
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Additive stabilization of SEI on graphite observed using cryo-electron microscopy†
Bing
Han
ab,
Yucheng
Zou
a,
Guiyin
Xu
c,
Shiguang
Hu
d,
Yuanyuan
Kang
d,
Yunxian
Qian
d,
Jing
Wu
e,
Xiaomin
Ma
e,
Jianquan
Yao
a,
Tengteng
Li
 a,
Zhen
Zhang
a,
Hong
Meng
b,
Hong
Wang
a,
Yonghong
Deng
*a,
Ju
Li
*c and
Meng
Gu
*a
a,
Zhen
Zhang
a,
Hong
Meng
b,
Hong
Wang
a,
Yonghong
Deng
*a,
Ju
Li
*c and
Meng
Gu
*a
aDepartment of Materials Science and Engineering, Guangdong Provincial Key Laboratory of Energy Materials for Electric Power, and Shenzhen Key Laboratory of Solid State Batteries, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: dengyh@sustech.edu.cn; gum@sustech.edu.cn
bAdvanced Materials, Peking University Shenzhen Graduate School, Shenzhen, 518055, China
cDepartment of Nuclear Science and Engineering and Department of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: liju@mit.edu
dShenzhen CAPCHEM Technology Co. Ltd, Pingshan District, Shenzhen, 518118, China
eCryo-TEM center, Southern University of Science and Technology, Shenzhen 518055, China
First published on 6th July 2021
Abstract
Revealing the atomic structures of the solid electrolyte interphase (SEI) is challenging due to its sensitivity to electron beam and environmental factors such as moisture and oxygen. Here, we unveiled the atomic structures and phase distribution of the fragile solid electrolyte interphase (SEI) on graphite using ultra-low-dosage and aberration-corrected cryo-transmission electron microscopy (cryo-TEM). It is known that propylene carbonate electrolyte can exfoliate a graphite anode and damage its structural integrity. Surprisingly, ethylene carbonate–diethyl carbonate can also damage the surface of the graphite anode by exfoliation even with an initial formation protocol of constant-current charging (0.05C) for three hours and then 0.1C for another 3 hours at 45 °C: we hypothesize that the exfoliated graphene layers embedded in the SEI enhance local electron channeling, which induces an ever-growing, thick SEI layer with randomly distributed graphene, Li2O, and Li2CO3 nano-crystals. Using the same formation protocol but with 1 wt% vinylene carbonate (VC), triphenyl phosphate (TPP), or ethylene sulfate (DTD) or 10 wt% monofluoroethylene carbonate (FEC) as the additive is found to cause solid deposition prior to the graphite exfoliation instability, which generates a stable and thin SEI (<90 nm) on the graphite surface which prevents further exfoliation of graphite and rapidly suppresses the decomposition of electrolyte in the later cycles. When using a slower formation protocol including 2 cycles between 3.0 and 4.2 V at a rate of 0.01C at room temperature, graphite exfoliation is dramatically reduced, but is still observable initially.
Broader contextGraphite is the industrially dominating commercial anode for rechargeable Li-ion batteries that has revolutionized electric transportation and the consumer electronics industry. The solid electrolyte interphase (SEI) plays a critical role in determining the cycling stability of the graphite anode. However, viewing the SEI in batteries remains challenging due to its sensitivity to electron beam under the microscope. We develop an ultra-low dosage cryo-TEM protocol to directly visualize the SEI at different charging states. It is known that propylene carbonate (PC) electrolyte can exfoliate the graphite and damage its structural integrity. However, here we show that not only PC but also ethylene carbonate (or EC-based) electrolyte can diffuse into the graphite layers and exfoliate the surface of graphite anode. Additives are needed to ensure a long-lived battery with a high capacity. The successful imaging and composition analysis at the atomic scale using cryo-TEM provide us with new details on the most delicate component in batteries. |
Introduction
Graphite is the prevalent commercial anode for rechargeable Li-ion batteries in electric vehicles and consumer electronics. Issues still exist in finding the best electrolyte for graphite anode. During the first cycle of the battery, reduction of the organic electrolyte takes place on the anode surface to form a passivating solid electrolyte interphase (SEI).1,2 Additives in the electrolyte can modify the reductive decomposition of the electrolyte through biasing the reaction direction and forming functional components in the SEI layer, enhancing the battery performance and cycle life. A stable SEI should be a good Li+ conductor, but must be sufficiently electronically insulating to prevent electron transport by tunneling/hopping. So far, the most widely-used anode in the industry is graphite with a very low open-circuit potential of ∼0.1 V versus Li metal and a volume change of less than 12% after Li+ insertion. Although being studied for several decades, the SEI structures on graphite still remain not well defined due to inaccessibility and vulnerability to radiation, air, and moisture.3How SEI forms has always been an important question. Many researchers obtained useful chemical information of the SEI through indirect testing methods, such as X-ray photoelectron spectroscopy, nuclear magnetic resonance, and synchrotron X-ray analysis. However, these techniques lack the spatial resolution to resolve the atomic lattices of the nanocrystals in the SEI. Aberration-corrected high resolution TEM (HRTEM) has sub-Angstrom resolution, which is capable of probing the detailed atomic structure of the inorganic and organic components in the SEIs. However, SEI is quickly damaged and modified by the large flux of electrons in the conventional TEM, resulting in false imaging of the SEI and other interfaces. Researchers showed that useful information by the direct imaging of the SEI4 or lithium dendrites can be obtained using a cryo-TEM holder setup.5,6 The SEI structure is so delicate that even a slightly larger electron dosage in the cryo-TEM can still damage the components. Critical dosage limit needs to be calibrated before quantitative HRTEM analysis of the chemical components in the native SEI is performed.
Here, we use a combination of direct-detection camera and aberration-corrected cryo-TEM to image the delicate SEI at the atomic scale with an ultra-low electron dosage (well below the damage threshold). We use cryo-TEM to systematically study the SEI on graphite using propylene carbonate C4H6O3 (abbreviated as PC) electrolyte, ethylene carbonate–diethyl carbonate (EC–DEC) electrolyte, and EC–DEC with additives, such as 1 wt% vinylene carbonate (VC), 1 wt% triphenyl phosphate (C6H5)3PO4 (TPP), 1 wt% ethylene sulfate C2H4O4S (DTD), or 10 wt% monofluoroethylene carbonate C3H3FO3 (FEC) as shown in Fig. S1, ESI.† Before cycling at larger currents, all graphite anodes are charged using a constant current (0.05C) for three hours and then 0.1C for another 3 hours at 45 °C (quoted as ‘formation process I’). A slower formation process at 0.01C for two cycles between 3.0 and 4.2 V at room temperature (quoted as ‘formation process II’) using EC–DEC without additional additives also shows effective suppression of graphite exfoliation. The formation process is generally critical to ensure the activation of the graphite and the creation of a relatively stable interphase between graphite and electrolyte to prolong its electrochemical cycle life.
Results and discussions
Li+–solvent interactions lead to the formation of Li+-solvation shell in the liquid electrolyte, Li+(PC)y or Li+(EC)y, with y ranging from 4 to 6. A large degree of graphite layer expansion has been observed due to the Li(PC)x intercalation into the graphite as shown by the schematic in Fig. S2-a (ESI†),7 where Li(PC)x stands for a Li+(PC)x cation complex with an electron attracted to the metallic graphene layers nearby, where x may be smaller than y as this cation may shed some solvent molecules as it inserts in between the graphene. For any x ≥ 1, such process will be harmful to the long-term cycling stability of the graphite anode, as the expanded c-spacing and residual stress due to the solvent molecule easily lead to the exfoliation of the graphene layers or chunks of graphite. As depicted in Fig. S2-b (ESI†), the exfoliated graphite layers are clearly observed. As observed in the electron energy loss spectroscopy (EELS) map shown in Fig. S2-c (ESI†), significant signals from oxygen, carbon, fluorine, and lithium were found inside the cracks of the graphite, indicating the content of Li(PC)x in between the carbon layers in the surface of the graphite. The graphite layers bend and crack, resulting in structural damage, loss of electron percolation, unstable SEI, and capacity loss of the battery. The original layer spacing expanded from 0.35 nm to 0.5 nm after electrochemical lithiation, shown in Fig. S2d and e (ESI†). Large amounts of graphite exfoliation are observed using PC electrolyte. This is the reason why PC is generally not preferred for graphite. Really only x = 0, that is naked Li+ cation without any solvent molecule, is an acceptable intercalant for the long-term cycling of graphite. The only way to achieve this is to form a compact, adherent and conformal SEI that acts as a filter that “filtrates” out any molecules and free electrons, and only allows naked Li+ cations to intercalate/deintercalate.The graphite anode with formation process I was cycled in EC–DEC at 45 °C, shown in Fig. 1a and b. Fresh graphite and graphite that has just rested in EC–DEC electrolyte are comparatively shown in Fig. S3 and S4 (ESI†), where no SEI or surface exfoliation is observed. Previously, researchers believe that no exfoliation happens during electrochemical lithiation in the EC–DEC electrolyte. However, a mild degree of graphite exfoliation can be clearly visualized in the SEI as pointed by the arrows in Fig. 1b. HRTEM in Fig. 1c highlights the exfoliated graphite layers after 200 cycles at 45 °C. As is well known, these graphitic layers are electronically conductive, which leads to a non-uniform electric field and tip-enhanced electron tunneling. At a low enough anode potential (most of the carbonate solvents have electrochemical stability window [Ulower, Uupper], where Ulower ∼1 V versus Li metal), these tunneling electrons can cause reductive decomposition of the electrolyte, further thickening the SEI. Simultaneously, mobile Li+ ions in the electrolyte may be trapped irreversibly in the newly formed SEI, reducing the Coulombic efficiency. We also detected the presence of Li2CO3 and Li2O inside the amorphous matrix in the SEI as shown in Fig. 1d. The atomic lattices of the graphite layer, Li2CO3, and Li2O are shown in Fig. 1e–g. The schematic clearly shows that the insertion of Li-electrolyte molecules leads to an increased layer distance, disrupted surface lattices, and exfoliated graphite thin layers on the graphite anode. Finally, the assembly of organic polymer, Li2CO3, Li2O, and exfoliated graphites forms the SEI layer on graphite in the EC–DEC electrolyte. The characteristics of SEI then dictate the rate of charge and discharge, and the life of the battery.
 | ||
Fig. 1 (a) The overall view of graphite after cycling in baseline electrolyte of 1.0 mol L−1 LiPF6 dissolved in a solution of EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEC (volume ratio of 30:70) after 200 cycles at 45 °C; (b) magnified surface region outlined in white in panel a; (c) region showing the exfoliated graphite layers and Li2O inside the amorphous SEI matrix; (d) region showing the Li2O, Li2CO3, and exfoliated graphite layers inside the amorphous SEI matrix; (e) HRTEM of the exfoliated graphite; (f) Li2CO3; (g) Li2O; and (h) scanning transmission electron microscopy (STEM) and EELS elemental maps of the SEI showing oxygen, carbon, and fluorine signals. (i) Schematic showing the insertion of Li(EC)x in the graphite leading to the disruption and exfoliation of near-surface graphite. :DEC (volume ratio of 30:70) after 200 cycles at 45 °C; (b) magnified surface region outlined in white in panel a; (c) region showing the exfoliated graphite layers and Li2O inside the amorphous SEI matrix; (d) region showing the Li2O, Li2CO3, and exfoliated graphite layers inside the amorphous SEI matrix; (e) HRTEM of the exfoliated graphite; (f) Li2CO3; (g) Li2O; and (h) scanning transmission electron microscopy (STEM) and EELS elemental maps of the SEI showing oxygen, carbon, and fluorine signals. (i) Schematic showing the insertion of Li(EC)x in the graphite leading to the disruption and exfoliation of near-surface graphite. | ||
EC electrolyte without additives still exfoliates graphite layers, rendering the solid SEI locally conductive. Therefore, electron transport along the exfoliated graphite layers and eventual tunneling into the liquid region can induce further decomposition of the electrolyte and even the growth of lithium metal dendrites. The exfoliation of graphite makes the SEI unstable, growing to a thickness of ∼450 nm at 200 cycles at 45 °C, as shown by the scanning transmission electron microscopy analysis (STEM) and electron energy loss spectroscopy (EELS) maps in Fig. 1h. Elemental mapping reveals oxygen, carbon, and fluorine inside the SEI, consistent with the high-resolution TEM analysis and electrolyte/salt compositions used in the battery. A schematic showing the disrupted graphite layers inside the SEI is shown in Fig. 1i. All the cells follow the formation and aging process that includes first-cycle charging using small current density (0.05C) for three hours and then 0.1C for another 3 hours. Detailed additional information on the formation process is described in the ESI.† In comparison, we carried cryo-TEM analysis of the graphite cycled in the EC–DEC electrolyte without such formation protocol. As seen, the exfoliation of graphite surface is much worse, showing large amounts of exfoliated graphite layers mixing in the SEI as shown in Fig. S5 and S6 (ESI†). What our cryo-TEM experiments show is that SEI formed with EC and EC–DEC is “marginal” at best, and this is indeed the reason why an elaborate formation process is needed industrially. The reason for using additives is to boost the speed and efficacy of SEI formation, and this would also lead to lower capital expenditures (CAPEX) and operating expenditures (OPEX) associated with the industrial battery formation protocol.
To evaluate the effectiveness of the formation process I, we performed battery cycling at room temperature. As shown in Fig. 2a, the capacity retention reaches 74% after 1000 cycles at room temperature in a 2000 mA h pouch cell with a graphite/NCM523 full cell. Cryo-TEM in Fig. 2b demonstrates mild graphite exfoliation after 1000 cycles at room temperature compared to the large degree of graphite exfoliation after cycling at 45 °C for 200 cycles, which indicates that temperature may play a vital role in the exfoliation of graphite during cycling. Both LiF and Li2O phases are observed to randomly distribute in the SEI; Fig. 2c and d capture the lattices of the LiF crystals and the exfoliated graphite layers. In addition, careful inspections indicate that Li metal dendrite can grow out of the graphite after charging (Fig. 2e), indicating that the graphite layers in the SEI may leak electrons through the SEI to induce Li metal dendrite formation during charging. The cryo-EELS confirms the metallic state of the Li dendrite at the graphite surface (Fig. 2f). The safety of electrical devices and vehicles using Li batteries is then likely compromised due to the possibility of short-circuit caused by Li metal dendrite growth.
 | ||
| Fig. 2 Room-temperature cycling performance at 1C rate for 1000 cycles after the formation process I (a) and cryo-TEM structural characterization of graphite anode in baseline electrolyte of 1.0 mol L−1 LiPF6 dissolved in a solution of EC:DEC (volume ratio of 30:70) after 1000 cycles at room temperature (b–d); cryo-TEM imaging of the Li metal dendrite deposited on graphite (e); and cryo-EELS identification of the Li metal growth on the graphite surface after cycling (f). | ||
In addition, we evaluated the exfoliation conditions of the graphite anode using EC–DEC with a much slower formation process II (2 cycles between 3.0 and 4.2 V at a rate of 0.01C) at room temperature. Using formation process II, the degree of graphite exfoliation is largely reduced compared to that of formation process I as shown in Fig. S7 (ESI†). A minor amount of exfoliated graphite layers and Li2O nanocrystals are randomly distributed in an amorphous matrix in the SEI. The results of HRTEM analysis, shown in Fig. S7b (ESI†), and fast Fourier transform (FFT), shown in Fig. S7c (ESI†), demonstrate clearly the lattice fringes of the graphite layers and Li2O crystals. The Li2O crystals may play critical roles in preserving the electronic insulation characteristic of the SEI layer.
With the quicker formation process I, we find clear evidence that the usage of additives in the liquid electrolyte has a large positive impact on the SEI morphology and battery performance. The additives are often designed to sacrifice themselves to form stable interphases in the initial formation of the battery. Here we have tested well-known additives such as DTD, TPP, VC, and FEC in EC–DEC electrolyte. The systematic studies show that these additive molecules are preferentially consumed in the reduction reactions at low U, leaving their signature by forming a highly protective conformal layer consisting of compact crystalline inorganic compounds and polymers. An effective SEI should have good adhesion to the graphite surface, resilience in deformation, resistance to solvent permeation and dissolution, and high Li+ conductivity. Thereby, further electrolyte–graphite interactions are blocked from graphite by the SEI. The usage of these additives largely lowered the extent of exfoliation of graphite layers, protecting the integrity of graphite anode.
DTD has a higher reduction potential (1.3 V versus Li+/Li metal) than EC (0.95 V),8 which means that DTD is easier to be reduced at the graphite anode surface to form SEI, which then blocks the entry of the Li-electrolyte solvation sheath complex and only allows naked Li+ ion to transport across it. Therefore, DTD usage results in an effective blockage of Li+(EC)x with x ≥ 1 and the subsequent exfoliation of graphite. Large quantities of Li2SO4 form the dominant part of the thin and compact SEI. Li2SO4 is nanocrystalline with crystal size ranging from a few nanometers to 50 nm, forming a dense compact layer on graphite as shown in Fig. 3a and b. A thin amorphous layer is also present on the top part of the SEI with Li2SO4 islands dispersed in it. The STEM and EELS analyses in Fig. 3c reveal the content of Li, O, S, and C in the SEI which is consistent with the HRTEM analysis. The as-formed Li2SO4 appears surprisingly stable against lithiated graphite at ∼0.1 V (perhaps with a very thin overlithiated-Li2SO4 underlayer),2 effectively blocking the transport of the Li-electrolyte molecular complex. Reports show that Li2SO4-treated graphite exhibits outstanding cycling performance and low interfacial impedance.9 Therefore, the formation of Li2SO4-dominating SEI using DTD is an effective method to improve the performance of graphite anode.
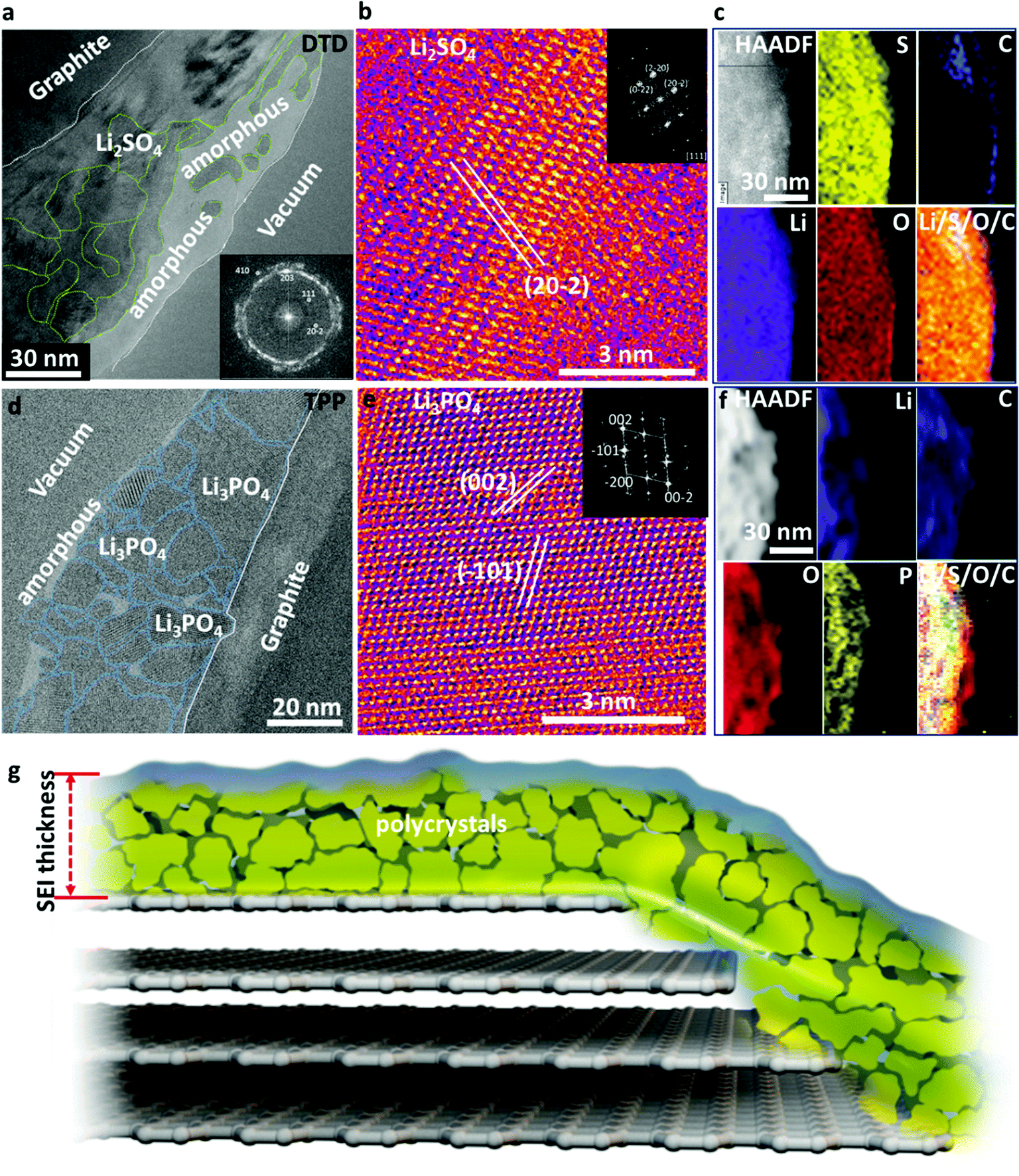 | ||
| Fig. 3 (a–c) Analysis of the graphite cycled using electrolyte solution containing 1.0 mol L−1 LiPF6 dissolved in EC:DEC (30:70 volume ratio) with 1 wt% DTD after two hundred cycles at 45 °C. (a) Large-scale TEM image showing the SEI; (b) HRTEM of Li2SO4 crystals; (c) HAADF STEM image and EELS elemental maps of the SEI using DTD/EC–DEC; (d–f) analysis of the graphite cycled using containing 1.0 mol L−1 LiPF6 dissolved in EC:DEC (30:70 volume ratio) with 1 wt% TPP after two hundred cycles at 45 °C; (d) large-scale TEM analysis of the SEI; (e) HRTEM of Li3PO4 crystals in the SEI; (f) HAADF STEM image and EELS elemental maps of Li, C, O, and P, composite maps using TPP/EC–DEC; and (g) schematic showing the SEI formed with DTD and TPP additive in EC–DEC electrolyte. | ||
Similarly, the use of TPP is effective in forming a dense and compact inorganic crystalline SEI with only a small amount of polymeric components. We see large quantities of Li3PO4 formed in the SEI. These Li3PO4 nanocrystals range from 2 nm to 40 nm (Fig. 3d). In most regions, they are closely packed together, similar to the case of Li2SO4, while some amorphous zones are present with Li3PO4 crystals dispersed inside. The Li3PO4 crystals show some stacking faults, as shown by the cryo-HRTEM and FFT results (Fig. 3e). STEM and EELS analyses (Fig. 3f) reveal the content of Li, C, O, and P in the SEI, in good agreement with the HRTEM analysis. The dense Li3PO4 inorganic layers also effectively prevent the transport of the Li-electrolyte complex and only allow naked Li+ to go in and out. The stability of Li3PO4 is well known and the nitrogen-doped glass-type Li3PO4 (LIPON) is a famous thin-film solid electrolyte. The Li-conductive Li3PO4 layer with a high Young's modulus or Li3PO4–polymer composite can even stabilize Li metal anode and restrain dendrite growth, enhancing the performance of the Li–metal batteries.10–12 The Li metal is slightly more electronegative than lithiated graphite; therefore, such an in situ formed Li3PO4 layer on graphite anode should be even more stable, enhancing the stability and life of batteries. The schematic in Fig. 3g illustrates the SEI architecture formed with DTD or TPP additive, where a dense layer of polycrystalline inorganic nanocrystals dominates the SEI functions, and the grain boundaries may also play important roles in Li+ transport across the SEI.
VC is the most successful representative of unsaturated additives, which polymerize under reductive conditions and form a thick polymer layer. Nanocrystalline LiF and Li2O islands of 2–15 nm sizes are randomly distributed inside the polymer matrix, as shown in Fig. 4a. The polymers are also an effective protection buffer that blocks the electrolyte molecules and allows only Li+ transport. The cryo-HRTEM of LiF is shown in Fig. 4b displaying a typical [111] zone atomic arrangement of LiF. Different from DTD and TPP, the dominant portion of the SEI with VC additive is the amorphous polymeric phase matrix; and only small amounts of nanocrystals form inside the amorphous matrix in the SEI. The elastic polymer matrix helps to protect the SEI from cracking during volumetric expansion and contraction.
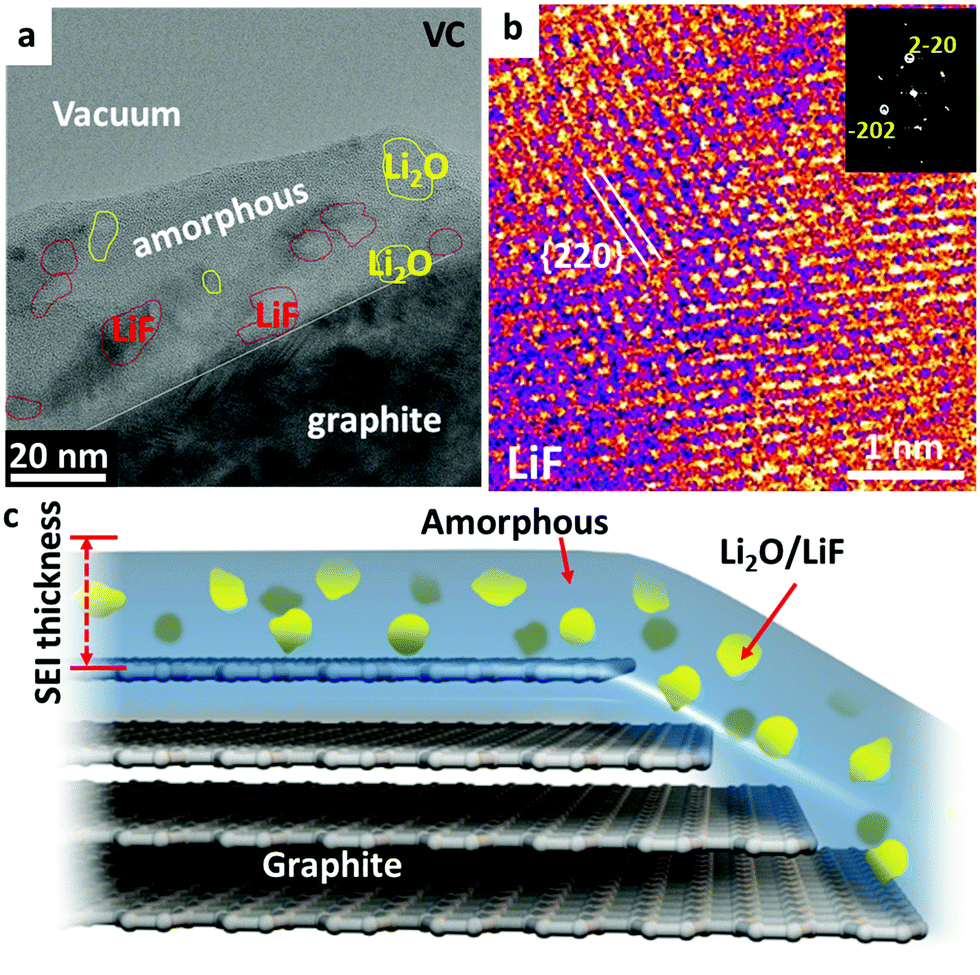 | ||
| Fig. 4 Analysis of the graphite electrode cycled after formation process I in an electrolyte solution containing 1.0 mol L−1 LiPF6 dissolved in EC:DEC (30:70 volume ratio) with 1 wt% VC after 200 cycles at 45 °C. (a) Large-scale image showing the SEI layer where small amounts of LiF and Li2O are distributed in an amorphous polymeric matrix; (b) HRTEM identification of LiF nanocrystals in the SEI; (c) schematic showing the SEI where the amorphous polymeric phase forms the dominating matrix. | ||
Fluorine-containing additives are another useful family of additives, which can result in a thin SEI layer. Researchers are still debating whether LiF was formed in the SEI and the role of LiF in electrochemical cycling.7 Studies show that the reduction potential (1.37 V versus Li+/Li metal) for FEC is higher than that of EC (0.95 V), implying that a FEC molecule can be reduced prior to EC.13 As shown in Fig. 5a, LiF-rich zones and Li2O-rich zones alternatingly stack on top of the graphite surface, which is the general signature of a FEC-induced SEI. An enlarged LiF-rich zone is displayed in Fig. 5b, which demonstrates that the LiF crystals in an amorphous polymer matrix form a percolating network. The Li ions can transport via the grain boundaries of LiF, phase boundaries or the polymer matrix. The size of the LiF crystals ranges from 1 nm to 35 nm, thus showing a wide size dispersion. The cryo-HRTEM of a Li2O-rich zone is shown in Fig. S8 of the ESI,† which also contains some dispersed LiF phases. The large-scale EELS elemental maps in Fig. 5c prove that the SEI contains alternating Li2O-rich and LiF-rich zones in the polymer matrix. Fig. 5d presents the HRTEM of the LiF crystal along the [001] zone projection, which shows the cubic FCC type lattices clearly. In addition, high-resolution EELS maps in Fig. 5e confirm the content of fluorine, oxygen, lithium, and carbon in the SEI. Fine structure analysis from the summed spectra of the whole map in Fig. 5f–i indicates that the amorphous polymeric matrix should contain C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–H, CO, and C–O bonds. In addition, the Li K and F K edge prove the content of LiF, which is consistent with the HRTEM analysis. This LiF-decorated organic–inorganic composite as illustrated by the schematic in Fig. 5l results in a stable SEI due to the chemical stability across a wide voltage window (0–5 V)14 and the electron-insulating characteristic of LiF, while still allowing facile Li+ transport. Going beyond the graphite anode, the literature also reports that a LiF-rich composite with artificial SEI coating or LiF-containing SEI is remarkably effective in stabilizing the cycling performance of silicon and phosphorous anodes.15,16 FEC is the representative example of the formation of LiF-rich SEI layers with a wide electrochemical stability voltage window and excellent chemical stability.17
C, C–H, CO, and C–O bonds. In addition, the Li K and F K edge prove the content of LiF, which is consistent with the HRTEM analysis. This LiF-decorated organic–inorganic composite as illustrated by the schematic in Fig. 5l results in a stable SEI due to the chemical stability across a wide voltage window (0–5 V)14 and the electron-insulating characteristic of LiF, while still allowing facile Li+ transport. Going beyond the graphite anode, the literature also reports that a LiF-rich composite with artificial SEI coating or LiF-containing SEI is remarkably effective in stabilizing the cycling performance of silicon and phosphorous anodes.15,16 FEC is the representative example of the formation of LiF-rich SEI layers with a wide electrochemical stability voltage window and excellent chemical stability.17
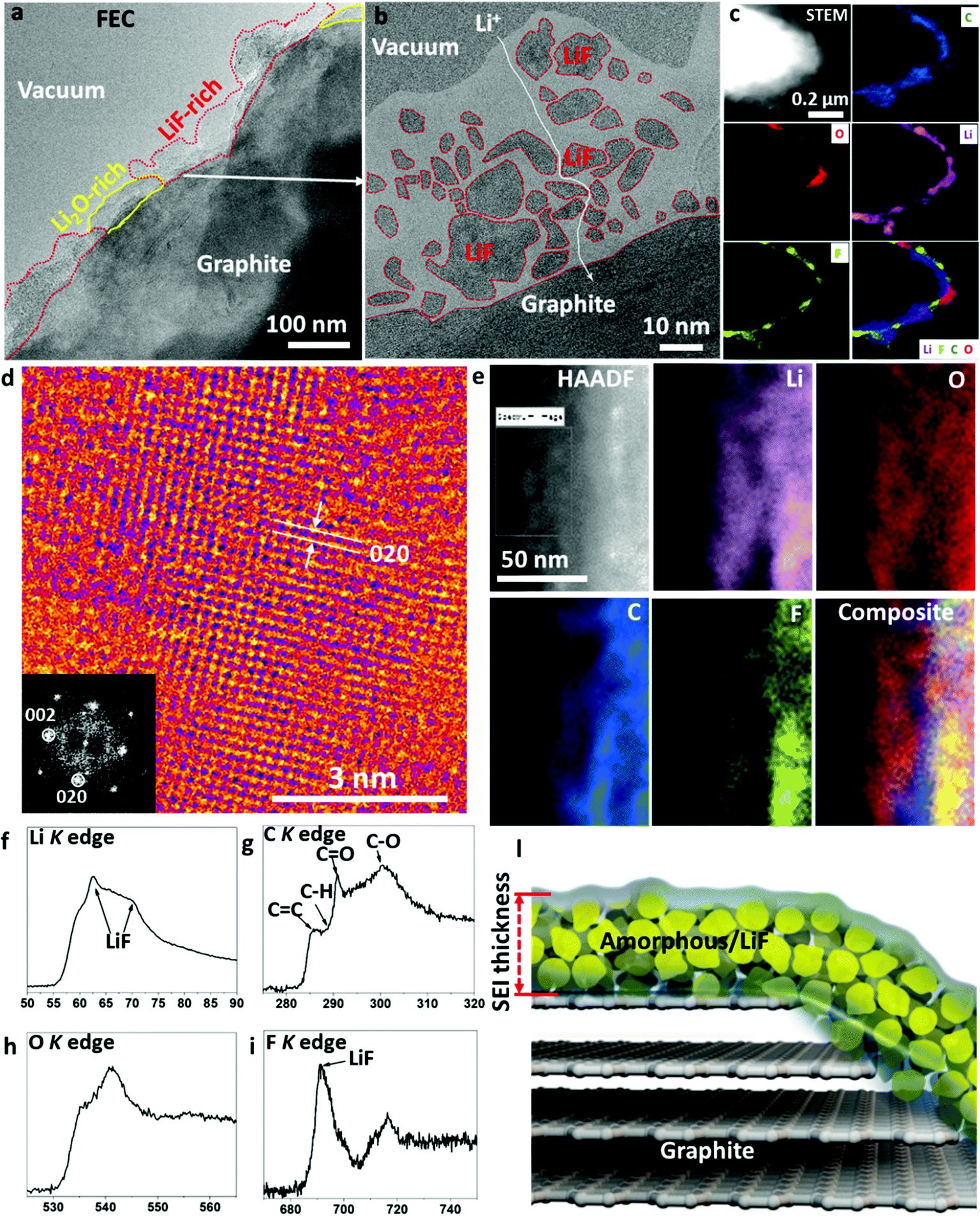 | ||
| Fig. 5 (a) Overall view of the SEI formed using formation process I in an electrolyte solution of 1.0 mol L−1 LiPF6 dissolved in EC:DEC:FEC with volume ratio of 20:70:10 after two hundred cycles at 45 °C; (b) SEI layer with identified LiF islands inside the amorphous matrix using HRTEM analysis; (c) large-scale STEM and EELS elemental maps of C, O, Li, and F, composite in the SEI using FEC/EC–DEC; (d) HRTEM showing the LiF atomic lattices along the [001] zone projection; (e) high-magnification HAADF STEM and EELS elemental maps of the LiF-rich region in the SEI; (f) Li K, (g) C K, (h) O K, and (i) F K edge EELS from the SEI; and (l) schematic showing the as-formed percolating SEI using FEC/EC-DEC. | ||
The cycling performances of the additive-modified electrolytes are compared with those of the blank EC-DEC electrolyte in pouch cells of NCM523||graphite in Fig. 6. Due to the exfoliation of graphite, the capacity drops quickly in the blank-EC–DEC full cell. Benefiting from the rapid formation of effective SEI layers in the VC, TPP, DTD and FEC modified cells, the cycling performances are outstanding compared to those of the blank EC–DEC. Significant battery capacity decay of the blank EC–DEC electrolyte cell takes place at about 20 cycles. In contrast, the capacity of cells cycled with VC, TPP, DTD and FEC electrolyte additives are maintained above 1500 mA h. At the 200th cycle, the capacity of the blank-EC–DEC cell drops to 800 mA h, while those of the VC, TPP, DTD and FEC additive modified batteries still remain above 1300 mA h. The FEC-containing full cell has the highest capacity of above 1500 mA h, which perhaps indicates slight superiority of the inorganic-dominant nanocomposite approach, where LiF and Li2O alternatingly stack on top of each other within the amorphous polymer matrix. The VC-containing cell exhibits the most decay to ∼1300 mA h among all additive-modified cells, while the DTD and TPP-containing cells retain a capacity of ∼1400 mA h at 200 cycles. This ranking of FEC > DTD, TPP > VC probably suggests that a polymer-dominant SEI is slightly less effective than inorganic-dominant composite SEIs for graphite.
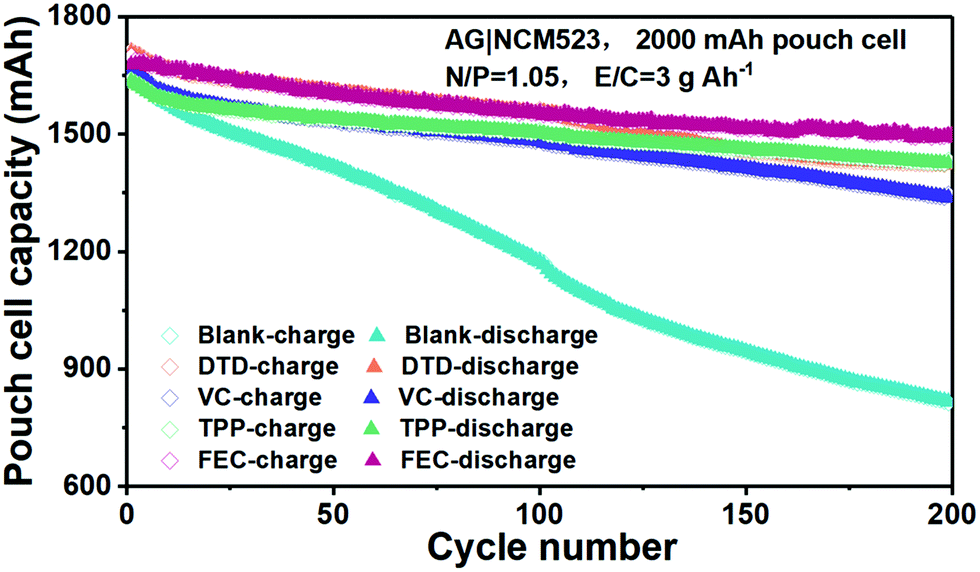 | ||
| Fig. 6 Comparison of the cycling performance of the blank EC–DEC and additive modified EC–DEC electrolytes in pouch cells of NCM523||graphite with formation process I. | ||
Modeling using DFT-based molecular dynamics predicts that the innermost SEI on a Li metal surface should contain predominantly binary compounds such as Li2O or LiF instead of less stable polyanion compounds such as Li2CO3.2 Here, experimental cryo-TEM results showed that the polyanion compounds such as Li2SO4 and Li3PO4 are rather stable in contact with graphite anode, at least in the interior of the SEI.2 The usage of diglyme electrolyte may lead to an even thinner SEI or SEI-free graphite anode, which may be a future system to explore using atomic-scale cryo-TEM.18
Conclusion
In summary, exfoliation of graphite is observed in PC and EC electrolytes with a 6 hours formation protocol (formation process I), which destabilizes the surface of the graphite anode and makes the SEI thick, thus reducing the cycle life and capacity of the battery. The exfoliated graphite layers form an electron tunneling network, which causes unstable SEI growth as well as depletion of cyclable lithium inventory. The additives can preferentially decompose to form a dense inorganic layer of Li2SO4 and Li3PO4 nanocrystals, a Li2O/LiF-decorated amorphous polymer composite layer, or a stable VC-reduced polymeric layer, which can prevent the permeation of dressed cations Li+(EC)x with x ≥ 1 and electron tunneling. This then prevents the exfoliation of graphite and the thickening of the SEI, and therefore extends the cycle life of the battery. Additive-decomposition modified SEIs are stabilized at a thickness of less than 90 nm as observed for all additives, without significant graphite exfoliation. In contrast, the slower formation process II can largely suppress the graphite exfoliation in EC–DEC without any additives, protecting the graphite anode. The chemical composition of the SEI components and how they are arranged in the SEI dictate the efficacy of the SEI. This study reveals a rich diversity of “structural solutions” using VC (polymer-dominant SEI), TPP (Li3PO4), DTD (Li2SO4) and FEC (LiF/Li2O), all of which offer good protection to the liquid electrolyte and lithiated graphite, even under an aggressive formation protocol. A clear structural phase analysis of the SEI using cryo-TEM and cryo-EELS provides valuable information for new additive and electrolyte design, as well as for optimizing the formation protocol in order to reduce the capital and operating expenditures of battery manufacturers, thus improving the competitiveness and environmental benefits of battery energy storage.Author contributions
M. G. leads the project; M. G., J. L. and Y. D. designed the experiments; M. G., and J. L. wrote the paper; B. H., Y. Z. performed the battery cycling and cryo-EM experiments; M. G., H. B., J. L., and G. X. analyzed the data; all authors edited the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from Key-Area Research and Development Program of Guangdong Province (2020B090919001), National Natural Science Foundation of China (21802065, 22078144), Shenzhen Clean Energy Research Institute (No. CERI-KY-2019-003), Shenzhen Key Laboratory of Solid State Batteries (ZDSYS20180208184346531), Guangdong Innovative and Entrepreneurial Research Team Program (2019ZT08C044), and Shenzhen Science and Technology Program (Grant No. KQTD20190929173815000), Shenzhen DRC project [2018]1433. The authors want to thank Dr Peiyi Wang, Dr Jun Lu and Dr Kang Xu for their insights and suggestions to improve the paper. The microscopy work was performed at the Pico and cryo-TEM Center at the SUSTech CRF that receives support from Presidential fund and Development and Reform Commission of Shenzhen Municipality.References
- S. Jurng, Z. L. Brown, J. Kim and B. L. Lucht, Energy Environ. Sci., 2018, 11, 2600–2608 RSC.
- B. Han, Z. Zhang, Y. Zou, K. Xu, G. Xu, H. Wang, H. Meng, Y. Deng, J. Li and M. Gu, Adv. Mater., 2021, 33, 2100404 CrossRef CAS PubMed.
- J. Asenbauer, T. Eisenmann, M. Kuenzel, A. Kazzazi, Z. Chen and D. Bresser, Sustainable Energy Fuels, 2020, 4, 5387–5416 RSC.
- M. J. Zachman, Z. Tu, S. Choudhury, L. A. Archer and L. F. Kourkoutis, Nature, 2018, 560, 345–349 CrossRef CAS PubMed.
- X. Zhang, S. Han, B. Zhu, G. Zhang, X. Li, Y. Gao, Z. Wu, B. Yang, Y. Liu, W. Baaziz, O. Ersen, M. Gu, J. T. Miller and W. Liu, Nat. Catal., 2020, 3, 411–417 CrossRef CAS.
- X. Wang, Y. Li and Y. S. Meng, Joule, 2018, 2, 2225–2234 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- X. Li, Z. Yin, X. Li and C. Wang, Ionics, 2013, 20, 795–801 CrossRef.
- X. Cui, F. Tang, C. Li, Y. Zhang, P. Wang, S. Li and X. Ye, Energy Technol., 2017, 5, 549–556 CrossRef CAS.
- S. Hao, Z. Ma, Y. Zhao, L. Kong, H. He, G. Shao, X. Qin and W. Gao, ACS Omega, 2020, 5, 8299–8304 CrossRef CAS PubMed.
- N. W. Li, Y. X. Yin, C. P. Yang and Y. G. Guo, Adv. Mater., 2016, 28, 1853–1858 CrossRef CAS PubMed.
- M. Bai, K. Xie, B. Hong, K. Yuan, Z. Li, Z. Huang, C. Shen and Y. Lai, Solid State Ionics, 2019, 333, 101–104 CrossRef CAS.
- U. Purushotham, N. Takenaka and M. Nagaoka, RSC Adv., 2016, 6, 65232–65242 RSC.
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Chem. Mater., 2015, 28, 266–273 CrossRef.
- J. Zhao, Z. Lu, H. Wang, W. Liu, H. W. Lee, K. Yan, D. Zhuo, D. Lin, N. Liu and Y. Cui, J. Am. Chem. Soc., 2015, 137, 8372–8375 CrossRef CAS PubMed.
- X. Han and J. Sun, Chem. Commun., 2020, 56, 6047–6049 RSC.
- L.-M. Suo, W.-J. Xue, M. Gobet, S. G. Greenbaum, C. Wang, Y.-M. Chen, W.-L. Yang, Y.-X. Li and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1156–1161 CrossRef CAS PubMed.
- M. Goktas, C. Bolli, E. J. Berg, P. Novák, K. Pollok, F. Langenhorst, M. v. Roeder, O. Lenchuk, D. Mollenhauer and P. Adelhelm, Adv. Energy Mater., 2018, 8, 1702724 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee01678d |
| This journal is © The Royal Society of Chemistry 2021 |