
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoscale interfacial engineering enables highly stable and efficient perovskite photovoltaics†
Anurag
Krishna‡
 *a,
Hong
Zhang‡
b,
Zhiwen
Zhou‡
b,
Thibaut
Gallet
c,
Mathias
Dankl
d,
Olivier
Ouellette
b,
Felix T.
Eickemeyer
b,
Fan
Fu
e,
Sandy
Sanchez
b,
Mounir
Mensi
f,
Shaik M.
Zakeeruddin
b,
Ursula
Rothlisberger
d,
G. N.
Manjunatha Reddy
g,
Alex
Redinger
c,
Michael
Grätzel
b and
Anders
Hagfeldt§
*a
*a,
Hong
Zhang‡
b,
Zhiwen
Zhou‡
b,
Thibaut
Gallet
c,
Mathias
Dankl
d,
Olivier
Ouellette
b,
Felix T.
Eickemeyer
b,
Fan
Fu
e,
Sandy
Sanchez
b,
Mounir
Mensi
f,
Shaik M.
Zakeeruddin
b,
Ursula
Rothlisberger
d,
G. N.
Manjunatha Reddy
g,
Alex
Redinger
c,
Michael
Grätzel
b and
Anders
Hagfeldt§
*a
aLaboratory of Photomolecular Science, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne, Lausanne 1015, Switzerland. E-mail: anurag.krishna@epfl.ch
bLaboratory of Photonics and Interfaces, École Polytechnique Fédérale de Lausanne, Lausanne 1015, Switzerland
cScanning Probe Microscopy Laboratory, Department of Physics and Materials Science, University of Luxembourg, Luxembourg
dLaboratory of Computational Chemistry and Biochemistry, École Polytechnique Fédérale de Lausanne, Lausanne 1015, Switzerland
eLaboratory for Thin Films and Photovoltaics, Empa-Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, CH-8600 Dübendorf, Switzerland
fInstitute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne, Valais Wallis, CH-1951 Sion, Switzerland
gUniv. Lille, CNRS, Centrale Lille Institut, Univ. Artois, UMR 8181-UCCS-Unité de Catalyse et Chimie du Solide, F-59000 Lille, France
First published on 16th September 2021
Abstract
We present a facile molecular-level interface engineering strategy to augment the long-term operational and thermal stability of perovskite solar cells (PSCs) by tailoring the interface between the perovskite and hole transporting layer (HTL) with a multifunctional ligand 2,5-thiophenedicarboxylic acid. The solar cells exhibited high operational stability (maximum powering point tracking at one sun illumination) with a stabilized TS80 (the time over which the device efficiency reduces to 80% after initial burn-in) of ≈5950 h at 40 °C and a stabilized power conversion efficiency (PCE) over 23%. The origin of high device stability and performance is correlated to the nano/sub-nanoscale molecular level interactions between ligand and perovskite layer, which is further corroborated by comprehensive multiscale characterization. These results provide insights into the modulation of the grain boundaries, local density of states, surface bandgap, and interfacial recombination. Chemical analysis of aged devices showed that molecular passivation suppresses interfacial ion diffusion and inhibits the photoinduced I2 release that irreversibly degrades the perovskite. The interfacial engineering strategies enabled by multifunctional ligands can expedite the path towards stable PSCs.
Broader contextMetal halide perovskites have demonstrated a certified power conversion efficiency (PCE) of 25.5% in single-junction solar cells. In perovskite solar cells, the ‘soft’ interface between the perovskite layer and charge transporting layer (CTL) is one of the major bottlenecks that limit operational stability and device longevity. The defects at this interface cause non-radiative recombination and thus lead to performance loss. In addition, these interfacial defects promote the diffusion of halide ions across the interface into the CTL, which adversely affect the long-term operational stability. Here we present a facile molecular-level interface engineering strategy using a multifunctional ligand, which augments the stability of single-junction solar cells. Chemical analysis of aged devices showed that molecular passivation suppresses the interfacial ion diffusion and inhibits the photoinduced I2 release that irreversibly degrades the perovskite. This study provides molecular-level insights into the modulation of the grain boundaries, local density of states, surface bandgap, and interfacial recombination. These results and understandings offer recommendations to expedite the design strategies of passivating ligands and are expected to stimulate further work on the fundamental understanding of interfacial ion diffusion mechanisms to facilitate stable and efficient PSCs. |
Introduction
Metal halide perovskites are poised to revolutionize next-generation optoelectronics, enabling economically viable and highly efficient photovoltaics,1–4 light-emitting diodes,5,6 and photodetectors.7,8 The leap forward in the power conversion efficiency (PCE) enabled by lead (Pb) halide perovskites is unprecedented, with PCEs emerging from 3.8% in its first study9 to a recent certified value of 25.5% in single-junction perovskite solar cells (PSCs).10 Nevertheless, the functional instability associated with these materials under operational conditions11–14 constrains their widespread applications in optoelectronic devices. While the molecular-level origins that contribute to the instability depend on the compositions, structures, processing, and operating conditions, the ionic nature and weak molecular interaction, and soft crystal lattice are believed to create point defects and mobile ions15,16 leading to the performance degradation of perovskites. Besides these point and electronic defects, perovskite materials often contain undercoordinated Pb2+ and iodide (I−) ions, which are susceptible to reduction or oxidation (redox) reactions.17 These undercoordinated ions are mostly present at the surface grains and grain boundaries (GBs).18 The reduction of Pb2+ ions to metallic Pb (Pb0) is central to the non-radiative recombination process.19,20 The I− ions can oxidize into volatile iodine, I2, which can cause irreversible decomposition of perovskites and trigger chemical chain reactions to accelerate the degradation processes.3,21,22 Furthermore, vacancies provide pathways for mobiles ions, particularly iodide species, which can migrate to interfaces under an electric field, thus impairing the charge transporting layer and compromising photovoltaic performance and long-term operational stability.15,23 The ionic nature of defects allows passivation via tailored molecular modulators based on Lewis acid–base chemistry, coordination bonding, and ionic bonding.24–29 Notably, small organic molecules containing electron lone pair donors such as nitrogen,28,30 sulfur,29,30 or oxygen16,31 have been shown to effectively passivate cationic defects and undercoordinated cation sites in perovskites. Recent studies lend credibility to the perovskite surface passivation by multifunctional molecules27 as a promising approach, leading to performance and stability enhancement. While molecular passivation is emerging as a credible and robust strategy for perovskite defect passivation, the molecular-level understanding of the passivation mechanism is still not fully understood. In addition, studies on the correlation between molecular passivation and long-term operational stability (>1000 h) of high-efficiency PSCs (>23%) under different aging conditions are also lacking. To this end, an accurate understanding of passivation mechanisms and the associated enhanced stabilities and optoelectronic properties will expedite the development of stable-efficient perovskite photovoltaic technologies.Here, we report a facile molecular design strategy that shows how 2,5-thiophenedicarboxylic acid ligand effectively targets interface defects and enhances the stability of the PSCs. The ligand is rigid, has high thermal stability, and contains carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and hydrogen bonding donors, thus sustaining the passivation of various cationic and anionic defects via coordination and hydrogen bonding. The strong binding of the ligand on the surface leads to suppression of interfacial recombination, halide migration into the hole transporting layer (HTL) and prevents I2 formation. The PSCs fabricated show stabilized PCEs over 23% and enhanced long-term operational stability at 40 °C and thermal stability at 85 °C.
O), and hydrogen bonding donors, thus sustaining the passivation of various cationic and anionic defects via coordination and hydrogen bonding. The strong binding of the ligand on the surface leads to suppression of interfacial recombination, halide migration into the hole transporting layer (HTL) and prevents I2 formation. The PSCs fabricated show stabilized PCEs over 23% and enhanced long-term operational stability at 40 °C and thermal stability at 85 °C.
Results and discussions
To test the potential of the 2,5-thiophenedicarboxylic acid ligand and its role in enhancing the thermal stability of perovskites, we employ perovskite with FAPbI3 rich composition of FA0.97MA0.03PbI2.91Br0.09 (FA+ = formamidinium = CH(NH2)2+; MA+ = methylammonium = CH3NH3+). Perovskite films were deposited by spin-coating onto the mesoporous TiO2 (mp-TiO2) layer by a one-step method using diethyl ether as an antisolvent, and then by spin-coating the passivating ligand on the top of the perovskite layer (see Methods in ESI†). Hereafter, the pristine composition (FA0.97MA0.03PbI2.91Br0.09) is referred to as the control, and perovskite coated with the ligand (FA0.97MA0.03PbI2.91Br0.09/ligand) is referred to as the target material. The impact of interface passivation is investigated using the multi-technique structure and electronic characterizations at different length scales, device physics, and complemented by stability assessments, as discussed below.Molecular design, interactions, and local structure
At the outset, we prepared a molecular design strategy focussed on developing a multifunctional molecule for interface passivation to reduce defects, passivate GBs, and suppress the ion migration across the interface. In the current strategy, we have combined the functionalities associated with different anchoring groups into a single molecule, which in turn can bind to the perovskite crystal through multiple non-covalent interactions. Given the importance of an aromatic core that can impart rigidity to the molecule and conjugated π-electrons are expected to improve the charge transport across grain boundaries, and the end groups that enhance the binding affinity to perovskite grains/GBs, we choose thiophene as an aromatic core and a carboxyl (COOH) as a functional group. The COOH contains a carbonyl group (CO) that can coordinate Pb2+ ions and it also has a hydroxyl (–OH) group that can participate in H-bonding with FA and or I.32 Thiophene itself can act as Lewis base by lone pair donation through sulfur and has been reported to improve the property of perovskites by passivating undercoordinated lead.30 By linking two such COOH groups on 2 and 5 positions of thiophene moiety (2,5 thiophenedicarboxylic acid), it has been feasible to synthesize a rigid bifunctional molecule that can potentially bind between adjacent surface grains thus passivating the deleterious surface grain boundaries and suppressing the interfacial ion diffusion. Our molecular design strategy is further corroborated by the electrostatic potential (ESP) analysis of thiophene, 2-thiophenecarboxylic acid, and 2,5 thiophenedicarboxylic acid (Fig. S1, ESI†), which allows us to compare the electrostatic properties of thiophene and carboxylic acid functional groups. As shown in Fig. S1 (ESI†), in the case of thiophene a negative ESP is observed over the thiophene ring whereas in 2,5 thiophenedicarboxylic acid a higher negative ESP was found to be on the two carbonyl end groups, which could enhance its passivation ability. Thus, 2,5 thiophenedicarboxylic acid has been chosen for the interface engineering in this work.
The crystallinity and structural properties of perovskite films were analyzed by X-ray diffraction, which is bulk sensitive (Fig. S2a, ESI†), and grazing incidence X-ray diffraction (GIXRD; Fig. S2b, ESI†), which is more surface sensitive. Both control and target films showed a well-crystallized photoactive α-phase of the 3D perovskite. In GIXRD of the target, the absence of any additional peak at lower angles indicates that surface passivation did not lead to the formation of low dimensional structures. The surfaces of both control and target films were found to be PbI2 rich, which is well known for perovskite formulations prepared with excess PbI2.33 Morphological features of the control and the target perovskite films were analyzed by scanning electron microscopy (SEM; Fig. S3, ESI†). The surface of both control and target perovskite films are uniform and highly crystalline with similar compact textures and grain sizes of hundreds of nanometers. In the target sample, we observe several spots with different contrast than the crystal grains and we speculate these could be the ligand molecules located in between the grains or over the grains. The surface composition was further studied by X-ray photoelectron spectroscopy (XPS). XPS spectra of both the control and the target films in the Pb 4f level range (Fig. 1a) reveals the two prominent peaks associated with Pb 4f7/2 and Pb 4f5/2 at 138.8 and 143.7 eV, respectively, attributed to Pb–I species. However, in target film, a small broadening of the signal is observed, which could be attributable to Pb–ligand interactions. Furthermore, the control film showed two smaller peaks located at 136.9 and 141.8 eV that arise from the presence of metallic Pb (Pb0). This Pb0 is known to form from the reduction of Pb2+ ions.17 The metallic Pb peaks do not appear in the target film, which suggests that the ligand binds to undercoordinated Pb2+ ions responsible for forming metallic Pb, thus inhibiting the formation of metallic Pb. Moreover, the presence of the S 2p level peak at 165.3 eV and O 1s level peak at 532.4 eV (Fig. S4, ESI†) in the target film suggests surface adsorption of the ligand. This was also confirmed computationally by density functional theory (DFT) calculations that identified a stable adsorbate structure with a surface binding energy of −0.85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) eV, in which the oxygen from one carbonyl group of the ligand binds to undercoordinated Pb2+ at the surface while a hydrogen bond is formed by the second carboxyl group with a neighboring surface iodide (Fig. 1b). In order to validate this structural model, the molecular interactions of the ligand with the perovskite surface were investigated with attenuated total reflection – Fourier-transform infrared spectroscopy (ATR-FTIR). The FTIR spectrum of ligand reveals peak characteristic of the CO stretching vibration at ∼1656 cm−1 (Fig. 1c and Fig. S5, ESI†), which shifted to 1664 cm−1 in the target film. This was not observed in the spectrum of the control perovskite film. This shift indicates interaction of the CO with the perovskite surface. The CO is Lewis base and was expected to interact with uncoordinated Pb ions. The FTIR spectrum of a film of PbI2 + ligand showed that CO stretching undergoes broadening and is blue-shifted to 1664 cm−1 compared to the pristine ligand, which shows CO stretching vibration at 1656 cm−1 (Fig. S6, ESI†). Thus, the ligand interacts with undercoordinated Pb2+ ions on the perovskite surface. Although FTIR results point to the intermolecular interactions between surface ligand and photoactive layer, the information on the atomic-level interfacial interactions cannot be fully obtained from this characterization tool. To gain deeper insights into atomic level site-specific interfacial contacts between ligand and perovskite layer, we employed 1D and 2D solid-state nuclear magnetic resonance (ss-NMR) spectroscopy.
eV, in which the oxygen from one carbonyl group of the ligand binds to undercoordinated Pb2+ at the surface while a hydrogen bond is formed by the second carboxyl group with a neighboring surface iodide (Fig. 1b). In order to validate this structural model, the molecular interactions of the ligand with the perovskite surface were investigated with attenuated total reflection – Fourier-transform infrared spectroscopy (ATR-FTIR). The FTIR spectrum of ligand reveals peak characteristic of the CO stretching vibration at ∼1656 cm−1 (Fig. 1c and Fig. S5, ESI†), which shifted to 1664 cm−1 in the target film. This was not observed in the spectrum of the control perovskite film. This shift indicates interaction of the CO with the perovskite surface. The CO is Lewis base and was expected to interact with uncoordinated Pb ions. The FTIR spectrum of a film of PbI2 + ligand showed that CO stretching undergoes broadening and is blue-shifted to 1664 cm−1 compared to the pristine ligand, which shows CO stretching vibration at 1656 cm−1 (Fig. S6, ESI†). Thus, the ligand interacts with undercoordinated Pb2+ ions on the perovskite surface. Although FTIR results point to the intermolecular interactions between surface ligand and photoactive layer, the information on the atomic-level interfacial interactions cannot be fully obtained from this characterization tool. To gain deeper insights into atomic level site-specific interfacial contacts between ligand and perovskite layer, we employed 1D and 2D solid-state nuclear magnetic resonance (ss-NMR) spectroscopy.
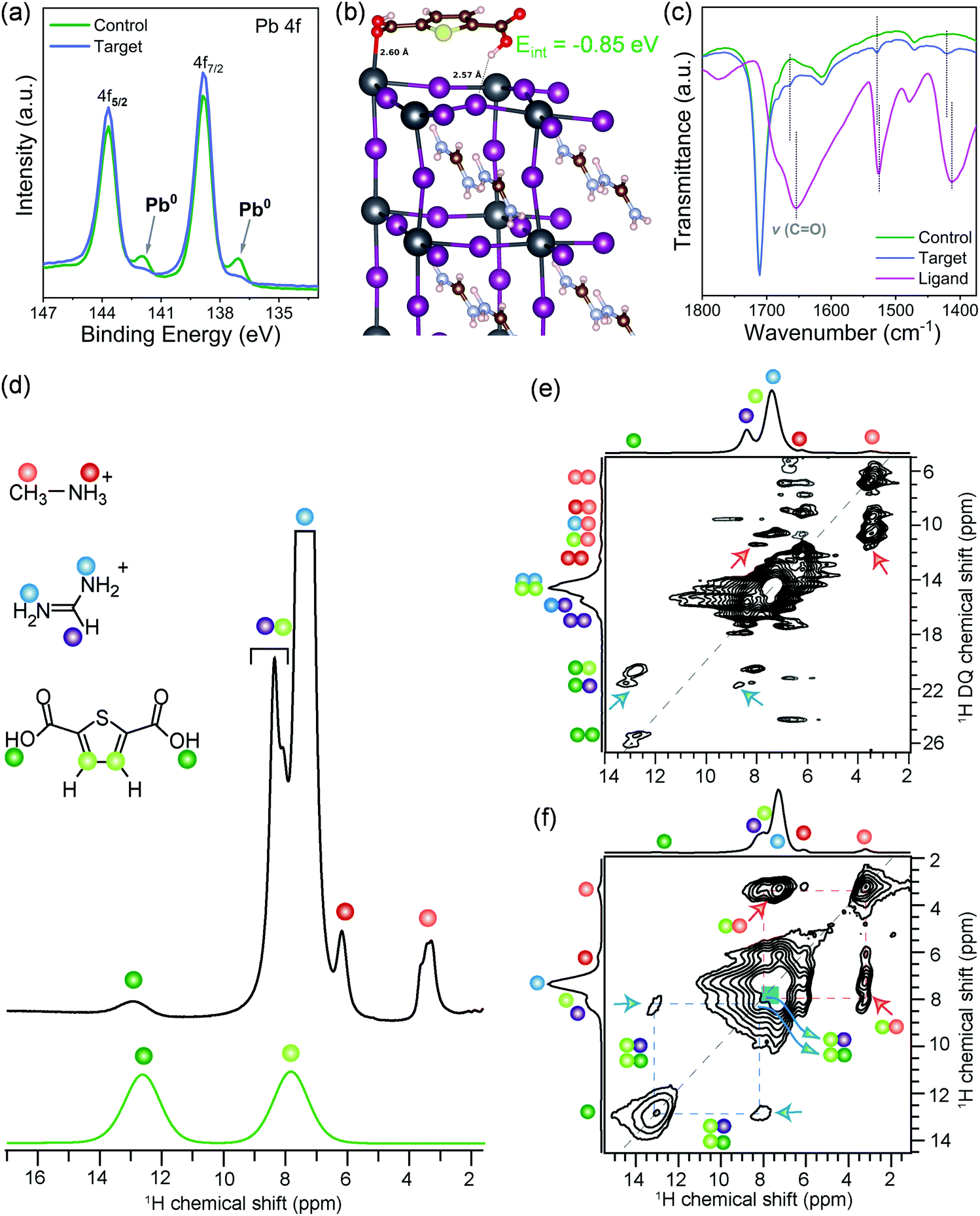 | ||
| Fig. 1 Molecular interactions and local structure. (a) XPS data for Pb 4f7/2 and Pb 4f5/2 core-level spectra in control and target perovskite films. (b) The optimized configuration of the 2,5-thiophenedicarboxylic acid molecule on the surface of FAPbI3. (c) FTIR spectra of the passivating ligand, control, and target films. Solid-state (d) 1D 1H MAS NMR spectra of the target sample and 2,5-thiophenedicarboxylic acid ligand, (e) 2D 1H–1H DQ–SQ correlation spectrum, and (f) 2D 1H–1H spin diffusion NMR spectrum acquired with 500 ms of mixing time for target sample. 1H signals associated MA+, FA+, and surface ligands are shown in color dots, as depicted in the schematic structures (d). Red and blue arrows in (e) and (f) indicate the through-space 1H–1H proximities between the surface passivating agent and MA+/FA+ organic cations in perovskite crystals. All ssNMR spectra were acquired at 18.8 T (1H, 800 MHz) and room temperature using 20 kHz MAS. | ||
The short-range structures, interfacial interactions, and site-specific dynamics in optoelectronic materials have previously been elucidated by ss-NMR spectroscopy techniques,34–42 owing to its nanoscale sensitivity and resolution that allows organic and inorganic interfaces to be identified and distinguished. A comparison of 1D 1H NMR spectra of the target film and ligand (Fig. 1d) indicates that the weak intensity 1H signals at 3.3 and 6.2 ppm corresponding to the methyl (orange dot) and ammonium protons (red dot) of MA+ cations, and strong intensity signals at 7.4 and 8.4 ppm corresponding to –NH2 (blue dots) and –CH (purple dot) proton sites in FA+ cations, respectively. Partially resolved signal at ∼7.9 ppm and a peak at 12.9 ppm are attributable to the thiophene (light green), and carboxylic acid protons (dark green dots) of the ligand. The assignment of this latter signal at 12.9 ppm (COOH) is corroborated by the 1H NMR spectrum of neat ligand, which is attributable to weak hydrogen-bonding interactions. Correspondingly, the thiophene 1H chemical shift at 7.9 ppm indicates well dispersed molecules. If the thiophene molecules were π−π stacked, this signal would be in the vicinity of 6–7 ppm due to ring current effects.43 The local structures of MA+, FA+ and ligand, and inter- and intramolecular interactions at sub-nanometer (<5 Å) distances were characterized by analyzing 2D 1H double-quantum (DQ)–single-quantum (SQ) correlation spectrum of the target sample (Fig. 1e). In the 2D 1H DQ–SQ spectrum, the on-diagonal 1H DQ peaks at 6.6 ppm and 12.4 ppm correspond to proton sites in methyl and ammonium groups of MA+ cations, respectively, and the DQ peak at 14.8 ppm corresponds to NH2 groups of FA+ cations. A weak 1H DQ signal at ∼25.8 ppm is attributed to intermolecular 1H–1H dipolar interactions between hydroxyl protons in the ligand, further indicating the surface dispersion of ligands. By comparison, the off-diagonal 1H DQ peak at 3.3 + 6.2 = 9.5 ppm (orange-red dots) originates from the intramolecular CH3–NH3 proximities in MA+, and at 7.4 + 8.4 = 15.8 ppm (blue-purple dots) originates from the intramolecular (NH2)2–CH proximities in FA+ cations, and the DQ peak at 7.9 + 12.9 = 20.8 ppm (light- and dark-green dots) originates from intramolecular 1H–1H proximities between thiophene and carboxylic protons in the surface ligand. In addition, an off-diagonal 1H DQ peak at 3.3 + 7.4 = 10.7 ppm (blue-red dots) indicate the intermolecular 1H–1H proximities between MA+ and FA+ cations within the perovskite framework. Of particular interest is the weak intensity 1H DQ signal at ∼11.2 ppm (red arrows), which could be attributed to the intermolecular interactions between thiophene protons (7.9 pm) of surface ligand and methyl protons (3.3–3.5 ppm) of MA+ cations. In addition, a DQ correlation peak at 21.3 ppm corresponding to the dipolar interactions between hydroxyl proton (12.9 ppm) of the ligand and –CH site (8.4 ppm) of the FA+ cation (blue arrows) confirms the molecular proximity between the ligand and FA+ cations. Although severely overlapped 1H signal intensities originating from chemically distinct protons in the vicinity of 1H DQ 14–18 ppm hinders the accurate identification of intermolecular interactions between FA+ cations and the ligand, 2D correlation peaks depicted in blue and red arrows confirm the molecular proximity between the ligand and FA+/MA+ cations, as suggested by the DFT modeled structure (Fig. 1b). Further insights into the intermolecular 1H–1H proximities between framework MA+/FA+ cations and ligand can be obtained by analyzing 2D 1H–1H spin diffusion (SD) spectra (Fig. 1f and Fig. S7, ESI†). In a 2D 1H spin-diffusion experiment, nuclear spin magnetization is allowed to exchange between neighboring protons as function of time (also referred to as spin diffusion mixing time). 2D 1H–1H SD spectrum acquired with 500 ms of mixing time (Fig. 1f) shows the off-diagonal correlation peaks at 3.3 and 7.9 ppm (red arrows), which indicate the proton spin magnetization exchange between closely proximate CH3 groups of MA+ and thiophene protons of the surface ligand. The through-space molecular proximities between the organic cations and ligands are best consolidated by the off-diagonal peaks (blue arrows) between FA+ protons (7.4 ppm) and thiophene protons (7.9 ppm) of the surface ligand. In addition, the off-diagonal 2D peaks associated with 7.9 ppm and 12.9 ppm (blue arrows) are attributable to intermolecular 1H–1H proximities between thiophene and FA+ as well as intramolecular thiophene and carboxylic protons within the surface ligand, which are detected with much higher intensities in the 2D 1H SD spectrum acquired with 1000 ms of mixing time (Fig. S7, ESI†). Overall, 2D ss-NMR results reveal the through-space intermolecular proximities between 2,5-thiophenedicarboxylic acid and organic cations in the perovskite framework at the at sub-nanometer to nanometer distances thus confirming the atomic level interaction at the perovskite–ligand interface.
Grain boundaries, surface potential, and local density of states
Having established the molecular-level interactions between surface ligand and perovskite layers in the target material, we next carried out a detailed surface analysis of grain boundaries (GBs) and grain interior/surface. Kelvin probe force microscopy measurements (KPFM) were carried out on both types of absorbers (topography images in Fig. S8, ESI†). Both surfaces exhibited variations in the work function of 150–200 meV, which was extracted from the average distribution of the work function maps as shown in Fig. S9 (ESI†). We attribute these electrical inhomogeneities to grain-to-grain variations induced by different crystallographic facets with distinct surface terminations, which is consistent with similar studies and analyses in the literature.44–46 The surface treatment with the ligand reduced the average work function Φ by approximately 200 meV, as indicated by the scale bar next to the calibrated work function map in Fig. 2(a and b). Furthermore, we observed a clear work function contrast at most of the GBs of both absorbers, which is characteristic of electric charge accumulation/depletion. A detailed statistical analysis of the GB band bending is presented in Fig. 2(c), where histograms of the difference between the work function at the grains ΦGrain and the adjacent GBs ΦGB are summarized and compared. For the control sample, we observed a uniform distribution of upward and downward band bending of approximately ±100 meV. In contrast, the target sample showed almost exclusively upward band bending, which is a characteristic of negative charges at the GBs, i.e., a small electron barrier. Since GBs in perovskite have been shown to be potential recombination centers,47,48 thus, by creating the potential barrier for electrons at the GBs, recombination can be strongly suppressed, while for holes, a sufficient driving force exists at the HTL interface which favors the hole extraction. Next, we carried out scanning tunneling microscopy (STM) and spectroscopy (STS) measurements to get insights into the surface bandgap and local density of states (LDOS) at the surface. Constant current imaging spectroscopy (CITS) maps, acquired at sample locations to extract a representative measure of the local density of states of both types of absorber layers, are shown together with STM images in ESI,† (Fig. S10). The average LDOS of both samples is summarized in Fig. 2(d). The data is plotted as in order to estimate the surface bandgap.46,49 For the control sample, we deduced a value of 1.6 eV ± 0.1 eV, which is close to the bulk bandgap of the perovskite absorber (1.56 eV, from the Tauc plot and IPCE inflection point in ESI,† Fig. S11 and S12). For the target absorber, we measured an increased surface bandgap of 1.9 eV ± 0.1 eV. Such a large surface bandgap can suppress the interfacial recombination at the perovskite/HTL interface. For the control sample, the Fermi-level was closer to the conduction band, in consonance with the previous reports,46,50,51 which points towards weak n-type doping or defect-induced surface band bending. The target sample exhibited a local density of states (LDOS) curve, closer to an intrinsic semiconductor. Compared to the control samples, most of the changes occurred at positive bias voltages, i.e., an upward shift of the conduction band. In the inset of Fig. 2(d), we plot the LDOS on a logarithmic scale to analyze the vanishing conductance at the band edges and within the bandgap. Interestingly, we observed an increased tunneling conductance at positive applied voltages for the control sample, which was conspicuously absent in the target absorber. The removal of a defect state at the interface to the extraction layer reduces the surface recombination velocity and thereby improves VOC. Although the origin of the defect response cannot be accurately identified directly from the STS measurements, defect formation energy (DFE) computations suggest that from a thermodynamical perspective, among the possible surface defects, lead-iodine antisite (PbI DFE = 0.59 eV) is likely to form on lead iodide rich FAPbI3 perovskite surface.16,52 In this respect, the most stable configurations of the molecule that interact with PbI defect was modelled using DFT calculations (Fig. S13, ESI†). The passivating molecule showed interaction energy of −0.98 eV thus confirming a favorable interaction with the antisite defect on the perovskite surface. Based on the KPFM/STM/STS measurements and DFT calculations, we reasoned that the surface passivation with ligand removed defects in the vicinity of the conduction band, increased the surface bandgap, homogenized the grain boundary band bending towards an electron barrier, and showed favorable interaction with PbI defect.
in order to estimate the surface bandgap.46,49 For the control sample, we deduced a value of 1.6 eV ± 0.1 eV, which is close to the bulk bandgap of the perovskite absorber (1.56 eV, from the Tauc plot and IPCE inflection point in ESI,† Fig. S11 and S12). For the target absorber, we measured an increased surface bandgap of 1.9 eV ± 0.1 eV. Such a large surface bandgap can suppress the interfacial recombination at the perovskite/HTL interface. For the control sample, the Fermi-level was closer to the conduction band, in consonance with the previous reports,46,50,51 which points towards weak n-type doping or defect-induced surface band bending. The target sample exhibited a local density of states (LDOS) curve, closer to an intrinsic semiconductor. Compared to the control samples, most of the changes occurred at positive bias voltages, i.e., an upward shift of the conduction band. In the inset of Fig. 2(d), we plot the LDOS on a logarithmic scale to analyze the vanishing conductance at the band edges and within the bandgap. Interestingly, we observed an increased tunneling conductance at positive applied voltages for the control sample, which was conspicuously absent in the target absorber. The removal of a defect state at the interface to the extraction layer reduces the surface recombination velocity and thereby improves VOC. Although the origin of the defect response cannot be accurately identified directly from the STS measurements, defect formation energy (DFE) computations suggest that from a thermodynamical perspective, among the possible surface defects, lead-iodine antisite (PbI DFE = 0.59 eV) is likely to form on lead iodide rich FAPbI3 perovskite surface.16,52 In this respect, the most stable configurations of the molecule that interact with PbI defect was modelled using DFT calculations (Fig. S13, ESI†). The passivating molecule showed interaction energy of −0.98 eV thus confirming a favorable interaction with the antisite defect on the perovskite surface. Based on the KPFM/STM/STS measurements and DFT calculations, we reasoned that the surface passivation with ligand removed defects in the vicinity of the conduction band, increased the surface bandgap, homogenized the grain boundary band bending towards an electron barrier, and showed favorable interaction with PbI defect.
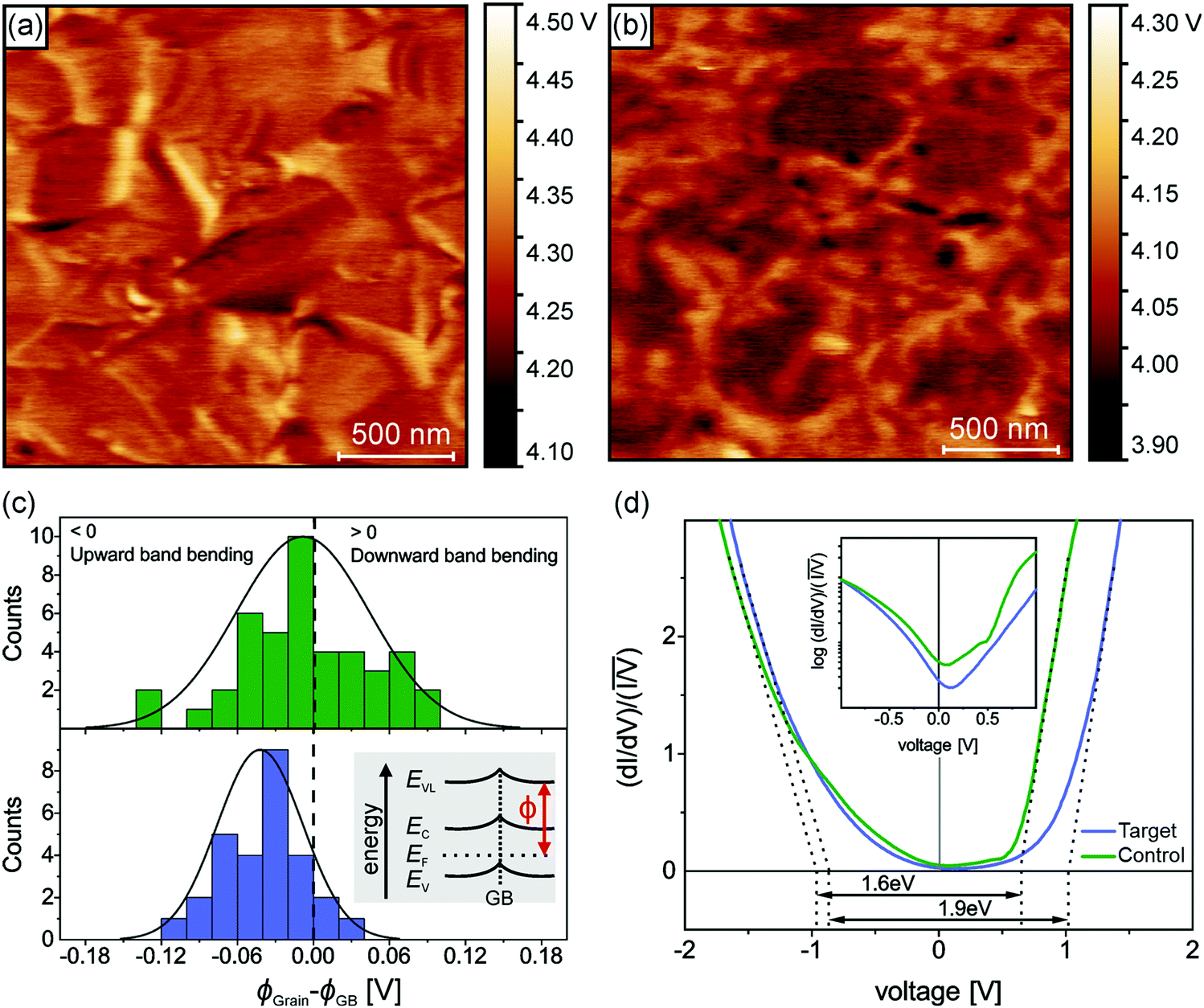 | ||
| Fig. 2 Surface potential and the local density of states. (a) Calibrated work function map of the control sample and (b) of the target sample as measured with Kelvin probe force microscopy measurements (KPFM) in ultrahigh vacuum (UHV). The contrast of the work function maps has been manually adjusted for visual clarity. (c) Grain boundary band bending for the control sample (upper graph) and the target sample (lower graph). The inset depicts a schematic band diagram of a grain boundary with an excess of negative electric charges (upward band bending). (d) Average scanning tunneling spectroscopy curves of the control and target sample. Linear extrapolation of the normalized LDOS curves (dotted lines) corroborated that the target sample exhibits an enlarged surface bandgap compared to the control sample. Inset: LDOS curves plotted on a logarithmic scale. The control sample shows a defect response at a positive applied voltage. | ||
Photovoltaic performance and interfacial recombination
The photovoltaic performance of the corresponding PSCs was investigated using the conventional n–i–p configuration of FTO/compact TiO2/Li doped mesoporous TiO2: perovskite composite layer/perovskite upper layer/spiro-OMeTAD/Au (Fig. S14, ESI†). The concentration of ligand was optimized with respect to the photovoltaic performance (see details in Fig. S15, ESI†). Current density–voltage (J–V) curves of the champion devices shown in Fig. 3a and Fig. S16 (ESI†) reveal that the target device exhibited a PCE of 23.4% empowered by a high VOC of 1.17 V, a short-circuit current density (JSC) of 25.2 mA cm−2, and, a fill factor (FF) of 79.1%, whereas the control device showed a PCE of 22.4% with a VOC of 1.13 V, a JSC of 25.2 mA cm−2, and a FF of 78.5%. We further substantiated these values by recording scan-speed-independent maximum power point tracking (MPP) measurements for 5 min (Fig. 3a, inset), corresponding to PCEs of 21.3% and 23.0% for the control and target device, respectively. Table S1 (ESI†) summarises the performance of the control and target champion device. The integrated current density derived from the internal photon conversion efficiency (IPCE; Fig. 3b) is within a 5% difference in the JSC value obtained from the J–V curve. Please note that for the champion devices we applied an anti-reflection (AR) coating on the front glass. The AR coating decreases the reflection losses of the incident light at the air/glass interface. The J–V curve of the champion target cell with and without AR coating in Fig. S17 (ESI†) shows that there is an improvement in the PCE upon application of AR coating, which is mainly due to the increase in the JSC. A comparison of different photovoltaic parameters for control and target devices is summarized in Fig. S18 (ESI†). The improvement in the optoelectronic properties upon surface passivation is particularly reflected in the enhanced VOC value from 1.12 ± 0.009 V to 1.16 ± 0.009 V and the enhanced fill factor (FF) from 77.68 ± 0.75% to 78.96 ± 0.45%. The role of surface passivation on the electronic properties of control films was further evaluated by the ideality factor (nid), which is related to the dependence of VOC on the intensity of the incident light (Fig. 3c). Post surface passivation, the nid decreases from 1.65 to 1.27, indicating the suppression of non-radiative recombination channels.53 To further discern processes leading to the VOC enhancement and to understand the interface losses, we measured absolute photoluminescence (PL) photon fluxes ΦPL(E) of pristine perovskite films (Fig. S19 and Table S2, ESI†) and complete devices (Fig. 3d). The photoluminescence quantum yield (PLQY) of the target device was 5 times higher than that of the control device, which translated to an increase of 43 meV in quasi-Fermi level splitting (ΔEF) for the target device with respect to the control (see calculations in ESI† Note 1). This value is in good agreement with the measured VOC difference of 40 mV, which confirms that the VOC improvement originates mainly from a ΔEF increase, and thus from a reduction in non-radiative interfacial recombination. Different types of non-radiative losses, as shown in Fig. 3e and Fig. S20 (ESI†), corroborate that molecular passivation significantly reduces the recombination at the perovskite/HTL interface.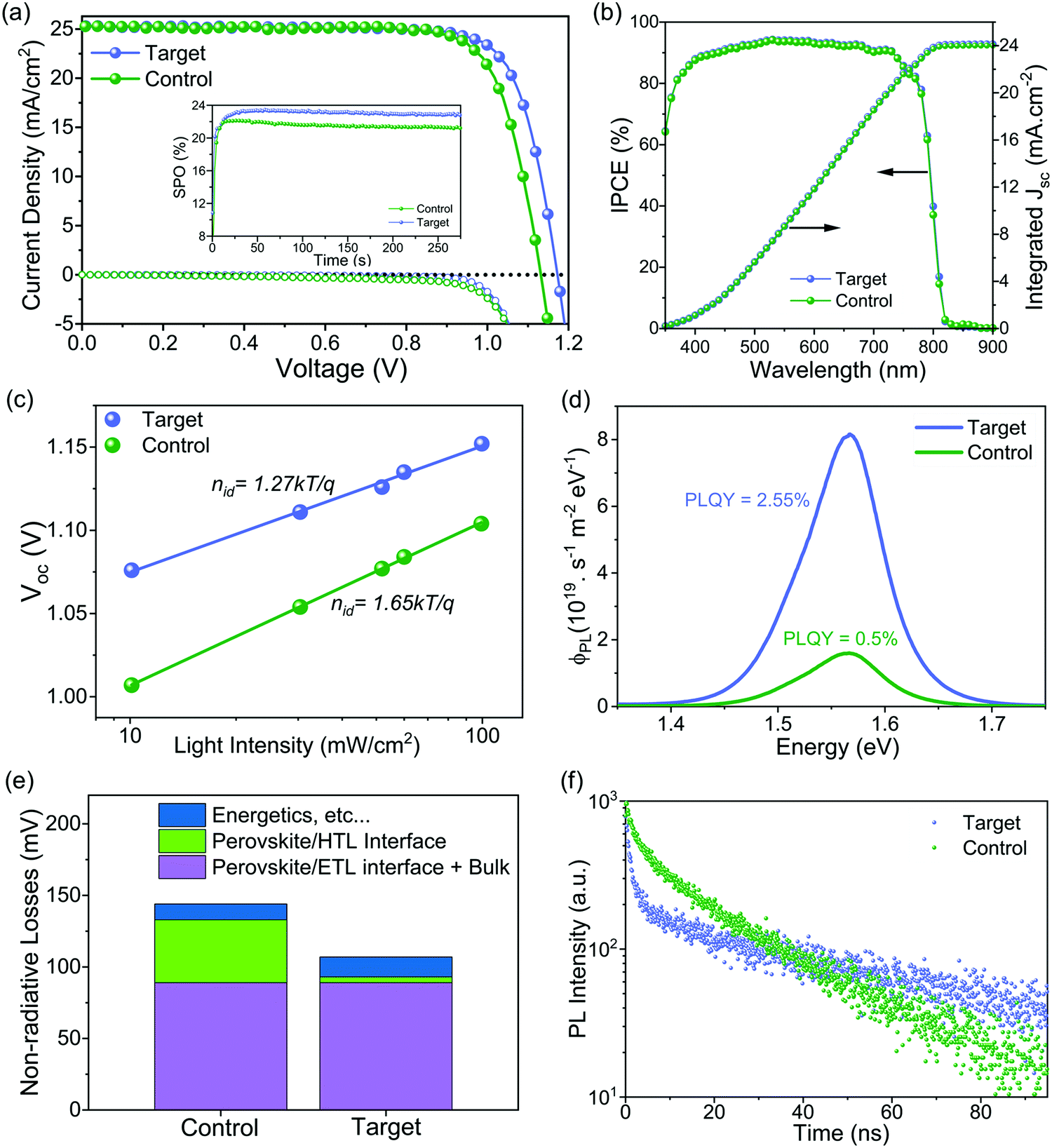 | ||
| Fig. 3 Device performance and interfacial recombination. (a) J–V curves of the champion solar cells under reverse scan. An antireflection coating on the glass/FTO was applied before these J–V measurements. The inset shows MPP tracking of the device within the first 250 s under ambient air. (b) The IPCE spectra of the control and target device. An antireflection coating was used. (c) VOC dependence of light intensity with ideality factors nid. (d) Absolute photon flux ΦPL(E) measurements of complete devices at 1 sun excitation from which the PLQY is determined. (e) Different types of energy losses are calculated from the PLQY of neat films and devices to compare the contribution of the different recombination processes for control and target films and devices. Loss (perovskite/ETL interface + Bulk) = VOC,rad. − VOC,PLQY,film; loss (perovskite/HTL interface) = VOC,PLQY,Film − VOC,PLQY,Device; loss (energetics, etc.) = VOC,PLQY, device − VOC,measured. (f) TRPL measurements of control and treated samples. The architecture is FTO glass/c-TiO2/mp-TiO2/perovskite/with and without interface treatment/spiro-OMeTAD. | ||
To get insights into the charge carrier dynamics we performed time-resolved photoluminescence spectroscopy (TRPL). The surface passivation has profound consequences on the charge carrier dynamics of the perovskite/HTL interface. To unravel this, we conducted TRPL measurements on the control and target perovskite films with HTL (doped spiro-OMeTAD) (as shown in Fig. 3f). For both films, we observe two features: a rapid decay within the first 5 ns followed by an almost monoexponential slower decay. The fast decay at early times has been attributed to initial carrier trapping,54 and hole transfer,55 whereas the following slower decay, is caused by non-radiative recombination.56 Note that with the low fluences used here, radiative bimolecular and non-radiative Auger recombination are negligible.56 For the target sample, we observe a slower decay at later times compared to the control sample indicating reduced non-radiative recombination, which is in good agreement with the steady-state PL measurements. At early times, for the target film, we observe a faster decay compared to the control one. Since we have fewer traps for the target film, this indicates a faster hole transfer compared to the control sample. From spatially resolved PL mapping (Fig. S21, ESI†) of the perovskite/HTL interface, we confirmed higher PL intensity with a more homogeneous distribution in the target sample compared to the control sample. These results can be reconciled with steady-state and TRPL measurements.
Operational and thermal stability, ion migration, and I2 formation
Long term operational stability at elevated temperature remains the main obstacle to the commercialization of PSCs.11,23 There are still only a few reports of long-term stability (>1000 h) under maximum power point tracking (MPPT) conditions with thermal stress for high-efficiency PSCs.57 We performed long-term operational stability tests at MPP for 1440 hours at 40 °C under continuous one sun illumination. We choose 40 °C for aging because spiro-OMeTAD was used as HTL, which is known to degrade over 50 °C; nevertheless, polymeric HTMs like PTAA has been used for stability test at higher temperatures, but they compromise the performance.57 Fig. S22 (ESI†) shows the statistical distribution of the device performance of the batches of cells used for the stability test. Fig. 4a shows the MPP tracking of unencapsulated control and the target device in a nitrogen atmosphere for 1440 h. The control device lost over 60% of the initial performance just after 600 h, while the target device retained ∼87% of the initial efficiency after 1440 h. The different device parameters during the MPP tracking are shown in Fig. S23 (ESI†), and the J–V of the target device before and after aging is shown in Fig. S24 (ESI†). For the target, there is a quick drop of ∼8% in the PCE after 50 h, which is a feature commonly observed in PSCs known as the burn-in effect.58 After the initial burn-in, there is only a 5% drop for the next 1350 h. In regards to the temporal stability of the PSCs, T80 is the time when devices retain 80% of the initial performance.58 By fitting the MPPT data (Fig. S25, ESI†), we obtain a T80 of ∼3570 h, and by fitting the MPPT after the burn-in region, we get a post-burn-in stabilized TS80 of ∼5950 h. To put our stability results in perspective, we summarized the best long-term operational stability (≥1000 h) of n–i–p based PSCs from the literature in Table S3 (ESI†), which shows that our target PSC compares favourably with other reported results. We also investigated the thermal stability by collecting J–V curves (one sun, ambient conditions) at various times for devices stored at 85 °C under dark in the nitrogen atmosphere. The best control device lost ∼40% of their initial PCE while the best target device lost only around ∼20% of their initial PCE after ∼1020 h (Fig. 4b). The J–V curves of the best target device before and after aging are shown in Fig. S26 (ESI†). The thermal and operational stability test results show that interface engineering significantly improves the stability.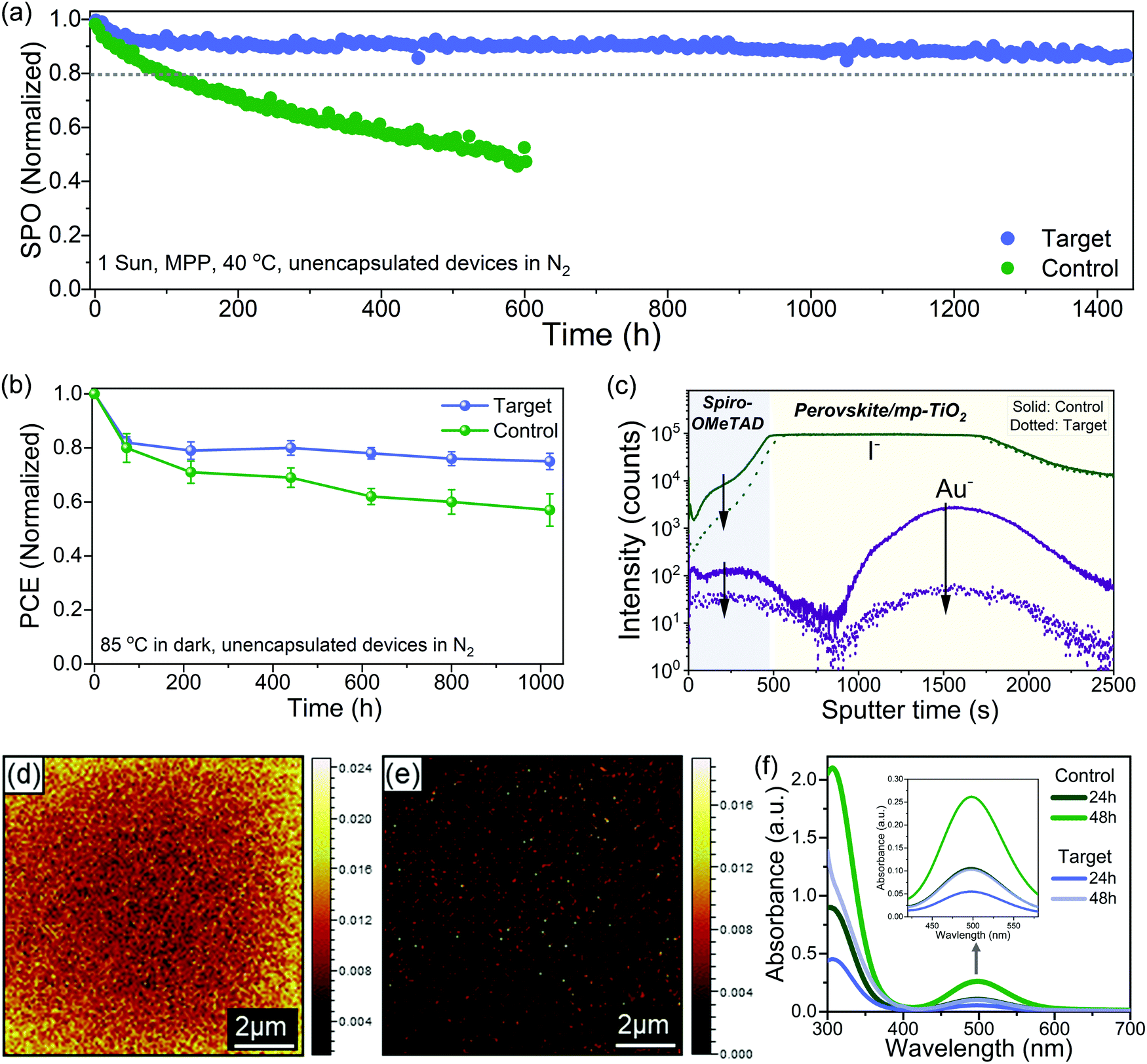 | ||
| Fig. 4 Stability measurements and degradation mechanism. (a) Maximum power point tracking measured with the unencapsulated device white LED light (100 mW cm−2 in N2, 40 °C). The initial stabilized power output (SPO) for control and target was 19.7% and 20.9%, respectively. (b) Thermal stability of devices at a fixed temperature of 85°C in a nitrogen atmosphere. Statistical data of four devices for each control and target. The data points (color-filled dots) were collected by measuring the J–V curves of the devices at different time intervals. The initial PCE for best control and target was 20.6% and 21.5%, respectively. (c) TOF-SIMS depth profile for the aged devices. (d) and (e) TOF-SIMS 2D images of I− ion signal on the spiro-OMeTAD surface (10 μm × 10 μm) for (d) control and (e) target devices. The color scale for each image is normalized intensity per pixel. For TOF-SIMS the samples were aged after 350 hours of light-soaking at the maximum power point at 40 °C, and after aging, gold was exfoliated by the scotch tape. (f) UV-vis absorbance spectra recorded for the toluene solution taken from the control and target vials at 24 and 48 h. The band at 500 nm corresponds to I2 and is zoomed in the inset. | ||
To understand the stability enhancement and to have a deeper insight into the mechanisms associated, we performed an analysis of the aged devices. It is known that under operational conditions at elevated temperature, the iodide ions can migrate into the HTL layer, thus deteriorating the device performance.15 We performed the time-of-flight secondary ion mass spectrometry (TOF-SIMS) depth profile to see the ion distribution across the interface into the layers in the aged devices (Fig. 4c). In the control device, we observed I− in the HTL layer and gold (Au) in the perovskite bulk and perovskite/TiO2. However, in the target devices, we observed a significantly lower intensity of I− in the HTL film and Au in the perovskite bulk. We further performed the TOF-SIMS surface mapping to investigate the spatial distribution of iodide ions (I−) on the HTL surface in the aged devices. In the control devices, we observed a high concentration of I− in the spiro-OMeTAD layer (Fig. 4d). However, in the target devices, the signal count associated with I− in the spiro-OMeTAD layer was significantly reduced (Fig. 4e). The interfacial ion diffusion is driven by vacancies and grain boundaries (GB) at the interface.47 Thus, effective passivation of grain boundaries and defects with ligand considerably can suppress this process. Generation of I2 under illumination has also been identified as one of the critical factors for the degradation.3,22,59 Typically for the I2 formation, I− need to capture a hole to form a neutral iodine atom, and two neutral interstitial iodine atoms need to diffuse together and combine to form I2. Since the surface of the perovskite has a higher defect density than the bulk, this process is more probable to occur at the former. To confirm this, we studied the formation of I2 in our control and target films by UV-vis spectroscopy of films immersed in toluene and subjected to one sun illumination at 40 °C (Fig. S27, ESI†). The toluene allows to form a complex with I2 with an absorption band at 500 nm (Fig. 4f). We observed a faster release of I2 from the control film as compared to the target film. These results showed that interface passivation with ligand substantially reduced the ion migration and suppressed I2 formation.
Conclusion
This study demonstrates a significant step forward in enhancing the longevity of perovskite solar cells by employing molecular engineering of the interfaces, a key approach to achieving high operational stability and high PCE in PSCs. Departing from the traditional wisdom of using organic ammonium salts for interface passivation, this work opens avenues for the supramolecular chemistry approach based on tailored non-covalent interactions for the interfacial passivation of these soft hybrid perovskites. In particular, we present a facile molecular-level interface engineering strategy using a multifunctional ligand, facilitating the remarkable stability of single-junction PSCs (stabilized TS80 ≈ 5950 h) with a high PCE of 23.4%. The origin of high device stability and performance can be linked to the nano/sub-nanoscale molecular level interactions between ligand and perovskite layer, which is corroborated by comprehensive multiscale characterization of local structure, grain/GB band bending modulation, change in the surface bandgap, and LDOS, interface defect reduction and interfacial ion diffusion. The results provide recommendations to expedite the design strategies of passivating ligands to facilitate interfacial engineering to enhance device stability and stimulate further work on the interfacial ion migration mechanisms. Overall, the scientific outcomes from this work are expected to have far researching consequences and initiate substantial further investigations in materials science, self-assembly, energy conversion, and optoelectronics, particularly to the development and understanding of functional nanoscale structures and interfaces of metal halide perovskites.Author contributions
A. K. conceived the idea, designed the project, carried out initial investigations of materials and devices, analyzed the data, and coordinated the project. A. H. supervised the study. H. Z., A. K. and Z. Z. optimized and fabricated high-efficiency devices and thin films. H. Z was also involved in scientific discussions and data analysis. A. K. performed the stability test and analysis. G. N. M. R. performed solid-state NMR studies and analysis. T. G. and A. R. performed KPFM, STM, and STS measurements and the analysis. D. M. and U. R. were responsible for the DFT calculations as well as their analysis. O. O. performed the steady-state PL measurements and analysis. A. K. performed the TRPL, and F. E. analyzed the TRPL data. F. F. performed TOF-SIMS measurements and analysis. S. S. performed SEM measurements. M. M. performed PL mapping and XPS measurements, and analysis. M. G. and S. M. Z. were involved in scientific discussions. A. K. wrote the manuscript through the contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 Research and Innovation program under the Marie Skłodowska-Curie Grant Agreement No. 843453. H. Z., S. M. Z., and M. G. acknowledge the funding from the European Union's Horizon 2020 research and innovation program GRAPHENE Flagship Core 3 grant agreement No. 881603. A. K. and A. H. acknowledge EUSMI Grant No. E200900442 for an access to 800 MHz spectrometer to conduct solid-state NMR measurements. G. N. M. R acknowledges financial support from University of Lille, France. T. G. and A. R. acknowledge funding from the Fonds National de la Recherche Luxembourg under the project “Sunspot” No 11244141. F. F. acknowledges funding from the Swiss Federal Office of Energy (SFOE)-BFE (project no. SI/501805-01). O. O. acknowledges funding from the National Sciences and Engineering Research Council of Canada. U. R. acknowledges Swiss National Science Foundation Grant No. 200020-165863 and the NCCR-MUST for funding as well as computational resources from the Swiss National Computing Centre CSCS. The authors acknowledge the help of Dr Wen Hua Bi for the assistance GIXRD measurements.References
- G. Kim, H. Min, K. S. Lee, D. Y. Lee, S. M. Yoon and S. Il Seok, Science, 2020, 370, 108–112 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, K. Domanski, J.-Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J.-P. Correa-Baena, W. R. Tress, A. Abate, A. Hagfeldt and M. Gratzel, Science, 2016, 354, 206–209 CrossRef CAS PubMed.
- Y. H. Lin, N. Sakai, P. Da, J. Wu, H. C. Sansom, A. J. Ramadan, S. Mahesh, J. Liu, R. D. J. Oliver, J. Lim, L. Aspitarte, K. Sharma, P. K. Madhu, A. B. Morales-Vilches, P. K. Nayak, S. Bai, F. Gao, C. R. M. Grovenor, M. B. Johnston, J. G. Labram, J. R. Durrant, J. M. Ball, B. Wenger, B. Stannowski and H. J. Snaith, Science, 2020, 369, 96–102 CrossRef CAS PubMed.
- X. Zheng, Y. Hou, C. Bao, J. Yin, F. Yuan, Z. Huang, K. Song, J. Liu, J. Troughton, N. Gasparini, C. Zhou, Y. Lin, D. J. Xue, B. Chen, A. K. Johnston, N. Wei, M. N. Hedhili, M. Wei, A. Y. Alsalloum, P. Maity, B. Turedi, C. Yang, D. Baran, T. D. Anthopoulos, Y. Han, Z. H. Lu, O. F. Mohammed, F. Gao, E. H. Sargent and O. M. Bakr, Nat. Energy, 2020, 5, 131–140 CrossRef CAS.
- Y. Cao, N. Wang, H. Tian, J. Guo, Y. Wei, H. Chen, Y. Miao, W. Zou, K. Pan, Y. He, H. Cao, Y. Ke, M. Xu, Y. Wang, M. Yang, K. Du, Z. Fu, D. Kong, D. Dai, Y. Jin, G. Li, H. Li, Q. Peng, J. Wang and W. Huang, Nature, 2018, 562, 249–253 CrossRef CAS PubMed.
- K. Lin, J. Xing, L. N. Quan, F. P. G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, D. Zhang, C. Yan, W. Li, X. Liu, Y. Lu, J. Kirman, E. H. Sargent, Q. Xiong and Z. Wei, Nature, 2018, 562, 245–248 CrossRef CAS PubMed.
- Y. C. Kim, K. H. Kim, D. Y. Son, D. N. Jeong, J. Y. Seo, Y. S. Choi, I. T. Han, S. Y. Lee and N. G. Park, Nature, 2017, 550, 87–91 CrossRef CAS PubMed.
- Y. Liu, Y. Zhang, X. Zhu, J. Feng, I. Spanopoulos, W. Ke, Y. He, X. Ren, Z. Yang, F. Xiao, K. Zhao, M. Kanatzidis and S. (Frank) Liu, Adv. Mater., 2021, 2006010 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- NREL efficiency chart, https://www.nrel.gov/pv/assets/images/efficiency-chart.png.
- C. C. Boyd, R. Cheacharoen, T. Leijtens and M. D. McGehee, Chem. Rev., 2019, 119, 3418–3451 CrossRef CAS PubMed.
- T. Leijtens, G. E. Eperon, N. K. Noel, S. N. Habisreutinger, A. Petrozza and H. J. Snaith, Adv. Energy Mater., 2015, 5, 1500963 CrossRef.
- W. Nie, J. Blancon, A. J. Neukirch, K. Appavoo, H. Tsai, M. Chhowalla, M. A. Alam, M. Y. Sfeir, C. Katan, J. Even, S. Tretiak, J. J. Crochet, G. Gupta and A. D. Mohite, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- J.-P. Correa-Baena, M. Saliba, T. Buonassisi, M. Grätzel, A. Abate, W. Tress and A. Hagfeldt, Science, 2017, 358, 739–744 CrossRef CAS PubMed.
- Y. Wang, T. Wu, J. Barbaud, W. Kong, D. Cui, H. Chen, X. Yang and L. Han, Science, 2019, 365, 687–691 CrossRef CAS PubMed.
- R. Wang, J. Xue, K. L. Wang, Z. K. Wang, Y. Luo, D. Fenning, G. Xu, S. Nuryyeva, T. Huang, Y. Zhao, J. L. Yang, J. Zhu, M. Wang, S. Tan, I. Yavuz, K. N. Houk and Y. Yang, Science, 2019, 366, 1509–1513 CrossRef CAS PubMed.
- L. Wang, H. Zhou, J. Hu, B. Huang, M. Sun, B. Dong, G. Zheng, Y. Huang, Y. Chen, L. Li, Z. Xu, N. Li, Z. Liu, Q. Chen, L. D. Sun and C. H. Yan, Science, 2019, 363, 265–270 CrossRef CAS PubMed.
- B. Chen, P. N. Rudd, S. Yang, Y. Yuan and J. Huang, Chem. Soc. Rev., 2019, 48, 3842–3867 RSC.
- H. Cho, S. H. Jeong, M. H. Park, Y. H. Kim, C. Wolf, C. L. Lee, J. H. Heo, A. Sadhanala, N. S. Myoung, S. Yoo, S. H. Im, R. H. Friend and T. W. Lee, Science, 2015, 350, 1222–1225 CrossRef CAS PubMed.
- Y. Li, X. Xu, C. Wang, B. Ecker, J. Yang, J. Huang and Y. Gao, J. Phys. Chem. C, 2017, 121, 3904–3910 CrossRef CAS.
- E. J. Juarez-Perez, L. K. Ono, M. Maeda, Y. Jiang, Z. Hawash and Y. Qi, J. Mater. Chem. A, 2018, 6, 9604–9612 RSC.
- F. Fu, S. Pisoni, Q. Jeangros, J. Sastre-Pellicer, M. Kawecki, A. Paracchino, T. Moser, J. Werner, C. Andres, L. Duchêne, P. Fiala, M. Rawlence, S. Nicolay, C. Ballif, A. N. Tiwari and S. Buecheler, Energy Environ. Sci., 2019, 12, 3074–3088 RSC.
- G. Tumen-Ulzii, C. Qin, T. Matsushima, M. R. Leyden, U. Balijipalli, D. Klotz and C. Adachi, Sol. RRL, 2020, 4, 2000305 CrossRef CAS.
- M. M. Tavakoli, W. Tress, J. V. Milić, D. Kubicki, L. Emsley and M. Grätzel, Energy Environ. Sci., 2018, 11, 3310–3320 RSC.
- S. Wu, J. Zhang, Z. Li, D. Liu, M. Qin, S. H. Cheung, X. Lu, D. Lei, S. K. So, Z. Zhu and A. K. Y. Jen, Joule, 2020, 4, 1248–1262 CrossRef CAS.
- S. Akin, N. Arora, S. M. Zakeeruddin, M. Grätzel, R. H. Friend and M. I. Dar, Adv. Energy Mater., 2020, 10, 1–29 Search PubMed.
- J. Xue, R. Wang and Y. Yang, Nat. Rev. Mater., 2020, 5, 809–827 CrossRef CAS.
- F. Zhang, D. Bi, N. Pellet, C. Xiao, Z. Li, J. J. Berry, S. M. Zakeeruddin, K. Zhu and M. Grätzel, Energy Environ. Sci., 2018, 11, 3480–3490 RSC.
- D. Bi, X. Li, J. V. Milić, D. J. Kubicki, N. Pellet, J. Luo, T. LaGrange, P. Mettraux, L. Emsley, S. M. Zakeeruddin and M. Grätzel, Nat. Commun., 2018, 9, 4482 CrossRef PubMed.
- N. K. Noel, A. Abate, S. D. Stranks, E. S. Parrott, V. M. Burlakov, A. Goriely and H. J. Snaith, ACS Nano, 2014, 8, 9815–9821 CrossRef CAS PubMed.
- C. Qin, T. Matsushima, T. Fujihara and C. Adachi, Adv. Mater., 2017, 29, 1603808 CrossRef PubMed.
- S. Yang, J. Dai, Z. Yu, Y. Shao, Y. Zhou, X. Xiao, X. C. Zeng and J. Huang, J. Am. Chem. Soc., 2019, 141, 5781–5787 CrossRef CAS PubMed.
- B. Wook Park, N. Kedem, M. Kulbak, D. Y. Lee, W. S. Yang, N. J. Jeon, J. Seo, G. Kim, K. J. Kim, T. J. Shin, G. Hodes, D. Cahen and S. Il Seok, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed.
- C. J. Dahlman, R. M. Kennard, P. Paluch, N. R. Venkatesan, M. L. Chabinyc and G. N. Manjunatha Reddy, Chem. Mater., 2021, 33, 642–656 CrossRef CAS.
- I. Spanopoulos, I. Hadar, W. Ke, P. Guo, E. M. Mozur, E. Morgan, S. Wang, D. Zheng, S. Padgaonkar, G. N. Manjunatha Reddy, E. A. Weiss, M. C. Hersam, R. Seshadri, R. D. Schaller and M. G. Kanatzidis, J. Am. Chem. Soc., 2021, 143, 7069–7080 CrossRef CAS PubMed.
- A. Krishna, M. A. Akhavan Kazemi, M. Sliwa, G. N. M. Reddy, L. Delevoye, O. Lafon, A. Felten, M. T. Do, S. Gottis and F. Sauvage, Adv. Funct. Mater., 2020, 30, 1909737 CrossRef CAS.
- L. Piveteau, V. Morad and M. V. Kovalenko, J. Am. Chem. Soc., 2020, 142, 19413–19437 CrossRef CAS PubMed.
- M. Seifrid, G. N. M. Reddy, B. F. Chmelka and G. C. Bazan, Nat. Rev. Mater., 2020, 5, 910–930 CrossRef CAS.
- E. A. Alharbi, A. Y. Alyamani, D. J. Kubicki, A. R. Uhl, B. J. Walder, A. Q. Alanazi, J. Luo, A. Burgos-Caminal, A. Albadri, H. Albrithen, M. H. Alotaibi, J.-E. Moser, S. M. Zakeeruddin, F. Giordano, L. Emsley and M. Grätzel, Nat. Commun., 2019, 10, 3008 CrossRef PubMed.
- W. M. J. Franssen and A. P. M. Kentgens, Solid State Nucl. Magn. Reson., 2019, 100, 36–44 CrossRef CAS PubMed.
- G. M. Bernard, R. E. Wasylishen, C. I. Ratcliffe, V. Terskikh, Q. Wu, J. M. Buriak and T. Hauger, J. Phys. Chem. A, 2018, 122, 1560–1573 CrossRef CAS PubMed.
- I. Schnell, A. Lupulescu, S. Hafner, D. E. Demco and H. W. Spiess, J. Magn. Reson., 1998, 133, 61–69 CrossRef CAS PubMed.
- D. Dudenko, A. Kiersnowski, J. Shu, W. Pisula, D. Sebastiani, H. W. Spiess and M. R. Hansen, Angew. Chem., Int. Ed., 2012, 51, 11068–11072 CrossRef CAS PubMed.
- S. Y. Leblebici, L. Leppert, Y. Li, S. E. Reyes-Lillo, S. Wickenburg, E. Wong, J. Lee, M. Melli, D. Ziegler, D. K. Angell, D. F. Ogletree, P. D. Ashby, F. M. Toma, J. B. Neaton, I. D. Sharp and A. Weber-Bargioni, Nat. Energy, 2016, 1, 1–7 Search PubMed.
- J. L. Garrett, E. M. Tennyson, M. Hu, J. Huang, J. N. Munday and M. S. Leite, Nano Lett., 2017, 17, 2554–2560 CrossRef CAS PubMed.
- T. Gallet, D. Grabowski, T. Kirchartz and A. Redinger, Nanoscale, 2019, 11, 16828–16836 RSC.
- Y. Shao, Y. Fang, T. Li, Q. Wang, Q. Dong, Y. Deng, Y. Yuan, H. Wei, M. Wang, A. Gruverman, J. Shield and J. Huang, Energy Environ. Sci., 2016, 9, 1752–1759 RSC.
- X. Zheng, B. Chen, J. Dai, Y. Fang, Y. Bai, Y. Lin, H. Wei, X. C. Zeng and J. Huang, Nat. Energy, 2017, 2, 17102 CrossRef CAS.
- R. M. Feenstra, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 4561–4570 CrossRef CAS PubMed.
- P. Schulz, E. Edri, S. Kirmayer, G. Hodes, D. Cahen and A. Kahn, Energy Environ. Sci., 2014, 7, 1377–1381 RSC.
- J. Endres, D. A. Egger, M. Kulbak, R. A. Kerner, L. Zhao, S. H. Silver, G. Hodes, B. P. Rand, D. Cahen, L. Kronik and A. Kahn, J. Phys. Chem. Lett., 2016, 7, 2722–2729 CrossRef CAS PubMed.
- N. Liu and C. Y. Yam, Phys. Chem. Chem. Phys., 2018, 20, 6800–6804 RSC.
- W. Tress, M. Yavari, K. Domanski, P. Yadav, B. Niesen, J. P. Correa Baena, A. Hagfeldt and M. Graetzel, Energy Environ. Sci., 2018, 11, 151–165 RSC.
- M. J. Trimpl, A. D. Wright, K. Schutt, L. R. V. Buizza, Z. Wang, M. B. Johnston, H. J. Snaith, P. Müller-Buschbaum and L. M. Herz, Adv. Funct. Mater., 2020, 30, 2004312 CrossRef CAS.
- J. Haddad, B. Krogmeier, B. Klingebiel, L. Krückemeier, S. Melhem, Z. Liu, J. Hüpkes, S. Mathur and T. Kirchartz, Adv. Mater. Interfaces, 2020, 7, 2000366 CrossRef CAS.
- T. Kirchartz, J. A. Márquez, M. Stolterfoht and T. Unold, Adv. Energy Mater., 2020, 10, 1904134 CrossRef CAS.
- E. H. Jung, N. J. Jeon, E. Y. Park, C. S. Moon, T. J. Shin, T. Y. Yang, J. H. Noh and J. Seo, Nature, 2019, 567, 511–515 CrossRef CAS PubMed.
- M. V. Khenkin, E. A. Katz, A. Abate, G. Bardizza, J. J. Berry, C. Brabec, F. Brunetti, V. Bulović, Q. Burlingame, A. Di Carlo, R. Cheacharoen, Y. B. Cheng, A. Colsmann, S. Cros, K. Domanski, M. Dusza, C. J. Fell, S. R. Forrest, Y. Galagan, D. Di Girolamo, M. Grätzel, A. Hagfeldt, E. von Hauff, H. Hoppe, J. Kettle, H. Köbler, M. S. Leite, S. (Frank) Liu, Y. L. Loo, J. M. Luther, C. Q. Ma, M. Madsen, M. Manceau, M. Matheron, M. McGehee, R. Meitzner, M. K. Nazeeruddin, A. F. Nogueira, Ç. Odabaşı, A. Osherov, N. G. Park, M. O. Reese, F. De Rossi, M. Saliba, U. S. Schubert, H. J. Snaith, S. D. Stranks, W. Tress, P. A. Troshin, V. Turkovic, S. Veenstra, I. Visoly-Fisher, A. Walsh, T. Watson, H. Xie, R. Yıldırım, S. M. Zakeeruddin, K. Zhu and M. Lira-Cantu, Nat. Energy, 2020, 5, 35–49 CrossRef.
- S. Wang, Y. Jiang, E. J. Juarez-Perez, L. K. Ono and Y. Qi, Nat. Energy, 2017, 2, 1–8 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee02454j |
| ‡ These authors contribute equally to this work. |
| § Present address: Department of Chemistry – Ångström Laboratory, Uppsala University, 75120 Uppsala, Sweden. E-mail: anders.hagfeldt@uu.se |
| This journal is © The Royal Society of Chemistry 2021 |