
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-controlled O2 diffusion enables air-tolerant solar hydrogen generation†
Michael G.
Allan
a,
Morgan J.
McKee
a,
Frank
Marken
b and
Moritz F.
Kuehnel
 *ac
*ac
aDepartment of Chemistry, Swansea University, Singleton Park, Swansea SA2 8PP, Wales, UK. E-mail: m.f.kuehnel@swansea.ac.uk
bDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK
cFraunhofer Institute for Microstructure of Materials and Systems IMWS, Walter-Hülse-Straße 1, 06120 Halle, Germany
First published on 31st August 2021
Abstract
Solar water splitting into H2 and O2 is a promising approach to provide renewable fuels. However, the presence of O2 hampers H2 generation and most photocatalysts show a major drop in activity in air without synthetic modification. Here, we demonstrate efficient H2 evolution in air, simply enabled by controlling O2 diffusion in the solvent. We show that in deep eutectic solvents (DESs), photocatalysts retain up to 97% of their H2 evolution activity and quantum efficiency under aerobic conditions whereas in water, the same catalysts are almost entirely quenched. Solvent-induced O2 tolerance is achieved by H2 generation outcompeting O2-induced quenching due to low O2 diffusivities in DESs combined with low O2 solubilities. Using this mechanism, we derive design rules and demonstrate that applying these rules to H2 generation in water can enhance O2 tolerance to >34%. The simplicity and generality of this approach paves the way for enhancing water splitting without adding complexity.
Broader contextGreen hydrogen production is a key process for the transition to a carbon–neutral economy, but oxygen, ubiquitous in air and generated during water splitting, interferes with hydrogen generation. Not only does the presence of O2 lower the hydrogen evolution efficiency, it can also degrade hydrogen evolution catalysts; in addition, O2 causes problems in other key energy technologies, such as Li–O2 batteries, fuel cells and in many other redox processes. The current approaches to improving O2 tolerance add complexity and often come at the expense of consuming redox equivalents for O2 removal, which lowers the overall efficiency. Here we show that by simply choosing solvents with a low O2 diffusivity and solubility, photocatalysts normally inefficient for H2 generation in air become highly O2 tolerant, with minimal loss in activity and efficiency in air, even for extended periods of time. By unravelling the mechanism of the solvent-induced O2 tolerance, we can translate it to achieve oxygen tolerance even in water, making it an important new concept with general applicability independent of the catalyst, solvent or process – a key step in making green H2 production simpler and more efficient on a global scale. |
Introduction
Solar hydrogen production from water is viewed as a viable method for generating clean renewable fuel to aid in combatting global energy challenges.1,2 Materials employed for solar-driven H2 production should be considered based on their cost, stability, toxicity and most importantly their practical applicability on a large scale. Real-world photocatalytic H2 production systems must be active in the presence of O2 generated in situ by water splitting and by exposure to air.3 However, H2 evolution in an aerobic environment is usually suppressed because of the more favourable oxygen reduction reaction.4 In addition, molecular O2 can inhibit H2 evolution co-catalysts via interaction with the active site or by forming reactive oxygen species (ROSs).5,6 Proton reduction in the presence of O2 has been achieved by developing electrocatalysts with selectivity for H2 evolution over O2 reduction7 or by creating a local anaerobic environment around the catalyst. Methods of lowering the effective O2 concentration at the catalyst include O2 reduction at catalysts capable of performing both O2 reduction and H+ evolution8–10 and at organic dyes,11 constructing layered architectures in which O2 is reduced before it reaches the active site,12–14 introducing antioxidant additives,15,16 and modifying catalytic sites with O2-blocking layers.17–19 However, these approaches require a costly re-design of the catalyst to enhance O2 tolerance and in many cases photons and charges are used to reduce O2, leading to a decrease in quantum and faradaic yield, respectively. Recent work has demonstrated O2-tolerant CO2 reduction enabled by controlling O2 diffusion to the electrode using selective membranes and coatings.20,21 Related approaches have been used in lithium–oxygen batteries.22 To date, research in hydrogen evolution has not exploited solvent effects for promoting O2 tolerance, even though O2 solubility and diffusivity in the reaction medium are the primary factors controlling the availability of O2 to the catalytically active site.In this work we demonstrate that using deep eutectic solvents (DESs) as a reaction medium enables O2-tolerant photocatalytic H2 production with O2-intolerant photocatalysts without making any catalyst modifications and without affecting the quantum efficiency. DESs are an alternative class of low-cost, highly tuneable ionic liquids23 that can be prepared from readily available precursors and possess lower toxicities than conventional ionic liquids.24 DESs have been employed for air-tolerant organic reactions involving highly reactive organolithium compounds25,26 and it has recently been shown they can stabilise O2-sensitive radicals in air.27 Using a carbon nitride photocatalyst, we now show that DESs create a near-anaerobic environment in which up to 97% of the photocatalytic H2 evolution activity is retained under air (Fig. 1). Mechanistic studies reveal a close interplay between O2 solubility and diffusivity and allow us to develop a quantitative model of the O2 tolerance. Based on this model we derive key design criteria for tailored reaction media that promote efficient and cost-effective O2 tolerance with established H2 generation photocatalysts without synthetic modification.
 | ||
| Fig. 1 Schematic representation of solvent-mediated oxygen-tolerant photocatalytic hydrogen production in deep eutectic solvents demonstrated in this work. | ||
Results and discussion
Deep eutectic solvents as a medium for solar H2 generation
To investigate solvent effects on the photocatalytic H2 evolution performance, we chose cyanamide-functionalised carbon nitride (NCNCNx) as a model photocatalyst (Fig. S1–S4, ESI†)28,29 and studied its activity in three well-known type-III DESs, namely![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) choline chloride–urea 1:2, choline chloride–glycerol 1:2, and choline chloride–ethylene glycol 1:2, termed reline, glyceline and ethaline, respectively. These solvents were chosen due to their facile preparation, low cost, low toxicity and infinite miscibility with water.23 Pt was used as a HER co-catalyst, in situ photodeposited from H2PtCl6 (Pt/NCNCNx). In reline, Pt/NCNCNx generated 138.3 ± 2.6 μmolH2 after 14 h irradiation with simulated solar light (AM 1.5G, 1 sun) at an activity of 8.9 ± 0.9 mmolH2 gCNx−1 h−1 using triethanolamine (TEOA) as a sacrificial electron donor (Fig. 2). Addition of water (12.5% by volume) was essential as in neat DESs, H2 evolution activity was negligible (Fig. S5, ESI†). The same conditions yielded an activity of 8.0 ± 0.6 and 4.1 ± 0.1 mmolH2 gCNx−1 h−1 for ethaline and glyceline, respectively with cumulative values of 105.1 ± 8.6 and 58.7 ± 3.5 μmolH2 after 14 hours (Table S1, ESI†). Depending on the solvent, a decay in activity was observed after 5–9 h which we attribute to the well-known decomposition of the redox mediator methyl viologen (MV2+)30 as with a further addition of MV2+, the rate increased again (Fig. S6, ESI†). In the absence of MV2+, H2 evolution was slower but no decay in activity was observed (Fig. S7, ESI†) proving that the DESs do not compromise the stability of the NCNCNx photocatalyst. In water, NCNCNx displayed a maximum activity of 6.5 ± 0.7 mmolH2 gCNx−1 h−1 and a cumulative production of 86.1 ± 5.4 μmolH2 after 14 h irradiation under optimised conditions (0.38 M TEOA, pH 7.O, no MV2+) which is on par with recent literature values.29 This was lower in comparison to reline and ethaline and higher than the activity in glyceline (see Fig. S8 and S9, ESI† for optimisation and controls). The external quantum efficiency for H2 evolution in reline was determined at 3.7 ± 1.5% and was stable even after 20 h of irradiation (Table S2, ESI†). We can therefore state that under the given conditions, DESs are a competitive solvent with water for solar H2 generation.
choline chloride–urea 1:2, choline chloride–glycerol 1:2, and choline chloride–ethylene glycol 1:2, termed reline, glyceline and ethaline, respectively. These solvents were chosen due to their facile preparation, low cost, low toxicity and infinite miscibility with water.23 Pt was used as a HER co-catalyst, in situ photodeposited from H2PtCl6 (Pt/NCNCNx). In reline, Pt/NCNCNx generated 138.3 ± 2.6 μmolH2 after 14 h irradiation with simulated solar light (AM 1.5G, 1 sun) at an activity of 8.9 ± 0.9 mmolH2 gCNx−1 h−1 using triethanolamine (TEOA) as a sacrificial electron donor (Fig. 2). Addition of water (12.5% by volume) was essential as in neat DESs, H2 evolution activity was negligible (Fig. S5, ESI†). The same conditions yielded an activity of 8.0 ± 0.6 and 4.1 ± 0.1 mmolH2 gCNx−1 h−1 for ethaline and glyceline, respectively with cumulative values of 105.1 ± 8.6 and 58.7 ± 3.5 μmolH2 after 14 hours (Table S1, ESI†). Depending on the solvent, a decay in activity was observed after 5–9 h which we attribute to the well-known decomposition of the redox mediator methyl viologen (MV2+)30 as with a further addition of MV2+, the rate increased again (Fig. S6, ESI†). In the absence of MV2+, H2 evolution was slower but no decay in activity was observed (Fig. S7, ESI†) proving that the DESs do not compromise the stability of the NCNCNx photocatalyst. In water, NCNCNx displayed a maximum activity of 6.5 ± 0.7 mmolH2 gCNx−1 h−1 and a cumulative production of 86.1 ± 5.4 μmolH2 after 14 h irradiation under optimised conditions (0.38 M TEOA, pH 7.O, no MV2+) which is on par with recent literature values.29 This was lower in comparison to reline and ethaline and higher than the activity in glyceline (see Fig. S8 and S9, ESI† for optimisation and controls). The external quantum efficiency for H2 evolution in reline was determined at 3.7 ± 1.5% and was stable even after 20 h of irradiation (Table S2, ESI†). We can therefore state that under the given conditions, DESs are a competitive solvent with water for solar H2 generation.
 | ||
| Fig. 2 Photocatalytic H2 generation in DESs at Pt/NCNCNx: (a) H2 production in different DESs and water; (b) max. H2 production rate in DESs vs. H2O. Conditions: NCNCNx (2.0 mg), H2PtCl6 (0.05 mg Pt), in 2.0 mL DES (12.5% v/v H2O, 0.38 M TEOA, 2 mM MV2+) or water (0.38 M TEOA, pH 7, no MV2+); AM 1.5G, 1 sun, 40 °C, constant N2 purge. | ||
A notable difference between water and DES is the effect of added MV2+ on the H2 evolution: While adding MV2+ increases H2 generation in DES, a decrease in activity is observed in water (Fig. S10, ESI†). A suppression of H2 evolution upon addition of the redox mediator MV2+ has been previously observed in cases where there is good electron transfer between the photocatalyst and the HER co-catalyst.31 In this case, adding MV2+ does not enhance HER but instead causes a visible accumulation of reduced MV+˙ in the solution which blocks light penetration to the photocatalyst due to its deep blue colour. The beneficial effects of adding MV2+ in DESs, in turn, suggest poor electron transfer between NCNCNx and Pt in DESs. To prove this, we performed recycling experiments in which we separated the photocatalyst after 4 h irradiation in the presence of H2PtCl6 from its supernatant and re-suspended it in a fresh solution without added Pt, before continuing irradiation. In water, the photocatalytic H2 evolution activity was not affected by this procedure, suggesting Pt is deposited on the NCNCNx photocatalyst (Fig S11, ESI†), in line with previous literature. In DES, however, the photocatalytic activity was almost completely quenched, corroborating poor immobilisation of Pt on NCNCNx in DES, possibly due to differences in solvation in DESs.
O2-tolerant H2 generation in DESs
Inspired by their application as solvents to perform air-sensitive syntheses under an aerobic atmosphere,25,26 we set out to achieve air-tolerant H2 evolution in DESs. It is well known that photocatalytic H2 evolution is suppressed in air even for highly active materials32 arising from the thermodynamically favourable O2 reduction and quenching of the photosensitiser. Fig. 3a indicates that Pt/NCNCNx generates only 0.8 ± 0.2 mmolH2 gCNx−1 h−1 upon irradiation in aerated water corresponding to a retention of only 8.8 ± 1.5% of its photocatalytic activity seen under inert conditions. When the redox mediator MV2+ was added the retention dropped to 1.7 ± 0.7%. However, in the DES reline, the same catalyst without any modification achieved an activity of up to 8.7 ± 0.9 mmolH2 gCNx−1 h−1 in air, corresponding to a remarkable activity retention of up to 97.3 ± 17.5% compared to anaerobic conditions (Fig. 3b and Table S3, ESI†). While the O2 tolerance in water decreases further over time with almost complete deactivation after 10 h, DESs maintain a high level of O2 tolerance over prolonged periods of time (Fig. S12, ESI†). After 14 h, 123.5 ± 8.1 μmolH2 were produced in air (Fig. 3c) corresponding to 89.3 ± 6.1% of the amount produced under N2 and the system remained active (Fig. 3d). Similarly, an activity of 5.7 ± 1.3 mmolH2 gCNx−1 h−1 was seen in aerobic ethaline (73.5 ± 9.0% retention) and 3.6 ± 0.3 mmolH2 gCNx−1 h−1 in aerobic glyceline (90.4 ± 7.9% retention). The external quantum efficiency for H2 evolution in aerobic reline was determined at 3.9 ± 0.3% after 20 h of irradiation (Table S4, ESI†) which is within error identical to the EQE observed in anaerobic conditions. The optimum O2 tolerance was observed at 12.5% water content. Increasing the water content led to a lower O2 tolerance (Fig. S13, ESI†), whereas without added water, H2 evolution activity was much lower, presumably for lack of available protons (Fig. S5, ESI†). | ||
| Fig. 3 Solvent-mediated oxygen-tolerant H2 generation at Pt/NCNCNx. Effect of aerobic conditions on H2 production in (a) H2O and (b) reline; (c) total H2 evolved after 14.0 h under anaerobic and aerobic conditions in different DESs and H2O; (d) relative H2 evolution activities under aerobic conditions depending on the solvent. Conditions: NCNCNx (2.0 mg) H2PtCl6 (0.05 mg Pt) in 2.0 mL DES (12.5% v/v H2O, 0.38 M TEOA, 2 mM MV2+) or water (0.38 M TEOA, pH 7); AM 1.5G, 1 sun, 40 °C, constant air purge. | ||
The O2 tolerance induced by DESs compares favourably with examples of O2-tolerant H2 evolution from the literature (Table S5, ESI†). A range of CdS-based photocatalysts33–35 achieve O2 tolerances between 40–80%; air can even increase the activity of CdS by suppressing photocorrosion.36 These studies typically operate at high H2 production rates due to high electron donor concentrations, closed photoreactors and often high light intensities, where O2 in the solution and the reactor headspace is rapidly depleted by reduction to H2O, effectively generating anaerobic conditions in situ. This is often indicated by an observed lag period before H2 evolution occurs. In contrast, H2 production in DESs shows no detectable lag period and a high O2 tolerance despite a continuous air purge maintaining a constant O2 concentration. The latter is particularly important to exploit O2 tolerance to enhance overall water splitting, where O2 is continuously generated and H2 production rates are much lower than in sacrificial systems. Photocatalysts operating at lower rates where O2 depletion is less effective have shown lower O2 tolerances, e.g. RuP/CoP/TiO2 (17% O2 tolerance),9 Ni2P/OH-GQD (64%)37 and PFBT polymer dots (37%).38 To the best of our knowledge, there is no literature on O2-tolerant H2 generation using carbon nitride-based photocatalysts.
The advantage of solvent-induced O2 tolerance lies in its applicability independent of the photocatalyst. When Pt/TiO2 was used as the photocatalyst instead of Pt/NCNCNx, the O2 tolerance similarly increased from 29.6 ± 6.5% in water to 86.1 ± 12.8% in reline after 12 h irradiation (Fig. S14, ESI†), proving this effect is not limited to a single photocatalyst. To further demonstrate the generality of this approach, we also studied H2 evolution at the homogeneous photocatalyst Pt/Eosin Y (Pt/EY).39 Even though the H2 evolution in ethaline and reline (17.5 ± 1.7 μmolH2 mmolEY−1 and 11.4 ± 1.7 mmolH2 molEY−1 after 5.5 h, respectively, non-optimised conditions, Fig. S15, ESI†) was slower than in water (81.1 ± 6.8 μmolH2 mmolEY−1), the DESs promote excellent retention of activity in air. Pt/EY in aerobic H2O produced 0.7 mmolH2 molEY−1 after 5.5 h (<1% activity retained), whereas 14.9 mmolH2 molEY−1 was generated in ethaline corresponding to 85.5% O2 tolerance. This, again, compares well with literature examples of aerobic H2 evolution at homogeneous photocatalysts,40–42e.g. CoP/EY retained 70 ± 4% activity in air, however activity was limited to 2 h.9
The mechanism of solvent-induced O2 tolerance
Having demonstrated that DESs promote O2 tolerance of photocatalytic H2 evolution independent of the photocatalyst, we sought to gain understanding of the underlying mechanism. Previous work on DESs enabling air-tolerant alkylation with organolithium and Grignard reagents suggested that the high halide concentration in DESs increases the reagents’ reactivity to levels where they can outcompete hydrolysis. However, no explanation for the observed insensitivity to O2 was given.25,43 To elucidate the mechanism by which DESs promote O2-tolerant H2 evolution, we first studied the formation and stability of reduced NCNCNx in both H2O and DESs in air. NCNCNx is known to form a turquoise-blue photoreduced state originating from charge accumulation in the material when irradiated in the presence of an electron donor and absence of a hydrogen evolution co-catalyst.29
originating from charge accumulation in the material when irradiated in the presence of an electron donor and absence of a hydrogen evolution co-catalyst.29 persists in an anaerobic environment but is quenched rapidly by reaction with O2. In water,
persists in an anaerobic environment but is quenched rapidly by reaction with O2. In water,  is therefore only formed under N2 and immediately quenched upon exposure to air as indicated by the blue material regaining its original yellow colour. However, when NCNCNx is irradiated in DESs,
is therefore only formed under N2 and immediately quenched upon exposure to air as indicated by the blue material regaining its original yellow colour. However, when NCNCNx is irradiated in DESs,  is quickly formed even in an aerated solution. Moreover, the blue colour is stable in air for several days, with a noticeable absorbance at ∼680 nm in the DR-UV spectrum, ascribed to the reduced photocatalyst (Fig. 4). This absorbance is not observed in an aerated aqueous solution, highlighting the solvent effect on limiting the quenching of the photoabsorber by reaction with O2. To further corroborate the absence of O2 quenching in aerated DESs, we investigated photocatalytic degradation of the organic dye methylene blue in aerated DESs using NCNCNx as a photocatalyst. Dye degradation relies on reactive oxygen species (ROSs) such as O2− to act as oxidants, generated by the quenching of the excited state of a photocatalyst by O2; it is therefore strongly dependant on dissolved O2.44 Consistently, we observed that the degradation of methylene blue was much slower in DESs than in water, which lends further evidence to a suppression of O2 quenching depending on the solvent (Fig. S16, ESI†).
is quickly formed even in an aerated solution. Moreover, the blue colour is stable in air for several days, with a noticeable absorbance at ∼680 nm in the DR-UV spectrum, ascribed to the reduced photocatalyst (Fig. 4). This absorbance is not observed in an aerated aqueous solution, highlighting the solvent effect on limiting the quenching of the photoabsorber by reaction with O2. To further corroborate the absence of O2 quenching in aerated DESs, we investigated photocatalytic degradation of the organic dye methylene blue in aerated DESs using NCNCNx as a photocatalyst. Dye degradation relies on reactive oxygen species (ROSs) such as O2− to act as oxidants, generated by the quenching of the excited state of a photocatalyst by O2; it is therefore strongly dependant on dissolved O2.44 Consistently, we observed that the degradation of methylene blue was much slower in DESs than in water, which lends further evidence to a suppression of O2 quenching depending on the solvent (Fig. S16, ESI†).
 | ||
Fig. 4 (a) Absorption spectra of  in DES TEOA solution (green trace) and aqueous TEOA solution (black trace) prior to irradiation with simulated solar light. in DES TEOA solution (green trace) and aqueous TEOA solution (black trace) prior to irradiation with simulated solar light.  absorption spectra in DES TEOA solution (blue) recorded in ambient air. (b) Photo of absorption spectra in DES TEOA solution (blue) recorded in ambient air. (b) Photo of  in DES solutions exposed to air (left) and in inert atmosphere (right). in DES solutions exposed to air (left) and in inert atmosphere (right). | ||
Further quantitative insight was sought from determining the saturation concentration and diffusion coefficient of O2 in DESs by studying the electrochemical O2 reduction at a Pt microwire electrode.45 Potential step chronoamperometry was performed in each solvent and the observed current transients for the electrocatalytic O2 reduction were fitted according to the Shoup–Szabo equation46 to simultaneously derive the O2 concentrations and the O2 diffusion coefficients in aerated DESs and water, under the conditions tested for photocatalytic H2 evolution (Table 1 and Fig. S17–S20, ESI†).47 All the DES-based solutions exhibited lower O2 solubilities than conventional organic solvents,48,49 presumably due to their high ionic strengths causing a salting-out effect.50,51 In addition, O2 diffusion coefficients were found to be lower than in most other solvents48,49 including water52 but varied strongly between the different DESs. This behaviour is likely a result of their high viscosities combined with their complex liquid structure,53 in which hydrogen bond donor dependent cluster formation presumably influences molecular diffusion in the liquid as well as causing large variations in viscosity.54
| Solvent | c(O2) [μM] | D(O2) [m2 s−1] | O2 tolerancea [%] |
|---|---|---|---|
| a O2 tolerance = total H2 produced under air relative to total H2 produced under N2 at Pt/NCNCNx after 14 h irradiation under otherwise identical conditions. b Without added MV2+. | |||
| Reline | 167.8 ± 9.1 | 2.93 ± 0.02 × 10−10 | 89.3 ± 6.1 |
| Ethaline | 250.7 ± 0.4 | 3.32 ± 0.01 × 10−10 | 73.5 ± 9.0 |
| Glyceline | 218.8 ± 2.0 | 9.52 ± 0.01 × 10−11 | 90.4 ± 7.9 |
| H2O | 223.5 ± 0.4 | 2.94 ± 0.01 × 10−9 | 8.8 ± 1.5b |
We expect O2 tolerance to be a function of the effective O2 concentration at the photocatalyst surface, which depends on both solubility and diffusivity of O2 in the reaction medium. Comparing the trends in these parameters for the different DES-based solutions to the trend in O2 tolerance shows a clear correlation between the observed retention of photocatalytic activity in air (glyceline ≈ reline > ethaline > water) and the O2 diffusivities (glyceline < reline < ethaline < water). As O2 in solution is being consumed due to O2 reduction at the photocatalyst, the steady-state O2 concentration at the catalyst surface depends on how rapidly more O2 is supplied to the photocatalyst, therefore O2 tolerance is primarily dominated by the O2 diffusivity. The O2 solubility of the solutions (reline < glyceline < ethaline < water) is of secondary importance: glyceline and reline solutions show comparable O2 tolerances despite them showing varying O2 solubilities and diffusivities – this is likely because the lower diffusivity in glyceline is compensated by a higher O2 solubility, and vice versa. Water shows poor O2 tolerance because it exhibits the highest O2 diffusion coefficient among the solvents studied here and a relatively high O2 solubility. Due to a combination of low O2 diffusivities and low O2 solubilities, DESs thus create pseudo-inert conditions by limiting O2 mass transport, which is outcompeted by H+ diffusion.
Having identified the combination of low O2 solubility and O2 diffusivity as key factors to O2 tolerance, we use these design criteria to promote O2 tolerance in other solvents. Saline water is an attractive feedstock for renewable H2 production since seawater is much more abundant than freshwater and its use avoids competition with drinking water supplies.55 While using seawater can be challenging, we show here that it can enable highly O2-tolerant H2 evolution. It is well known that high salt concentrations lower the O2 solubility in water as well as the O2 diffusion coefficients.47,56 We therefore determined the O2 solubility and diffusivity in brines under photocatalysis conditions (40 °C, 0.38 M TEOA, pH 7) by microwire electrochemistry. Table 2 shows that the O2 solubility and diffusivity both decrease by approx. 50% upon increasing the NaCl concentration from 0 to 4 M. Consistently, Fig. 5 demonstrates that in line with our identified design criteria, the O2 tolerance in water increases with increasing NaCl concentrations. In 4 M aqueous NaCl a cumulative O2 tolerance of 34.2 ± 4.4% is observed after 14 h (Fig. S21 and Table S6, ESI†), more than 10 times higher than without added NaCl (Table 2). However, despite lower O2 solubilities, the O2 tolerance never reaches the levels observed in DESs consistent with the higher O2 diffusion coefficient in water. This demonstrates that the O2 diffusivity is decisive for the overall O2 tolerance, ideally when paired with a low O2 solubility. Furthermore, we studied the direct use of seawater collected from Swansea Beach as a solvent for H2 evolution (Fig. S22, ESI†). While the H2 generation activity was lower than in pure brines, presumably due to its brownish colour, the observed O2 tolerance of 7.2 ± 4.4% was higher than in pure DI water. Considering the local salinity of 0.41–0.53 M,57 this data is in good agreement with Table 2 and demonstrates the usefulness of using non-potable water for solar H2 generation.
| Solventa | c(O2) [μM] | D(O2) [m2 s−1] | O2 toleranceb [%] |
|---|---|---|---|
| a Solubilities were determined under the same conditions as the photocatalysis experiments were performed. b O2 tolerance = total H2 produced under air relative to total H2 produced under N2 at Pt/NCNCNx after 14 h irradiation under otherwise identical conditions. | |||
| 0 M NaCl | 223 ± 0.4 | 2.94 ± 0.01 × 10−9 | 3.1 ± 1.7 |
| 1 M NaCl | 265 ± 0.6 | 2.30 ± 0.01 × 10−9 | 13.9 ± 3.3 |
| 2 M NaCl | 165 ± 0.2 | 1.55 ± 0.01 × 10−9 | 19.0 ± 11.4 |
| 4 M NaCl | 128 ± 0.3 | 1.13 ± 0.01 × 10−9 | 34.2 ± 4.4 |
 | ||
| Fig. 5 Enhanced oxygen tolerance by control of O2 diffusion. Effect of aerobic conditions on H2 production in (a) H2O and (b) 4 M aqueous NaCl; (c) total H2 evolved after 14.0 h depending on the NaCl concentration and atmosphere; (d) O2 tolerance of H2 evolution depending on the NaCl concentration. Conditions: NCNCNx (2.0 mg) H2PtCl6 (0.05 mg Pt) in 2.0 mL saline water (0.38 M TEOA, pH 7, 2 mM MV2+); AM 1.5G, 1 sun, 40 °C, constant N2 or air purge. | ||
Design rules from a quantitative model for O2 tolerance
To explain the effect of O2 diffusivity and solubility on the O2 tolerance quantitatively, we have developed a mechanistic model based on fluxes to and from the photocatalyst particles (Fig. 6a). The rate of charge carrier generation, R(hν), depends on light intensity and quantum efficiency and is assumed largely independent of the solvent. O2 is expected to quench charge carriers with consuming O2, expressed as the rate R(O2). Approximating the O2-dependent quenching as O2 reduction at a spherical particle at the limit of diffusional control gives eqn (1):| R(O2) = 4π × r × n × D(O2) × c(O2) | (1) |
 | (2) |
 | (3) |
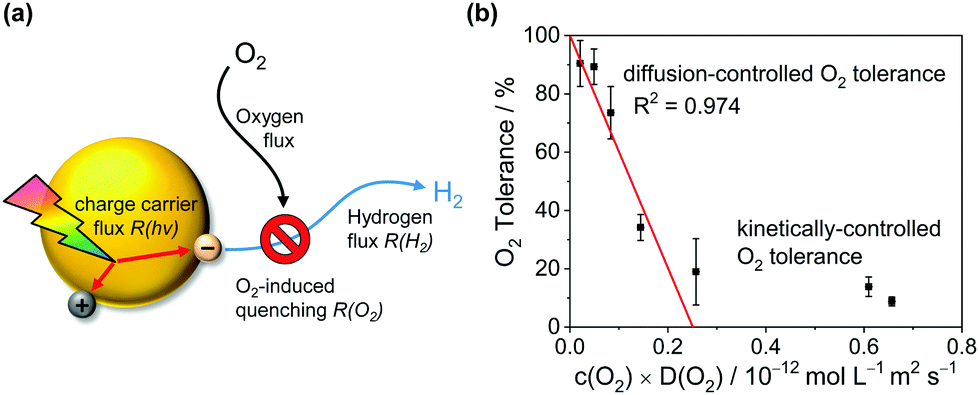 | ||
| Fig. 6 Mechanistic model for the solvent-induced O2 tolerance. (a) Schematic illustration of fluxes to and from the photocatalyst particle. (b) Plot of the O2 tolerance for H2 evolution versus the product of D(O2) and c(O2) in the respective reaction medium fitted according to eqn (3). | ||
Fig. 6 shows that the experimentally observed O2 tolerances fit well to this model (see ESI† for details). At high O2 tolerances, the slope represents the consumption of photo-generated charge carriers by O2 in diffusion-limited quenching. When the O2 flux increases with higher O2 solubility and diffusivity, the quenching process is no longer diffusion limited but instead kinetically limited by the rate of O2 reduction, resulting in O2 tolerance gradually tailing towards zero at a much lower slope. From this model, we can infer a set of design rules for improving O2 tolerance through further solvent design:
1. Minimise the c × D parameter (low O2 solubility and diffusivity, high viscosity).
2. Decrease particle size (large particles increase O2 flux).
3. Increase light intensity (outcompete O2 flux which is independent of light).
4. Increase photon-to-charge carrier conversion.
Future work should focus on exploring all variables of the model to further verify and refine its predictive ability and achieve sustained, fully O2-tolerant H2 generation.
Conclusions
We have shown that O2-tolerant H2 evolution can be achieved by controlling O2 diffusion and solubility in the reaction medium. We introduced DESs as a versatile medium for solar H2 generation with both heterogenous and homogenous light absorbers and showed that DESs induce a high O2 tolerance to otherwise O2-intolerant photocatalysts without compromising the quantum efficiency. We demonstrated this effect results from their low O2 solubilities and diffusivities. Exploiting these properties as design criteria enables a 10-fold increase in O2 tolerance in water by controlling O2 diffusion and solubility. Through developing a quantitative model for oxygen tolerance, we believe this investigation paves the way for further solvent-enhanced solar fuel production and, owing to the tuneable nature of DESs, allows for a wide scope of solvents to be examined. The fact that a relatively small change in the solvent constituents (replacing ethylene glycol with glycerol) causes a considerable change in the O2 diffusivity and thus in the O2 tolerance demonstrates the enormous potential of solvent design for solar water splitting, yet it highlights the need for establishing structure–function relationships to allow a rational solvent design. Future studies will expand the concept of O2 diffusion control to fully explore all parameters of our model with the potential to massively enhance solar water splitting without adding complexity.Data availability
Raw experimental data is openly available via dx.doi.org/10.5281/zenodo.5236823.Author contributions
MFK conceived, supervised and led the project. MGA conducted most of the experimental work and analysed the data. MJM performed dye degradation experiments. FM conceived and supervised electrochemical measurements. MFK, MGA and FM wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by EPSRC through a DTA studentship to MGA (EP/R51312X/1), and a capital investment grant to MFK (EP/S017925/1). We thank Swansea University for providing start-up funds to MFK. We thank Prof. Alex Cowan and Dr Gaia Neri (University of Liverpool) for recording DR-UV spectra. We wish to thank Julian Kivell and Phil Hopkins in the Swansea University mechanical workshop for their assistance with the experimental setup. MFK is grateful to the Ernst Schering Foundation for support.References
- Y. Tachibana, L. Vayssieres and J. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2015, 8, 2283–2295 RSC.
- P. M. Wood, Biochem. J., 1988, 253, 287–289 CrossRef CAS PubMed.
- M. T. Stiebritz and M. Reiher, Chem. Sci., 2012, 3, 1739–1751 RSC.
- B. Mondal and A. Dey, Chem. Commun., 2017, 53, 7707–7715 RSC.
- S. Chatterjee, K. Sengupta, S. Dey and A. Dey, Inorg. Chem., 2013, 52, 14168–14177 CrossRef CAS PubMed.
- N. Kaeffer, A. Morozan and V. Artero, J. Phys. Chem. B, 2015, 119, 13707–13713 CrossRef CAS PubMed.
- F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381–9384 CrossRef CAS PubMed.
- Z.-H. Pan, Y.-W. Tao, Q.-F. He, Q.-Y. Wu, L.-P. Cheng, Z.-H. Wei, J.-H. Wu, J.-Q. Lin, D. Sun, Q.-C. Zhang, D. Tian and G.-G. Luo, Chem. – Eur. J., 2018, 24, 8275–8280 CrossRef CAS PubMed.
- T. Sakai, D. Mersch and E. Reisner, Angew. Chem., Int. Ed., 2013, 52, 12313–12316 CrossRef CAS PubMed.
- H. Li, D. Buesen, S. Dementin, C. Léger, V. Fourmond and N. Plumeré, J. Am. Chem. Soc., 2019, 141, 16734–16742 CrossRef CAS PubMed.
- A. Ruff, J. Szczesny, N. Marković, F. Conzuelo, S. Zacarias, I. A. C. Pereira, W. Lubitz and W. Schuhmann, Nat. Commun., 2018, 9, 3675 CrossRef PubMed.
- C. Tapia, R. D. Milton, G. Pankratova, S. D. Minteer, H.-E. Åkerlund, D. Leech, A. L. De Lacey, M. Pita and L. Gorton, ChemElectroChem, 2017, 4, 90–95 CrossRef CAS.
- S. Dey, A. Rana, D. Crouthers, B. Mondal, P. K. Das, M. Y. Darensbourg and A. Dey, J. Am. Chem. Soc., 2014, 136, 8847–8850 CrossRef CAS PubMed.
- Z. Li, B. Tian, W. Zhen, Y. Wu and G. Lu, Appl. Catal., B, 2017, 203, 408–415 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806–7809 CrossRef CAS PubMed.
- A. T. Garcia-Esparza, T. Shinagawa, S. Ould-Chikh, M. Qureshi, X. Peng, N. Wei, D. H. Anjum, A. Clo, T.-C. Weng, D. Nordlund, D. Sokaras, J. Kubota, K. Domen and K. Takanabe, Angew. Chem., Int. Ed., 2017, 56, 5780–5784 CrossRef CAS PubMed.
- W. J. Jo, G. Katsoukis and H. Frei, Adv. Funct. Mater., 2020, 30, 1909262 CrossRef CAS.
- H. Li, U. Münchberg, A. A. Oughli, D. Buesen, W. Lubitz, E. Freier and N. Plumeré, Nat. Commun., 2020, 11, 920 CrossRef CAS PubMed.
- Y. Xu, J. P. Edwards, J. Zhong, C. P. O’Brien, C. M. Gabardo, C. McCallum, J. Li, C.-T. Dinh, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2020, 13, 554–561 RSC.
- X.-D. Lin, Y. Gu, X.-R. Shen, W.-W. Wang, Y.-H. Hong, Q.-H. Wu, Z.-Y. Zhou, D.-Y. Wu, J.-K. Chang, M.-S. Zheng, B.-W. Mao and Q.-F. Dong, Energy Environ. Sci., 2021, 14, 1439–1448 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- A. K. Halder and M. N. D. S. Cordeiro, ACS Sustainable Chem. Eng., 2019, 7, 10649–10660 CrossRef CAS.
- C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2014, 53, 5969–5973 CrossRef CAS PubMed.
- D. Arnodo, S. Ghinato, S. Nejrotti, M. Blangetti and C. Prandi, Chem. Commun., 2020, 56, 2391–2394 RSC.
- J. A. McCune, M. F. Kuehnel, E. Reisner and O. A. Scherman, Chem, 2020, 6, 1819–1830 CAS.
- V. W.-H. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. Lotsch, Nat. Commun., 2016, 7, 12165 CrossRef CAS PubMed.
- W. Yang, R. Godin, H. Kasap, B. Moss, Y. Dong, S. A. J. Hillman, L. Steier, E. Reisner and J. R. Durrant, J. Am. Chem. Soc., 2019, 141, 11219–11229 CrossRef CAS PubMed.
- M. Heyrovský, J. Chem. Soc., Chem. Commun., 1987, 1856–1857 RSC.
- E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef CAS PubMed.
- L. Ma, M. Liu, D. Jing and L. Guo, J. Mater. Chem. A, 2015, 3, 5701–5707 RSC.
- Z. Sun, H. Zheng, J. Li and P. Du, Energy Environ. Sci., 2015, 8, 2668–2676 RSC.
- D. A. Reddy, H. Park, S. Hong, D. P. Kumar and T. K. Kim, J. Mater. Chem. A, 2017, 5, 6981–6991 RSC.
- S. Cao, Y. Chen, C.-J. Wang, X.-J. Lv and W.-F. Fu, Chem. Commun., 2015, 51, 8708–8711 RSC.
- D. W. Wakerley, K. H. Ly, N. Kornienko, K. L. Orchard, M. F. Kuehnel and E. Reisner, Chem. – Eur. J., 2018, 24, 18385–18388 CrossRef CAS PubMed.
- L. Zhu, Q. Yue, D. Jiang, H. Chen, R. M. Irfan and P. Du, Chin. J. Catal., 2018, 39, 1753–1761 CrossRef CAS.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS PubMed.
- L. Wang, H. Zhao, Y. Chen, R. Sun and B. Han, Opt. Commun., 2016, 370, 122–126 CrossRef CAS.
- T. R. Canterbury, S. M. Arachchige, K. J. Brewer and R. B. Moore, Chem. Commun., 2016, 52, 8663–8666 RSC.
- L. Petermann, R. Staehle, M. Pfeifer, C. Reichardt, D. Sorsche, M. Wächtler, J. Popp, B. Dietzek and S. Rau, Chem. – Eur. J., 2016, 22, 8240–8253 CrossRef CAS PubMed.
- A. Call, Z. Codolà, F. Acuña-Parés and J. Lloret-Fillol, Chem. – Eur. J., 2014, 20, 6171–6183 CrossRef CAS PubMed.
- C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2016, 55, 16145–16148 CrossRef CAS PubMed.
- J. Schneider, D. Bahnemann, J. Ye, G. L. Puma and D. D. Dionysiou, Photocatalysis: Fundamentals and Perspectives, Royal Society of Chemistry, Cambridge, 2016 Search PubMed.
- A. Neudeck and L. Kress, J. Electroanal. Chem., 1997, 437, 141–156 CrossRef CAS.
- A. Szabo, D. K. Cope, D. E. Tallman, P. M. Kovach and R. M. Wightman, J. Electroanal. Chem. Interfacial Electrochem., 1987, 217, 417–423 CrossRef CAS.
- J. Weber, A. J. Wain and F. Marken, Electroanalysis, 2015, 27, 1829–1835 CrossRef CAS.
- J. D. Wadhawan, P. J. Welford, H. B. McPeak, C. E. W. Hahn and R. G. Compton, Sens. Actuators, B, 2003, 88, 40–52 CrossRef CAS.
- A. Schürmann, R. Haas, M. Murat, N. Kuritz, M. Balaish, Y. Ein-Eli, J. Janek, A. Natan and D. Schröder, J. Electrochem. Soc., 2018, 165, A3095–A3099 CrossRef.
- K. Onda, E. Sada, T. Kobayashi, S. Kito and K. Ito, J. Chem. Eng. Jpn., 1970, 3, 18–24 CrossRef CAS.
- W. Lang and R. Zander, Ind. Eng. Chem. Fundam., 1986, 25, 775–782 CrossRef CAS.
- W. Xing, M. Yin, Q. Lv, Y. Hu, C. Liu and J. Zhang, in Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, ed. W. Xing, G. Yin and J. Zhang, Elsevier, Amsterdam, 2014, pp. 1–31 Search PubMed.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS PubMed.
- A. Y. M. Al-Murshedi, H. F. Alesary and R. Al-Hadrawi, J. Phys.: Conf. Ser., 2019, 1294, 052041 CrossRef CAS.
- W. Tong, M. Forster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- S. L. Clegg and P. Brimblecombe, Geochim. Cosmochim. Acta, 1990, 54, 3315–3328 CrossRef CAS.
- J. A. M. Abbas, PhD thesis, Swansea University, 1986.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional tables and figures. See DOI: 10.1039/d1ee01822a |
| This journal is © The Royal Society of Chemistry 2021 |