
c,
Robert B.
Hull
b,
Elsie M.
Sunderland
ad and
Chad D.
Vecitis
*a
aHarvard John A. Paulson School of Engineering and Applied Sciences, Cambridge, MA 02138, USA. E-mail: vecitis@seas.harvard.edu
bU.S. Geological Survey, Northborough, MA 01532, USA
cU.S. Geological Survey, Boulder, CO 80303, USA
dDepartment of Environmental Health, Harvard T. H. Chan School of Public Health, Harvard University, Boston, MA 02115, USA
First published on 15th November 2021
Abstract
Elevated concentrations of per- and polyfluoroalkyl substances (PFAS) in drinking-water supplies are a major concern for human health. It is therefore essential to understand factors that affect PFAS concentrations in surface water and groundwater and the transformation of perfluoroalkyl acid (PFAA) precursors that degrade into terminal compounds. Surface-water/groundwater exchange can occur along the flow path downgradient from PFAS point sources and biogeochemical conditions can change rapidly at these exchange boundaries. Here, we investigate the influence of surface-water/groundwater boundaries on PFAS transport and transformation. To do this, we conducted an extensive field-based analysis of PFAS concentrations in water and sediment from a flow-through lake fed by contaminated groundwater and its downgradient surface-water/groundwater boundary (defined as ≤100 cm below the lake bottom). PFAA precursors comprised 45 ± 4.6% of PFAS (PFAA precursors + 18 targeted PFAA) in the predominantly oxic lake impacted by a former fire-training area and historical wastewater discharges. In shallow porewater downgradient from the lake, this percentage decreased significantly to 25 ± 11%. PFAA precursor concentrations decreased by 85% between the lake and 84–100 cm below the lake bottom. PFAA concentrations increased significantly within the surface-water/groundwater boundary and in downgradient groundwater during the winter months despite lower stable concentrations in the lake water source. These results suggest that natural biogeochemical fluctuations associated with surface-water/groundwater boundaries may lead to PFAA precursor loss and seasonal variations in PFAA concentrations. Results of this work highlight the importance of dynamic biogeochemical conditions along the hydrological flow path from PFAS point sources to potentially affected drinking water supplies.
Environmental significanceGroundwater contamination by per- and polyfluoroalkyl substances (PFAS) poses risks for drinking-water supplies across the United States. Surface-water/groundwater boundaries are frequently found along flow paths downgradient from point sources. Transport through these boundaries results in rapid changes in aqueous biogeochemistry, but the impact of these changes on PFAS transport and precursor transformation is poorly understood. We examined PFAS transport through surface-water/groundwater boundaries and discovered order-of-magnitude seasonal fluctuations in perfluoroalkyl acid (PFAA) concentrations and loss of ∼85% of influent precursors. PFAA concentrations were significantly and inversely associated with temperature and nitrate concentrations. This work highlights the possibility of substantial spatial and temporal variability in PFAS concentrations originating in boundary regions of high biogeochemical reactivity. |
Introduction
Human exposure to per- and polyfluoroalkyl substances (PFAS) has been linked to many health effects such as adverse effects on metabolism, endocrine disruption, and immunotoxicity.1–3 Aqueous film-forming foams (AFFF) containing PFAS and used during fire emergencies and fire-training activities have contaminated water supplies across the United States of America (USA).4–7 Processes such as sediment-water sorption and perfluoroalkyl acid (PFAA) precursor transformation (herein referred to as precursors) influence PFAS mobility and potential contamination of drinking-water supplies near PFAS source zones.8 Thus, understanding processes that control PFAS transport and transformation is essential for developing strategies for protecting groundwater and surface-water supplies.High PFAS concentrations and diverse precursors have been reported at AFFF-impacted sites.9–11 Some precursors degrade in the environment into terminal PFAA known to be a concern for human health.12 Biotransformation of precursors can also lead to the formation of persistent intermediate precursors,13,14 with altered mobility and transformation rates compared to the primary compounds. Prior work has suggested PFAA precursor biotransformation rates are slower in anaerobic environments compared to oxic conditions, and that biotransformation under anaerobic and oxic conditions produces different end-products.14–16 However, impacts of redox gradients and enhanced biological activity, as found at surface-water/groundwater boundaries, on precursor transformation and transport are poorly understood.
Numerous laboratory studies have illustrated that PFAS sorption at the sediment-water interface affects mobility in groundwater.17–19 The perfluorocarbon chain length (ηpfc), head group, and a variety of environmental properties (e.g., sediment organic carbon content, mineral and grain coating composition, pH, aqueous calcium and humic acid concentration) have all been shown to influence PFAS partitioning in laboratory experiments.17–21 However, it is not clear whether PFAS sorption in the field is affected by the gradients in redox and biological activity frequently observed across surface-water/groundwater boundaries. Experimental data on sorption of precursors is more limited due to difficulties in detecting and quantifying the diverse compounds that occur at AFFF-impacted sites, making new field-based observations especially useful.
Globally, lakes and rivers are commonly hydraulically connected to groundwater. PFAS-contaminated groundwater has been shown to contaminate surface waters, and vice versa in such systems.22–24 For example, our prior work on Cape Cod, Massachusetts (MA), USA, characterized a contaminated groundwater plume that discharges to a groundwater-flow-through glacial kettle lake (Ashumet Pond).6 Such glacial kettle lakes are common in northern latitudes and formed when blocks of glacial ice left behind by retreating ice sheets melted resulting in depressions (kettle holes) in the land surface that subsequently filled with water.25,26 Upgradient from Ashumet Pond, we detected PFAS and precursors throughout a 1.2 km-long longitudinal transect of the suboxic groundwater plume, highlighting the mobility of precursors in groundwater at this site.6 PFAS from the upgradient groundwater plume discharges to Ashumet Pond, and the PFAS-contaminated lake water then passes through a downgradient surface-water/groundwater boundary and recharges the downgradient aquifer. The surface-water/groundwater recharge boundary is a dynamic region of increased biological and chemical activity.25,27–29
Here we hypothesize that biogeochemical fluctuations such as oxygen content within the downgradient surface-water/groundwater boundary layer (defined for this study as the top 100 cm of the lake-bottom sediments) affects PFAS transport and transformation on both daily and seasonal timescales. To test this hypothesis and determine if there are significant changes in PFAS and precursor concentrations across the surface-water/groundwater boundary, we sampled groundwater, surface water, and sediment downgradient from the PFAS groundwater plume, with a focus on Ashumet Pond, and measured changes in 23 targeted PFAS, inferred precursor concentrations from the total oxidizable precursor (TOP) assay, and extractable organofluorine (EOF). The results of this work are used to better understand how shifts in biogeochemical conditions along the hydrological flow path affect PFAS transport and precursor persistence.
Materials and methods
Site overview
This study was conducted on western Cape Cod, MA, USA (Fig. 1). We measured PFAS concentrations along the hydrological pathway of the contaminated groundwater plume as it discharges to Ashumet Pond, mixes with the lake water, and subsequently recharges back into downgradient groundwater (Fig. 1B and C), thereby passing through groundwater/surface-water discharge and surface-water/groundwater recharge boundaries. Exchange between groundwater and surface water occurs predominantly in the shallow, near-shore zones of lakes, and seepage velocity decreases with increasing distance from the shoreline.30 At the study site, groundwater generally flows from northwest to southeast (average flow velocity ∼0.4 m d−1 (ref. 31–33)) and is intersected by two kettle lakes (Ashumet Pond and Johns Pond) along the flow path. The term “lake” is used to describe Ashumet Pond herein because of its large volume and surface area. Ashumet Pond is the focus of this study, unless otherwise stated. Ashumet Pond is the first kettle lake along the hydrological flow path of groundwater contaminated by PFAS from a former-fire training area (FTA) and former wastewater infiltration beds (Fig. 1A).6 Regular AFFF use at the FTA ended in 1985, and one additional application occurred in 1997 due to a fire emergency.6 The use of wastewater infiltration beds at the site was discontinued in 1995.6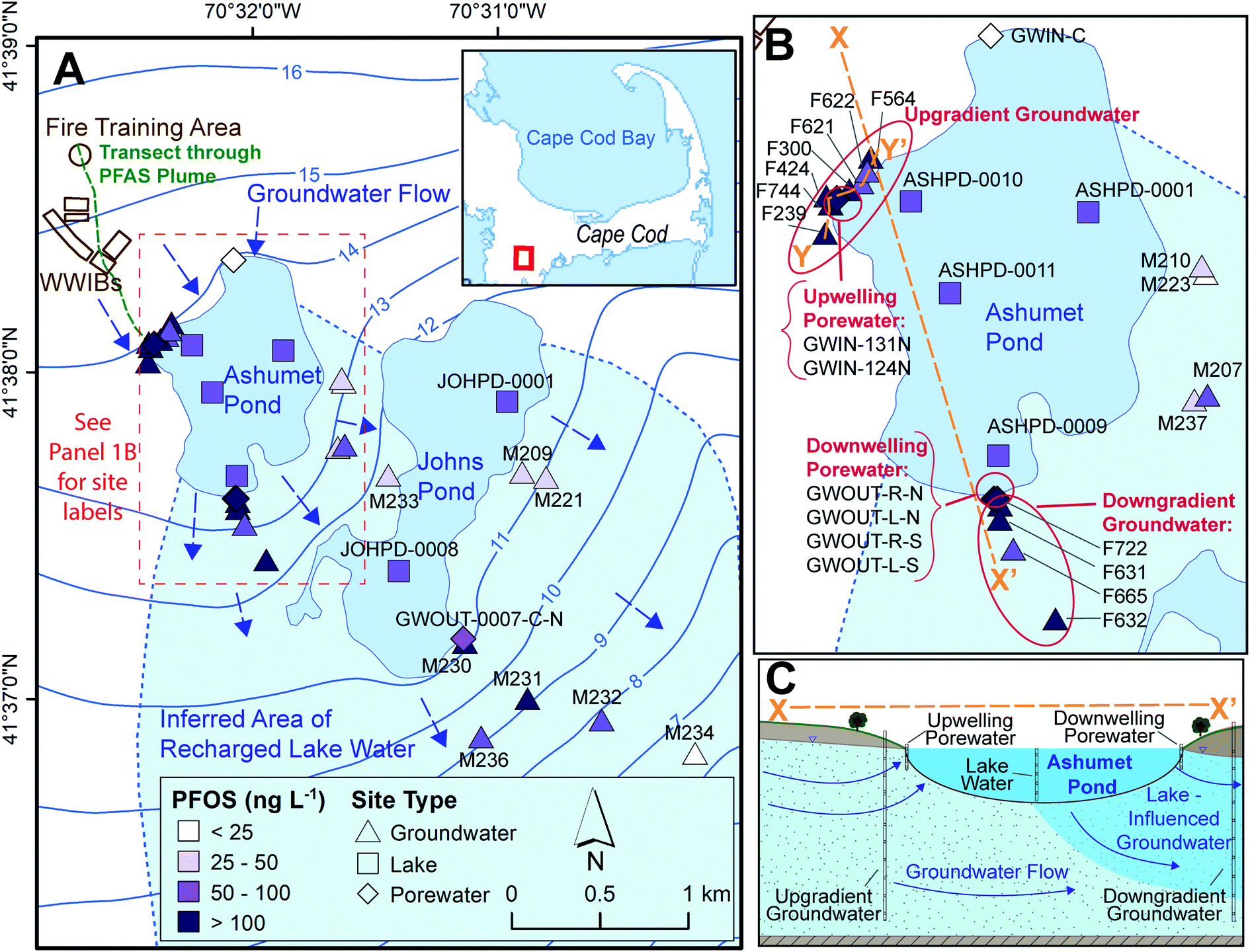 | ||
| Fig. 1 Study sampling locations. (A) Map of locations sampled between 2016 and 2019. WWIBs denotes wastewater infiltration beds. Most sampling locations include multiple sampling depths. Blue arrows indicate groundwater flow directions. Contours show elevation of water table in m above sea level, referenced to the National Geodetic Vertical Datum of 1929 (NGVD 29).38 The plume of per- and polyfluoroalkyl substances (PFAS)6 primarily discharges to Ashumet Pond near the westernmost extent of the lake. The maximum aqueous perfluorooctane sulfonate (PFOS) concentration is shown for each location.41 The locator map is from the U. S. Geological Survey National Hydrography Dataset, data refreshed April 2019.66 (B) Enlarged view of Ashumet Pond and associated sampling sites. (C) Cross-sectional representation of Ashumet Pond (not to scale) along section line X–X′ depicted in (B). | ||
This study focuses on the downwelling surface-water/groundwater recharge boundary rather than the upwelling boundary, because Ashumet Pond provides a spatially and temporally consistent source of PFAS concentrations to the downwelling groundwater. PFAS concentrations at the upwelling surface-water/groundwater boundary are highly influenced by location relative to the discharging groundwater contamination plume. This means that in the upwelling groundwater PFAS concentration fluctuations due to biogeochemical effects are not easily distinguishable from those due to the dynamic plume position.
Ashumet Pond is a mesotrophic groundwater-flow-through kettle lake with no permanent surface-water inflow or outflow.34 It has an area of 0.82 km2, an estimated hydraulic residence time of 1.6 years, and a maximum depth of 26 m.34,35 The lake level varied by about 1.1 m during this study (Fig. S1†).36 Residences with septic systems surround Ashumet Pond (Fig. 1A). The potential areal extent of recharged lake water from Ashumet Pond in the downgradient aquifer was determined using the groundwater-flow directions inferred from water-table maps prepared from field water-level measurements37 and simulated flow paths from calibrated groundwater-flow models38,39 (see “Inferred Area of Recharged Lake Water”, Fig. 1A, blue shading). The ESI† contains additional site details.
Sampling overview
We collected 289 water samples and 13 deionized (DI) water equipment blanks) between 2016 and 2019 following U.S. Geological Survey (USGS) field protocols.40 Ancillary data collected in the field during sampling included temperature, specific conductance, pH, dissolved oxygen (DO), and nitrate. Data are provided in the associated data release,41 and additional details on sample collection are provided in the ESI.† Water samples were collected from depth profiles in Ashumet Pond and Johns Pond (Fig. 1) in September 2017 (n = 40 samples, 4 profiles) and November 2017 (n = 10, 1 profile).Chemicals and materials
Table S2† contains the full names and abbreviations for all PFAS compounds analyzed in this study. PFAS standards (24 native compounds and 19 isotopically labeled compounds, Table S2†) were obtained from Wellington Laboratories (Guelph, Canada). Other chemicals used in the analyses are listed in the ESI.†PFAS extraction and analysis
Water samples were spiked with 40 μL of a 0.03 ng μL−1 internal standard solution before offline solid phase extraction (SPE) using Oasis® WAX cartridges (6 mL, 150 mg, 30 μm particle size, Waters, Milford, MA, USA) following established methods.6,42,43 Oasis WAX SPE cartridges were preconditioned with 4 mL of 0.1% ammonium hydroxide in methanol, 4 mL of methanol, and 4 mL of DI water, and then the 20 mL sample was added to the cartridge and placed under vacuum to yield a flow rate of ∼1 drop per second followed by a 4 mL DI water rinse before drying the cartridge under vacuum. The samples were eluted with 4 mL of methanol followed by 4 mL of 0.1% ammonium hydroxide in methanol, and the collected eluent was evaporated to dryness using an ultra-high-purity nitrogen gas stream, reconstituted in 0.75 mL of methanol, heated to 40 °C for 30 min, and vortexed. Finally, 0.75 mL of water was added, the sample was transferred to a polypropylene microcentrifuge tube and centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min, and the supernatant was transferred to a polypropylene autosampler vial for analysis. Samples were analyzed with an Agilent (Santa Clara, CA) 6460 triple quadrupole liquid chromatograph-tandem mass spectrometer (LC-MS/MS), as detailed in the ESI† and previously described.6 6:2 Fluorotelomer sulfonate (6:2 FtS) was removed from the reported results due to periodic blank contamination. Precursors that were quantified included 4:2 FtS, 8:2 FtS, N-methyl perfluorooctane sulfonamidoacetate (N-MeFOSAA), N-ethyl perfluorooctane sulfonamidoacetate (N-EtFOSAA), and perfluorooctane sulfonamide (FOSA). Further details including quality control, recovery, and precision results (Table S3) are presented in the ESI.†
000 rpm for 20 min, and the supernatant was transferred to a polypropylene autosampler vial for analysis. Samples were analyzed with an Agilent (Santa Clara, CA) 6460 triple quadrupole liquid chromatograph-tandem mass spectrometer (LC-MS/MS), as detailed in the ESI† and previously described.6 6:2 Fluorotelomer sulfonate (6:2 FtS) was removed from the reported results due to periodic blank contamination. Precursors that were quantified included 4:2 FtS, 8:2 FtS, N-methyl perfluorooctane sulfonamidoacetate (N-MeFOSAA), N-ethyl perfluorooctane sulfonamidoacetate (N-EtFOSAA), and perfluorooctane sulfonamide (FOSA). Further details including quality control, recovery, and precision results (Table S3) are presented in the ESI.†
TOP assay
The TOP assay44,45 was applied to 79 water samples (8 upwelling porewater, 16 lake water, 5 lake water near shore above porewater locations, 12 downwelling porewater, and 38 downgradient groundwater) from July 2017 to February 2018. All lake-water and porewater samples subjected to the TOP assay were from Ashumet Pond. Oxidation was completed by combining a 20 mL water sample with 20 mL of an aqueous potassium persulfate sodium hydroxide solution. Samples were placed in a heated (85 °C) water bath overnight, cooled, neutralized with hydrochloric acid, and extracted with offline SPE. For method validation, groundwater and lake-water samples were spiked, in triplicate, with 3 ng of 6:2 FtS, 8:2 FtS, N-MeFOSAA, N-EtFOSAA, and FOSA. These precursor concentrations were all reduced by >95%, and the molar recovery calculated from the produced perfluoroalkyl carboxylates (PFCA) was between 93% and 104%, indicating near-quantitative recovery. Details are available in the ESI,† and recovery and precision results are presented in Table S4.†The total concentration of oxidizable precursors (Σ precursors) for aqueous samples was inferred from the measured increases in PFCA with ηpfc = 3 to ηpfc = 8 produced by the TOP assay using a previously developed Bayesian inference method that has been applied to both AFFF and AFFF-impacted freshwater46,47 for all samples with measured increases in PFCA following the TOP assay. The model produces a distribution of inferred precursor concentrations using measurements of the PFCA produced upon oxidation and representative precursor yields from the literature, and their respective uncertainties.47 This inference method provides a more realistic estimate of precursor concentrations than the analytically detected changes in individual PFCA concentrations because it accounts for method and instrumentation uncertainties as well as incomplete recovery due to potential losses during oxidation to fluoride and ultra-short chain length PFCA.47 We report the median of inferred precursor concentrations. Details of the model and the criteria used to perform inference are provided in the ESI.†
Sediment PFAS and metals analysis
Sediment samples were extracted for PFAS analysis in triplicate with 0.1% ammonium hydroxide in methanol following previously developed methods.6,44,48 Selected sediment sample extracts were subjected to the TOP assay to estimate sediment precursor concentrations. Separate aliquots of sediment were extracted with 0.5 M hydrochloric acid, and extractable metal concentrations, including aluminum, iron, and manganese, were measured by inductively coupled plasma mass spectrometry (ICP-MS). See ESI† for sediment extraction and analysis details, and Tables S5–S12† for results and quality assurance and quality control.Extractable organofluorine (EOF)
EOF analysis was performed on a subset of samples from Ashumet Pond and downwelling groundwater following the procedure described in prior work.46 Briefly, weak anion exchange cartridges (Oasis® WAX) were preconditioned and then loaded with sample. Then, 10 mL of 0.01% v/v ammonium hydroxide (ACS grade, BDH® VWR International, Radnor, PA) in deionized water was passed through the cartridges to remove inorganic fluorine and the sample was eluted with LC-MS grade methanol (Honeywell, Charlotte, NC) and 0.1% v/v ammonium hydroxide in methanol, evaporated to dryness, and finally reconstituted into 1.5 mL methanol. Extracts were split 50:50 between the LC-MS/MS and the combustion ion chromatograph (CIC) with a combustion unit from Analytik Jena (Jena, Germany) and a 920 Absorber Module and 930 Compact IC Flex ion chromatograph from Metrohm (Herisau, Switzerland). Isotopically labeled internal standards (MPFAC-24 ES, Wellington Laboratories, Guelph, Ontario, Canada) were added to the LC-MS/MS fraction after the extract was split. See ESI† for details and results (Table S13†).
Statistics
All statistical tests were performed in Python version 3.6 using scipy.stats or scikit_posthocs. We used nonparametric statistics for our analysis as data frequently failed Shapiro–Wilk tests for normality (p < 0.05). The Mann–Whitney U-test (a non-parametric t-test) was used to evaluate significant differences between two populations. For comparisons of more than two populations of nonparametric data, the Kruskal–Wallis test with Dunn's post hoc test was used to identify groups that differed significantly.Results and discussion
Homogenous PFAS concentrations in the groundwater-fed kettle lake
Our results showed the upgradient groundwater PFAS plume primarily discharges to Ashumet Pond (Fig. 1 and S2†). This was confirmed by elevated PFAS concentrations in upwelling porewater at sites GWIN-131N and GWIN-124N (Fig. 1B). These results are consistent with previously reported groundwater discharge patterns.34,35,49–51In Ashumet Pond, mean PFAS concentrations were highest for perfluorohexane sulfonate (PFHxS) (74 ± 6.5 ng L−1), perfluorooctane sulfonate (PFOS) (50 ± 4.1 ng L−1), and perfluorooctanoate (PFOA) (29 ± 2.8 ng L−1). No significant temporal differences in concentrations of the sum of 23 targeted PFAS (Σ23PFAS) were observed between September and November 2017 (Mann–Whitney U test, p > 0.05), despite the collapse of the thermocline between the two sampling dates (Fig. S3†).41 Non-parametric Kruskal–Wallis with post hoc Dunn's test performed on all vertical profile sampling stations and for all seasons in Ashumet Pond revealed Σ23PFAS concentrations showed no significant spatial differences (Dunn's test, p > 0.05, Fig. S3†) except between two profiles in Ashumet Pond (ASHPD-0010 and ASHPD-0011, Dunn's test, p < 0.05). Inspection of the data reveals the median Σ23PFAS is 200 ng L−1 for ASHPD-0010 and 230 ng L−1 for ASHPD-0011, which is a 14% difference and within analytical uncertainty. These data suggest PFAS concentrations were spatially and temporally consistent in Ashumet Pond.
Elevated PFAS concentrations in wells downgradient from both Ashumet and Johns Pond confirm that PFAS are present in groundwater in the inferred area of recharged lake water.41 Mixing of the upgradient shallow groundwater PFAS plume (<30 m below land surface,6 Fig. S2†) of limited lateral dimensions (<200 m wide) with a surface-water body creates a much wider zone (>3 km; Fig. 1A) of PFAS-contaminated groundwater upon recharge of lake water to the downgradient groundwater. This general hydrological dispersion mechanism has important implications for other PFAS-contaminated regions with significant groundwater/lake interactions. For example, when a groundwater plume of limited lateral and vertical dimensions enters a well-mixed surface-water body, the spatial extent of contamination will disperse to the extent of the surface-water body. Further, if the surface-water body discharges downgradient to groundwater (as is the case here) and/or river outlets, the PFAS contamination will be transported to the entire area that receives and transmits the discharging water. Although dilution of PFAS from a more concentrated groundwater contamination plume will occur upon interaction with a surface-water body, the spatial footprint of contamination of PFAS has the potential to increase substantially, as observed here.
Precursor degradation within the surface-water/groundwater boundary layer
We hypothesized that precursors would biodegrade during transport through the biogeochemically active surface-water/groundwater boundary layer. Consistent with this hypothesis, the sum of total inferred precursors (Fig. 2, center) decreased by 85% and the inferred precursor mole fraction (Fig. 3) decreased by 59% between Ashumet Pond lake water sampled above the downwelling zone and samples taken 84–100 cm below the lake bottom (n = 5) (Fig. 2, center). In Ashumet Pond water (n = 19), the mole fraction of inferred precursors measured was statistically greater (non-parametric Kruskal–Wallis with post hoc Dunn's test) than in downwelling porewater (n = 12, p < 0.05) and downgradient groundwater (n = 37, p < 0.05) (Fig. 3). The presence of precursors in the downgradient groundwater indicates some precursors persist and are transported intact (or transformed to intermediate precursors) across the surface-water/groundwater boundary.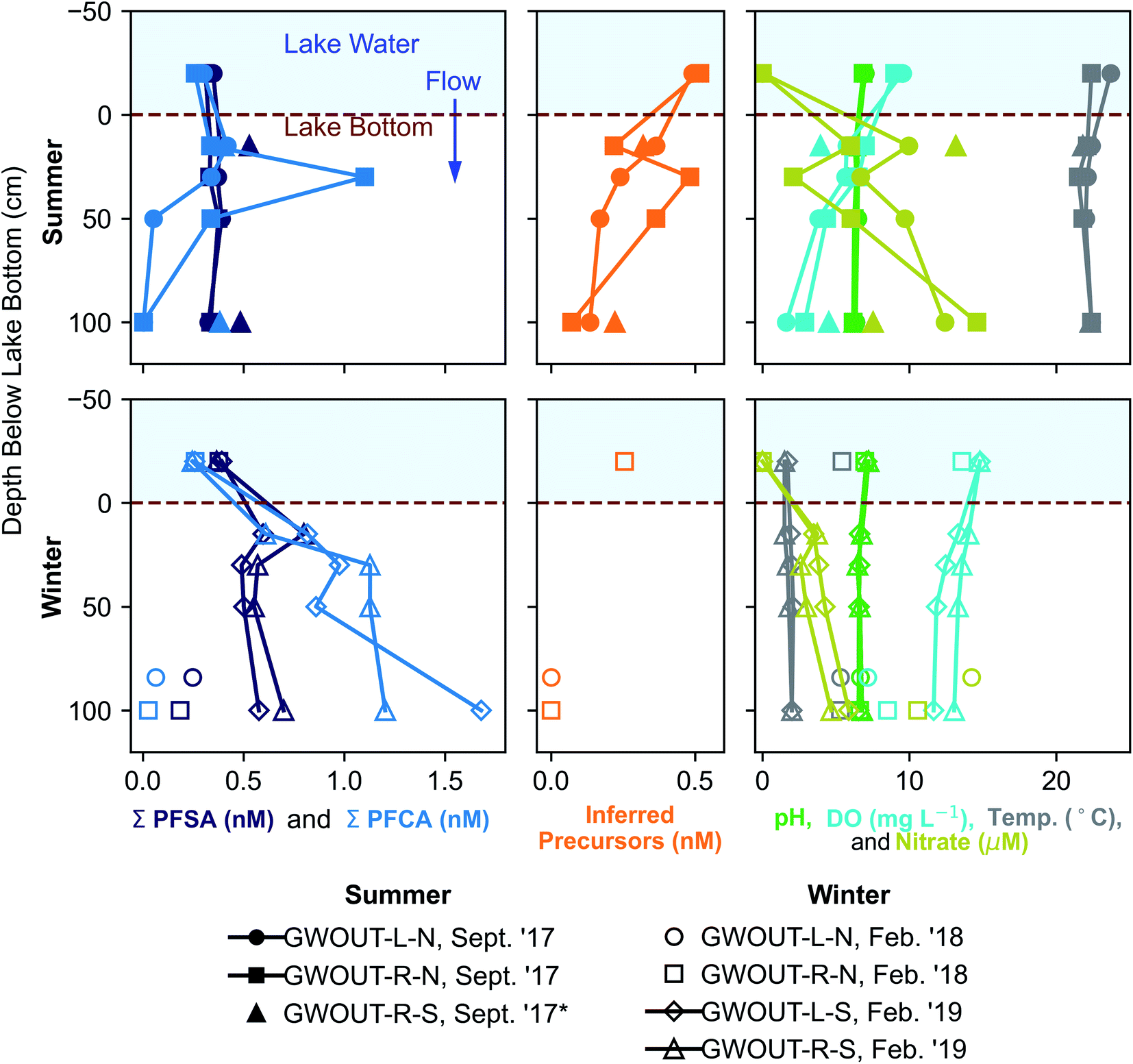 | ||
| Fig. 2 Seasonal fluctuations in perfluoroalkyl acids (PFAA) at the surface-water/groundwater boundary. Vertical depth profiles of the sum of perfluoroalkyl sulfonates (PFSA), the sum of perfluoroalkyl carboxylates (PFCA), inferred precursors, pH, dissolved oxygen (DO), temperature, and nitrate at the surface-water/groundwater boundary at downwelling sites in September 2017, February 2018, and February 2019. *Site GWOUT-R-S sampled in September 2017 (see legend) was landward of the lake shore due to low water level in Ashumet Pond; the samples are from 15 and 100 cm below the water table, located 40 cm below the ground surface. See Fig. S4† for profiles of individual PFAA concentrations. | ||
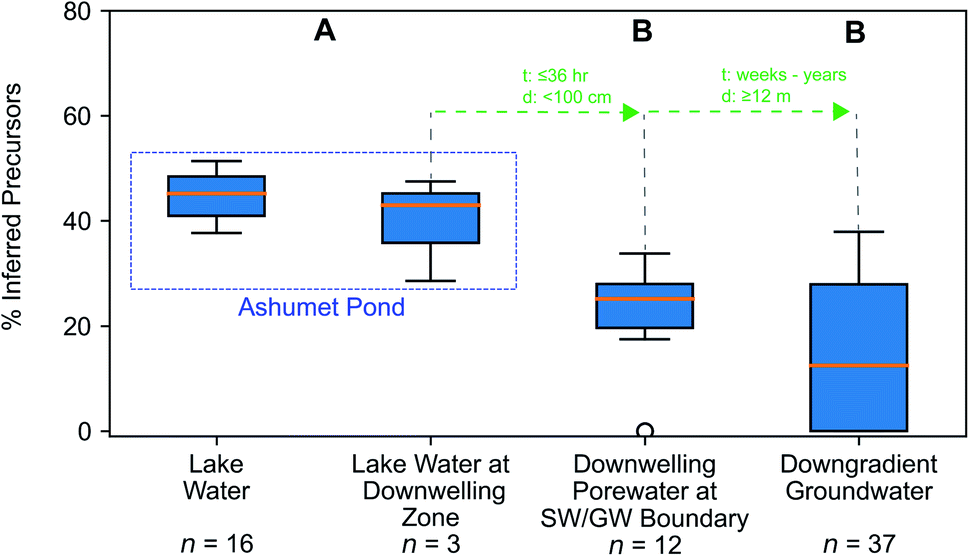 | ||
| Fig. 3 Precursor fraction along the hydrological flow path. The inferred molar fraction of precursors out of the total molar mass of per- and polyfluoroalkyl substances (PFAS) (inferred precursors, perfluoroalkyl carboxylates (PFCA), and perfluoroalkyl sulfonates (PFSA)) along the hydrological flow path. The box encompasses the 1st and 3rd quartiles of the data and the orange line denotes the median. Whiskers represent the 3rd quartile plus 1.5 times the interquartile range (upper bound) or the 1st quartile minus 1.5 times the interquartile range (lower bound) and extend only to the highest (upper bound) or lowest (lower bound) data point within that range. Circles represent data outside of the whisker range. Common letters above each boxplot indicate no significant difference (p < 0.05) between group comparisons using the non-parametric Kruskal–Wallis test and post hoc Dunn's test for multiple comparisons. For statistical tests, the three lake-water samples from the second boxplot from the left were combined with the 16 lake-water samples. The travel time (t) and distance (d) from one sample type to the next is indicated in green. Porewater velocities at the downwelling sites at Ashumet Pond have been observed to range from 67 to 440 cm d−1, assuming a porosity of 0.39,28,32,34,67 which equates to a maximum transport time of 36 hours from the lake bottom to 100 cm below the lake bottom. SW/GW stands for surface-water/groundwater. | ||
Both sorption and biotransformation could cause the observed reduction in precursors across the surface-water/groundwater boundary. Estimated sediment precursor concentrations (Table S7†) indicate only small increases in terminal PFAAs following the TOP assay compared to pre-TOP PFAA concentrations suggesting minimal precursor sorption. A statistically significant inverse correlation (Spearman, r = −0.62, p < 0.05) was observed between inferred precursor concentrations and nitrate in porewater downwelling from Ashumet Pond, suggesting the decrease in concentration is related to biotransformation. Nitrate concentrations increased below the lake bottom in the downwelling zone, reaching a maximum of 15 μM at 100 cm below the lake bottom in the September 2017 samples (temperature >20 °C, Fig. 2). This increase in nitrate is consistent with previous observations at this field site and has been hypothesized to reflect biomineralization of organic nitrogen coupled to nitrification.34 These results suggest an association between conditions that lead to increased nitrate concentrations and precursor loss. Prior work has shown that the surface-water/groundwater boundary is a zone of increased microbial activity relative to the overlying water column owing to the greater nutrient and microbial abundance in the sediments compared to lake water.52,53 Nitrification is microbially driven and typically requires oxygen, ammonia, and redox gradients to proceed.54,55 Nitrification dominates over denitrification in areas with short porewater residence times, as found at this site.56 The production of nitrate (Fig. 2) indicates an active microbial community, which likely also facilitates biodegradation of PFAS precursor compounds during passage through the surface-water/groundwater boundary and would explain the inverse correlation between nitrate and inferred precursor concentrations.
Seasonal changes in PFAA concentrations at the surface-water/groundwater boundary
PFCA and perfluoroalkyl sulfonates (PFSA) concentrations in downwelling porewater (Fig. 2 and 4) and downgradient groundwater were significantly different (Mann–Whitney U test, p < 0.05) between seasons, with lower average concentrations in the summer (September) and higher average concentrations in the winter (February). These seasonal differences are supported by a strong inverse correlation (highest concentrations at low temperature) between temperature and the sum of PFCA concentrations (Spearman, r = −0.50, p < 0.05, n = 102) and the sum of PFSA concentrations (Spearman, r = −0.38, p < 0.05, n = 102) for all porewater and groundwater directly downgradient from Ashumet Pond (Fig. 4). Ashumet Pond is the source water for downwelling porewater and downgradient groundwater. Temporal trends in the porewater and groundwater are not linked to PFAA concentration changes in the source water because PFAA concentrations in Ashumet Pond water were consistent across all sampling dates (Fig. 4).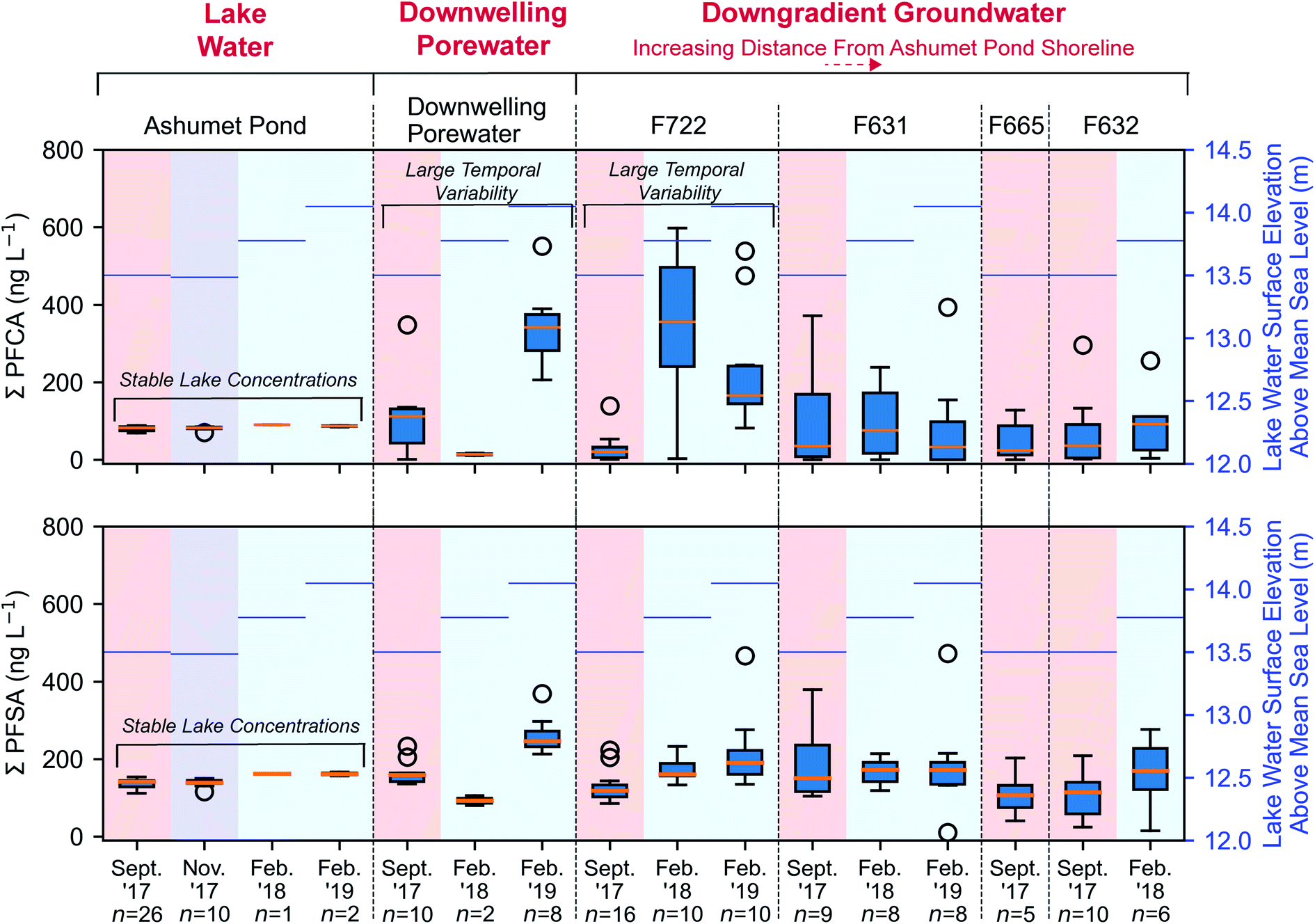 | ||
| Fig. 4 Perfluoroalkyl acid (PFAA) concentrations along the hydrological flow path. The sum of measured concentrations of perfluoroalkyl carboxylates (PFCA) and perfluoroalkyl sulfonates (PFSA) along the hydrological flow path (from left to right). The box encompasses the 25th and 75th percentiles and the orange line represents the 50th percentile. Whiskers represent the 3rd quartile plus 1.5 times the interquartile range (upper bound) or the 1st quartile minus 1.5 times the interquartile range (lower bound). Circles represent outliers. Lake-water samples from February 2018 and 2019 were collected 20 cm above the lake bottom at the downwelling-porewater sampling locations. The mean lake water surface elevation above mean sea level (National Geodetic Vertical Datum of 1929) in Ashumet Pond36 associated with each sampling event is shown on the right axis in blue. Downwelling-zone profiles can be found in Fig. 2. | ||
At the downwelling surface-water/groundwater boundary (GWOUT) of Ashumet Pond, the mean concentration in porewater increased from September (Σ23PFAS = 270 ng L−1, Σ11PFCA = 110 ng L−1, Σ7PFSA = 160 ng L−1) to February (Σ23PFAS = 510 ng L−1, Σ11PFCA = 280 ng L−1, Σ7PFSA = 230 ng L−1). Concentrations differed significantly between the September and February samples for Σ23PFAS, Σ11PFCA, and Σ7PFSA (Mann–Whitney U test, p < 0.05). Porewater PFCA concentrations varied with depth over the 100 cm porewater profile. In September 2017, the lowest PFCA concentrations were observed in the deepest, 100 cm samples (Fig. 2, upper left), suggesting loss of aqueous PFCA with depth. In contrast, PFCA concentrations increased with increasing depth for the February 2019 samples (Fig. 2, lower left). This is a large (>3-fold) increase in aqueous PFCA concentrations, considering the shallow depth of the boundary-layer porewater samples (<100 cm below the lake bottom) and short hydraulic residence times (<36 h) relative to other regions along the hydrological flow path such as the upgradient groundwater and Ashumet Pond lake water. The two samples of downwelling porewater from February 2018 appeared to be more consistent with the trends seen in September 2017 samples (Fig. 2). However, the general water-quality parameters (high nitrate concentrations, an intermediate temperature of 5.3 °C, and intermediate DO concentrations, Fig. 2, lower right, circles and squares)41 indicate the February 2018 porewater had not completely transitioned to wintertime conditions. In contrast, February 2019 conditions were indicative of a complete wintertime transition, with mean water temperatures of 1.8 °C and high DO concentrations (Fig. 2, lower right, diamonds and triangles).
Similar to the porewater at the surface-water/groundwater boundary, PFCA concentrations in F722, the first groundwater well downgradient from Ashumet Pond, were also significantly different than in the lake water at all sampling dates (Mann–Whitney, p < 0.05) and varied by an order of magnitude across sampling times. Specifically, well F722 displayed low PFCA concentrations (mean ± std. dev.: 27 ± 34 ng L−1) in September 2017 and high concentrations in February 2018 (340 ± 200 ng L−1) and February 2019 (230 ± 150 ng L−1) (Fig. 4). PFSA concentrations followed similar, but less pronounced, trends than PFCA, with low September 2017 concentrations (130 ± 38 ng L−1) and increased concentrations in February 2018 (170 ± 30 ng L−1) and February 2019 (220 ± 100 ng L−1) (Fig. 4). September 2017 PFCA and PFSA concentrations were statistically different from February 2018 and February 2019 concentrations at well F722 (Mann–Whitney, p < 0.05). PFCA and PFSA concentrations were not significantly different for February 2018 and February 2019 (Mann–Whitney, p > 0.05), supporting the seasonal pattern in concentrations. Similar trends are observed for both PFCA and PFSA when considering nanomolar units (Fig. S5†) and individual PFSA compounds (Fig. S6–S10†).
PFCA and PFSA concentrations in wells with increasing distance from Ashumet Pond (F631, F665, F632) were not statistically different (Mann–Whitney, p > 0.05) from those in Ashumet Pond except for PFSA concentrations in the February 2018 samples from well F631 and the September 2017 samples from well F632. PFAA concentrations in wells aligned along the hydrological flow path downgradient from Ashumet Pond were more variable than those sampled within the lake (Fig. 4). This reflects the observed variability in PFAA concentrations at the surface-water/groundwater boundary. Processes such as mixing, vertical layering of seasonal waters, and accumulation of groundwater recharge at the water table along the flow path likely dampen the signal of seasonal fluctuations in PFAA groundwater concentrations farther away from the lake.
Temporal changes in PFAA concentration cannot be explained by precursor transformations alone because the inferred total precursor concentration in Ashumet Pond water is less than the fluctuation in PFAA concentration. Increases in median PFAA (0.73 nM) concentrations in downwelling porewater between September and February exceeded the median estimate for the concentrations of all inferred precursors in Ashumet Pond samples (0.43 ± 0.09 nM). Additionally, biotransformation of precursors would be expected to be more rapid in summer when there is high biological activity due to increased temperatures.57 This would lead to higher downgradient aqueous PFAA concentrations in summer, not in winter (as observed here). Temporal changes in EOF concentrations (Table S13†) further support the conclusion that temporal differences in PFAA are not driven by precursor transformation. Concentrations of EOF decrease by 73% from Ashumet Pond (32 ± 3.6 nM F) to MA-FSW 722-05BKT (8.7 nM F) in September 2017. Conversely, concentrations of EOF at MA-FSW 722-05BKT in February 2018 (60.7 nM F) and 2019 (34.2 nM F) are similar to or exceed concentrations in Ashumet Pond. EOF captures both terminal compounds and precursors, thus total EOF concentrations would not be expected to change temporally if transformation of precursors into PFAAs at the surface-water/groundwater boundary drove PFAA fluctuations.
Adsorption at the air–water interface cannot explain the observed temporal trends at the surface water/groundwater boundary either. For example, GWOUT-L-N was fully submerged throughout the sampling campaign, yet PFAA loss with depth was still observed in the summer. Also, temporal trends were strongest for PFCA, including short chain-length compounds like perfluoropentanoate (PFPeA) that would be expected to sorb less strongly at the air–water interface (Fig. S6–S10†).58,59 While neither precursor transformation nor adsorption at the air–water interface can explain the observed seasonal trends, the reduction of porewater PFAA concentrations in summertime and increased porewater PFAA concentrations in wintertime indicates a reversible phenomenon. The strong inverse correlation between nitrate and the sum of PFCA concentrations (Spearman, r = −0.55, p < 0.05, n = 102) and sum of PFSA concentrations (r = −0.28, p < 0.05, n = 102) for all porewater and groundwater directly downgradient from Ashumet Pond suggests biological activity is driving the temporal variations, as further discussed in the following section.
The groundwater and porewater results indicate that: (1) the surface-water/groundwater boundary is the source of fluctuations in downgradient groundwater concentrations, (2) concentrations in downwelling porewater and nearby downgradient groundwater change temporally (seasonally), with lower concentrations in the summer (September) and higher concentrations in the winter (February), and (3) these temporal changes are not driven by precursor transformation or the air–water interface. There have been few studies investigating temporal trends of PFAS in groundwater or PFAS transport across surface-water/groundwater boundaries. Steele et al.60 found no statistically significant temporal trend in groundwater data from a site in Alaska, but did find (weakly) statistically significant temporal trends for groundwater at Pease Air Force Base in New Hampshire. Our results indicate stronger seasonal trends, likely owing to the surface-water/groundwater boundary investigated in this work. Another study investigated transport of PFAS between groundwater and surface water, but was primarily focused on mass transfer rates.24 The data presented here provide new information about the importance of the surface-water/groundwater boundary layer and its potential to drive temporal fluctuations in downgradient groundwater.
Elevated PFCA sediment/water Kd values at lake downwelling sites
To investigate the processes driving the PFAA (particularly PFCA) temporal variability, PFAS concentrations in sediment from the surface-water/groundwater boundary layer were analyzed at upwelling and downwelling sites on Ashumet Pond (Fig. S11–S13†). The sediment PFAS composition at the surface-water/groundwater boundary at downwelling sites (GWOUT) was dominated by C4–C7 PFCA (21–76% of the total molar mass, n = 8), whereas at upwelling sites (GWIN) the sediment PFAS composition had only a small molar fraction of C4–C7 PFCA (0.80–3.2%, n = 4).Field-derived sediment-water partition coefficients (Kd) were determined from porewater and solid-phase measurements (Fig. 1B, S12 and Table S8†). In the upwelling zone (GWIN), PFCA and PFSA homologues of equivalent perfluorocarbon chain length had similar Kd values, as expected, and varied by a factor of ≤5.3 within each sample (Fig. S12 and Table S8†). The samples from the upwelling zone also followed expected trends of increasing Kd values with increasing perfluorinated chain length (for ηpfc > 5, Fig. S12†). Kd values typically decreased with increasing sediment depth (Table S8†).
By contrast, in the downwelling zone (GWOUT), field-derived PFCA Kd values were larger than PFSA Kd values for homologues of equivalent ηpfc (for ηpfc < 8, Fig. S12†) and varied by up to a factor of 28 within each sample (Fig. S12 and Table S8†). Additionally, downwelling-zone PFCA Kd values were not always dependent on chain length, as observed for site GWOUT-R-N 15–30 cm (Sept. 2017), where PFCA Kd values had no chain-length dependence for ηpfc ≤ 8 (Fig. S12†). Kd values typically decreased with depth (Table S8†) for ηpfc > 6 and increased with depth for ηpfc ≤ 6. The high PFCA Kd values compared to PFSA Kd values was unexpected but is consistent with the temporal trends observed in porewater and downgradient groundwater that were particularly pronounced for PFCA.
Prior work, based primarily on laboratory studies, has shown perfluorocarbon chain length is a primary factor mediating sorption to sediments, with each additional perfluorinated carbon increasing the Kd by 0.50–0.60 log units.5,17 Chain-length effects are less prominent for PFAA with ηpfc < 5.61,62 PFSA are reported to adsorb more strongly (+0.23 log units) than PFCA with the same perfluorocarbon chain length due to headgroup (sulfonate vs. carboxylate) effects.5,17 Sorption to solids has also been shown to increase with increasing sediment organic carbon content, iron and aluminum oxide grain coatings, and divalent cation concentrations.17,19,63 However, these studies find that the trends observed for chain-length and headgroup are typically preserved. Our field-derived Kd results display important differences compared to the lab-based results. Specifically, chain-length dependent relationships are not always observed, and PFCA sorbed more strongly than PFSA in the downwelling surface-water/groundwater boundary examined during this field study.
There are important geochemical and hydrologic differences between the upgradient and downgradient sides of Ashumet Pond that may help explain the observed differences in PFAA sorption. Measured sediment concentrations of iron (Fe) and manganese (Mn) (Table S9†) were typically higher at the upwelling sites (Fe: 360 ± 440 μg g−1; Mn: 968 ± 1000 μg g−1) compared to downwelling sites (136 ± 45 μg g−1 for Fe and 32 ± 24 μg g−1 for Mn), where increased sorption for PFCA was observed. Total carbon in sediment near the downwelling site is higher (92 ± 1 μmol g−1) than in the sediment near the upwelling sites (∼43 μmol g−1).34 Higher sediment organic carbon would lead to higher expected Kd values at the downgradient site, as reported here (Fig. S12 and Table S8†), but PFAS sorption by organic carbon is also expected to result in a larger Kd for PFSA than for PFCA, which contrasts with the field observations presented here.17 Porewater velocity measurements indicate flow rates are typically higher in the downwelling zone compared to the upwelling zone (Table S14†). Higher flow rates favor non-equilibrium conditions. However, non-equilibrium conditions and/or sorption nonlinearity cannot explain the large PFCA Kd values and temporal aqueous PFAA trends, especially given the homogenous concentrations in the Ashumet Pond source water.
The field evidence suggests that PFAA are reversibly sequestered in summer (during times of high biogeochemical activity) and subsequently released in the winter (during times when biological activity is diminished).52,57 Biologically mediated sorption (such as sorption to microbial biofilms or incorporation into lipid bilayers) would explain both the temporal trends in downgradient porewater and groundwater and also the strong PFCA Kd values observed in sediment. On the upgradient side of the lake, suboxic PFAS-containing groundwater containing recalcitrant dissolved organic carbon concentrations of 0.78–1.1 mgC L−1 discharges to the largely oxic lake water.34,41 On the downgradient side of the lake, oxic lake water containing more labile and higher concentrations of dissolved organic carbon (∼2 mgC L−1)34,41 recharges the groundwater and stimulates microbial growth (e.g. the porewater DO concentrations progressively decrease with depth below the lake bottom in the summer (Fig. 2), indicating biological consumption). In winter, the porewater within 100 cm of the lake bottom remains oxygen-rich (Fig. 2), likely owing to a decrease in biological activity during the winter. Observed temporal fluctuations in aqueous PFCA and PFSA concentrations are associated with cyclical changes in biogeochemical conditions (DO, temperature, nitrate) in the downgradient porewater (Fig. 2 and 4). There is a statistically significant inverse correlation (Spearman, r = −0.69, p < 0.05) between PFCA and nitrate concentrations at well F722 for all available sample data, suggesting sorption may be related to biological processes. Sorption to bacteria, lipid bilayers, and proteins has been shown to be stronger for PFSA than PFCA with similar ηpfc,64,65 in contrast to observations presented here. However, it has also been reported that live Gram-negative bacteria may accumulate more PFAS (particularly PFOA) than dead bacteria.64 This is consistent with high porewater and groundwater aqueous PFCA concentrations in winter, when the sediment is less biologically active and thus may sorb less PFCA mass and/or potentially release PFCA mass back to the aqueous phase as the local algal and microbial activity is reduced. Similarly, the increased porewater and groundwater aqueous PFCA concentrations in winter coincide with a general decrease in downwelling PFCA Kd values from September 2017 to February 2019 (Fig. S12†). The findings here suggest that typical oxic laboratory partitioning experiments using sterilized, dried sediments may not reflect partitioning in a natural environment where a range of dynamic processes and biogeochemical conditions occur, such as those found at the surface-water/groundwater recharge boundary.
Conclusion
This study investigated variability in PFAS concentrations between a surface-water body and the region where recharge of surface water to the downgradient groundwater occurs and disperses PFAS over an area that is much larger than the original upgradient groundwater plume. This work demonstrates that sorption and transformation mechanisms at the surface-water/groundwater boundary are important in reducing transport of precursors into downgradient groundwater. A fraction of the precursors persists in the downgradient groundwater, indicating a need for better accounting of their contribution to the total PFAS burden in the environment, even at locations distant (>km) from sites of direct AFFF application. PFAS concentrations in source water from Ashumet Pond were constant over time and space, but fluctuated significantly at the surface-water/groundwater boundary at the downgradient side of the lake, which can impact downgradient PFAS concentrations in groundwater that may be used as a drinking-water supply. The spatial and temporal variability in aqueous PFAS concentrations found at this site, where the hydrology is well known and the well-characterized source water has consistent PFAS concentrations, indicate that single space-time-point sampling is insufficient to fully characterize PFAS concentrations in groundwater and may neglect substantial temporal concentration fluctuations. The temporal variability in groundwater PFCA concentrations (and, to a lesser degree, PFSA concentrations) and large field-derived Kd values for PFCA suggest that there are processes that have not been accounted for and need to be examined further to allow for accurate modeling and prediction of PFAS fate, transport, and risk. The combined evidence of temporal trends and strong inverse correlations with nitrate suggests that future work should investigate biologically driven sorption mechanisms and whether rapid biotransformation of precursors affects field Kd values. Specifically, future studies should incorporate analysis of microbial community and abundance and investigate the interconnection between redox conditions, biological activity, and PFAS sorption.Data availability
For U.S. Geological Survey data release see: http://doi.org/10.5066/P9HPBFRT.Author contributions
Conceptualization, A. K. T. and D. R. L.; formal analysis, A. K. T.; funding acquisition, E. M. S. and C. D. V.; investigation, A. K. T., D. R. L, H. M. P, B. J. R., and R. B. H; methodology, A. K. T., D. R. L., and B. J. R.; resources, D. R. L., E. M. S., and C. D. V.; supervision, E. M. S., and C. D. V.; validation, A. K. T.; visualization, A. K. T.; writing – original draft, A. K. T., E. M. S., and C. D. V.; writing – review and editing, A. K. T., D. R. L., H. M. P., L. B. B., B. J. R., R. B. H., E. M. S., and C. D. V.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Institute of Environmental Health Sciences Superfund Research Program (P42ES027706) and the U.S. Geological Survey (USGS) Toxic Substances Hydrology Program. We thank John Karl Böhlke, Richard Smith, and Douglas Kent (USGS) for scientific feedback; and Timothy McCobb, Deborah Repert, Jennifer Underwood, Stephen O'Brien, Paul Bliznik, and Colin Sweeney (USGS) for assistance in sample collection; and Prentiss Balcom (Harvard) for sediment ICP-MS analyses. Any use of trade, firm, or product names is for description purposes only and does not imply endorsement by the U.S. Government.References
- P. Grandjean, E. W. Andersen, E. Budtz-Jørgensen, F. Nielsen, K. Mølbak, P. Weihe and C. Heilmann, Serum vaccine antibody concentrations in children exposed to perfluorinated compounds, JAMA, J. Am. Med. Assoc., 2012, 307, 391–397 CrossRef CAS PubMed.
- J. W. Nelson, E. E. Hatch and T. F. Webster, Exposure to polyfluoroalkyl chemicals and cholesterol, body weight, and insulin resistance in the general U.S. population, Environ. Health Perspect., 2010, 118, 197–202 CrossRef CAS PubMed.
- Q. Sun, G. Zong, D. Valvi, F. Nielsen, B. Coull and P. Grandjean, Plasma concentrations of perfluoroalkyl substances and risk of Type 2 diabetes: A prospective investigation among U.S. women, Environ. Health Perspect., 2018, 126, 037001 CrossRef PubMed.
- R. H. Anderson, G. C. Long, R. C. Porter and J. K. Anderson, Occurrence of select perfluoroalkyl substances at U.S. Air Force aqueous film-forming foam release sites other than fire-training areas: Field-validation of critical fate and transport properties, Chemosphere, 2016, 150, 678–685 CrossRef CAS PubMed.
- M. E. McGuire, C. Schaefer, T. Richards, W. J. Backe, J. A. Field, E. Houtz, D. L. Sedlak, J. L. Guelfo, A. Wunsch and C. P. Higgins, Evidence of remediation-induced alteration of subsurface poly- and perfluoroalkyl substance distribution at a former firefighter training area, Environ. Sci. Technol., 2014, 48, 6644–6652 CrossRef CAS PubMed.
- A. K. Weber, L. B. Barber, D. R. LeBlanc, E. M. Sunderland and C. D. Vecitis, Geochemical and hydrologic factors controlling subsurface transport of poly- and perfluoroalkyl substances, Cape Cod, Massachusetts, Environ. Sci. Technol., 2017, 51, 4269–4279 CrossRef CAS PubMed.
- X. C. Hu, D. Q. Andrews, A. B. Lindstrom, T. A. Bruton, L. A. Schaider, P. Grandjean, R. Lohmann, C. C. Carignan, A. Blum, S. A. Balan, C. P. Higgins and E. M. Sunderland, Detection of poly- and perfluoroalkyl substances (PFASs) in U.S. drinking water linked to industrial sites, military fire training areas, and wastewater treatment plants, Environ. Sci. Technol. Lett., 2016, 3(10), 344–350 CrossRef CAS PubMed.
- M. Filipovic, A. Woldegiorgis, K. Norström, M. Bibi, M. Lindberg and A.-H. Österås, Historical usage of aqueous film forming foam: A case study of the widespread distribution of perfluoroalkyl acids from a military airport to groundwater, lakes, soils and fish, Chemosphere, 2015, 129, 39–45 CrossRef CAS PubMed.
- B. J. Place and J. A. Field, Identification of novel fluorochemicals in aqueous film-forming foams used by the US military, Environ. Sci. Technol., 2012, 46, 7120–7127 CrossRef CAS PubMed.
- K. A. Barzen-Hanson, S. C. Roberts, S. Choyke, K. Oetjen, A. McAlees, N. Riddell, R. McCrindle, P. L. Ferguson, C. P. Higgins and J. A. Field, Discovery of 40 classes of per- and polyfluoroalkyl substances in historical aqueous film-forming foams (AFFFs) and AFFF-impacted groundwater, Environ. Sci. Technol., 2017, 51, 2047–2057 CrossRef CAS PubMed.
- L. A. D'Agostino and S. A. Mabury, Identification of novel fluorinated surfactants in aqueous film forming foams and commercial surfactant concentrates, Environ. Sci. Technol., 2014, 48, 121–129 CrossRef PubMed.
- E. M. Sunderland, X. C. Hu, C. Dassuncao, A. K. Tokranov, C. C. Wagner and J. G. Allen, A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFASs) and present understanding of health effects, J. Expo. Sci. Environ. Epidemiol., 2019, 29, 131–147 CrossRef CAS PubMed.
- K. C. Harding-Marjanovic, E. F. Houtz, S. Yi, J. A. Field, D. L. Sedlak and L. Alvarez-Cohen, Aerobic biotransformation of fluorotelomer thioether amido sulfonate (Lodyne) in AFFF-amended microcosms, Environ. Sci. Technol., 2015, 49, 7666–7674 CrossRef CAS PubMed.
- S. Yi, K. C. Harding-Marjanovic, E. F. Houtz, Y. Gao, J. E. Lawrence, R. V. Nichiporuk, A. T. Iavarone, W.-Q. Zhuang, M. Hansen, J. A. Field, D. L. Sedlak and L. Alvarez-Cohen, Biotransformation of AFFF component 6:2 fluorotelomer thioether amido sulfonate generates 6:2 fluorotelomer thioether carboxylate under sulfate-reducing conditions, Environ. Sci. Technol. Lett., 2018, 5, 283–288 CrossRef CAS PubMed.
- S. Zhang, X. Lu, N. Wang and R. C. Buck, Biotransformation potential of 6:2 fluorotelomer sulfonate (6:2 FTSA) in aerobic and anaerobic sediment, Chemosphere, 2016, 154, 224–230 CrossRef CAS PubMed.
- S. Zhang, B. Szostek, P. K. McCausland, B. W. Wolstenholme, X. Lu, N. Wang and R. C. Buck, 6:2 and 8:2 Fluorotelomer alcohol anaerobic biotransformation in digester sludge from a WWTP under methanogenic conditions, Environ. Sci. Technol., 2013, 47, 4227–4235 CrossRef CAS PubMed.
- C. P. Higgins and R. G. Luthy, Sorption of perfluorinated surfactants on sediments, Environ. Sci. Technol., 2006, 40, 7251–7256 CrossRef CAS PubMed.
- H. Campos Pereira, M. Ullberg, D. B. Kleja, J. P. Gustafsson and L. Ahrens, Sorption of perfluoroalkyl substances (PFASs) to an organic soil horizon – Effect of cation composition and pH, Chemosphere, 2018, 207, 183–191 CrossRef CAS PubMed.
- F. Li, X. Fang, Z. Zhou, X. Liao, J. Zou, B. Yuan and W. Sun, Adsorption of perfluorinated acids onto soils: Kinetics, isotherms, and influences of soil properties, Sci. Total Environ., 2019, 649, 504–514 CrossRef CAS PubMed.
- C. Y. Tang, Q. Shiang Fu, D. Gao, C. S. Criddle and J. O. Leckie, Effect of solution chemistry on the adsorption of perfluorooctane sulfonate onto mineral surfaces, Water Res., 2010, 44, 2654–2662 CrossRef CAS PubMed.
- F. Wang, K. Shih and J. O. Leckie, Effect of humic acid on the sorption of perfluorooctane sulfonate (PFOS) and perfluorobutane sulfonate (PFBS) on boehmite, Chemosphere, 2015, 118, 213–218 CrossRef CAS PubMed.
- M. A. Briggs, A. K. Tokranov, R. B. Hull, D. R. LeBlanc, A. B. Haynes and J. W. Lane, Hillslope groundwater discharges provide localized stream ecosystem buffers from regional per- and polyfluoroalkyl substances contamination, Hydrol. Processes, 2020, 34, 2281–2291 CrossRef CAS.
- B. M. Sharma, G. K. Bharat, S. Tayal, T. Larssen, J. Bečanová, P. Karásková, P. G. Whitehead, M. N. Futter, D. Butterfield and L. Nizzetto, Perfluoroalkyl substances (PFAS) in river and ground/drinking water of the Ganges River basin: Emissions and implications for human exposure, Environ. Pollut., 2016, 208, 704–713 CrossRef CAS PubMed.
- M.-A. Pétré, D. P. Genereux, L. Koropeckyj-Cox, D. R. U. Knappe, S. Duboscq, T. E. Gilmore and Z. R. Hopkins, Per- and polyfluoroalkyl substance (PFAS) transport from groundwater to streams near a PFAS manufacturing facility in North Carolina, USA, Environ. Sci. Technol., 2021, 55, 5848–5856 CrossRef PubMed.
- T. C. Winter, J. W. Harvey, O. L. Franke and W. M. Alley, Ground water and surface water—A single resource, U.S. Geological Survey Circular 1139, U.S. Geological Survey, Reston, VA, 1998 Search PubMed.
- R. M. Thorson, Beyond Walden: The Hidden History of America's Kettle Lakes and Ponds, Walker and Company, New York, 2009 Search PubMed.
- J. Lewandowski, K. Meinikmann, G. Nützmann and D. O. Rosenberry, Groundwater – the disregarded component in lake water and nutrient budgets. Part 2: Effects of groundwater on nutrients, Hydrol. Processes, 2015, 29, 2922–2955 CrossRef.
- R. W. Harvey, D. W. Metge, D. R. LeBlanc, J. C. Underwood, G. R. Aiken, K. D. Butler, T. D. McCobb and J. Jasperse, Importance of the colmation layer in the transport and removal of cyanobacteria, viruses, and dissolved organic carbon during natural lake-bank filtration, J. Environ. Qual., 2015, 44, 1413–1423 CrossRef CAS PubMed.
- R. W. Lee and P. C. Bennett, Reductive dissolution and reactive solute transport in a sewage-contaminated glacial outwash aquifer, Ground Water, 1998, 36, 583–595 CrossRef CAS.
- M. S. McBride and H. O. Pfannkuch, The distribution of seepage within lakebeds, J. Res. U.S. Geol. Surv., 1975, 3, 505–512 Search PubMed.
- S. P. Garabedian, D. R. LeBlanc, L. W. Gelhar and M. A. Celia, Large-scale natural gradient tracer test in sand and gravel, Cape Cod, Massachusetts: 2. Analysis of spatial moments for a nonreactive tracer, Water Resour. Res., 1991, 27, 911–924 CrossRef.
- D. R. LeBlanc, S. P. Garabedian, K. M. Hess, L. W. Gelhar, R. D. Quadri, K. G. Stollenwerk and W. W. Wood, Large-scale natural gradient tracer test in sand and gravel, Cape Cod, Massachusetts: 1. Experimental design and observed tracer movement, Water Resour. Res., 1991, 27, 895–910 CrossRef.
- D. A. Walter, T. D. McCobb, J. P. Masterson and M. N. Fienen, Potential effects of sea-level rise on the depth to saturated sediments of the Sagamore and Monomoy flow lenses on Cape Cod, Massachusetts, U.S. Geological Survey Scientific Investigations Report 2016-5058, U.S. Geological Survey, Reston, VA, 2016 Search PubMed.
- D. L. Stoliker, D. A. Repert, R. L. Smith, B. Song, D. R. LeBlanc, T. D. McCobb, C. H. Conaway, S. P. Hyun, D.-C. Koh, H. S. Moon and D. B. Kent, Hydrologic controls on nitrogen cycling processes and functional gene abundance in sediments of a groundwater flow-through lake, Environ. Sci. Technol., 2016, 50, 3649–3657 CrossRef CAS PubMed.
- T. D. McCobb, D. R. LeBlanc, D. A. Walter, K. M. Hess, D. B. Kent and R. L. Smith, Phosphorus in a Ground-Water Contaminant Plume Discharging to Ashumet Pond, Cape Cod, Massachusetts, 1999, U.S. Geological Survey Water-Resources Investigations Report 02-4306, U. S. Geological Survey, Reston, VA, 2003 Search PubMed.
- National Water Information System, National Water Information System: Mapper, https://maps.waterdata.usgs.gov/mapper/index.html, accessed May 12, 2020, 2020 Search PubMed.
- J. Savoie, Altitude and Configuration of the Water Table, Western Cape Cod Aquifer, Massachusetts, March 1993, U.S. Geological Survey Open File Report 94-462, 1 Plate, U.S. Geoloigical Survey, Reston, VA, 1995 Search PubMed.
- D. A. Walter, T. D. McCobb and M. N. Fienen, Use of a numerical model to simulate the hydrologic system and transport of contaminants near Joint Base Cape Cod, western Cape Cod, Massachusetts, U.S. Geological Survey Scientific Investigations Report 2018-5139, U.S. Geoloigical Survey, Reston, VA, 2019 Search PubMed.
- D. A. Walter, J. P. Masterson and K. M. Hess, Ground-water recharge areas and traveltimes to pumped wells, ponds, streams, and castal water bodies, Cape Cod, Massachusetts, U.S. Geological Survey Scientific Investigations Map I-2857, 1 Sheet; U.S. Geoloigical Survey: Reston, VA, 2004 Search PubMed.
- U.S. Geological Survey, National field manual for the collection of water-quality data: U.S. Geological Survey Techniques of Water-Resources Investigations, Book 9, Ch. A1–A10, U. S. Geological Survey, Reston, VA, variously dated, http://pubs.water.usgs.gov/twri9A, 2018 Search PubMed.
- A. K. Tokranov, H. M. Pickard, D. R. LeBlanc, B. J. Ruyle, R. B. Hull, L. B. Barber, D. A. Repert, E. M. Sunderland, and C. D. Vecitis, Concentrations of per- and polyfluoroalkyl substances (PFAS) and related chemical and physical data at and near surface-water/groundwater boundaries on Cape Cod, Massachusetts, 2016-19, U.S. Geological Survey data release, U.S. Geological Survey, Reston, VA, 2021, https://doi.org/10.5066/P9HPBFRT Search PubMed.
- E. F. Houtz, R. Sutton, J.-S. Park and M. Sedlak, Poly- and perfluoroalkyl substances in wastewater: Significance of unknown precursors, manufacturing shifts, and likely AFFF impacts, Water Res., 2016, 95, 142–149 CrossRef CAS PubMed.
- S. Taniyasu, K. Kannan, L. W. Y. Yeung, K. Y. Kwok, P. K. S. Lam and N. Yamashita, Analysis of trifluoroacetic acid and other short-chain perfluorinated acids (C2–C4) in precipitation by liquid chromatography–tandem mass spectrometry: Comparison to patterns of long-chain perfluorinated acids (C5–C18), Anal. Chim. Acta, 2008, 619, 221–230 CrossRef CAS PubMed.
- E. F. Houtz, C. P. Higgins, J. A. Field and D. L. Sedlak, Persistence of perfluoroalkyl acid precursors in AFFF-impacted groundwater and soil, Environ. Sci. Technol., 2013, 47, 8187–8195 CrossRef CAS PubMed.
- E. F. Houtz and D. L. Sedlak, Oxidative conversion as a means of detecting precursors to perfluoroalkyl acids in urban runoff, Environ. Sci. Technol., 2012, 46, 9342–9349 CrossRef CAS PubMed.
- B. J. Ruyle, H. M. Pickard, D. R. LeBlanc, A. K. Tokranov, C. P. Thackray, X. C. Hu, C. D. Vecitis and E. M. Sunderland, Isolating the AFFF Signature in Coastal Watersheds Using Oxidizable PFAS Precursors and Unexplained Organofluorine, Environ. Sci. Technol., 2021, 55, 3686–3695 CrossRef CAS PubMed.
- B. J. Ruyle, C. P. Thackray, J. P. McCord, M. J. Strynar, K. A. Mauge-Lewis, S. E. Fenton and E. M. Sunderland, Reconstructing the composition of per- and polyfluoroalkyl substances in contemporary aqueous film-forming foams, Environ. Sci. Technol. Lett., 2021, 8, 59–65 CrossRef CAS PubMed.
- C. P. Higgins, J. A. Field, C. S. Criddle and R. G. Luthy, Quantitative determination of perfluorochemicals in sediments and domestic sludge, Environ. Sci. Technol., 2005, 39, 3946–3956 CrossRef CAS PubMed.
- D. R. LeBlanc, Sewage Plume in a Sand and Gravel Aquifer, Cape Cod, Massachusetts, U.S. Geological Survey Water-Supply Paper 2218, U.S. Geological Survey, Reston, VA, 1984 Search PubMed.
- R. L. Smith, D. A. Repert, D. L. Stoliker, D. B. Kent, B. Song, D. R. LeBlanc, T. D. McCobb, J. K. Böhlke, S. P. Hyun and H. S. Moon, Seasonal and spatial variation in the location and reactivity of a nitrate-contaminated groundwater discharge zone in a lakebed, J. Geophys. Res.: Biogeosci., 2019, 124, 2186–2207 CrossRef.
- T. D. McCobb, M. A. Briggs, D. R. LeBlanc, F. D. Day-Lewis and C. D. Johnson, Evaluating long-term patterns of decreasing groundwater discharge through a lake-bottom permeable reactive barrier, J. Environ. Manage., 2018, 220, 233–245 CrossRef CAS PubMed.
- B. C. Sander and J. Kalff, Factors controlling bacterial production in marine and freshwater sediments, Microb. Ecol., 1993, 26, 79–99 CrossRef CAS PubMed.
- T. B. Hampton, J. P. Zarnetske, M. A. Briggs, K. Singha, J. W. Harvey, F. D. Day-Lewis, F. MahmoodPoor Dehkordy and J. W. Lane, Residence time controls on the fate of nitrogen in flow-through lakebed sediments, J. Geophys. Res.: Biogeosci., 2019, 124, 689–707 CrossRef CAS.
- J. P. Zarnetske, R. Haggerty, S. M. Wondzell, V. A. Bokil and R. González-Pinzón, Coupled transport and reaction kinetics control the nitrate source-sink function of hyporheic zones, Water Resour. Res., 2012, 48, W11508 Search PubMed.
- R. L. Smith, L. K. Baumgartner, D. N. Miller, D. A. Repert and J. K. Böhlke, Assessment of nitrification potential in ground water using short term, single-well injection experiments, Microb. Ecol., 2006, 51, 22–35 CrossRef CAS PubMed.
- J. P. Zarnetske, R. Haggerty, S. M. Wondzell and M. A. Baker, Dynamics of nitrate production and removal as a function of residence time in the hyporheic zone, J. Geophys. Res.: Biogeosci., 2011, 116, G01025 Search PubMed.
- S. Bertilsson, A. Burgin, C. C. Carey, S. B. Fey, H.-P. Grossart, L. M. Grubisic, I. D. Jones, G. Kirillin, J. T. Lennon, A. Shade and R. L. Smyth, The under-ice microbiome of seasonally frozen lakes, Limnol. Oceanogr., 2013, 58, 1998–2012 CrossRef.
- Y. Lyu, M. L. Brusseau, W. Chen, N. Yan, X. Fu and X. Lin, Adsorption of PFOA at the air–water interface during transport in unsaturated porous media, Environ. Sci. Technol., 2018, 7745–7753, DOI:10.1021/acs.est.8b02348.
- M. L. Brusseau, Assessing the potential contributions of additional retention processes to PFAS retardation in the subsurface, Sci. Total Environ., 2018, 613–614, 176–185 CrossRef CAS PubMed.
- M. Steele, C. Griffith and C. Duran, Monthly variations in perfluorinated compound concentrations in groundwater, Toxics, 2018, 6, 56 CrossRef CAS PubMed.
- J. L. Guelfo and C. P. Higgins, Subsurface transport potential of perfluoroalkyl acids at aqueous film-forming foam (AFFF)-impacted sites, Environ. Sci. Technol., 2013, 47, 4164–4171 CrossRef CAS PubMed.
- C. Zhang, H. Yan, F. Li, X. Hu and Q. Zhou, Sorption of short- and long-chain perfluoroalkyl surfactants on sewage sludges, J. Hazard. Mater., 2013, 260, 689–699 CrossRef CAS PubMed.
- A. C. Umeh, R. Naidu, S. Shilpi, E. B. Boateng, A. Rahman, I. T. Cousins, S. Chadalavada, D. Lamb and M. Bowman, Sorption of PFOS in 114 well-characterized tropical and temperate soils: Application of multivariate and artificial neural network analyses, Environ. Sci. Technol., 2021, 55, 1779–1789 CrossRef CAS PubMed.
- N. J. M. Fitzgerald, A. Wargenau, C. Sorenson, J. Pedersen, N. Tufenkji, P. J. Novak and M. F. Simcik, Partitioning and accumulation of perfluoroalkyl substances in model lipid bilayers and bacteria, Environ. Sci. Technol., 2018, 52, 10433–10440 CrossRef CAS PubMed.
- F. Li, X. Fang, Z. Zhou, X. Liao, J. Zou, B. Yuan and W. Sun, Adsorption of perfluorinated acids onto soils: Kinetics, isotherms, and influences of soil properties, Sci. Total Environ., 2019, 649, 504–514 CrossRef CAS PubMed.
- USGS The National Map: National Hydrography Dataset. Data refreshed April, 2019. Map services and data available from U.S. Geological Survey, National Geospatial Program, https://basemap.nationalmap.gov, accessed April 2019, 2019.
- R. B. Hull, M. A. Briggs, D. R. LeBlanc, D. A. Armstrong and T. D. McCobb, Temperature and Seepage Data from the Nearshore Bottom Sediments of Five Groundwater Flow-Through Glacial Kettle Lakes, Western Cape Cod, Massachusetts, 2015–18, U.S. Geological Survey data release, U.S. Geological Survey, Reston, VA, 2019 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1em00329a |
| This journal is © The Royal Society of Chemistry 2021 |