
Fate of transition metals in PO4-based in vitro assays: equilibrium modeling and macroscopic studies†
Brian. E.
Reed
 *,
Jayashree
Yalamanchili
,
Jennie. B.
Leach
and
Christopher. J.
Hennigan
*,
Jayashree
Yalamanchili
,
Jennie. B.
Leach
and
Christopher. J.
Hennigan
Department of Chemical, Biochemical and Environmental Engineering, University of Maryland, Baltimore County, 1000 Hilltop Circle, Baltimore, Maryland, USA 21250. E-mail: reedb@umbc.edu
First published on 21st December 2020
Abstract
Transition metals are thought to be among the most toxic components in atmospheric particulate matter (PM) due to their role in catalyzing reactive oxygen species (ROS) formation. We show that precipitation of the transition metals Fe(II), Fe(III), and Mn(II) are thermodynamically favored in phosphate-based assays used to measure the oxidative potential (OP) – a surrogate for toxicity – of PM. Fe and Mn precipitation is likely to occur at extremely low metal concentrations (<0.5 μM), levels that are imperceptible to the naked eye. The concentration of each metal (other than Cu) in aqueous PM filter extracts often exceeds the solubility limit in OP assays, indicating favorable thermodynamic conditions for precipitation. Macroscopic experimental results at higher metal concentrations (>100 μM) with visible precipitates provide quasi-validation of the thermodynamic modeling. Oxidation of Fe(II) to Fe(III) is likely to be rapid in all in vitro OP assays, transforming Fe to a much less soluble form. Fe precipitates are likely to increase the rate of precipitation of other metals and possibly induce co-precipitation. These results have direct relevance for all PO4-based assays; the implications for studies of PM toxicity are discussed.
Environmental significanceIn vitro assays are used to approximate the oxidative stress that results from particulate matter (PM) exposure by measuring oxidative potential (OP, a proxy for PM toxicity). While these assays have been used extensively to study organic compounds, they are also being used to determine the OP of transition metals. Several OP assays employ phosphate to buffer the system at pH = 7.4 for biological relevance. In this work, we show that oxidation and precipitation of many transition metals, including those present at high concentrations in atmospheric PM, occurs in this buffered phosphate matrix. This raises important questions about the use of such assays to assess PM toxicity. |
Introduction
Particulate matter (PM) exerts numerous deleterious effects on human health,1,2 which contributes significantly to the global burden of disease.3,4 There is strong evidence that PM induces oxidative stress, and this is hypothesized as a critical link between PM exposure and many adverse health outcomes.5–8 If true, this implies that the chemical species present in PM have different toxicities, since individual compounds have different reactivities and catalyze or form variable amounts of reactive oxygen species (ROS).Acellular methods have been widely deployed to approximate the oxidative stress that may result from PM exposure through measurements of oxidative potential (OP). The general approach is to select a compound that reacts with cellular oxidants, and measure its time-dependent decay when exposed to PM extracts. The OP of the sample or matrix scales linearly with the blank-corrected reaction extent in a given time. Because oxidative stress is definitively linked to many disease outcomes, the acellular OP measurements are taken as a proxy for PM toxicity.9 A variety of OP assays have been developed, including dithiothreitol (DTT),10 ascorbic acid (AA),11 and glutathione.12
In order to mimic conditions within human lungs, most of the assays are run at 37 °C and pH = 7.4, often using a phosphate buffer to keep the pH constant.10,11,13 However, under these conditions, precipitation, co-precipitation, complexation with PO4, adsorption and oxidation of metals present in PM (and PM extracts) are thermodynamically possible. The catalytic properties of metals change drastically with phase (solid vs. aqueous), solids composition (e.g., Fe2O3(s) vs. Fe3O4(s)), and oxidation state.14,15 Fe, the transition metal present in ambient PM at the highest concentrations,16,17 can form iron-oxides that have a well-known ability to adsorb other soluble metals.18–20 These phenomena may contribute to some of the inconsistency in studies characterizing the health effects of specific PM components.21,22 DTT consumption rates for organic compounds are widely reported to be linear with the contaminant concentration23 but for metals the relationship is often reported to be a power law response – the DTT consumption rate levels off at high metal concentrations.23,24 Chemical and physical transformations of transition metals may represent a previously unidentified artifact associated with OP assays used as proxies for PM toxicity.
The purpose of this study is to characterize the behavior of several PM metals in PO4-based in vitro incubation fluids using MINEQL thermodynamic software25 and quasi-validate modeling results using visual observations and metal fate experiments. Cu(II) and Mn(II) were chosen because of their high reactivity in acellular OP assays23,24,26 while Fe(II)/Fe(III) were chosen because they are often present in PM at concentrations an order of magnitude higher than other transition metals.16,17 Equilibrium modeling results guide future experimental work on the fate of metals in PO4-based acellular incubation fluids and the effect of metal precipitation on ROS generation.
Materials and methods
Modeling approach
The behavior of the studied metals were assessed as a function of precipitate type, pH, [TOTPO4], [TOTCO3], partial pressure of O2(g) (PO2) and iron-oxide concentration (for the adsorption of metals by iron oxide precipitate). Thermodynamic data (Tables S1–S4, ESI†) contained in MINEQL were supplemented with data from the NIST database as needed.27 Unless otherwise noted model runs were conducted at pH = 7.4, ionic strength (I) = 0.22 M, total TOTPO4 = 0.1 M, TOTCO3 = 1.4 × 10−4 M (closed system), and T = 37 °C. MINEQL corrects equilibrium constants for ionic strength and temperature using the Davies and Van't Hoff equations, respectively. An ionic strength of 0.22 M is based on the addition of 0.08 M of KH2PO4 and 0.02 M of KH2PO4 – a common recipe for PO4-based assays. The effect of TOTCO3 on metal precipitation/complexation was studied by assuming that the phosphate buffer and other reagents had equilibrated with the atmosphere (PCO2 = 10−3.5 atm) at pH = 7.4 during DI water/reagent storage, and then introduced to the assay incubation tubes (closed system with minimal headspace, T = 37 °C), resulting in TOTCO3 = 1.4 × 10−4 M. For the effect of dissolved oxygen, we assumed conditions ranging from very low levels (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) PO2 = −0.69 atm) to saturation (logPO2 = −50 atm). In Table S5,† modeling procedures for each task are presented based on the steps employed in MINEQL. The behavior of antioxidants (e.g., DTT) in the metal-PO4 mixtures was not modeled because thermodynamic data (e.g., redox equilibria, stability constants) were not available. In addition, we acknowledge the uncertainty that arises with equilibrium modeling due to: (1) the inherent difference in reported constants, (2) assuming equilibrium conditions for systems that may not be at equilibrium and (3) applying constants determined under specific experimental conditions (metal concentration, temperature and ionic strength) to systems having different conditions.
PO2 = −0.69 atm) to saturation (logPO2 = −50 atm). In Table S5,† modeling procedures for each task are presented based on the steps employed in MINEQL. The behavior of antioxidants (e.g., DTT) in the metal-PO4 mixtures was not modeled because thermodynamic data (e.g., redox equilibria, stability constants) were not available. In addition, we acknowledge the uncertainty that arises with equilibrium modeling due to: (1) the inherent difference in reported constants, (2) assuming equilibrium conditions for systems that may not be at equilibrium and (3) applying constants determined under specific experimental conditions (metal concentration, temperature and ionic strength) to systems having different conditions.
Macroscopic experiments
Precipitation and oxidation experiments were conducted to help assess which solids would form in the phosphate buffer, thus quasi-validating the thermodynamic modeling. Stock solutions were prepared using reagent-grade metal salts stored in 2% nitric acid solutions. The metal concentrations in the stock solutions were quantified by ICP-MS (NexION 300D, PerkinElmer). Experiments were conducted at metal concentrations between 5 and 5000 μM in the presence of phosphate buffer (10−5 to 0.1 M) and in DI water. PO4 solutions and DI water were oxygenated prior to use. Samples were prepared and placed in a thermomixer for 30 min at 37 °C and 400 rpm – conditions used in the DTT assay.10–12,23 Samples were allowed to settle for 10 min after which pictures were taken and a 30 mL aliquot was passed through a 0.45 μm filter. Aliquots were immediately (<5 min) analyzed for Cu(II), Mn(II), Fe(II), total Fe, and Fe(III) (by difference) using colorimetric methods (HACH, TOTFe: 8008; TOTFe(II): 8146; TOTFe(III) by difference; TOTCu(II): 8143; TOTMn(II): 8149).28 Interference from the high phosphate levels precluded the use of ICP-MS for metals analysis. Calibration curves were developed for a range of PO4 concentrations and all curves were linear (Fig. S1, ESI†). Most of the macroscopic experiments were conducted in triplicate and all showed a very high degree of reproducibility. The standard deviations of the amount precipitated for each metal are as follows: Fe(III): 2.2%; Fe(II): 2.7%; Cu(II): 1.0%; Mn(II): 1.8 (Table S7, ESI†). In Fe(II) oxidation experiments the average standard deviation for Fe(II) and Fe(III) measurements were 0.5 and 2.8, respectively. Macroscopic visual experiments were conducted in the presence of DTT (100 μM) and the absence. DTT did not inhibit precipitation for any of the metals studied (Fig. S2–S5†).Results and discussion
Precipitate type
Fe, Cu, and Mn can each form a number of precipitates. Our first goal was to identify the precipitates most likely to form and their corresponding solubility under conditions present in PO4-based acellular assays. Each precipitate was considered separately in MINEQL – other precipitates were prohibited. In Fig. 1, the metal solubility results (total concentration of all aqueous phase metal species) are presented for each precipitate type. All metals formed hydroxides, oxides or PO4-based precipitates. Aqueous metal concentrations in PM oxidative potential studies typically range from 0.5 μM to ∼50 μM23,24,26 and all studied metals had at least one precipitate with a solubility lower than 5 μM. For subsequent modeling tasks we selected one PO4-based (if one formed) and one non-PO4 precipitate for each metal/oxidation state. The chosen precipitates are represented by the cross-hatched bars in Fig. 1. For Fe(II) and Mn(II), only PO4-based precipitates were considered because the solubilities of the oxide/hydroxide precipitates were much higher than the concentration of these metals in PM extracts. Cu(I), Mn(III) and Mn(IV) only form oxide type solids. For Mn(III), manganite (MnO(OH)(s)) was selected because iron is a component of bixbyite29 and Mn(IV) only forms pyrolusite (MnO2(s)). For Cu(II), Cu3(PO4)2(s) and tenorite (CuO2(s)) were chosen because at 5 μM TOTCu a precipitate formed (the remaining Cu precipitates have solubilities >5 μM). Selecting which nonPO4-based Fe(III) solid was most likely to form was more difficult because of the large number of potential precipitates. For reasons discussed in detail below, we chose strengite (FePO4·2H2O) and ferrihydrite (Fe(OH)3(s)). Rather than use the terms “iron-oxide-like” or “iron-PO4-like” (or “Mn/Cu-oxide-like” or “Mn/Cu-PO4-like”) to denote newly formed precipitates, we choose to use the crystalline name with the caveat that the precipitates are likely in the amorphous form. In addition, solubility products (KS0) for amorphous solids are not readily available and we acknowledge that using KS0 for crystalline precipitates will result in a lower predicted solubility compared to the amorphous solids. For all metals, carbonate-based solids and complexes were not present. The behavior of Fe(II) and Fe(III) will be discussed together because Fe(II) is predicted to rapidly oxidize to Fe(III) under these conditions.
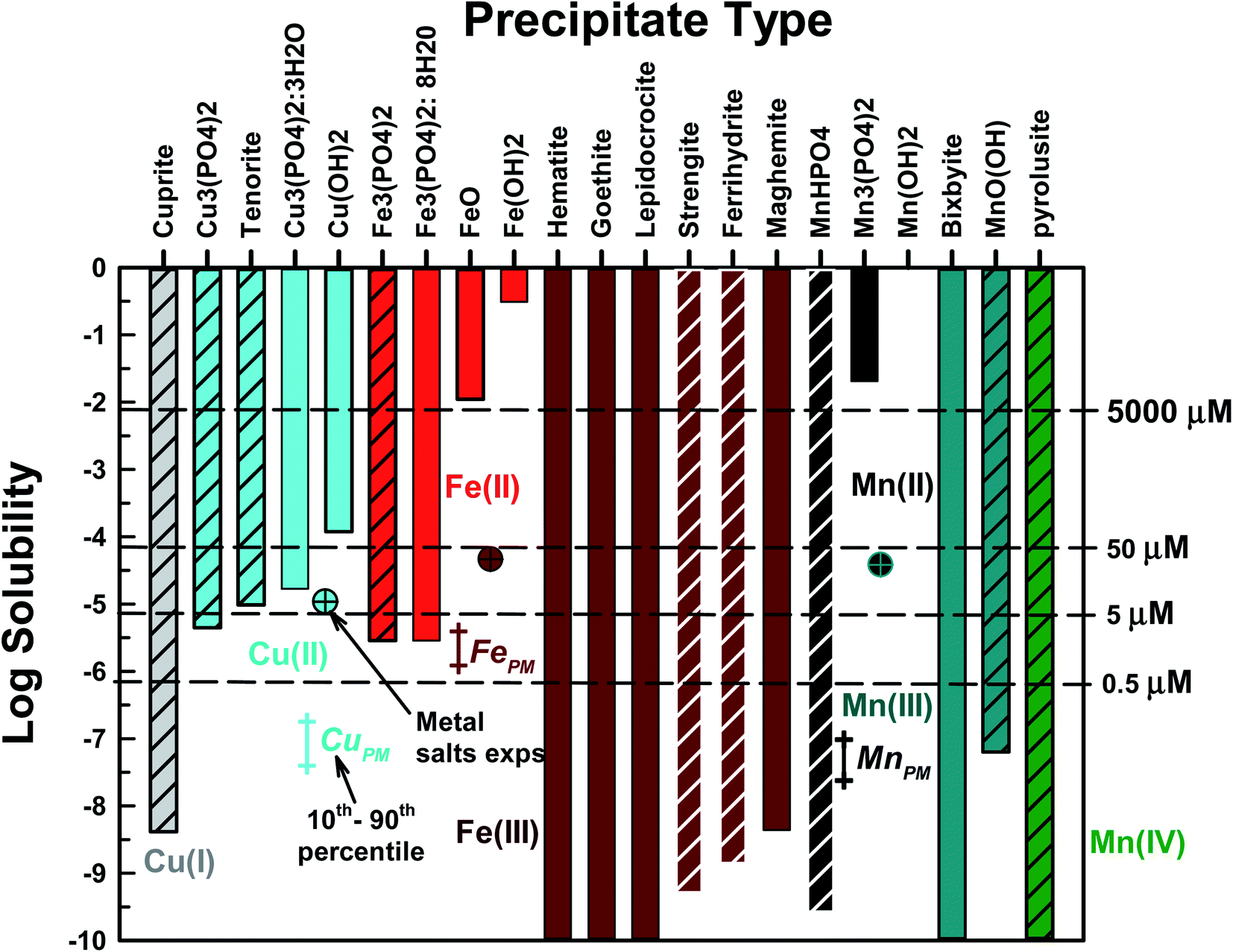 | ||
| Fig. 1 Thermodynamic predictions of metal solubility at pH = 7.4, TOTPO4 = 0.1 M, TOTCO3 = 1.4 × 10−4 M, I = 0.22 M and T = 37 °C. Cross-hatched bars are solids that were selected for further study. MePM data represent the 10th to 90th percentile average of urban PM, in conditions of aqueous PM filter extracts. Single cross-hatched data points represent the highest metal concentrations used in metal salt experiments. | ||
MINEQL runs were conducted with precipitation allowed and prevented – conditions that represent the two extreme possible behaviors in PO4-based assays (Fig. S7†). For the runs where precipitation was allowed, the precipitates described above (hatched bars in Fig. 1) were included in the model runs allowing for the most thermodynamically favored solid to form. The dominant species predicted for all simulation conditions are given in Table S6.†
The MePM data in Fig. 1 represent the 10th to 90th percentile average annual metal concentrations measured for ten urban areas across the US17 determined using well-established PM filter sampling and extraction protocols (e.g., following the approach of Verma et al., 2014).30 Note MePM data represent total metal concentrations, i.e., there was no differentiation between metal oxidation states. The single cross-hatched data points represent some metal concentrations examined in studies where metals were added as salts, shown for reference.23,24,26 Total FePM data averaged 2.3 μM and 10th to 90th percentile concentrations were 1.24 and 4.0 μM, respectively. The highest Fe(III) and Fe(II) concentrations used in metal salt studies were 47 and 10 μM, respectfully. As discussed above, any aqueous Fe(II) is likely to be rapidly oxidized to Fe(III), and all five Fe(III) compounds considered here have solubilities that are orders of magnitude lower than the range of concentrations in filter extracts from PM sampled in urban areas. This strongly suggests that aqueous Fe in PM extracts, even for samples that represent low atmospheric concentrations as well as those used in metal salt studies, will rapidly precipitate in PO4-based assays. CuPM data averaged 0.06 μM while the 10th to 90th percentile concentrations were 0.04 and 0.18 μM, respectively. The highest Cu(II) concentrations used in metal salt studies was 11 μM. CuPM is well below the solubility limit for all Cu(II) species considered; therefore, precipitation of aqueous Cu in PO4-based assays of PM toxicity is not likely. However, Cu(II) precipitation could occur in metal salt experiments if TOTCu(II) > 4.47 μM (solubility of Cu3(PO4)2(s)). While Cu(I) is highly insoluble, the redox conditions in the OP assays are oxidizing due to the presence of dissolved oxygen, suggesting that reduction of Cu(II) to Cu(I) and subsequent precipitation is unlikely. However, if Cu was present as Cu(I), precipitation is possible if Cu(I) is not oxidized to Cu(II). MnPM data averaged 0.058 μM while the 10th to 90th percentile concentrations were 0.024 to 0.098 μM, respectively. The highest Mn(II) concentration from metal salt studies was 39 μM. If Mn exists as Mn(II) and is not oxidized, Mn(II) precipitation is not predicted to occur. However, if Mn is present as Mn(III) or Mn(IV), or if Mn(II) is oxidized, then precipitation of aqueous Mn in PO4-based assays is thermodynamically favored.
The effect of Kso (logKso ± 2) on metal solubility was conducted for the selected precipitates to access the uncertainty in published values of Kso (Fig. S6†). Tenorite was most affected by the value of Kso, followed by Cu3(PO4)2(s), Fe3(PO4)2(s) and MnO(OH)(s). The remaining precipitates had solubilities well below 5 μM (the lowest metal concentration investigated).
In summary, precipitation of Fe and Mn is likely in PM filter extracts originating from urban areas and in metal salt studies. Cu(II) precipitation in PO4-based assays is unlikely for PM extracts, but may occur in metal salt experiments if the total Cu(II) concentration exceeds 4.47 μM.
In the following sections, we discuss in detail the modeling results for each metal, focusing on the effects of pH, TOTPO4, carbonate, and dissolved O2. We present the macroscopic experimental results, including the visual presence/absence of precipitates and chemical analysis of dissolved metals. We acknowledge that the high metal concentrations used to visually observe precipitation do not represent metal concentrations in PM extracts unless samples were taken from heavily polluted areas or for extended periods of time. However, detection of precipitates at concentrations typical of PM extracts is challenged by the extremely low mass present. For example, 15 mL of Hi-Vol filter extract following the procedure of Verma et al.30 contains approximately ∼2–8 × 10−8 g of Mn (range representing urban concentrations in the US).16 Creative experimental approaches combined with advanced analytical techniques are required to physically detect precipitates at such low levels: this is the topic of ongoing work by our group. Therefore, we believe that the macroscopic experimental results are useful in understanding what can occur thermodynamically in PO4-based assays in regards to precipitation, oxidation and changing assay conditions (e.g., effect of TOTPO4).
Iron
Fe(II) formed Fe3(PO4)2(s) and Fe(III) formed strengite or ferrihydrite depending on the magnitude of TOTPO4 (Fig. 2a). Based on visual observations and the aqueous phase Fe(III) analysis, we are confident that strengite formed at higher TOTPO4/TOTFe ratios (Fig. 2). Hsu (1982)31 reported that phosphate induces nearly immediate formation of a fluffy “whitish” precipitate, with ∼98% of the Fe(III) in the precipitate at equilibrium. In a similar aqueous matrix, Senn et al. (2015)32 reported amorphous FePO4(s) formation, which appeared beige in color. Senn et al. (2015)32 also observed shifting precipitate identity as a function of the TOTPO4/TOTFe molar ratio: the Fe(III) suspension changed from “whitish” to an “orange” or “brownish” as TOTPO4/TOTFe decreased, indicating the formation of lepidocrocite (orange) or ferrihydrite (brown). We observed similar behavior in our experiments (Fig. 2b) – the type of solid clearly changed from beige (FePO4(s)) to orange-brown between 0.01 and 0.001 M TOTPO4 (initial Fe constant at 5 mM). When lepidocrocite and strengite are both allowed to form in MINEQL, 100% of the Fe(III) exists as lepidocrocite (Fig. S8†), which does not match what we observed visually. Ferrihydrite and maghemite form at TOTPO4 less than 0.037 M and 0.011 M, respectively. While maghemite formation matched the visual pattern better than ferrihydrite (Fig. S9†), we selected ferrihydrite because Singh et al. (2010)33 reported that the precipitation of ferrihydrite is favored when hydrolysis occurs rapidly and ferrihydrite formation in the presence of PO4 was reported by Senn et al. (2015).32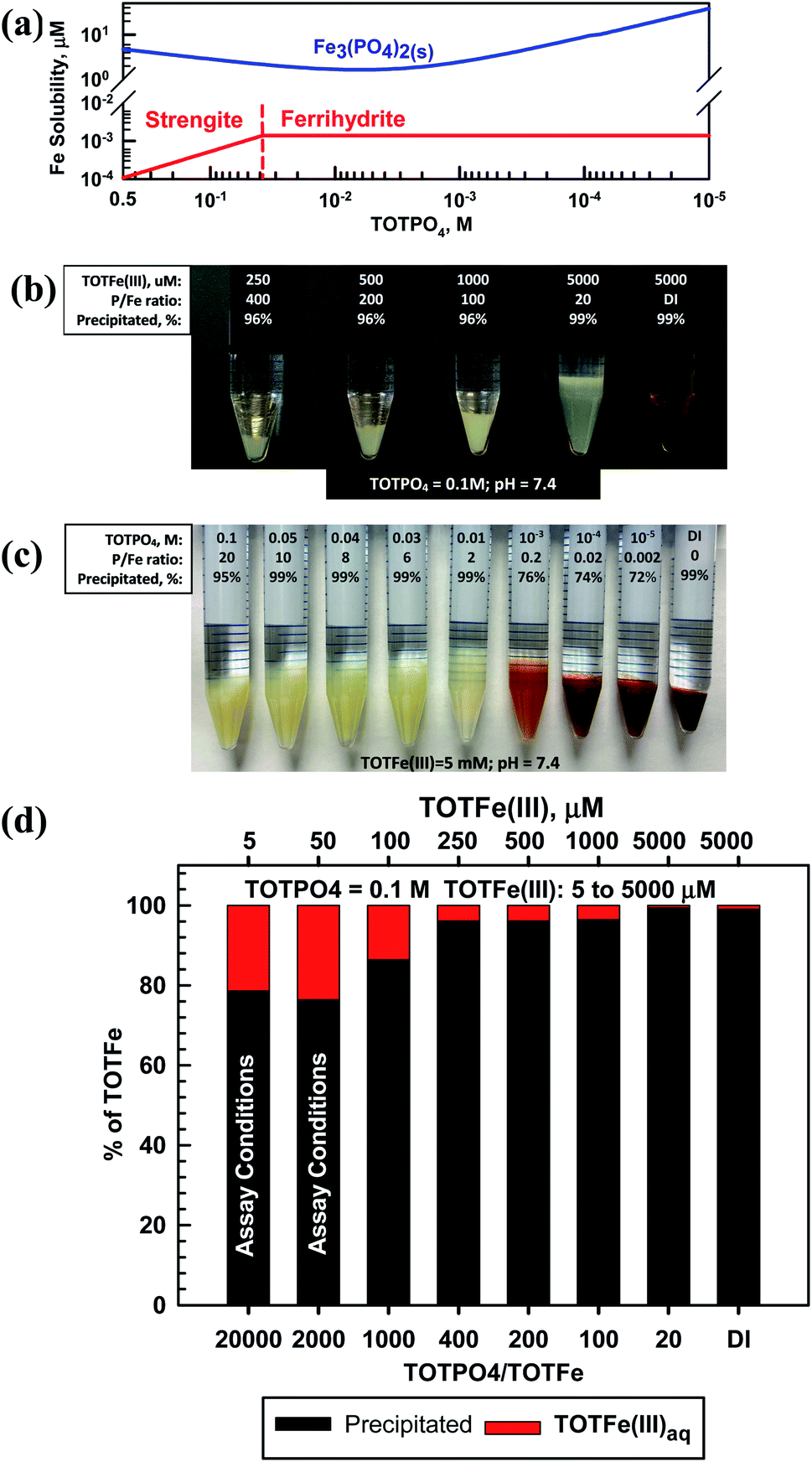 | ||
| Fig. 2 Equilibrium modeling and macroscopic results: (a) thermodynamic predictions of Fe(III) and Fe(II) solubility as function of TOTPO4; (b) macroscopic experimental results, including visual observations at TOTPO4 = 0.1 M, with varying TOTFe(III); (c) macroscopic experimental results, with visual observations at TOTFe(III) = 5 mM, varying TOTPO4 and (d) precipitation results for varying TOTFe(III), TOTPO4 = 0.1 M. Note, in (b), (c), and (d) the percent of Fe precipitated is based upon chemical analysis. | ||
At 0.1 M TOTPO4/pH = 7.4 (conditions used in most PO4-based acellular assays) precipitation of Fe(III) was observed visually over a wide range of TOTFe(III) (Fig. 2c). While we did not observe visible solid formation at low TOTFe(III) (a 10 mL solution of 50 μM Fe contains only 2.8 × 10−5 g of total Fe), precipitation was confirmed based on chemical analysis of filtered samples (Fig. 2d). Note, we confirmed that neither PO4 nor DTT interfere with the Fe measurements. At 5 and 50 μM TOTFe(III), about 80% of added Fe(III) precipitated, while 86–99% of Fe precipitated at higher TOTFe(III). Decreased precipitation at lower TOTFe(III) could be due to the presence of small strengite-type particles that escape filtration.34 The remaining aqueous Fe(III) exists as Fe(OH)2+ and Fe(OH)3(aq) (Table S6†).
Fe(III) (as well as Fe(II), Cu(II) and Mn(II)) precipitation occurred experimentally at all TOTFe(III)–TOTPO4 combinations within the 30 min duration of acellular assays. Precipitation is a two-step process of nucleation and crystal growth, with nucleation typically the rate-limiting step.35 Nucleation can be considered homogenous (slow, random particle growth) and heterogeneous (fast, nucleation occurs on existing surfaces). To date, there have been no kinetic studies of FePO4(s) growth under PO4-based acellular conditions. However, Senn et al.32 and Madsen and Koch34 investigated the formation of FePO4(s) following the oxidation of Fe(II) to Fe(III). Senn et al.32 conducted macroscopic studies and reported that within 6 min, a beige “Fe(III)-phosphate precipitate” formed after Fe(II) addition. At TOTPO4/TOTFe <0.52, the suspension color changed from initially beige to orange over 240 min indicating that the formation of FePO4(s) occurred first, followed by FePO4(s) dissolution and then the formation of Fe(III)-oxide precipitate. While we did not observe this time-dependent transition, we did observe the apparent shift from Fe(III)-phosphate precipitate to Fe(III)-oxide precipitate at a similar P/Fe ratio (between 0.2–2 in our work).
It is highly likely that Fe(III)-oxide solid would form in acellular assays at lower TOTPO4, suggesting that decreasing the phosphate buffer concentration would not prevent Fe(III) precipitation. Grundl and Delwiche36 reported that ferric iron precipitation in the absence of phosphate was first order with respect to Fe(OH)3(aq): dFe(III)/dt = −k[Fe(OH)3(aq)]. The precipitation rate decreased with increasing TOTFe(III) concentration (half-lives: 5 min and 15 min at TOTFe(III) = 500 and 1000 μM, respectively) due to the poisoning of the surface by Fe3+ and to a lesser extent by Fe(OH)2+. Grundl and Delwiche's work was conducted at pH 2.5–3 where Fe3+/Fe(OH)2+ are dominant, while at pH = 7.4 Fe3+/Fe(OH)2+ are essentially nonexistent. As with the other metals, our goal with this work is to characterize the predicted metal phase state and speciation in the PO4 matrix. Connecting the formation of metal-complexes and precipitates with the generation of ROS or the response of OP assays is beyond the scope of this study, but warrants attention. Metals can catalyze ROS formation in solid or aqueous form37 therefore, it is likely that the formation of precipitates could also affect the DTT consumption rate.
The effect of [O2(aq)] on metal oxidation was investigated by varying the O2 partial pressure from 0.21 to essentially zero (at 25 °C and PO2 = 0.21 atm, [O2(g)] ≈ 275 μM, 8.9 mg L−1). Fe(II) is oxidized to Fe(III) at very low [O2] concentrations (Fig. S10†). Fe(II) and Fe(III) either form Fe3(PO4)2(s) or strengite, respectively, depending on the PO2 level. Sung and Morgan38 summarized results from multiple studies on the kinetics of Fe(II) to Fe(III) oxidation in the natural environment. Half-lives ranged from ∼16–47 min at pH levels between 6.3–6.52 and ∼3.3–5.5 min at pH = 8.0. Madsen and Koch34 reported that Fe(II) was oxidized in water with very low dissolved oxygen (DO) concentrations (H2O purged with N2(g)) and that iron phosphate solid catalyzed Fe(II) oxidation. These results indicate the likely rapid transformation of Fe(II) to Fe(III) during any in vitro measures of PM toxicity with phosphate in the assay matrix.
In our experiments, Fe(II) formed a whitish precipitated at TOTPO4 ≥ 0.01 M, transitioned to a grayish-white precipitate at TOTPO4 = 10−3 M, and then to an orange/brown precipitate at TOTPO4 ≤ 10−4 M (Fig. 3a). For TOTPO4/TOTFe(II) combinations in which a whitish precipitate was observed, it is not possible to say if Fe3(PO4)2(s) or strengite formed. However, when the orange/brown precipitates were present, it is clear that Fe(II) was oxidized to Fe(III) with subsequent Fe(III)-oxide solid formation. FeO(s) or Fe(OH)2(s) solid formation was not possible given their very high solubilities (FeO2(s) – 0.011 M; Fe(OH)2(s) – 0.31 M, Fig. 1). Fe(II) oxidation to Fe(III) is further demonstrated in the Fe(II)-DI water experiments, where ferrihydrite formed following Fe(II) oxidation to Fe(III). Chemical analysis of filtered samples also reinforces this observation – at TOTPO4/TOTFe(II) ≤ 0.2 and in DI water there was a mixture of Fe(II)/Fe(III) in the aqueous phase.
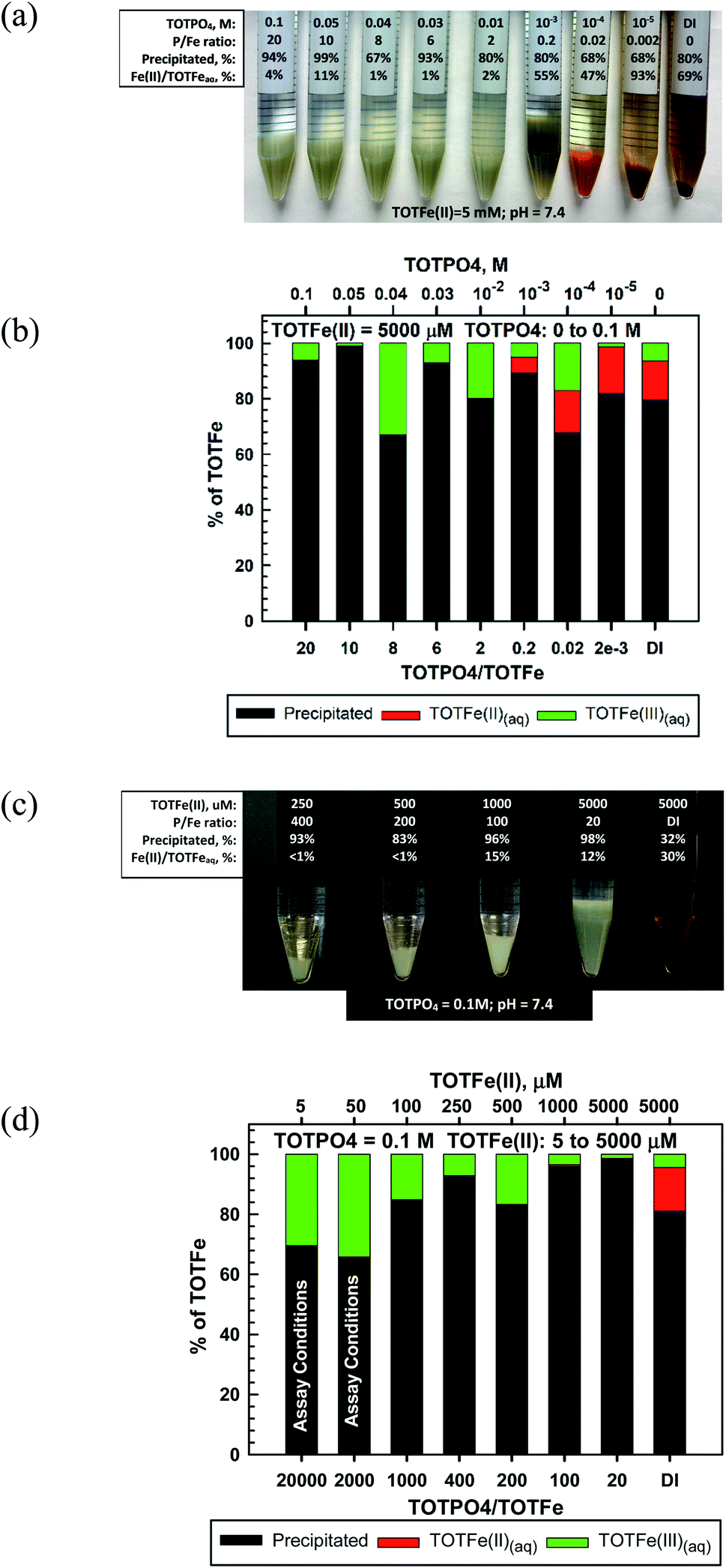 | ||
| Fig. 3 Fe(II) macroscopic results: (a) visual observations at TOTFe(II) = 5 mM, varying TOTPO4; (b) precipitation/oxidation results at TOTFe(II) = 5 mM, varying TOTPO4; (c) visual observations at TOTPO4 = 0.1 M, varying TOTFe(II) and (d) and precipitation/oxidation results at TOTPO4 = 0.1 M, varying TOTFe(II). Note all results in figures are based on chemical analyses. | ||
For TOTPO4 = 0.1 M a whitish precipitate formed at TOTFe(II) = 250 to 5000 μM (Fig. 3c). At 5 and 50 μM TOTFe(II) there was no visual precipitation but 70% and 65% of the total iron precipitated as either Fe3(PO4)2(s) or strengite. At the lower TOTFe(II) there was significant oxidation of Fe(II) to Fe(III) based on analysis of filtered samples (Fig. 3d). Under similar conditions for TOTFe(III) = 5 to 100 μM there was slighly more precipitation of Fe(III) but still a significant amount of Fe(III) in solution relative to higher TOTFe(III) concentrations (Fig. 2d). It is possible that the combination of Fe(II) oxidation to Fe(III) and Fe(III) precipitation to strengite were subject to slower kinetics than that observed at higher iron concentrations. Under PO4-based in vitro assay conditions (indicated in Fig. 3d) Fe(II) was oxidized and there was precipitation of either Fe3(PO4)2(s) or strengite (or both). Oxidation of Fe(II) to Fe(III) is highly likely to occur, though the identity of the specific solids is based upon specific conditions within the assay and we can speculate that these transformations will likely affect the generation of ROS in PO4-based assays.
Copper
At pH = 7.4 Cu(II) exists predominantly as CuHPO4(aq) (Fig. 4a and Table S7†). At slightly higher pH (>7.6), tenorite is predicted to form over Cu3(PO4)2(s) (Fig. S7†). For TOTCu(II) ≤ 250 μM at TOTPO4 = 0.1 M/pH = 7.4 there was no visible precipitate formation (Fig. 4b); however, the aqueous Cu measurements indicate that precipitation did occur (Fig. 4d). At TOTPO4 ≥ 0.14 M all Cu(II) exists as CuHPO4(aq); for TOTPO4 between 0.14 and 0.03 M Cu3(PO4)2(s) forms, while tenorite forms below 0.03 M TOTPO4 (Fig. 4a). Matching visual observations to equilibrium modeling results is more difficult with Cu(II) because there were no changes in precipitate appearance (the deeper blue precipitates could be due to density differences of the settled solids). Cu(II) precipitation occurred experimentally at all TOTCu(II)–TOTPO4 combinations within the 30 min duration of in vitro assays. For all macroscopic experiments at TOTPO4 = 0.1 M/pH = 7.4, the measured TOTCu(aq) was always greater than the predicted Cu(II) solubility (4.47 μM). Cu(I) in the acellular assay matrices would exist as aqueous Cu(I) or CuO2(s) (Fig. S10†) or be oxidized to Cu(II). It is interesting to note that Cu(II) is reported to be the most active of the transition metals in generating ROS,23 which could in part be due to its high solubility.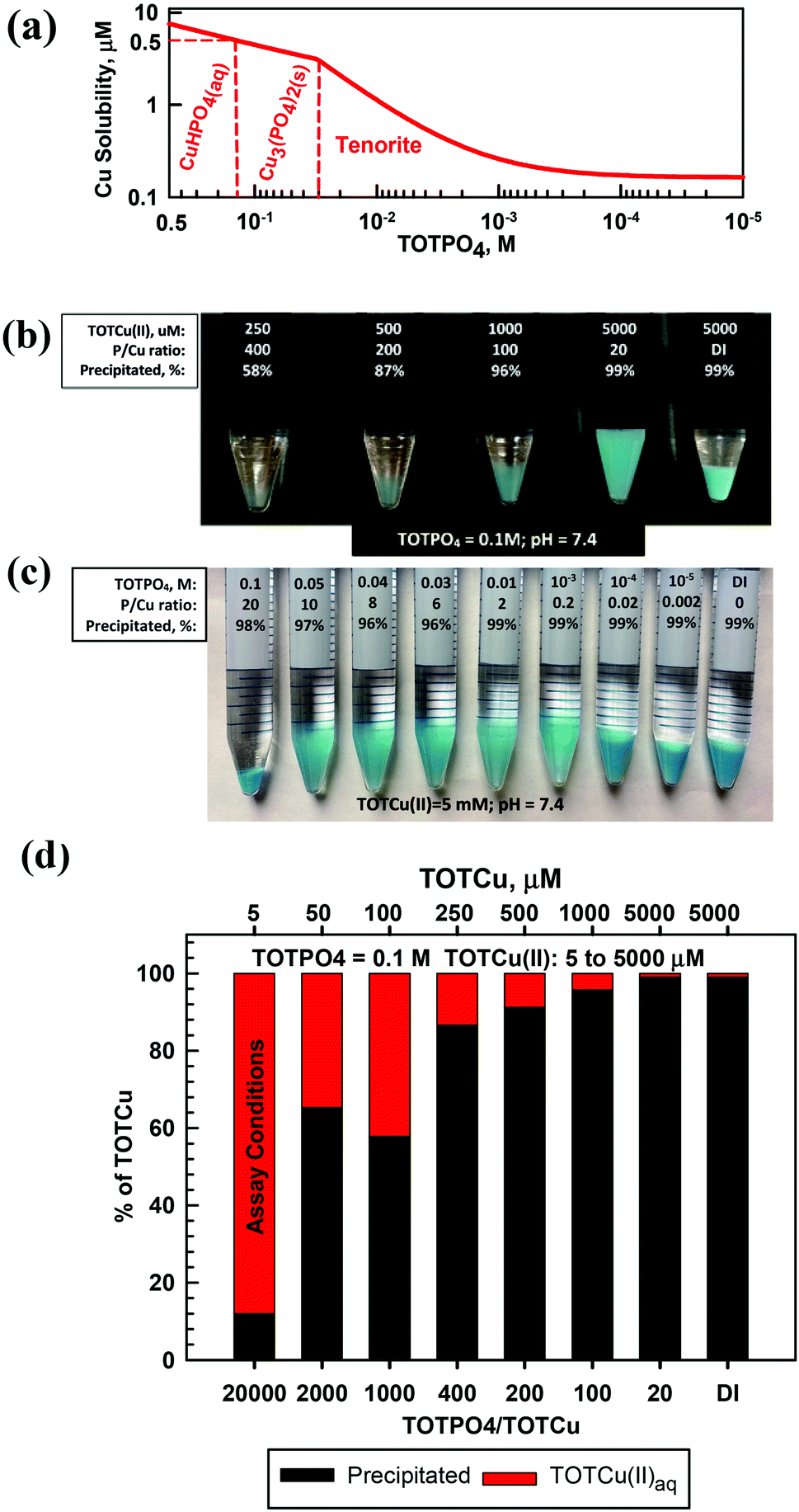 | ||
| Fig. 4 Cu(II) equilibrium modeling and macroscopic results: (a) thermodynamic predictions of Cu(II) solubility as function of TOTPO4; (b) macroscopic experimental results, including visual observations at TOTPO4 = 0.1 M, varying TOTCu(II); (c) macroscopic experimental results, including visual observations at TOTCu(II) = 5000 μM, varying TOTPO4 and (d) precipitation results for varying TOTCu(II), TOTPO4 = 0.1 M. Note, in (b), (c), and (d) the percent of Cu precipitated is based upon chemical analysis. | ||
Manganese
Mn proved to be the most complicated of the metals studied because of its multiple oxidation states and precipitates. At TOTPO4 = 0.1 M/pH = 7.4 Mn(II) exists as MnHPO4(aq) (95%; no precipitation, Table S7†) or MnHPO4(s) (Fig. 5a). Mn(III) exists as Mn3+ or MnO(OH)(s) (Table S7†). Mn(IV), a highly insoluble species (MnO2(s) solubility = 2.3 × 10−13 M), does not form complexes with OH− or PO4. At TOTMn(II) ≥ 500 μM and TOTPO4 = 0.1 M a whitish precipitate formed (Fig. 5b) while at lower TOTMn(II) precipitation was demonstrated by measuring the Mn concentration in filtered samples (Fig. 5d). When precipitation was studied as a function of TOTPO4, a whitish precipitate formed at TOTPO4 ≥ 0.01 M and a brownish/black precipitate at TOTPO4 ≤ 10−3 M (Fig. 5c). If we assume the whitish precipitate is MnHPO4(s) then clearly a different solid formed at lower TOTPO4. If MnHPO4(s) formation does not occur, the remaining two Mn(II) precipitate solubilities Mn3(PO4)2(s) (0.02 M) and Mn(OH)2(s) (2.07 M) (Fig. 1) are far higher than what we measured.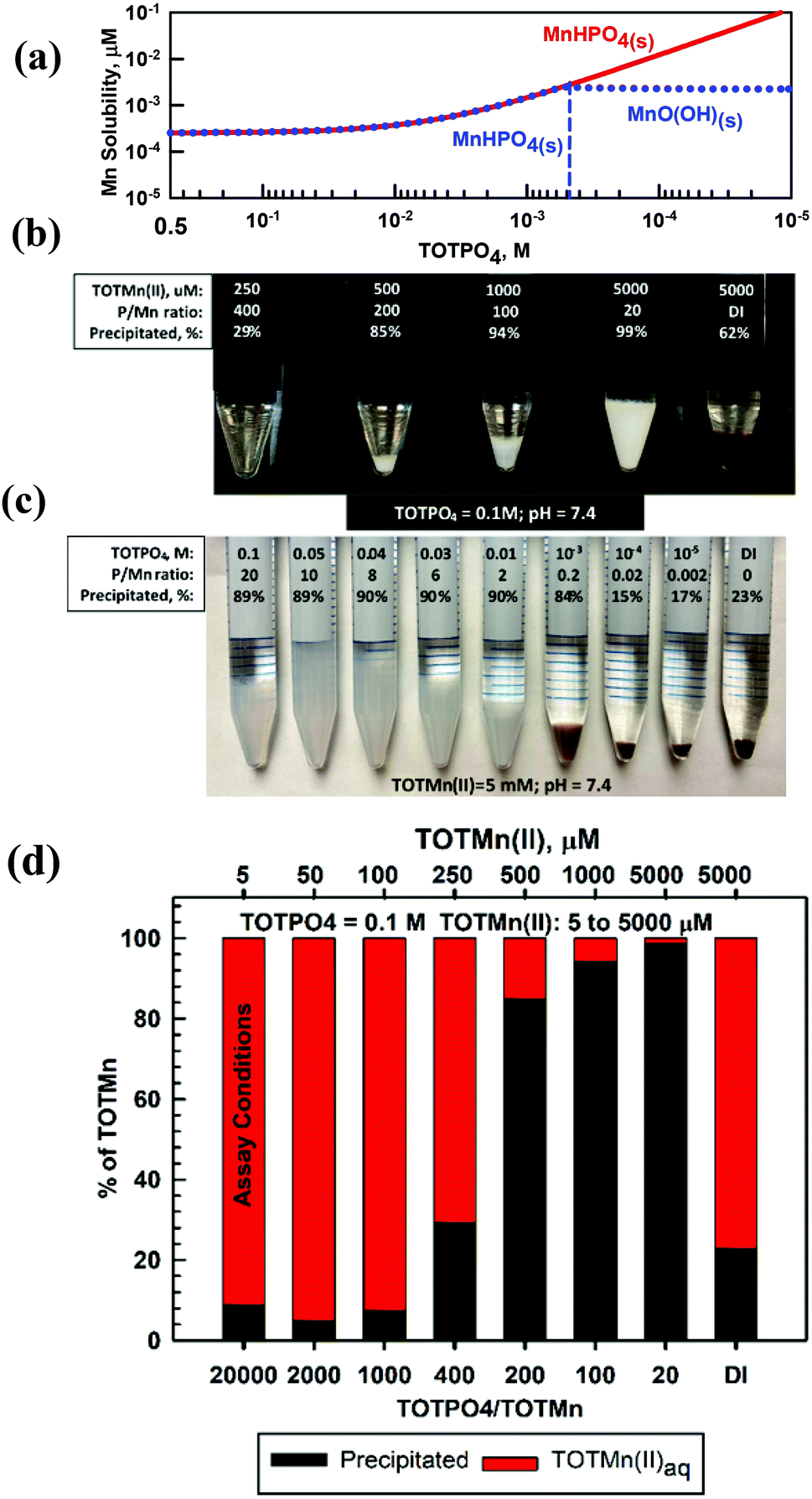 | ||
| Fig. 5 Mn(II) equilibrium modeling and macroscopic results: (a) thermodynamic predictions of Mn(II) solubility as function of TOTPO4; (b) macroscopic experimental results, including visual observations at TOTPO4 = 0.1 M, varying TOTMn(II); (c) macroscopic experimental results, including visual observations at TOTMn(II) = 5000 μM, varying TOTPO4 and (d) precipitation results varying TOTMn(II), TOTPO4 = 0.1 M. Note, in (b), (c), and (d) the percent of Mn precipitated is based upon chemical analysis. | ||
Redox transformations of dissolved Mn are likely in PO4-based matrices, though the speciation of Mn is uncertain. Mn(II) can be oxidized to Mn(III) or Mn(IV) under lower DO concentrations with the subsequent formation of MnO(OH)(s) or MnO2(s) (Fig. S10†). Stumm and Morgan35 reported an Fe(II)-type oxidation relationship for Mn(II) to Mn(IV): d[Mn2+]/dt = −k[OH−]PO2[Mn2+] with the Mn(II) oxidation rate very low at pH < 9. The oxidation rate may be higher in PO4-based assays due to the higher temperature and higher [O2] (assuming reagents are in equilibrium with the atmosphere). However, the formation of MnHPO4(aq) complexes could slow the Mn(II) oxidation rate. Faust and Aly39 reported that Mn(II) oxidization is catalyzed by MnO2(s) and it is possible that MnHPO4(s) may catalyze the Mn(II) → Mn(IV) reaction but there are no literature results to confirm this mechanism. A more likely scenario is that Mn(II) is oxidized to Mn(III) with the formation of MnO(OH)(s) – at lower TOTPO4 we observe that the formation of the brown/blackish precipitate occurs experimentally at TOTPO4 ≈ 10−3 M (Fig. 5b) and MnO(OH)(s) is predicted to form at TOTPO4 = 5 × 10−4 M (dotted blue line in Fig. 5a).
Adsorption by ferric iron precipitates
Adsorption of metals by iron oxides is a well-known phenomenon. In this task we investigated the adsorption of Cu(II) and Mn(II) onto hydrous ferric oxide (HFO) using the modeling approach of Dzombak and Morel.20 Fe(III) only forms ferrihydrite at TOTPO4 < 0.037 M (Fig. 2a), thus HFO adsorption modeling is only applicable at phosphate concentrations much lower than what are currently used in OP assays. At 50 μM Fe(III) (all of which was assumed to form HFO) there was minimal adsorption of Cu(II) and Mn(II) (Fig. S11†). Senn et al.32 reported that freshly formed FePO4(s) is capable of adsorbing metals, although there is no experimental data available to quantify FePO4(s) adsorption capacity. Iron oxide can adsorb orthophosphate40 and it is possible that FePO4(s) will also adsorb PO4. Given the high TOTPO4 used in OP assays we believe that PO4 will out-compete PM metals for adsorption sites and thus metal adsorption is likely of minimal concern.Conclusions
Overall, the processes we report likely have profound importance for in vitro assays of PM toxicity. The results also inform prior studies of transition metal toxicity. Charrier et al.23 and Fujitani et al.24 reported DTT consumption rate series as:| Cu(II) > Mn(II) ≫ Fe(III) ∼ Fe(II)23 |
| Ni(II) > Cu(II) > Mn(II)> Fe(III)24 |
It is likely that Fe(III) and Fe(II), either added as a reagent or through collected PM, will precipitate in PO4-based assays: Fe(III) precipitates as strengite and Fe(II) precipitates as Fe3(PO4)2(s). Fe(II) is readily oxidized to Fe(III) with subsequent precipitation of Fe(III). Mn(II) precipitates as MnPO4(s) but the precipitation rate appears to be slow at Mn(II) concentrations present in PM extracts (<5 μM), a possible reason for the high ROS generation potential of Mn(II). Mn(II) oxidation to Mn(IV) is slow at pH < 9 so a more likely scenario is that Mn(II) is oxidized to Mn(III) with subsequent formation of MnO(OH)(s). It is notable that Cu is – by far – the most active metal in catalyzing DTT reaction.24 The ascorbic acid assay is also highly sensitive to Cu.11,41 If these measurements are surrogates for ROS generation in human lungs, then this implies that Cu possesses an enhanced toxicity compared to other metals present in PM.42 However, toxicological43 and epidemiological studies44,45 generally do not support this assessment. This apparent disconnect is possibly due to its high solubility (∼90% solubility at pH = 7.4, TOTPO4 = 0.1 M, TOTCu(II) = 5 μM).
Metal precipitation is highly likely in most assay matrices due to the high TOTPO4 (∼0.1–0.5 M) and near-neutral pH (7.4) used to mimic biological systems. Decreasing TOTPO4 in acellular assays (while still maintaining pH = 7.4) shifts the precipitate identity to favor the formation of oxide/hydroxide solids, while still allowing oxidation of Fe(II) and Mn(II). When actual PM material is evaluated in PO4-based assays, multiple metals will be present increasing the likelihood of co-precipitation and increased precipitation rates (e.g., Fe(III) readily precipitates and then is a source of nucleation sites for other metal precipitates).
On the one hand, this behavior is unsurprising since a defining characteristic of metals is their pH-dependent solubility and their known complexation chemistry with PO4. However, this phenomenon has not been reported previously, likely because precipitate formation occurs even at low concentrations where solids are most likely colloidal in size and imperceptible to the human eye. Factors such as PM sample matrix, metal concentrations, and metal oxidation state can produce variability in metal precipitation (both rate and amount formed) suggesting that the process we report is quite variable within and between assays. Future studies should extend our results to include thermodynamic and experimental characterization of other PM metals, e.g., V and Ni, in PO4-based matrices.
The current study informs the fate and transformation of metals in OP assays that achieve pH control (typically pH = 7.4) using phosphate buffers. These include DTT, glutathione, and ascorbic acid. Our results likely extend to simulated (or surrogate) lung fluid (SLF), an acellular medium used to represent conditions in the human respiratory tract.13,46 The behavior and fate of metals in cellular assays is unknown, due to interactions between metals and cell culture media, especially organic molecules likely to form complexes with metals (e.g., proteins).
It is important to note that the effects of this phenomenon on assay response (e.g., ROS generation or antioxidant depletion) are beyond the scope of the present study and have not been characterized. Metals in solid and aqueous form can induce ROS formation,15,37 but it is likely that these would elicit quite different responses due to their different chemical forms and the surface effects associated with precipitates. For example the presence of antioxidants could facilitate rapid redox cycling of Fe(II)/Fe(III), Mn(II)/Mn(III)/Mn(IV), etc. Additionally, the formation of precipitates could affect the formation of hydroxyl radicals, superoxide and H2O2. We recommend that the effects of metal precipitation on ROS formation and differences in assay response to different chemical forms of each metal represent urgent research needs in understanding the health effects of PM and toxicity of individual PM constituents.
Data availability
All model inputs and outputs have been published, and are freely available at http://hdl.handle.net/11603/18201.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was funded by the National Science Foundation through award #1802474. We acknowledge Bill Schecher, Chief Programmer, MINEQL, for technical assistance with MINEQL, and Aidan Mucci and Carlos Castillo Lara: Undergraduate Research Assistants, UMBC Department of Chemical, Biochemical and Environmental Engineering for assistance with laboratory assays.References
- C. Davidson, R. Phalen and P. Solomon, Airborne Particulate Matter And Human Health: A Review, Aerosol Sci. Technol., 2005, 39(8), 737–749 CrossRef CAS.
- C. Pope and D. Dockery, Health Effects of Fine Particulate Air Pollution: Lines That Connect, J. Air Waste Manage. Assoc., 2006, 56(6), 709–742 CrossRef CAS.
- Institute, H.E., Health Effects Institute, State of Global Air, 2019 Search PubMed.
- C. Liu, R. Chen, F. Sera, A. Vicedo-Cabrera, Y. Guo, S. Tong, M. Coelho, P. Saldiva, E. Lavigne, P. Matus, N. Valdes Ortega, S. Osorio Garcia, M. Pascal, M. Stafoggia, M. Scortichini, M. Hashizume, Y. Honda, M. Hurtado-Díaz, J. Cruz, B. Nunes, J. Teixeira, H. Kim, A. Tobias, C. Íñiguez, B. Forsberg, C. Åström, M. Ragettli, Y. Guo, B. Chen, M. Bell, C. Wright, N. Scovronick, R. Garland, A. Milojevic, J. Kyselý, A. Urban, H. Orru, E. Indermitte, J. Jaakkola, N. Ryti, K. Katsouyanni, A. Analitis, A. Zanobetti, J. Schwartz, J. Chen, T. Wu, A. Cohen, A. Gasparrini and H. Kan, Ambient Particulate Air Pollution And Daily Mortality In 652 Cities, N. Engl. J. Med., 2019, 381(8), 705–715 CrossRef CAS.
- B. Maher, I. Ahmed, V. Karloukovski, D. MacLaren, P. Foulds, D. Allsop, D. Mann, R. Torres-Jardón and L. Calderon-Garciduenas, Magnetite Pollution Nanoparticles In The Human Brain, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(39), 10797–10801 CrossRef CAS.
- T. Wang, E. Chiang, L. Moreno-Vinasco, G. Lang, S. Pendyala, J. Samet, A. Geyh, P. Breysse, S. Chillrud, V. Natarajan and J. Garcia, Particulate Matter Disrupts Human Lung Endothelial Barrier Integrity Via ROS- And P38 MAPK-Dependent Pathways, Am. J. Respir. Cell Mol. Biol., 2010, 42(4), 442–449 CrossRef CAS.
- F. Schaumann, P. Borm, A. Herbrich, J. Knoch, M. Pitz, R. Schins, B. Luettig, J. Hohlfeld, J. Heinrich and N. Krug, Metal-Rich Ambient Particles (Particulate Matter 2.5) Cause Airway Inflammation In Healthy Subjects, Am. J. Respir. Crit. Care Med., 2004, 170(8), 898–903 CrossRef.
- L. Wood, P. Gibson and M. Garg, Biomarkers Of Lipid Peroxidation, Airway Inflammation And Asthma, Eur. Respir. J., 2003, 21(1), 177–186 CrossRef CAS.
- N. Li, C. Sioutas, A. Cho, D. Schmitz, C. Misra, J. Sempf, M. Wang, T. Oberley, J. Froines and A. Nel, Ultrafine Particulate Pollutants Induce Oxidative Stress And Mitochondrial Damage, Environ. Health Perspect., 2003, 111(4), 455–460 CrossRef CAS.
- A. Cho, C. Sioutas, A. Miguel, Y. Kumagai, D. Schmitz, M. Singh, A. Eiguren-Fernandez and J. Froines, Redox Activity Of Airborne Particulate Matter At Different Sites In The Los Angeles Basin, Environ. Res., 2005, 99(1), 40–47 CrossRef CAS.
- N. Janssen, A. Yang, M. Strak, M. Steenhof, B. Hellack, M. Gerlofs-Nijland, T. Kuhlbusch, F. Kelly, R. Harrison, B. Brunekreef, G. Hoek and F. Cassee, Oxidative Potential Of Particulate Matter Collected At Sites With Different Source Characteristics, Sci. Total Environ., 2014, 472, 572–581 CrossRef CAS.
- M. Strak, N. Janssen, K. Godri, I. Gosens, I. Mudway, F. Cassee, E. Lebret, F. Kelly, R. Harrison, B. Brunekreef, M. Steenhof and G. Hoek, Respiratory Health Effects Of Airborne Particulate Matter: The Role Of Particle Size, Composition, And Oxidative Potential—The RAPTES Project, Environ. Health Perspect., 2012, 120(8), 1183–1189 CrossRef CAS.
- A. Calas, G. Uzu, J. Martins, D. Voisin, L. Spadini, T. Lacroix and J. Jaffrezo, The Importance Of Simulated Lung Fluid (SLF) Extractions For A More Relevant Evaluation Of The Oxidative Potential Of Particulate Matter, Sci. Rep., 2017, 7(1), 1161 CrossRef.
- A. Teja and P. Koh, Synthesis, Properties, And Applications Of Magnetic Iron Oxide Nanoparticles, Prog. Cryst. Growth Charact. Mater., 2009, 55(1–2), 22–45 CrossRef CAS.
- P. Nico, B. Kumfer, I. Kennedy and C. Anastasio, Redox Dynamics Of Mixed Metal (Mn, Cr, And Fe) Ultrafine Particles, Aerosol Sci. Technol., 2009, 43(1), 60–70 CrossRef CAS.
- C. Hennigan, A. Mucci and B. Reed, Trends In PM 2.5 Transition Metals In Urban Areas Across The United States, Environ. Res. Lett., 2019, 14(10), 104006 CrossRef CAS.
- E. Vidrio, H. Jung and C. Anastasio, Generation Of Hydroxyl Radicals From Dissolved Transition Metals In Surrogate Lung Fluid Solutions, Atmos. Environ., 2008, 42(18), 4369–4379 CrossRef CAS.
- B. Venkataramani, K. Venkateswarlu and J. Shankar, Sorption Properties Of Oxides, J. Colloid Interface Sci., 1978, 67(2), 187–194 CrossRef CAS.
- B. Dempsey and P. Singer, The Effect Of Calcium On The Adsorption Of Zinc By Mnox (S) And Fe(OH)3 (Am), J. Colloid Interface Sci., 1980, 79, 209–221 Search PubMed.
- D. Dzombak and F. Morel, Surface complexation modeling, Wiley, New York, 1990 Search PubMed.
- L. Chen and M. Lippmann, Effects Of Metals Within Ambient Air Particulate Matter (PM) On Human Health, Inhalation Toxicol., 2009, 21(1), 1–31 CrossRef CAS.
- J. Mirowsky, C. Hickey, L. Horton, M. Blaustein, K. Galdanes, R. Peltier, S. Chillrud, L. Chen, J. Ross, A. Nadas, M. Lippmann and T. Gordon, The Effect Of Particle Size, Location And Season On The Toxicity Of Urban And Rural Particulate Matter, Inhalation Toxicol., 2013, 25(13), 747–757 CrossRef CAS.
- J. Charrier and C. Anastasio, On Dithiothreitol (DTT) As A Measure Of Oxidative Potential For Ambient Particles: Evidence For The Importance Of Soluble Transition Metals, Atmos. Chem. Phys., 2012, 12(19), 9321–9333 CrossRef CAS.
- Y. Fujitani, A. Furuyama, K. Tanabe and S. Hirano, Comparison Of Oxidative Abilities Of PM 2.5 Collected At Traffic And Residential Sites In Japan. Contribution Of Transition Metals And Primary And Secondary Aerosols, Aerosol Air Qual. Res., 2017, 17(2), 574–587 CrossRef CAS.
- E. Software, MINEQL+ Chemical Equilibrium Modeling System, https://mineql.com/ Search PubMed.
- M. Lin and J. Yu, Dithiothreitol (DTT) Concentration Effect And Its Implications On The Applicability Of DTT Assay To Evaluate The Oxidative Potential Of Atmospheric Aerosol Samples, Environ. Pollut., 2019, 251(0269–7491), 938–944 CAS.
- NIST Chemistry WebBook, https://webbook.nist.gov/chemistry/, accessed Apr 18, 2020 Search PubMed.
- USA H, DR/890 Portable Colorimeter, Hach USA – Downloads – Obsolete, https://www.hach.com/dr-890-portable-colorimeter/product-downloads?id=7640439041 Search PubMed.
- D. Barthelmy, Bixbyite Mineral Data, http://webmineral.com/data/Bixbyite.shtml#.XpmuMpl7lPY Search PubMed.
- V. Verma, T. Fang, H. Guo, L. King, J. Bates, R. Peltier, E. Edgerton, A. Russell and R. Weber, Reactive Oxygen Species Associated With Water-Soluble PM2.5 In The Southeastern United States: Spatiotemporal Trends And Source Apportionment, Atmos. Chem. Phys., 2014, 14(23), 12915–12930 CrossRef.
- P. Hsu, Crystallization Of Iron(III) Phosphate At Room Temperature, Soil Sci. Soc. Am. J., 1982, 46(5), 928–932 CrossRef CAS.
- A. Senn, R. Kaegi, S. Hug, J. Hering, S. Mangold and A. Voegelin, Composition And Structure Of Fe(III)-Precipitates Formed By Fe(II) Oxidation In Water At Near-Neutral Ph: Interdependent Effects Of Phosphate, Silicate And Ca, Geochim. Cosmochim. Acta, 2015, 162, 220–246 CrossRef CAS.
- B. Singh and M. Gräfe, Synchrotron-based techniques in soils and sediments, Elsevier, 2010 Search PubMed.
- L. Madsen and C. Koch, Kinetics Of Solution Crystal Growth Of Strengite, FePo4 2H2O, J. Cryst. Growth, 2018, 482, 9–14 CrossRef.
- J. Morgan and W. Stumm, Aquatic chemistry, Wiley, New York, 1981 Search PubMed.
- T. Grundl and J. Delwiche, Kinetics Of Ferric Oxyhydroxide Precipitation, J. Contam. Hydrol., 1993, 14(1), 71–87 CrossRef CAS.
- M. Schoonen, C. Cohn, E. Roemer, R. Laffers, S. Simon and T. O'Riordan, Mineral-Induced Formation Of Reactive Oxygen Species, Rev. Mineral. Geochem., 2006, 64(1), 179–221 CrossRef CAS.
- W. Sung and J. Morgan, Kinetics And Product Of Ferrous Iron Oxygenation In Aqueous Systems, Environ. Sci. Technol., 1980, 14(5), 561–568 CrossRef CAS.
- S. Faust and O. Aly, Chemistry of Water Treatment, Taylor & Francis, 2nd edn, 1998 Search PubMed.
- T. Angele Ngantcha-Kwimi and B. Reed, As(V) And PO4 Removal By An Iron-Impregnated Activated Carbon In A Single And Binary Adsorbate System: Experimental And Surface Complexation Modeling Results, J. Environ. Eng., 2016, 142(1), 04015046 CrossRef.
- E. DiStefano, A. Eiguren-Fernandez, R. Delfino, C. Sioutas, J. Froines and A. Cho, Determination Of Metal-Based Hydroxyl Radical Generating Capacity Of Ambient And Diesel Exhaust Particles, Inhalation Toxicol., 2009, 21(9), 731–738 CrossRef CAS.
- P. Lakey, T. Berkemeier, H. Tong, A. Arangio, K. Lucas, U. Pöschl and M. Shiraiwa, Chemical Exposure-Response Relationship Between Air Pollutants And Reactive Oxygen Species In The Human Respiratory Tract, Sci. Rep., 2016, 6(1), 32916 CrossRef CAS.
- U. Kodavanti, R. Hauser, D. Christiani, Z. Meng, J. McGee, A. Ledbetter, J. Richards and D. Costa, Pulmonary Responses To Oil Fly Ash Particles In The Rat Differ By Virtue Of Their Specific Soluble Metals, Toxicol. Sci., 1998, 43(2), 204–212 CAS.
- M. Bell, K. Ebisu, R. Peng, J. Samet and F. Dominici, Hospital Admissions And Chemical Composition Of Fine Particle Air Pollution, Am. J. Respir. Crit. Care Med., 2009, 179(12), 1115–1120 CrossRef CAS.
- B. Ostro, W. Feng, R. Broadwin, S. Green and M. Lipsett, The Effects Of Components Of Fine Particulate Air Pollution On Mortality In California: Results From CALFINE, Environ. Health Perspect., 2007, 115(1), 13–19 CrossRef CAS.
- D. Gonzalez, C. Cala, Q. Peng and S. Paulson, HULIS Enhancement Of Hydroxyl Radical Formation From Fe(II): Kinetics Of Fulvic Acid–Fe(II) Complexes In The Presence Of Lung Antioxidants, Environ. Sci. Technol., 2017, 51(13), 7676–7685 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0em00405g |
| This journal is © The Royal Society of Chemistry 2021 |