
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two birds with one stone: dual grain-boundary and interface passivation enables >22% efficient inverted methylammonium-free perovskite solar cells†
Saba Gharibzadeh‡
 ab,
Paul Fassl‡ab,
Ihteaz M. Hossainab,
Pascal Rohrbeckc,
Markus Frericksde,
Moritz Schmidtabf,
The Duongg,
Motiur Rahman Khana,
Tobias Abziehera,
Bahram Abdollahi Nejandab,
Fabian Schackmarab,
Osbel Almorah,
Thomas Feeneya,
Roja Singhab,
Dirk Fuchsi,
Uli Lemmera,
Jan P. Hofmannde,
Stefan A. L. Weberc and
Ulrich W. Paetzold*ab
ab,
Paul Fassl‡ab,
Ihteaz M. Hossainab,
Pascal Rohrbeckc,
Markus Frericksde,
Moritz Schmidtabf,
The Duongg,
Motiur Rahman Khana,
Tobias Abziehera,
Bahram Abdollahi Nejandab,
Fabian Schackmarab,
Osbel Almorah,
Thomas Feeneya,
Roja Singhab,
Dirk Fuchsi,
Uli Lemmera,
Jan P. Hofmannde,
Stefan A. L. Weberc and
Ulrich W. Paetzold*ab
aLight Technology Institute, Karlsruhe Institute of Technology, Engesserstrasse 13, 76131 Karlsruhe, Germany. E-mail: ulrich.paetzold@kit.edu
bInstitute of Microstructure Technology, Karlsruhe Institute of Technology, Hermann-von Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cMax Planck Institute for polymer research, department physics at interfaces, Ackermannweg 10, 55128 Mainz, Germany
dTechnical University of Darmstadt, Department of Materials and Earth Sciences, Surface Science Laboratory, Otto-Berndt-Strasse 3, 64287 Darmstadt, Germany
eInnovationLab GmbH, Speyerer Strasse 4, 69115 Heidelberg, Germany
fCenter for Nanophotonics, AMOLF, 1098 XG Amsterdam, The Netherlands
gSchool of Engineering, The Australian National University, Canberra, 2601, Australia
hInstitute of Advanced Materials, Universitat Jaume I, 12006 Castelló, Spain
iInsitute for Quantum Materials and Technologies, Karlsruhe Institute of Technology, Herrmann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 9th August 2021
Abstract
Advancing inverted (p–i–n) perovskite solar cells (PSCs) is key to further enhance the power conversion efficiency (PCE) and stability of flexible and perovskite-based tandem photovoltaics. Yet, the presence of defects at grain boundaries and in particular interfacial recombination at the perovskite/electron transporting layer interface induce severe non-radiative recombination losses, limiting the open-circuit voltage (VOC) and fill factor (FF) of PSCs in this architecture. In this work, we introduce a dual passivation strategy using the long chain alkylammonium salt phenethylammonium chloride (PEACl) both as an additive and for surface treatment to simultaneously passivate the grain boundaries and the perovskite/C60 interface. Using [2-(9H-carbazol-9-yl)ethyl]phosphonic acid (2PACz) as a hole transporting layer and a methylammonium (MA)-free Cs0.18FA0.82PbI3 perovskite absorber with a bandgap of ∼1.57 eV, prolonged charge carrier lifetime and an on average 63 meV enhanced internal quasi-Fermi level splitting are achieved upon dual passivation compared to reference p–i–n PSCs. Thereby, we achieve one of the highest PCEs for p–i–n PSCs of 22.7% (stabilized at 22.3%) by advancing simultaneously the VOC and FF up to 1.162 V and 83.2%, respectively. Using a variety of experimental techniques, we attribute the positive effects to the formation of a heterogeneous 2D Ruddlesden–Popper (PEA)2(Cs1−xFAx)n−1Pbn(I1−yCly)3n+1 phase at the grain boundaries and surface of the perovskite films. At the same time, the activation energy for ion migration is significantly increased, resulting in enhanced stability of the PSCs under light, humidity, and thermal stress. The presented dual passivation strategy highlights the importance of defect management both in the grain boundaries and the surface of the perovskite absorber layer using a proper passivation material to achieve both highly efficient and stable inverted p–i–n PSCs.
Broader contextDue to the rapid increase in power conversion efficiency of perovskite solar cells (PSCs), they are considered an emerging area of research in photovoltaic technologies. While inverted p–i–n PSCs have demonstrated great potential for flexible and perovskite-based tandem photovoltaics, key challenges still need to be addressed as compared to their n–i–p counterparts. In particular, severe non-radiative recombination losses induced by the presence of defects at grain boundaries (GBs) and interfacial recombination at the perovskite/electron transporting layer interface limit the open-circuit voltage (VOC) and fill factor (FF) of PSCs in this architecture. To address this issue, we demonstrate that utilizing a dual passivation strategy using phenethylammonium chloride both as an additive and for surface treatment simultaneously passivates defects at the GBs and the perovskite/C60 interface. We show that this is due to the formation of a heterogeneous 2D Ruddlesden-Popper phase, leading to a significant improvement in both the VOC and FF. In view of the urge to advance p–i–n PSCs for flexible and perovskite-based tandem photovoltaics, our findings stress the importance of defect management both at the GBs and the surface of the perovskite absorber layer in order to achieve both highly efficient and stable inverted p–i–n PSCs. |
Introduction
Single-junction organic–inorganic metal halide perovskite solar cells (PSCs) have demonstrated outstanding performance in laboratory-scale devices, closing the gap to the highest reported power conversion efficiencies (PCEs) of the market-dominating Si solar cells.1,2 While PCEs above 23% have been demonstrated using the mesoporous3–12 and planar13–22 n–i–p architecture (up to 25.5% certified23), inverted planar p–i–n PSCs still lag behind despite several recent studies reporting PCEs above 22% (up to 22.75% certified24) (see Fig. S1, ESI†).20,24–30 Further increasing the PCE of p–i–n PSCs is crucial given (1) their compatibility with p-type Si bottom solar cells for monolithic perovskite/Si tandem photovoltaics (PV),31,32 (2) their low-temperature processability (≤100 °C), and (3) their promising operational stability along with negligible hysteresis.33The most relevant bottleneck limiting the PCE of p–i–n PSCs is the apparent non-radiative recombination losses at the interface between the perovskite and the charge transport layers (CTLs).33–39 As a result, the open-circuit voltage (VOC) of p–i–n PSCs relative to the Shockley–Queisser (S–Q) limit for a given bandgap has long been significantly lower as compared to their n–i–p counterparts (Fig. S2a, ESI†), while recently specifically the VOC × fill factor (FF) product is lagging behind (Fig. S2b, ESI†).31,33,34 Considering that the novel self-assembled monolayer (SAM) hole transport layers (HTLs) 2PACz ([2-(9H-carbazol-9-yl)ethyl]phosphonic acid) and Me-4PACz ([2-(3,6-dimethoxy-9H-carbazol-9-yl)ethyl]phosphonic acid) developed by Albrecht and coworkers form a practically lossless interface,31,32 the remaining challenge is interfacial recombination at the electron transport layer (ETL), which is commonly the fullerene C60 or phenyl-C61-butyric acid methyl ester (PCBM). The second most relevant bottleneck is bulk defects in conjunction with the abundance of grain boundaries in perovskite films. Although the electronic properties of grain boundaries are still debated,40–42 they are commonly associated with an increased defect density, facilitated ion migration and an accelerated degradation under light and thermal stress.43–47 The latter aspect is particularly important considering that stability is one of the main concerns for the future commercialization of perovskite PVs.48–51 For these reasons, effective strategies to reduce both, (1) interfacial recombination at surface/interface defects and (2) bulk recombination at bulk or grain boundary defects are pivotal to maximize both the VOC and FF as well as the stability of planar p–i–n PSCs.
Post-treatment of perovskite films is a widely established strategy to suppress interfacial recombination and optimize the performance of PSCs.49,52–58 Prominent examples for tailored passivation schemes are the use of (alkyl)ammonium salts,3,4,6,10,11,13,14,20,22,59–80 other organic compounds16,19,24,45,81–87 and fluoride-containing materials.31,35,38,74,86,88,89 Alongside established chemical passivation that reduces the density of surface/interface defects,53,63,83,85 this strategy also encompasses performance enhancements by the formation of 2D/3D heterostructures10,59–61,63,64,70,76–78 and/or wide-bandgap interface layers.6,66,90,91 The latter enhancements can be the result of improved energy level alignment that promotes selectivity and carrier transport across perovskite/CTL interfaces and/or a reduced probability for interfacial recombination due to charge blocking.19,25,35,53,67,77,92 Recently, lithium fluoride (LiF) has been identified as an interlayer at the perovskite/ETL interface that significantly enhances the performance of p–i–n PSCs.31,35,38,88,89,93 However, PSCs with LiF undergo severe long-term degradation which limits the applicability of this approach.31,93
In order to reduce non-radiative recombination in the bulk and grain boundaries, the use of non-stoichiometric precursors94–96 or incorporation of different additives into the perovskite precursor solution or antisolvent such as metal cations,97 anions,98 chloride (Cl) or thiocyanate (SCN),4–6,10,14,15,17,19,88 (alkyl)ammonium salts,3,25,26,64,98–105 other organic compounds,74,84,106 and fluoride-containing materials74,107 have been proposed. Given that these additives directly assist in perovskite film formation, changes in crystallization dynamics as well as a reduced defect density are commonly observed.55,108,109 For instance, Xu et al. demonstrated that by alloying MAPbCl3 into the perovskite film, the VOC of the wide-bandgap p–i–n PSCs significantly improved due to a reduced bulk defect density.88 In other works, the addition of various long chain alkylammonium cations was shown to self-assemble into a wide-bandgap 2D perovskite phase passivating the surface and/or grain boundaries of the 3D perovskite film.25,58,64,99–105 However, due to the insulating nature of such 2D phases, adding too large amounts typically results in an overall lower PCE compared to control devices.25,64,99,101,103,104,107
Despite the apparent wide range of strategies suggested to reduce non-radiative recombination losses in PSCs,49,54,57,110,111 to our knowledge there is only one recent report that used the same passivation material both as an additive and for surface treatment to improve the performance of p–i–n PSCs.84 In this work, we report on an effective dual passivation approach using the long chain alkylammonium salt phenethylammonium chloride (PEACl) to simultaneously passivate the grain boundaries and the perovskite/C60 interface by using PEACl:PbCl2 as the additive and PEACl for surface treatment, respectively. Employing time-resolved photoluminescence (TRPL) and photoluminescence quantum yield (PLQY) measurements, dual passivation is proven to be most effective in reducing non-radiative recombination compared to either of the individual passivation strategies. By analyzing cathodoluminescence (CL), scanning electron microscopy (SEM), X-ray/ultraviolet photoelectron spectroscopy (XPS/UPS), X-ray diffraction (XRD) and Kelvin probe force microscopy (KPFM) measurements, we attribute the positive effects to the formation of a heterogeneous 2D Ruddlesden–Popper (RP) (PEA)2(Cs1−xFAx)n−1Pbn(I1−yCly)3n+1 perovskite phase with n ∼ 1–2 at the surface and grain boundaries of the film that exhibits a lower work function (WF) and hole blocking properties. Finally, thermal admittance spectroscopy (TAS) reveals that the activation energy for ion migration is strongly increased upon dual passivation, which is reflected in an enhanced device stability under maximum power point (MPP) tracking and heat treatment for 1000 h. In summary, by using PEACl both as the additive and for surface treatment, we could not only effectively reduce interfacial recombination at the perovskite/C60 interface, but simultaneously passivate the grain boundary defects. Employing dual passivation for methylammonium (MA)-free p–i–n PSCs with a bandgap of ∼1.57 eV leads to a very high PCE of 22.7% (stabilized at 22.3%) with a remarkable VOC and FF of up to 1.162 V and 83.2%, respectively. In view of the urge to advance the p–i–n structure for flexible and perovskite-based tandem photovoltaics, this development is pivotal.
Results and discussion
The dual passivation strategy developed in this work is based on combining the incorporation of PEACl:PbCl2 into the perovskite precursor solution and PEACl surface treatment (Fig. 1). Since using long chain alkylammonium salts as additive mainly passivates the grain boundaries, as will be shown later and has been proposed in previous works,26,98,101–103 it is for simplicity referred to as grain boundary passivation (GBP) in the following, while surface treatment is referred to as surface passivation (SP). For the reference perovskite films (referred to as Ref), we adapt an established fabrication route,112 yielding high-quality MA-free films with a composition of Cs0.18FA0.82PbI3 (10% excess PbI2) and a bandgap of ∼1.57 eV. In case of GBP, PEACl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PbCl2 (1:1 molar ratio) dissolved in dimethyl sulfoxide (DMSO) is added into the Ref precursor solution (optimized at a concentration of 2 mol%; see Fig. S3, ESI†). The Ref and GBP precursor solutions are spin-coated on top of ITO/2PACz and annealed at 150 °C for 30 min (Fig. 1a). In case of SP, PEACl dissolved in isopropanol (optimized at a concentration of 1.5 mg ml−1; see Fig. S4, ESI†), is dynamically spin-coated on the surface of Cs0.18FA0.82PbI3 films and subsequently annealed at 100 °C for 10 min (Fig. 1b). Finally, both individual passivation strategies are exploited together, referred to as GBP&SP (Fig. 1c). Further details are provided in the Experimental Section (ESI†).
:PbCl2 (1:1 molar ratio) dissolved in dimethyl sulfoxide (DMSO) is added into the Ref precursor solution (optimized at a concentration of 2 mol%; see Fig. S3, ESI†). The Ref and GBP precursor solutions are spin-coated on top of ITO/2PACz and annealed at 150 °C for 30 min (Fig. 1a). In case of SP, PEACl dissolved in isopropanol (optimized at a concentration of 1.5 mg ml−1; see Fig. S4, ESI†), is dynamically spin-coated on the surface of Cs0.18FA0.82PbI3 films and subsequently annealed at 100 °C for 10 min (Fig. 1b). Finally, both individual passivation strategies are exploited together, referred to as GBP&SP (Fig. 1c). Further details are provided in the Experimental Section (ESI†).
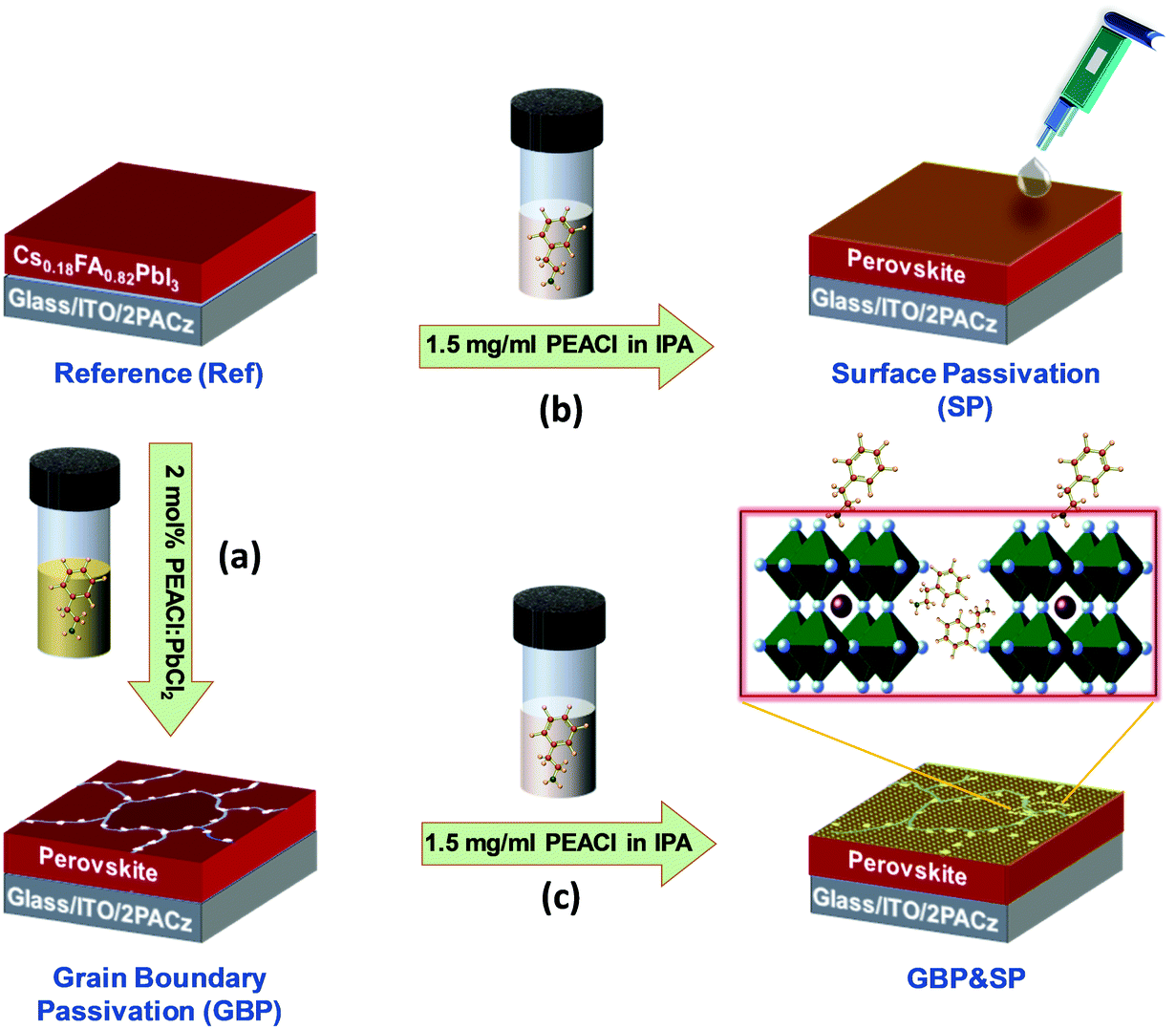 | ||
| Fig. 1 Schematic of the perovskite absorber deposition process employing the dual passivation strategy developed in this work: (a) grain boundary passivation (GBP) by incorporation of PEACl:PbCl2 into the perovskite precursor solution, (b) surface passivation (SP) by treatment of PEACl in IPA on top of the perovskite absorber layer and (c) combination of grain boundary and surface passivation (GBP&SP). | ||
Photovoltaic performance
In order to demonstrate the trend in performance of planar p–i–n PSCs upon employing either individual passivation (GBP or SP) as well as dual passivation (GBP&SP) compared to Ref PSCs, devices in the layer stack ITO/2PACz/perovskite/C60/BCP/Ag with an active area of 12.3 mm2 were prepared (Fig. 2a). Fig. 2b and Table S1 (ESI†) summarize the current density–voltage (J–V) characteristics and PV parameters of the best-performing p–i–n PSCs. The corresponding statistics (in total 147 devices) that emphasize the very high yield and good reproducibility of the key trends are shown in Fig. 2c. The best Ref PSC exhibits a PCE of 20.4% with a short-circuit current density (JSC) of 23.9 mA cm−2, a VOC of 1.086 V, and a FF of 78.6%. This denotes a very respectable starting point in performance for p–i–n PSCs compared to literature (see Fig. S1, ESI†). The PCE of the best GBP PSC is slightly improved to 20.7%, which is mainly associated with a 26 mV enhancement in VOC as well as a slightly improved FF. The small decrease in JSC to 23.6 mA cm−2 is attributed to the formation of a 2D RP perovskite (see discussion in the following) and, thus, a slight decrease in 3D perovskite absorber volume (see ultraviolet-visible (UV-vis) measurements in Fig. S5, ESI†).64,99 The best SP PSC already exhibits a very high PCE of 22.1% with a significant improvement in both the VOC (1.131 V) and FF (82.3%) as compared to the Ref PSC.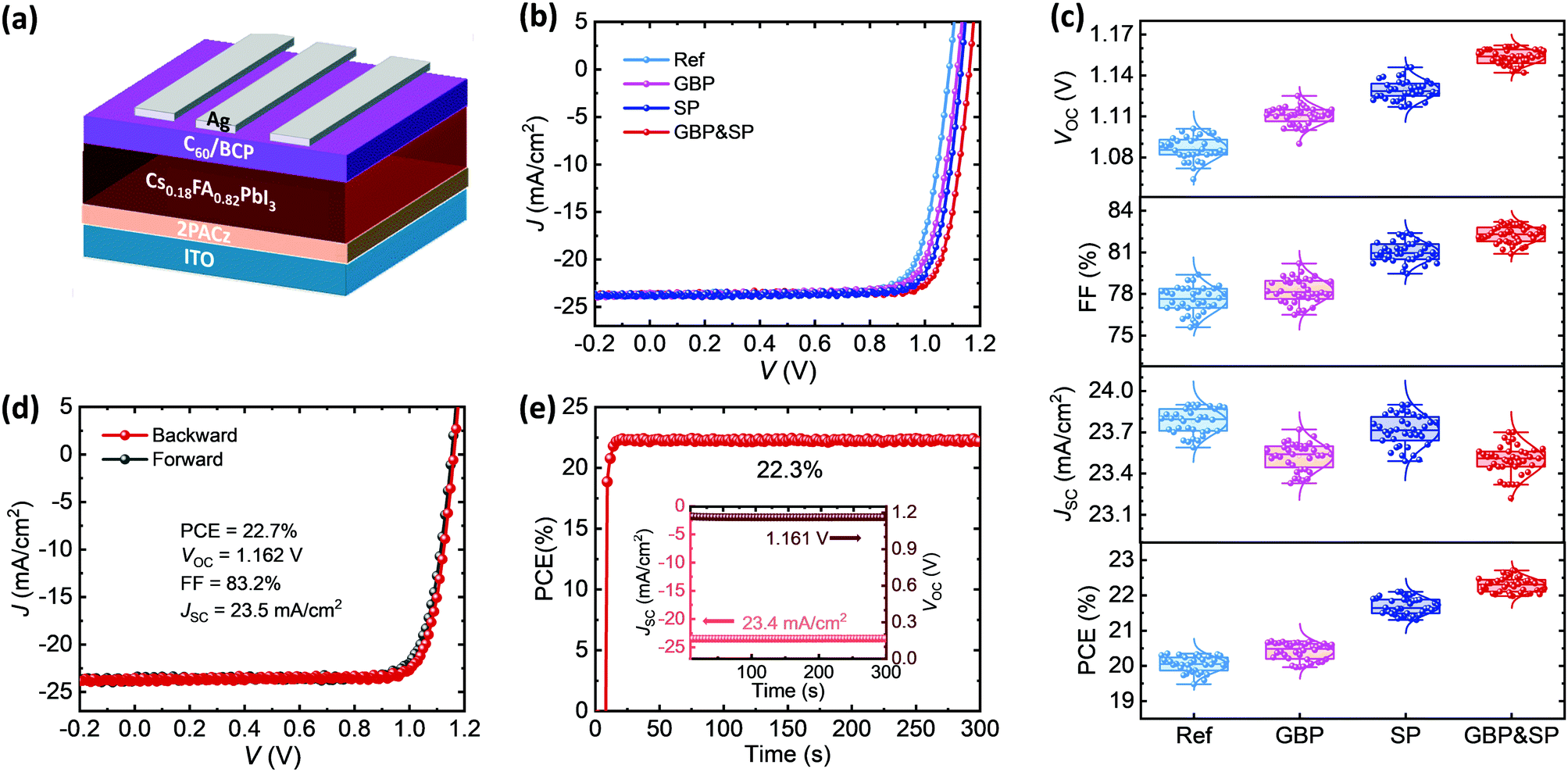 | ||
| Fig. 2 (a) Schematic of the employed perovskite solar cell configuration with a layer stack sequence of ITO/2PACz/perovskite/C60/BCP/Ag. (b) Current density versus voltage (J–V) characteristics and (c) statistical distribution of the open-circuit voltage (VOC), fill factor (FF), short-circuit current density (JSC), and power conversion efficiency (PCE) of perovskite solar cells without any modification (Ref), with surface passivation (SP), grain boundary passivation (GBP) and combined grain boundary and surface passivation (GBP&SP). (d) J–V characteristics and (e) maximum power point (MPP) tracking of the best-performing GBP&SP perovskite solar cell, demonstrating a stabilized PCE of 22.3%. The inset shows the stabilized JSC and VOC. | ||
Strikingly, upon dual passivation, the VOC and FF are further enhanced to 1.162 V and 83.2% respectively, which leads to a remarkable PCE of 22.7% for the best GBP&SP PSC (see Fig. 2d). This corresponds to a VOC × FF product of 0.891 with respect to the S–Q limit, the highest reported for p–i–n PSCs with a PCE above 21% (compare Fig. S1 and S2b, ESI†). Furthermore, the GBP&SP PSC also exhibits a remarkable stabilized PCE (under MPP tracking), VOC and JSC of 22.3%, 1.161 V and 23.4 mA cm−2 under continuous AM1.5G illumination for 5 min, respectively (Fig. 2e). It should be noted that the VOC enhancements are not governed by an increase in the bandgap, as shown by analysis via the Tauc plot method and the inflection point of the EQE spectra (Fig. S6a and b, ESI†),113 but relate to reduced non-radiative recombination, as we will later elaborate on in detail. We note that when increasing the concentration of PEACl to 3 mg ml−1 (beyond the optimum concentration), the VOC of GBP&SP PSCs increases further up to 1.184 V (see Fig. S4, ESI†), which represents a voltage deficit of only 393 mV and 104 mV with respect to the bandgap and radiative limit respectively (90.9% of the S–Q limit),113 that are among the lowest reported for p–i–n PSCs (Fig. S2a, ESI†). However, since the JSC and FF decline at the same time, possibly due to the insulating nature of a thicker 2D RP passivation layer at the surface,63,64,81,82,99 the PCE of the best GBP&SP PSC drops to 21.8%. It should be noted that the reported JSC for all PSCs is corrected using the ratio of JSC derived from the external quantum efficiency (EQE) and J–V measurements of the best PSCs (Fig. 2b and Fig. S7, ESI†). This results in a rather conservative determination of PCE in this work. To summarize, the combined enhancements in VOC and FF are highest for GBP&SP PSCs, which highlights the necessity of the simultaneous passivation of the perovskite/C60 interface, and – as we will show later – the grain boundaries of the perovskite thin film.
Next to dual passivation by PEACl, we first evaluated the effect on the VOC upon employing PEAI and PEABr, since they have been used in numerous previous reports for passivation of perovskite films.62,65,69,77,99,103 As shown in Fig. S8 (ESI†), PEACl-based GBP&SP PSCs show a much higher average of VOC of ∼1.15 V as compared to ∼1.12 V in case of PEAI and PEABr. Therefore, we focussed in more detail on alternative chloride-based long chain alkylammonium salts namely n-butylammonium chloride (BACl) and n-octylammonium chloride (OACl), since BAI,63,64,71,72,76,102 BABr,10,59,60,64 OAI,4,72,75,76,101 and OABr10,71 have previously been reported to serve as efficient passivation molecules as additive as well as for surface treatment.
While SP PSCs all exhibit an enhanced VOC and FF as compared to Ref PSCs, the enhancements are most pronounced in case of PEACl (Fig. S9, ESI†). Employing the dual passivation strategy leads to a∼30 mV VOC enhancement in case of OACl and BACl, which is much lower compared to ∼70 mV for PEACl-based GBP&SP PSCs (see Fig. S10, ESI†). Furthermore, while for BACl-based GBP&SP PSCs the FF remains similar and only a slight drop in JSC is observed compared to Ref PSCs, these parameters are even reduced in case of OACl, as expected based on previous reports employing too large amounts of alkylammonium salts as additive.25,64,99,102 Therefore, an overall lower average PCE of only 18.6% is obtained for OACl-based GBP&SP PSCs compared to 20.1% for Ref PSCs, while the average PCE is slightly higher at 20.9% in case of BACl. These results highlight that our dual GBP&SP passivation strategy in principle is compatible with other Cl-based long chain alkylammonium salts, but reduced charge carrier transport (i.e., lower JSC and/or FF) can easily impede any positive effects from reduced interfacial recombination (i.e., higher VOC). Hence, careful optimization of the fabrication parameters is required. Targeting high efficiency and reproducibility, we identified PEACl as the superior choice for the dual passivation strategy of p–i–n PSCs studied in this work.
Photophysical properties
To discriminate the effect of GBP, SP and GBP&SP on non-radiative recombination of the PSCs, we first show representative TRPL transients measured for ITO/2PACz/perovskite/C60 layer stacks in Fig. 3a. While the detailed interpretation of such transients can be challenging,114 a longer monomolecular lifetime at low-level injection can be attributed to reduced non-radiative recombination either within the bulk (including the grain boundaries) or the perovskite/CTL interfaces.114–117 The lifetime increases considerably by more than one order of magnitude in the order ref (19 ns) → GBP (48 ns) → SP (113 ns) → GBP&SP (256 ns) (dashed lines Fig. 3a). This indicates that non-radiative recombination is effectively suppressed in the same order as the observed VOC enhancement of the PSCs.114–117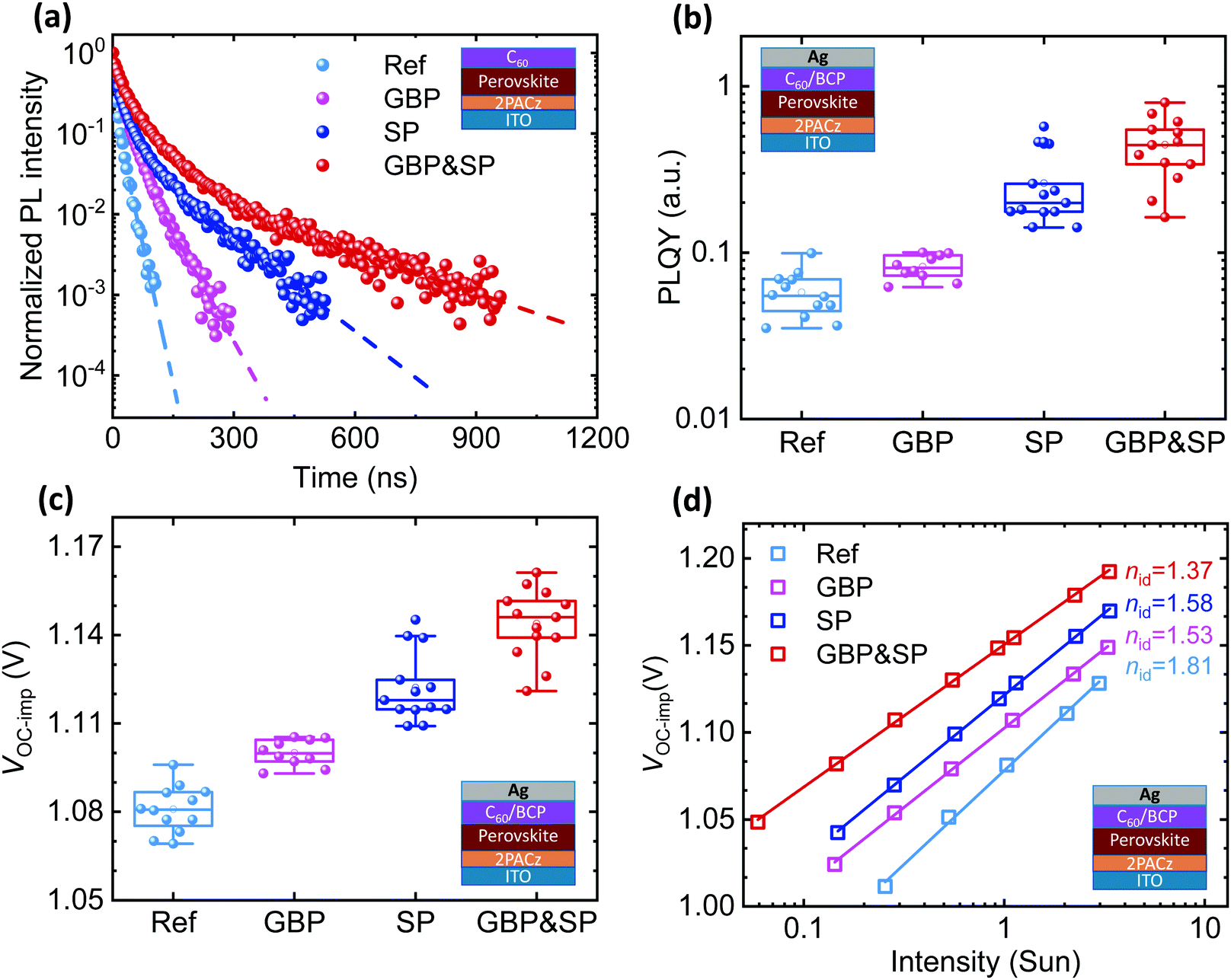 | ||
| Fig. 3 (a) Time-resolved photoluminescence (TRPL), (b) photoluminescence quantum yield (PLQY), (c) the obtained implied VOC (VOC-imp), and (d) ideality factor (nid) extracted from a fit to the intensity-dependent VOC-imp of the perovskite films prepared on ITO/2PACz substrates for the reference (Ref), surface passivation (SP), grain boundary passivation (GBP) and grain boundary & surface passivation (GBP&SP) films. TRPL in (a) is measured with a C60 layer on top, while (b–d) are measured for a full device stack. | ||
To quantify the reduction of non-radiative recombination, PLQY measurements along with the internal quasi-Fermi level splitting (EF), that are attributed to the ‘implied VOC’ via VOC_imp = ΔEF/q = VOC_rad + kBT/qln(PLQY), are discussed next.34,118 Analysing PLQY and VOC_imp for the stack ITO/2PACz/perovskite without C60 allows identification of whether non-radiative recombination at the HTL/perovskite limits the VOC of our PSCs.34,35,39 For the Ref films, we find an already very high average PLQY (VOC_imp) of 7.2% (1.206 V) which only slightly increases to 7.9% (1.218 V), 9.8% (1.218 V) and 9.7% (1.225 V) for GBP, SP and GBP&SP films, respectively (Fig. S11, ESI†), with VOC_imp being well above the obtained VOC of all PSCs presented in Fig. 2c. This shows that our perovskite films are of very high quality and that the nearly lossless 2PACz/perovskite interface does not limit the VOC, in line with previous reports.31,32,35 Upon addition of C60/BCP/Ag, the PLQY for Ref films severely drops to a low average value of 0.058% correlating to VOC_imp of 1.081 V, clearly showing that the perovskite/C60 interface limits the VOC (Fig. 2b and c). Impressively, the average PLQY increases by roughly one order of magnitude to 0.083%, 0.26% and 0.45% for GBP, SP, and GBP&SP films respectively (Fig. 3b), correlating to an enhanced VOC_imp of 1.100 V, 1.122 V and 1.144 V, respectively (Fig. 3c). Notably, the values of VOC_imp closely match with the average VOC of the respective PSCs (compare Fig. 2c), implying that all PSCs have a proper energetic alignment that does not result in an offset between VOC_imp and VOC.34,35,92,118,119 We note that increasing the PEACl concentration to 3 mg ml−1 for GBP&SP films further increases the PLQY and VOC_imp to remarkable values of up to 2.21% and 1.190 V, respectively, which is in line with the results discussed above for the respective PSCs (see Fig. S12, ESI†).
At first sight, the role of the PEACl:PbCl2 additive for the improved device performance remains unclear, since VOC_imp for the half layer stack without C60 only slightly increases for all passivation strategies as compared to Ref films. To shed more light on this aspect, we show representative TRPL transients for the stack ITO/2PACz/perovskite in Fig. S13 (ESI†). Here, we find a clear trend with the monomolecular lifetime for GBP films (∼1624 ns) and GBP&SP films (∼1497 ns) being considerably longer as compared to Ref films (∼335 ns). Interestingly, for perovskite films with surface passivation only, the lifetime is solely slightly increased to ∼464 ns. At this point, we hypothesize that the enhanced lifetimes in case of GBP relate to the passivation of shallow grain boundary traps via the self-assembly of PEA+ molecules and/or the formation of a PEACl-based 2D RP phase.25,26,36,41,46,102,104–106,120,121 We note that shallow traps are typically filled at high illumination intensities around 1 Sun, possibly explaining why the values of PLQY and VOC_imp for stacks without C60 are only slightly enhanced by all three passivation strategies. This explanation is in line with the common implication that grain boundaries are not necessarily detrimental to device performance at solar illumination intensities.40,41,120 To shed more light on this, we evaluate the trap-state density (nt) and charge carrier mobility (μ) of electrons and holes for the different passivation strategies via space charge limited current (SCLC) measurements. We fabricated both electron- and hole-only devices with the configuration of ITO/SnO2/perovskite/C60/BCP/Ag and ITO/2PACz/perovskite/Spiro-MeOTAD/Ag, respectively. The dark J–V characteristics of the devices are plotted in Fig. S14 and S15 (ESI†) and are analyzed according to the SCLC method (see further details in the ESI†). The electron mobility of the Ref device is 4.9 × 10−3 cm2 V−1 s−1, while both SP and GBP devices demonstrate a comparable increase in mobility to 6.3 × 10−3 and 7.2 × 10−3 cm2 V−1 s−1 (see Table S2, ESI†), respectively. Applying our dual passivation strategy, the electron mobility further increases to 10.0 × 10−3 cm2 V−1 s−1. Furthermore, the trap-filled limit voltage (VTFL), which is linearly proportional to the trap-state density, demonstrates a substantial decrease in the order Ref → GBP → SP → GBP&SP, correlating to a reduction in electron trap density from 9.2 × 1015 cm−3 to 6.5 × 1015, 5.4 × 1015 and 3.7 × 1015 cm−3, respectively (see Fig. S14 and Table S2, ESI†). A very similar trend is also observed for the calculated hole mobilities, while the reduction in the hole trap density is apparently slightly less pronounced (see Fig. S15 and Table S3, ESI†). Therefore, the TRPL and SCLC results indicate that both GBP and SP independently contribute to enhancing both the electron- and hole mobilities by roughly a factor of 2, while at the same time specifically reducing the electron trap density at the grain boundaries and surface of the perovskite film.
To assess the impact of the reduced trap-state density on device performance, we perform intensity-dependent PLQY measurements to obtain the internal ideality factor (nid) from a fit to the calculated VOC_imp.34 The ideality factor has been proven to be governed by bulk as well as interfacial recombination properties.34,122,123 For high-performing PSCs that are not limited by failures at either of the perovskite/CTL interfaces, a reduction of nid towards a value of 1 is typically associated with a predominant bimolecular radiative and reduced trap-assisted Shockley–Read Hall recombination and there is a direct correlation between nid and FF.31,34,122,124 Again, we first analyse half-layer stacks without C60 and find a considerable reduction of nid from 1.71 for Ref films to 1.48 and 1.60 for GBP and SP films, respectively, while GBP&SP films show by far the lowest nid of 1.30 (Fig. S16, ESI†). The observation that nid in case of GBP is slightly lower as compared to SP is in line with the enhanced monomolecular lifetime for GBP films without a C60 layer. Yet, despite the apparently improved bulk recombination properties, GBP PSCs remain severely limited by interfacial recombination at the perovskite/C60 interface, as exemplified by the lower device VOC and FF. Interestingly, nid of complete PSC layer stacks shows a very similar trend, with Ref PSCs exhibiting nid of 1.81, which considerably reduces to 1.53 and 1.58 for GBP and SP PSCs, respectively (Fig. 3d). Critically, GBP&SP PSCs again show by far the lowest nid of 1.37, only slightly higher as for the half-layer stack without C60. This is among the lowest nid values reported in the literature for p–i–n PSCs with a PCE > 20% and slightly below the value reported by Al Ashouri et al. for 2PACz-based wide-bandgap PSCs.26,31,34,38 We note that when increasing the PEACl concentration to 3 mg ml−1, nid of SP and GBP&SP PSCs increases again to 1.49 and 1.42 (Fig. S17, ESI†), respectively, showing that while the VOC of the respective PSCs further increases (Fig. S4, ESI†), the recombination behaviour does not further benefit from too thick passivation layers, to some extent contributing to the reduced FF.
Finally, using the ideality factor and PLQY values we can determine the implied PCE and FF without and with C60 and compare it with an ideal device (i.e., nid = 1) that has the same bandgap and JSC.34 This allows us to estimate the remaining losses in our devices, i.e. the FF losses due to series/shunt resistance, non-ideal nid and non-radiative recombination.34,125 As shown in Fig. S18 (ESI†), the ideal device exhibits a PCE of 27.8% with a FF of 90.3% for both Ref and GBP&SP. Without and with C60, Ref (GBP&SP) exhibits an implied FF of 84.8% (87.7%) and 82.8% (86.6%), respectively, while the respective best PSCs show a FF of 78.6 (83.2%). This relates to a FF loss due to series/shunt resistance of roughly 4.2% (3.4%), while non-ideal nid and non-radiative recombination account for another 2% (1.1%) from C60 and 5.5% (2.6%) from the bulk. This analysis reveals that, in addition to strongly enhancing the VOC, our dual passivation approach (GBP&SP) reduces the FF losses by 4.6% absolute as compared to Ref PSCs due to a reduced series/shunt resistance (0.8%) as well as simultaneous passivation of the perovskite/C60 interface (0.9%) and grain boundaries (2.9%).
In summary, the TRPL and PLQY results show that (i) the perovskite/C60 interface limits the VOC of our PSCs and (ii) that grain boundary passivation becomes specifically crucial in the case where the perovskite/CTL interfaces are already well passivated. Therefore, in line with the device data presented, we find that dual passivation is required to reach both the lowest non-radiative recombination losses and the lowest nid that only together result in the highest VOC and FF.
Material characterization
Having demonstrated that the superior performance of our dual passivation strategy stems from reduced non-radiative recombination at the grain boundaries as well as at the perovskite/C60 interface, the question arises: How do SP and GBP modify the perovskite film morphology, structure or composition? To start with, we examine the films by SEM and identify a similar surface morphology and grain size distribution for the Ref and SP perovskite films (see Fig. S19a and b, ESI†). This observation is in line with the literature as surface treatment with low concentration long chain alkylammonium salts commonly does not alter the perovskite film morphology; more distinct changes to the morphology are only observed for higher concentrations (see Fig. S20, ESI†).59,60,62,64,65,90 Further analysis of atomic force microscopy (AFM) images reveals a slight reduction in the root-mean-square surface roughness, which we attribute to the fact that PEACl preferentially fills regions close to the grain boundaries (Fig. S21, ESI†).10,14 For all perovskite films, the grains that appear brighter in SEM are attributed to PbI2-rich crystallites as will be discussed later in more detail.17,95By incorporating PEACl:PbCl2 in the film for GBP films, the size of the perovskite grains remains largely unchanged, however, the number and size of the PbI2-rich grains slightly increase (Fig. S19c, ESI†). Interestingly, notable small bright crystallites appear on the surface of the perovskite film which are specifically embedded close to the grain boundaries. This indicates that PEACl:PbCl2 leads to passivation mainly near the grain boundaries. Upon additional surface treatment with PEACl for GBP&SP films, the size of these small bright crystallites is reduced and they appear more dispersed all over the surface, growing with a plate-like appearance perpendicular to the perovskite grains (Fig. S19d, ESI†). This implies that some reaction with these crystallites occurs when PEACl is deposited on top of GBP films.
To gain a better understanding about the phase or composition of the small and large bright grains observed in SEM at the surface of the perovskite films as well as their potential relevance in the context of this work, we carry out CL measurement.126 For the Ref film, the grains which appear darker in SEM (highlighted by a green circle in Fig. 4) exhibit higher CL intensities (Fig. 4b) with the emission peak located at ∼774 nm (Fig. S22, ESI†) that correlates with the 3D perovskite phase with a bandgap of ∼1.57 eV (note that the CL setup is not spectrally calibrated). The grains which appear brighter and exhibit a different texture in the SEM images (highlighted by a yellow circle in Fig. 4; see further top-view SEM images in Fig. S23, ESI†) demonstrate lower CL intensities (dark spots in Fig. 4b) with a CL emission peak around 500 nm (Fig. S22, ESI†). By applying a 500 nm ± 40 nm bandpass filter to record the CL image, these grains can be clearly distinguished from the 3D perovskite grains (Fig. 4c). These regions are therefore attributed to PbI2-rich crystallites, in line with previous reports,17,64 and as seen from cross-sectional SEM images, they appear to be located on top of 3D perovskite grains (Fig. S24, ESI†). Looking specifically at the CL signal from individual large PbI2-rich grains, there is indeed still a signal from the (underlying) 3D perovskite phase (Fig. S22, ESI†). For the SP film, we observe a slight charging of the SEM images (Fig. 4d), which we attribute to the insulating nature of a 2D RP phase forming at the surface. No noticeable change in the CL images of the SP film without and with 500 nm ± 40 nm bandpass filter is observed compared to the Ref film (Fig. 4e and f). This indicates that the large PbI2-rich grains are not completely chemically reacting upon PEACl surface treatment, which is in contrast to previous observations that bright PbI2 related grains vanish upon treatment with various organic halides.11,20,66,77,78,84 Nevertheless, due to the passivation effect, the CL signal of the 3D perovskite phase exhibits a much higher intensity compared to the Ref film (Fig. S25, ESI†), in line with the PLQY results. For the GBP film, the grains which appear as small bright grains close to the grain boundaries in SEM (highlighted by a red circle in Fig. 4g) are detected as dark small spots around the perovskite grains in the CL image (Fig. 4h). This stresses that these small grains exhibit lower CL intensities similar to the large PbI2-rich grains. Interestingly, when we apply the 500 nm ± 40 nm bandpass filter, only the features related to PbI2-rich grains can be observed, whereas no signal or feature correlated with the small bright grains is traceable (Fig. 4i). This indicates that these are not related to PbI2 or PbCl2. Finally, for GBP&SP film no small grains are visible in SEM anymore due to their dispersion after surface treatment (Fig. 4j) and thus can no longer be identified in the CL image either without or with the bandpass filter (Fig. 4k and l). Consistent with the PLQY and TRPL results, the GBP&SP film (Fig. 4k) exhibits the highest CL intensity at a wavelength of ∼774 nm (Fig. S25, ESI†). This enhancement is attributed to the passivation of various recombination centres in the grain boundaries and/or at the surface of the perovskite layer.126 Importantly, an additional CL peak at ∼620 nm appears, which cannot be related to either the 3D perovskite phase or the PbI2-rich phase. We find similar peaks in PL measurements of GBP&SP films as well as for SP films when using a higher PEACl concentration of 3 mg ml−1 (see Fig. S26, ESI†). We correlate this observation to the formation of a thin emissive 2D (PEA)2(CsyFA1−y)n−1Pbn(I1−xClx)3n+1 RP phase with n = 2 at the surface of the films, as will be discussed in the following.75,127
 | ||
| Fig. 4 Scanning electron microscopy (SEM) images, cathodoluminescence (CL) images recorded without any filter and CL images with bandpass (500 ± 40 nm) filter for perovskite absorbers prepared (a–c) without any modification (Ref), (d–f) with surface passivation (SP), (g–i) grain boundary passivation (GBP) and (j–l) grain boundary & surface passivation (GBP&SP), respectively. The green encircled grains represent the expected 3D perovskite phase with a bandgap of ∼1.57 eV. The yellow encircled grains are attributed to PbI2-rich crystallites. The small red encircled grains appear close to the grain boundaries for GBP films. | ||
To analyse the crystal structure of our films, we perform X-ray diffraction (XRD) measurements (Fig. S27a, ESI†). All perovskite films exhibit the expected peaks at 14.2°, 20.1°, 24.6° and 28.4° from the (100), (110), (111) and (200) crystal planes of the 3D cubic α-Cs0.18FA0.82PbI3 phase,25,112,128 as well as a peak at ∼12.9° related to PbI2. The peak positions, intensities and FWHM of the XRD peaks are largely unchanged for SP films as observed in our previous work when using BABr for surface treatment.59 For GBP films the intensity of the PbI2 peak slightly increases (Fig. S27a, ESI†), in agreement with the larger number of PbI2-rich crystallites observed in SEM and CL. Furthermore, the ratio of the (100) to (111) peak slightly decreases for GBP films as compared to Ref and SP films, implying a slightly less preferred (100) orientation of the perovskite grains (Fig. S27b, ESI†).25,99 Similar to SP films, GBP&SP films exhibit a largely unchanged XRD spectrum as compared to GBP films. We do not observe a signal related to a 2D RP phase for the SP and GBP&SP films, which could be related to either the passivation layer being too thin to be detected by XRD, the presence of a heterogeneous distribution of (PEA)2(CsyFA1−y)n−1Pbn(I1−xClx)3n+1 phases with various n, or the presence of a non-crystalline PEA-based passivation layer.14,59,68,69 Yet, upon further increasing the PEACl concentration to 4.5 mg ml−1 or 10 mg ml−1, peaks at ∼5.3°, 10.5°, 15.7°, 20.9°, 26.1° and 31.8° start to appear. These can be attributed to a pure 2D (n = 1) (PEA)2Pb(I1−xClx)4 RP phase with a superlattice spacing of ∼1.7 nm that forms at the surface of the perovskite films (Fig. S28, ESI†).77,129–132
We perform XPS measurements of ITO/2PACz/perovskite stacks to get a better understanding of the elemental composition at the surface of our perovskite films and prove the presence of a PEACl-based passivation layer. For the Ref film, the XPS core-level spectra in Fig. 5a–c and Fig. S29a–c (ESI†) for the different elements in the 3D Cs0.18FA0.82PbI3 absorber show the expected peaks with binding energies of ∼138.7 eV (Pb 4f7/2), ∼400.8 eV (N 1s), ∼288.6 eV (C 1s from FA's N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonding), ∼619.6 eV (I 3d5/2) and ∼725.3 eV (Cs 3d5/2).133 The additional C 1s peak at ∼284.8 eV is attributed to adventitious carbon (sp3 C–C bonding) at the film surface.17,95 There is no signal related to a Cl 2p doublet at a binding energy of ∼198.6 eV and ∼200.2 eV despite using CsCl in the precursor solution which can be explained by the sublimation of FACl during the annealing process.16,112,134 We note that we do not observe a peak related to metallic lead (Pb0) at ∼137 eV which emphasizes the high quality of our reference perovskite films.19,20,74,87
N bonding), ∼619.6 eV (I 3d5/2) and ∼725.3 eV (Cs 3d5/2).133 The additional C 1s peak at ∼284.8 eV is attributed to adventitious carbon (sp3 C–C bonding) at the film surface.17,95 There is no signal related to a Cl 2p doublet at a binding energy of ∼198.6 eV and ∼200.2 eV despite using CsCl in the precursor solution which can be explained by the sublimation of FACl during the annealing process.16,112,134 We note that we do not observe a peak related to metallic lead (Pb0) at ∼137 eV which emphasizes the high quality of our reference perovskite films.19,20,74,87
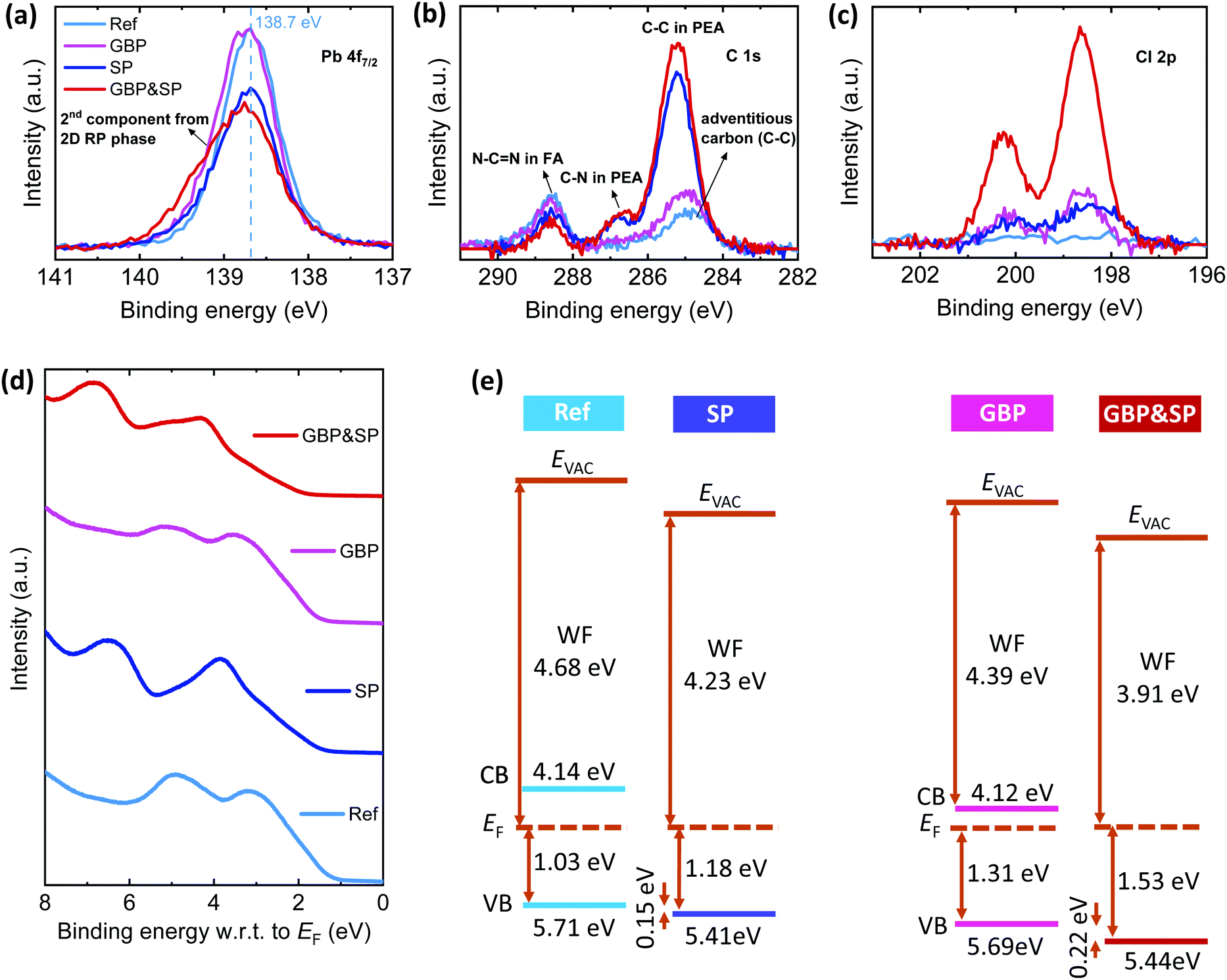 | ||
| Fig. 5 X-ray photoelectron spectroscopy (XPS) spectra of (a) Pb 4f7/2, (b) C 1s and (c) Cl 2p core levels for perovskite films prepared on ITO/2PACz substrates without (Ref), with surface passivation (SP), grain boundary passivation (GBP), and grain boundary & surface passivation (GBP&SP) processes. (d) Proposed energy-level scheme based on ultraviolet photoelectron spectroscopy (UPS) measurements and (e) the respective spectra of the region close to the valence band onset. EF is the Fermi level, Evac is the vacuum level, and CB and VB show the conduction and valence band, respectively. The CB position of the 3D perovskite was calculated from the corresponding value of the optical gap. | ||
Upon PEACl:PbCl2 incorporation and/or PEACl surface treatment (GBP, SP and GBP&SP films) there are three key observations as compared to the Ref film. Firstly, for BP films the Pb 4f7/2 core level slightly shifts toward higher binding energies and exhibits an increased FWHM, while for SP and GBP&SP films the signal intensity at ∼138.7 eV decreases and the peak becomes broader and asymmetric toward the high-energy side. This asymmetry is stronger for GBP&SP films. Secondly, a Cl signal at ∼198.6 eV (Cl 2p3/2) and ∼200.2 eV (Cl 2p1/2) appears for all films, which is by far strongest for GBP&SP films. Thirdly, two new peaks at ∼285.2 eV and 286.7 eV appear which are similar in intensity for SP and GBP&SP films and only very weak for GBP films. These peaks can be related to the C 1s emission from PEA (C–C and C–N bonds, respectively) with the expected stoichiometric ratio of 7:1.
Comparing SP and GBP&SP films using a higher PEACl concentration of 3 mg ml−1, we find even more pronounced changes in the Pb 4f core levels (Fig. S30, ESI†). We stress that these are not accompanied by changes in the peak position or shape of the I 3d5/2, Cs 3d5/2 and C 1s core levels, which excludes the possibility of a shift due to electronic doping of the perovskite bulk.87,95,135,136 We therefore relate the damping of the signal at ∼138.7 eV for SP and GBP&SP, together with the appearance of a second Pb component at ∼139.1 eV as well as a Cl 2p and PEA signal, to the formation of a thin PEACl-based passivation layer on the surface of the films that has a different chemical environment. The fact that for GBP the damping of the signal at ∼138.7 eV is less pronounced and the peak becomes broader fits with our observation from SEM and CL that passivation happens mainly close to the grain boundary regions. To further test this interpretation, we show XPS measurements of SP and GBP&SP films with a much thicker passivation layer (10 mg ml−1) in Fig. S31 (ESI†). A single Pb 4f7/2 peak at ∼139.4 eV can be observed with no remaining signal at ∼138.7 eV. In addition, no signals related to FA and Cs are observed anymore, which is an indication for the formation of a 2D RP phase with n = 1 and the composition (PEA)2Pb(I1−xClx)4, at the film surface that completely damps the Pb signal from the underlying 3D perovskite phase. This is in line with the appearance of the related XRD peaks discussed above. We note that the calculated atomic ratio of (I + Cl)/Pb is 4.71 (SP) and 5.14 (GBP&SP) and thus even larger than the expected 4, which could be related to excess PEAI or PEACl at the film surface possibly forming an amorphous phase as reported recently (see Table S4, ESI†).14,68 Similar shifts of the Pb 4f binding energies have previously been observed when changing the halide, i.e. for MAPbCl3 and PbCl2 as compared to MAPbI3 and PbI2,137,138 possibly due to the higher electronegativity of Cl as compared to I.
The formation of an n = 1 2D RP phase has been proposed in several studies that employed PEAI or OAI for surface treatment.14,61,65,67,70,77,79,80 However, for the thin passivation layer thicknesses studied in this work, the atomic ratio of Cs/Pb for SP and GBP&SP films first increases above the respective values for the Ref and GBP films (1.5 mg ml−1), and only starts to slightly decrease for a higher PEACl concentration of 3 mg ml−1 (Table S4 and Fig. S32, ESI†). This indicates that Cs is taking part in the formation of the thin 2D RP passivation layer as otherwise the signal should be strictly decreasing because of the damping overlayer. This strict decrease in intensity with the formation of a 2D passivation layer can be seen for the atomic ratio of FA/Pb. This points towards a 2D RP (PEA)2(CsyFA1−y)n−1Pbn(I1−xClx)3n+1 phase with y → 1 at the surface of our GBP, SP and GBP&SP films.10,64,78,80,119,127,132 Another clear observation is that the intensity of the Cl 2p doublet is considerably higher for GBP&SP as compared to SP films (Fig. 5b), while the I 3d5/2 peak shows the reversed trend (Fig. S29c, ESI†). Accordingly, we note larger atomic ratios of Cl/Pb for GBP&SP vs. SP while the atomic ratio of I/Pb is smaller (Table S4 and Fig. S32, ESI†). This suggests that the 2D RP (PEA)2(CsyFA1−y)n−1Pbn(I1−xClx)3n+1 phase in case of GBP&SP films is more chloride-rich as compared to SP films (x → 1). The hypothesis that an n > 1 2D RP (PEA)2(CsyFA1−y)n−1Pbn(I1−xClx)3n+1 phase exist for all thin passivation layers is further corroborated by our observation of a CL and PL peak at ∼620 nm for the SP (3 mg ml−1) and GBP&SP films (see Fig. S25 and S26, ESI†) which points toward the existence of n of presumably 2.75,127 To proof the existence of n > 1, we performed additional XRD measurements using a more sensitive setup. We specifically analyzed the low-angle region of the XRD spectrum (2θ < 12°) of GBP&SP films employing various concentrations of PEACL for surface treatment (3, 3.5, 4 and 4.5 mg ml−1) and used the (100) peak of a GBP film as reference point. As shown in Fig. S33 (ESI†), for all GBP&SP films we observe a clear peak at ∼5.1° and a corresponding one at ∼10.2° that correlate to an n = 1 2D RP phase, as well a small peak at ∼3.8° that correlates to n = 2. Upon increasing the PEACL concentration, the intensity of the n = 1 peak monotonically increases, while that for n = 2 is comparable in intensity for the lower concentrations and only slightly in-creases for 4.5 mg ml−1. Based on the XPS and XRD results together with the existence of a PL signal at ∼620 nm as discussed above, we conclude that for thin passivation layers the surface consists of a mixture of n = 1 and n = 2 2D RP phases, while for thicker passivation layers n = 1 becomes dominant toward the film surface. In order to give a rough estimate for the film thickness and n of the passivation layers, we take the Pb 4f7/2 peak which shows a clear indication for two phases and fit it with two components (see Fig. S34, ESI†): one for the 3D bulk phase (I) and one for the 2D RP surface phase (II). Here, for simplicity, we assume that the Pb 4f7/2 binding energy related to the 2D RP phase is situated at 139.1 eV. This allows to separate the relative contribution of the 2D (II) and 3D (I) material to the XPS signal and to evaluate the signal damping of the 3D bulk phase caused by the 2D RP overlayer. From here, we estimate a thickness of the 2D RP phase for a PEACl concentration of 1.5/3 mg ml−1 of ∼0.7/2.4 nm for SP films and ∼1.5/4.3 nm for GBP&SP films (see Table S5 and further information in the XPS/UPS section in the ESI†). Finally, for simplicity assuming that the complete PEA signal measured in XPS is bound in a 2D RP phase, we can make a rough estimation of the respective n (averaged over the measured XPS spot) by analysing the atomic ratio of PEA to the 2D Pb (II) signal for 1.5/3 mg ml−1 PEACl (see Table S5, ESI†). We find that n for GBP&SP films (∼2.6/2.3) is larger than for SP films (∼1.2/1.7).
To assess the effect of the thin (PEA)2(CsyFA1−y)n−1Pbn(I1−xClx)3n+1 surface layer on the energetics of our films we perform UPS measurements. From the onsets of the secondary electron cut-off and valence band spectra (Fig. S35, ESI† and Fig. 5d) we derive the energy band diagrams shown in Fig. 5e. The Ref films exhibit a WF of 4.68 eV and an ionization potential (IP) of 5.71 eV which represents a slightly n-type perovskite film, in line with previous observations.24,25,66,95,139 For GBP films, the WF considerably decreases to 4.39 eV while the IP stays roughly constant at 5.69 eV, implying that the perovskite becomes more n-type, which could be attributed to a reduced electron trap density, in line with our SCLC results, or a different surface termination.24,25,66,106,140 The SP and GBP&SP films exhibit a further reduction in WF to 4.23 eV and 3.91 eV, respectively, together with a similar IP of 5.41 eV and 5.44 eV. These changes we attribute to the formation of the thin 2D RP phase with a larger bandgap and different chemical environments at the film surface as observed by CL and XPS. This is further supported by measurements of SP and GBP&SP films with a PEACl concentration of 3 mg ml−1, for which the WF further decreases to 3.89 eV and 3.82 eV, while the IP exhibits similar values of 5.45 eV and 5.47 eV, which implies a valence band onset of 1.57 eV and 1.66 eV, respectively, the latter being larger than the 3D perovskite bandgap (Fig. S36, ESI†). We also note that the shape of the VB density of states clearly is affected by surface treatment (Fig. 5e), showing that the electronic properties of the 2D RP surface layer are different compared to the 3D bulk perovskite.141 Therefore, in addition to the expected chemical passivation,53 we speculate that the 2D RP phase at the film surface with an increased distance of the VB to the Fermi level results in hole blocking and thus a reduced probability for holes in the 3D perovskite absorber to recombine with electrons in the C60 layer.25,53,72,86,142 In our case, electrons can effectively tunnel through the very thin (∼0.7–1.5 nm) surface layer into C60,25,38,142 resulting in still efficient charge extraction that allows very high FF for low PEACl concentrations of 1.5 mg ml−1. For higher PEACl concentration of 3 mg ml−1 the passivation layers become too thick (∼2.4–4.3 nm), making tunneling less likely which results in a decreased device performance due to a lower FF (Fig. S4, ESI†). Finally, we note that our results indicate band bending at the narrow 2D/3D interface as has recently been shown experimentally for the n–i–p architecture,139,143 which also could contribute to the enhanced device performance.10
So far, our analyses clearly show that our dual passivation strategy effectively reduces non-radiative recombination mainly at the perovskite/C60 interface, but also the grain boundaries. Moreover, it is evident that a very thin 2D RP interlayer with a lower WF forms on the surface. The remaining questions are whether the PEACl:PbCl2 additive passivates mainly defects in the grain interior and/or at the grain boundaries and how heterogeneous the surface passivation is for the different strategies. To shed light on this, we did frequency modulated Kelvin probe force microscopy (FM-KPFM) in the heterodyne KPFM implementation. KPFM measures the local contact potential difference (CPD) between a metallic tip and the sample surface and thus is directly related to the WF.144,145 We used KPFM to map the effects of passivation on the CPD distribution of ITO/2PACz/perovskite/C60 layer stacks, especially looking at grain boundaries, PbI2-rich grains and extent of heterogeneity (see Fig. 6). We want to stress that KPFM is prone to crosstalk from topography, often leading to KPFM contrast in strongly curved surface regions, such as grain boundaries. To minimize crosstalk artefacts, we use FM-KPFM146 and carefully analysed the images, comparing the KPFM signal with the topography at the grain boundaries.
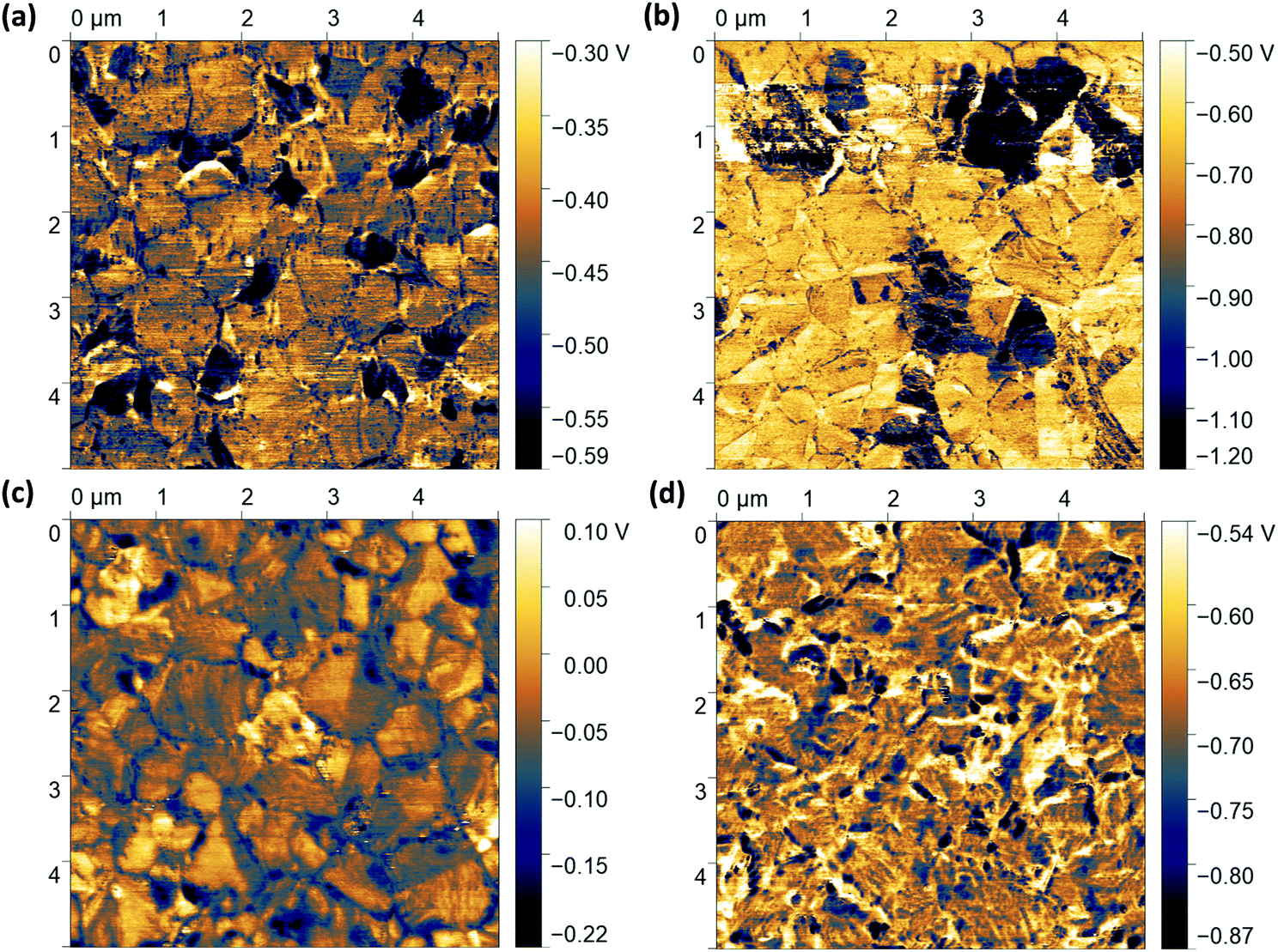 | ||
| Fig. 6 Kelvin pobe force microscopy (KPFM) images for perovskite films with the structure ITO/2PACz/perovskite/C60 prepared (a) without any modification (Ref), (b) with grain boundary passivation (GBP), (c) surface passivation (SP), and (d) grain boundary & surface passivation (GBP&SP) processes. | ||
The map for the Ref sample (Fig. 6a) shows perovskite grains with a rather uniform CPD of (−430 ± 40) mV interrupted by grains with a less uniform and ∼110–230 mV lower CPD. The size and the surface distribution of the darker regions correspond to the PbI2-rich grains as observed in SEM and CL (Fig. 4 and Fig. S19, ESI†). Here, the negative CPD contrast could be explained by a higher WF in PbI2 as compared to the 3D perovskite due to its larger bandgap and p-type characteristics,137,147–149 in line with previous observations.150,151 The map of the GBP sample (Fig. 6b) shows a similar trend in CPD contrast between more homogeneous perovskite grains with a CPD of (−700 ± 120) mV and less uniform spots with a CPD of (−1300 ± 280) mV. We note that the absolute value of the CPD depends on the tip's WF, which is sensitive to tip wear and contamination during the scanning. This could explain the overall lower absolute CPD in the GBP films. Assuming that the PbI2 covered regions for the Ref and GBP sample have a comparable WF, we can use these “dark” regions as internal reference surface. Therefore, we first compare the relative change of the CPD value at the 3D perovskite grains with that at the dark PbI2-rich grains for the Ref as compared to the GBP samples, i.e. ΔCPD3D-PbI2 (CPD3D-grains–CPDPbI2-grains). For the Ref sample ΔCPD3D-PbI2 is ∼170 mV, while it is ∼600 mV for the GBP sample (note the different scales in Fig. 6a and b). This relative difference of ΔCPD3D-PbI2 of ∼430 mV is comparable with the reduction in WF of ∼290 mV for GBP films as determined by UPS. Therefore, we attribute this observation to the fact that the PEACl:PbCl2 additive mainly lowers the WF of the 3D perovskite grains, while the PbI2-rich grains are not modified by this strategy.
To analyse if the CPD at the grain boundaries with respect to the grain interior is modified for the GBP sample as compared to the Ref sample, i.e., ΔCPDGB (CPDgrain boundary–CPDgrain), we show line profiles across representative grain boundaries in Fig. S37a and b (ESI†). To allow for a direct comparison, we shifted the CPD values of the grain interior at different positions to zero. For the Ref sample, many of the grain boundaries show a ∼50–100 mV lower CPD compared to the grains, similarly as previously reported.152–155 Such observations have been generally attributed to an enhanced ion and/or defect density, possibly due to a lower energy for defect formation at grain boundary regions.42,46,154 In contrast, for the GBP sample we observed on average less contrast between grain boundaries and grain interior such that some of them cannot be clearly distinguished in the CPD map and line profiles anymore. We analysed in total a larger number of grain boundaries (∼60) (Fig. S38a and b, ESI†) for better statistics and found an average reduction from ∼93 mV to ∼74 mV for the GBP as compared to the Ref sample together with a large number of grain boundaries not showing any CPD contrast (see histogram in Fig. S38c, ESI†). A reduction in ΔCPDGB for the GBP as compared to the Ref sample indicates that the PEACl:PbCl2 additive could specifically passivate the grain boundaries, resulting in a slightly more pronounced reduction of the WF with respect to the PbI2-rich grains as compared to the grain interior. We hypothesize that this is due to the formation of a PEACl-based 2D RP (PEA)2(CsyFA1−y)n−1Pbn(I1−xClx)3n+1 phase specifically close to the grain boundaries, in line with the observations from CL and XPS.
We further analyse the effects of PEACl surface treatment on the CPD (Fig. 6c and d). The SP Sample shows a heterogeneous CPD of (−52 ± 50) mV with no clear distinction between high- and low-CPD grains anymore; however, we still observe a slight grain boundary contrast. We attribute this to the very thin 2D RP phase on top of the perovskite film – as identified by XPS – that changes the electrical properties of the film surface, including the regions of the PbI2-rich grains. We speculate that PEACl cannot completely penetrate and thus passivate the grain boundary regions, explaining why we still observe a CPD contrast. The enhanced heterogeneity of the CPD over different grains as compared to the Ref sample could be explained by the fact that the 2D RP (PEA)2(CsyFA1−y)n−1Pbn(I1−xClx)3n+1 phase exhibits various n, i.e. a mixture of n = 1 and n = 2 phases with slightly different WF at different regions of the films. For the GBP&SP samples, we observe an even more heterogeneous CPD of (−670 ± 70) mV compared to all other samples (Fig. 6d and Fig. S39, ESI†). Critically, no grain boundaries and PbI2-rich grains can be identified in the CPD map anymore. Taking the CL, XPS and KPFM results together, this indicates that employing both PEACl:PbCl2 as additive and PEACl for surface treatment leads to a thin heterogeneous 2D RP (PEA)2(CsyFA1−y)n−1Pbn(I1−xClx)3n+1 phase at the grain boundaries and surface of the perovskite film with n of ∼1–2. Finally, we note that the CPD value can be affected both by the facet orientation as well as the existence of RP phases with various n, which makes it difficult to disentangle these effects in detail.146,153,156–158
Stability of passivated PSCs
Defect sites at grain boundaries and surfaces accelerate the degradation of perovskite thin films, since defects facilitate the migration of charged defects and mobile ions.25,73,159,160 Having demonstrated that our dual passivation strategy reduces defects at the grain boundaries and the surface of the perovskite film, the question arises whether the concept also serves to increase the stability, i.e. slows down the degradation of the perovskite films. For this purpose, we compare the activation energy for ion migration for Ref, SP, GBP and GBP&SP PSCs by thermal admittance spectroscopy (TAS). Fig. S40a–d (ESI†) depicts the TAS spectra of the lateral devices measured from 278 to 318 K in the dark. The activation energy (Ea) is obtained from the Arrhenius plot using the equation ωpeak = βT2exp(−Ea/kBT), where β is temperature independent prefactor, T is the absolute temperature, kB is the Boltzmann constant, and ωpeak is the angular frequency obtained by the maxima of the capacitance logarithmic derivative (Fig. S40e–h, ESI†). In line with the previously discussed trends, the GBP&SP PSCs exhibit by far the highest Ea of 696 meV, surpassing the activation energy for either single passivation strategy (GBP: 580 meV, SP: 551 meV) as well as the Ref PSC (502 meV) (Fig. 7a and Fig. S41, ESI†). The trend in Ea indicates that the simultaneous passivation of grain boundaries and the surface of the perovskite film yields by far the highest energy barrier for ion migration. As a consequence, the accumulation of ionic defects is most effectively suppressed for GBP&SP PSCs. To verify that the reduced ion migration also implies enhanced device stability under illumination,90,100 the operational stability of GBP&SP and Ref PSCs is examined under constant illumination (100 mW cm−2, AM1.5G, 14 h, room temperature) and MPP tracking conditions. The PCE of the Ref PSC decreases to around 80% of the initial value after only 8 h, whereas the GBP&SP PSC retained almost 98% of the initial PCE after 14 h (Fig. 7b). Furthermore, we investigate the operational stability of PSCs for which the perovskite/ETL interface is passivated using a thin evaporated LiF layer, which is often employed in high-efficiency p–i–n PSCs.31,35,38,88,89,93 In comparison with our GBP&SP PSCs (Fig. S42, ESI†) the p–i–n PSC with LiF passivation layer degrades much faster, reaching 80% of the initial PCE already after 7 h of constant illumination. Similar reports on the fast degradation of LiF containing p–i–n PSC can be found in literature.31,93 Next to improved operational stability under constant AM1.5G illumination, the GBP&SP PSCs demonstrate improved thermal stability as compared to Ref PSCs by tracking the photovoltaic performance of the devices after aging under 85 °C heating in the dark over 1000 h (see Fig. 7c). To further evaluate the stability with regard to moisture, we exposed unencapsulated Ref PSCs and GBP&SP PSCs to a relative humidity of ∼50% in ambient atmosphere and at room temperature for 1 day. The photographs and absorption data exhibit no changes for the GBP&SP film, while the Ref film is entirely decomposed to PbI2 (Fig. S43a, ESI†). The increased contact angle of water droplets from 55.9° for the Ref film to 78° for the GBP&SP film (Fig. S43b, ESI†) confirms the better moisture resistance capability in the case of GBP&SP, which is attributed to the presence of hydrophobic PEA+ cations at the surface and grain boundaries of the perovskite films that acts as a hydrophobic barrier.25,71,76 We note that the formation of shallow iodine interstitials upon passivation with iodide-based passivation molecules has recently been proposed to cause accelerated degradation of FAPbI3 perovskites.71 Our chloride-based dual passivation approach might potentially mitigate this issue. In summary, the presented fundamental assessment of stability for PSCs employing our dual passivation strategy highlights the importance of passivating defects at both the surface and the grain boundaries of perovskite films for achieving PCSs exhibiting both high efficiency and stability.
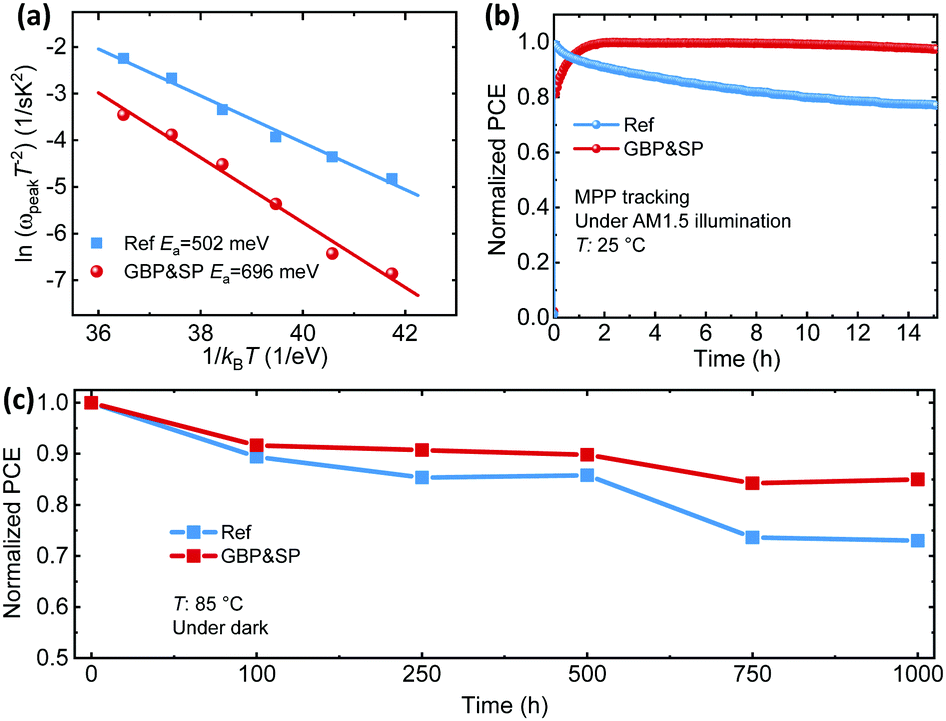 | ||
| Fig. 7 (a) Arrhenius plots determined from the derivative of admittance spectra to determine the activation energy (Ea) for reference (Ref) and grain boundary & surface passivation (GBP&SP) perovskite solar cells. (b) Maximum power point (MPP) tracking of the Ref and GBP&SP perovskite solar cells under continuous solar illumination (100 mW cm−2) in a nitrogen atmosphere. (c) Thermal stability of devices heated at temperature of 85 °C in dark condition inside of a glovebox. Data points were extracted from J–V curves at various time intervals. | ||
Conclusion
In summary, we demonstrate a dual passivation strategy for p–i–n PSCs that simultaneously passivates defects at the perovskite/C60 interface as well as in the grain boundaries using the long chain alkylammonium salt phenethylammonium chloride. We achieve a substantial enhancement in charge carrier lifetime and quasi-Fermi level splitting compared to reference films as well as to films with either individual grain boundary or surface passivation. The best PSC with dual passivation achieves a significant absolute enhancement in both VOC (76 mV) and FF (4.6%) compared to the best reference device. As a result, a remarkable stabilized PCE of 22.3% is demonstrated, one of the highest reported for p–i–n PSCs. We attribute this improvement in performance to the formation of a heterogeneous 2D RP (PEA)2(CsyFA1−y)n−1Pbn(I1−xClx)3n+1 phase with n∼1–2 at the surface and grain boundaries of the films, which leads to (1) efficient chemical passivation of grain boundary and surface/interface defects and (2) additional hole blocking at the perovskite/C60 interface. Finally, we demonstrate that the activation energy for ion migration is strongly increased upon dual passivation, which is reflected by an enhanced device stability under maximum power point (MPP) tracking and prolonged heat treatment. This work highlights the importance of defect management by employing a proper material both for grain boundary as well as surface passivation for achieving high-efficiency and stable inverted p–i–n PSCs. Thereby this work makes a relevant contribution to the advance of perovskite-based flexible and tandem photovoltaics.Author contributions
S. G. conceived the initial idea for this study and developed it further with support of P. F., I. M. H. and U. W. P. Furthermore, S. G. designed the experiments, fabricated the perovskite films and performed the J–V, EQE and long-term stability measurements. P. F. performed the absolute and intensity-dependent PLQY measurements and data analysis. I. M. H. performed the transient photoluminescence measurements and data analysis. S. G. performed the XRD measurements. M. F. performed the XPS/UPS measurements and data analysis and P. F. and J. P. H. supported in data interpretation. M. S. helped for device fabrication. P. R. performed the KPFM measurements, supervised by S. A. L. W., and the data was analysed by P. R., S. G., P. F. and S. A. L. W. The CL measurements were performed by T. D. The TAS measurements were performed by S. G. and M. R. K., and O. A. and M. R. K. analysed the data. T. A. and B. A. N. performed the SEM measurements. F. S. performed the contact angle measurement. T. F. applied and optimized the antireflection coating. R. S. helped for long-term stability measurement. B. A. N. helped with designing the schematics. D. F. performed additional high-resolution XRD measurements. U. L. and U. W. P. supervised the whole project. S. G., P. F. and U. W. P. drafted the manuscript. All authors reviewed and commented on the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. A. N. acknowledges the financial support from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No [840937]. T. D. acknowledges the financial support from Australian Centre for Advanced Photovoltaics (ACAP). O. A. acknowledges the funding from the European Union's Horizon 2020 research and innovation program under the Photonics Public Private Partnership (http://www.photonics21.org) with the project PEROXIS (grant no. 871336). M. S. acknowledges funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant agreement No. 947221). Financial support by the Initiating and Networking funding of the Helmholtz Association (HYIG of U. W. P. (VHNG-1148)), the Helmholtz Energy Materials Foundry (HEMF), PEROSEED (ZT0024), the German Federal Ministry for Economic Affairs (BMWi) through the project 27Plus6 (03EE1056B) as well as the project CAPITANO (03EE1038B), and the Karlsruhe School of Optics & Photonics (KSOP) is gratefully acknowledged. Furthermore, this project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No 850937 (PERCISTAND). The financial support of this research through the Baden-Württemberg Foundation is acknowledged (project acronym: ANU-KIT-PV).References
- NREL Best Research-Cell Efficiency Chart, https://www.nrel.gov/pv/cell-efficiency.html, accessed: March 2021.
- O. Almora, D. Baran, G. C. Bazan, C. Berger, C. I. Cabrera, K. R. Catchpole, S. Erten-Ela, F. Guo, J. Hauch, A. W. Y. Ho-Baillie, T. J. Jacobsson, R. A. J. Janssen, T. Kirchartz, N. Kopidakis, Y. Li, M. A. Loi, R. R. Lunt, X. Mathew, M. D. McGehee, J. Min, D. B. Mitzi, M. K. Nazeeruddin, J. Nelson, A. F. Nogueira, U. W. Paetzold, N. Park, B. P. Rand, U. Rau, H. J. Snaith, E. Unger, L. Vaillant-Roca, H. Yip and C. J. Brabec, Adv. Energy Mater., 2021, 11, 2002774 CrossRef CAS
.
- G. Kim, H. Min, K. S. Lee, D. Y. Lee, S. M. Yoon and S. Il Seok, Science, 2020, 370, 108–112 CrossRef CAS PubMed
- M. Jeong, I. W. Choi, E. M. Go, Y. Cho, M. Kim, B. Lee, S. Jeong, Y. Jo, H. W. Choi, J. Lee, J. Bae, S. K. Kwak, D. S. Kim and C. Yang, Science, 2020, 1620, 1615–1620 CrossRef PubMed
- M. Kim, G. Kim, T. K. Lee, I. W. Choi, H. W. Choi, Y. Jo, Y. J. Yoon, J. W. Kim, J. Lee, D. Huh, H. Lee, S. K. Kwak, J. Y. Kim and D. S. Kim, Joule, 2019, 3, 2179–2192 CrossRef CAS
- E. H. Jung, N. J. Jeon, E. Y. Park, C. S. Moon, T. J. Shin, T. Y. Yang, J. H. Noh and J. Seo, Nature, 2019, 567, 511–515 CrossRef CAS
- H. Zhu, Y. Ren, L. Pan, O. Ouellette, F. T. Eickemeyer, Y. Wu, X. Li, S. Wang, H. Liu, X. Dong, S. M. Zakeeruddin, Y. Liu, A. Hagfeldt and M. Grätzel, J. Am. Chem. Soc., 2021, 143, 3231–3237 CrossRef CAS PubMed
- M. A. Mahmud, T. Duong, Y. Yin, J. Peng, Y. Wu, T. Lu, H. T. Pham, H. Shen, D. Walter, H. T. Nguyen, N. Mozaffari, G. D. Tabi, Y. Liu, G. Andersson, K. R. Catchpole, K. J. Weber and T. P. White, Small, 2020, 16, 2005022 CrossRef CAS PubMed
- T.-S. Su, F. T. Eickemeyer, M. A. Hope, F. Jahanbakhshi, M. Mladenović, J. Li, Z. Zhou, A. Mishra, J.-H. Yum, D. Ren, A. Krishna, O. Ouellette, T.-C. Wei, H. Zhou, H.-H. Huang, M. D. Mensi, K. Sivula, S. M. Zakeeruddin, J. V. Milić, A. Hagfeldt, U. Rothlisberger, L. Emsley, H. Zhang and M. Grätzel, J. Am. Chem. Soc., 2020, 142, 19980–19991 CrossRef CAS PubMed
- J. J. Yoo, S. Wieghold, M. C. Sponseller, M. R. Chua, S.
N. Bertram, N. T. P. Hartono, J. S. Tresback, E. C. Hansen, J.-P. Correa-Baena, V. Bulović, T. Buonassisi, S. S. Shin and M. G. Bawendi, Energy Environ. Sci., 2019, 12, 2192–2199 RSC
- B. Yang, J. Suo, E. Mosconi, D. Ricciarelli, W. Tress, F. De Angelis, H. Kim and A. Hagfeldt, ACS Energy Lett., 2020, 5, 3159–3167 CrossRef CAS
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381–385 CrossRef CAS
- J. J. Yoo, G. Seo, M. R. Chua, T. G. Park, Y. Lu, F. Rotermund, Y. Kim, C. S. Moon, N. J. Jeon, J.-P. Correa-Baena, V. Bulović, S. S. Shin, M. G. Bawendi and J. Seo, Nature, 2021, 590, 587–593 CrossRef CAS
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS
- P. Wang, R. Li, B. Chen, F. Hou, J. Zhang, Y. Zhao and X. Zhang, Adv. Mater., 2020, 32, 1905766 CrossRef CAS PubMed
- C. Ma and N.-G. Park, ACS Energy Lett., 2020, 5, 3268–3275 CrossRef CAS
- X. Yang, Y. Fu, R. Su, Y. Zheng, Y. Zhang, W. Yang, M. Yu, P. Chen, Y. Wang, J. Wu, D. Luo, Y. Tu, L. Zhao, Q. Gong and R. Zhu, Adv. Mater., 2020, 32, 2002585 CrossRef CAS
- E. H. Jung, B. Chen, K. Bertens, M. Vafaie, S. Teale, A. Proppe, Y. Hou, T. Zhu, C. Zheng and E. H. Sargent, ACS Energy Lett., 2020, 5, 2796–2801 CrossRef CAS
- Q. Jiang, Z. Ni, G. Xu, Y. Lin, P. N. Rudd, R. Xue, Y. Y. Li, Y. Y. Li, Y. Gao and J. Huang, Adv. Mater., 2020, 32, 2001581 CrossRef CAS
- X. Yang, D. Luo, Y. Xiang, L. Zhao, M. Anaya, Y. Shen, J. Wu, W. Yang, Y. H. Chiang, Y. Tu, R. Su, Q. Hu, H. Yu, G. Shao, W. Huang, T. P. Russell, Q. Gong, S. D. Stranks, W. Zhang and R. Zhu, Adv. Mater., 2021, 33, 2006435 CrossRef CAS
- J. Zhuang, P. Mao, Y. Luan, N. Chen, X. Cao, G. Niu, F. Jia, F. Wang, S. Cao and J. Wang, Adv. Funct. Mater., 2021, 31, 2010385 CrossRef CAS
- H. Lu, Y. Liu, P. Ahlawat, A. Mishra, W. R. Tress, F. T. Eickemeyer, Y. Yang, F. Fu, Z. Wang, C. E. Avalos, B. I. Carlsen, A. Agarwalla, X. Zhang, X. Li, Y. Zhan, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, L. Zheng, A. Hagfeldt and M. Grätzel, Science, 2020, 370, eabb8985 CrossRef CAS PubMed
- M. Green, E. Dunlop, J. Hohl-Ebinger, M. Yoshita, N. Kopidakis and X. Hao, Prog. Photovoltaics, 2021, 29, 3–15 Search PubMed
- F. Li, X. Deng, F. Qi, Z. Li, D. Liu, D. Shen, M. Qin, S. Wu, F. Lin, S. Jang, J. Zhang, X. Lu, D. Lei, C. Lee, Z. Zhu and A. K.-Y. Jen, J. Am. Chem. Soc., 2020, 142, 20134–20142 CrossRef CAS PubMed
- X. Zheng, Y. Hou, C. Bao, J. Yin, F. Yuan, Z. Huang, K. Song, J. Liu, J. Troughton, N. Gasparini, C. Zhou, Y. Lin, D.-J. Xue, B. Chen, A. K. Johnston, N. Wei, M. N. Hedhili, M. Wei, A. Y. Alsalloum, P. Maity, B. Turedi, C. Yang, D. Baran, T. D. Anthopoulos, Y. Han, Z.-H. Lu, O. F. Mohammed, F. Gao, E. H. Sargent and O. M. Bakr, Nat. Energy, 2020, 5, 131–140 CrossRef CAS
- W. Wu, P. N. Rudd, Q. Wang, Z. Yang and J. Huang, Adv. Mater., 2020, 32, 2000995 CrossRef CAS
- S. Wu, J. Zhang, Z. Li, D. Liu, M. Qin, S. H. Cheung, X. Lu, D. Lei, S. K. So, Z. Zhu and A. K.-Y. Jen, Joule, 2020, 4, 1248–1262 CrossRef CAS
- X. Sun, Z. Li, X. Yu, X. Wu, C. Zhong, D. Liu, D. Lei, A. K.-Y. Jen, Z. Li and Z. Zhu, Angew. Chem., Int. Ed., 2021, 60, 7227–7233 CrossRef CAS PubMed
- X. Hu, C. Liu, Z. Zhang, X. Jiang, J. Garcia, C. Sheehan, L. Shui, S. Priya, G. Zhou, S. Zhang and K. Wang, Adv. Sci., 2020, 7, 2001285 CrossRef CAS PubMed
- A. Y. Alsalloum, B. Turedi, K. Almasabi, X. Zheng, R. Naphade, S. D. Stranks, O. F. Mohammed and O. M. Bakr, Energy Environ. Sci., 2021, 14, 2263–2268 RSC
- A. Al-Ashouri, E. Köhnen, B. Li, A. Magomedov, H. Hempel, P. Caprioglio, J. A. Márquez, A. B. Morales Vilches, E. Kasparavicius, J. A. Smith, N. Phung, D. Menzel, M. Grischek, L. Kegelmann, D. Skroblin, C. Gollwitzer, T. Malinauskas, M. Jošt, G. Matič, B. Rech, R. Schlatmann, M. Topič, L. Korte, A. Abate, B. Stannowski, D. Neher, M. Stolterfoht, T. Unold, V. Getautis and S. Albrecht, Science, 2020, 370, 1300–1309 CrossRef CAS PubMed
- A. Al-Ashouri, A. Magomedov, M. Roß, M. Jošt, M. Talaikis, G. Chistiakova, T. Bertram, J. A. Márquez, E. Köhnen, E. Kasparavičius, S. Levcenco, L. Gil-Escrig, C. J. Hages, R. Schlatmann, B. Rech, T. Malinauskas, T. Unold, C. A. Kaufmann, L. Korte, G. Niaura, V. Getautis and S. Albrecht, Energy Environ. Sci., 2019, 12, 3356–3369 RSC
- X. Lin, D. Cui, X. Luo, C. Zhang, Q. Han, Y. Wang and L. Han, Energy Environ. Sci., 2020, 13, 3823–3847 RSC
- M. Stolterfoht, M. Grischek, P. Caprioglio, C. M. Wolff, E. Gutierrez-Partida, F. Peña-Camargo, D. Rothhardt, S. Zhang, M. Raoufi, J. Wolansky, M. Abdi-Jalebi, S. D. Stranks, S. Albrecht, T. Kirchartz and D. Neher, Adv. Mater., 2020, 32, 2000080 CrossRef CAS
- M. Stolterfoht, P. Caprioglio, C. M. Wolff, J. A. Márquez, J. Nordmann, S. Zhang, D. Rothhardt, U. Hörmann, Y. Amir, A. Redinger, L. Kegelmann, F. Zu, S. Albrecht, N. Koch, T. Kirchartz, M. Saliba, T. Unold and D. Neher, Energy Environ. Sci., 2019, 12, 2778–2788 RSC
- D. Luo, R. Su, W. Zhang, Q. Gong and R. Zhu, Nat. Rev. Mater., 2020, 5, 44–60 CrossRef CAS
- C. M. Wolff, P. Caprioglio, M. Stolterfoht and D. Neher, Adv. Mater., 2019, 31, 1902762 CrossRef CAS
- M. Stolterfoht, C. M. Wolff, J. A. Márquez, S. Zhang, C. J. Hages, D. Rothhardt, S. Albrecht, P. L. Burn, P. Meredith, T. Unold and D. Neher, Nat. Energy, 2018, 3, 847–854 CrossRef CAS
- S. Zhang, P. E. Shaw, G. Zhang, H. Jin, M. Tai, H. Lin, P. Meredith, P. L. Burn, D. Neher and M. Stolterfoht, ACS Appl. Mater. Interfaces, 2020, 12, 37647–37656 CrossRef CAS
- J.-W. Lee, S.-H. Bae, N. De Marco, Y.-T. Hsieh, Z. Dai and Y. Yang, Mater. Today Energy, 2018, 7, 149–160 CrossRef
- A. Castro-Méndez, J. Hidalgo and J. Correa-Baena, Adv. Energy Mater., 2019, 9, 1901489 CrossRef
- J.-S. Park and A. Walsh, Annu. Rev. Condens. Matter Phys., 2021, 12, 042020 Search PubMed
- Y. Shao, Y. Fang, T. Li, Q. Wang, Q. Dong, Y. Deng, Y. Yuan, H. Wei, M. Wang, A. Gruverman, J. Shield and J. Huang, Energy Environ. Sci., 2016, 9, 1752–1759 RSC
- J. S. Yun, J. Seidel, J. Kim, A. M. Soufiani, S. Huang, J. Lau, N. J. Jeon, S. Il Seok, M. A. Green and A. Ho-Baillie, Adv. Energy Mater., 2016, 6, 1600330 CrossRef
- L. Liu, S. Huang, Y. Lu, P. Liu, Y. Zhao, C. Shi, S. Zhang, J. Wu, H. Zhong, M. Sui, H. Zhou, H. Jin, Y. Li and Q. Chen, Adv. Mater., 2018, 30, 1800544 CrossRef
- N. Phung, A. Al-Ashouri, S. Meloni, A. Mattoni, S. Albrecht, E. L. Unger, A. Merdasa and A. Abate, Adv. Energy Mater., 2020, 10, 1903735 CrossRef CAS
- L. McGovern, I. Koschany, G. Grimaldi, L. A. Muscarella and B. Ehrler, J. Phys. Chem. Lett., 2021, 2423–2428 CrossRef CAS PubMed
- S. P. Dunfield, L. Bliss, F. Zhang, J. M. Luther, K. Zhu, M. F. A. M. Hest, M. O. Reese and J. J. Berry, Adv. Energy Mater., 2020, 10, 1904054 CrossRef CAS
- L. Fu, H. Li, L. Wang, R. Yin, B. Li and L.-W. Yin, Energy Environ. Sci., 2020, 13, 4017–4056 RSC
- Q. Sun, P. Fassl, D. Becker-Koch, A. Bausch, B. Rivkin, S. Bai, P. E. Hopkinson, H. J. Snaith and Y. Vaynzof, Adv. Energy Mater., 2017, 7, 1700977 CrossRef
- J. P. Bastos, G. Uytterhoeven, W. Qiu, U. W. Paetzold, D. Cheyns, S. Surana, J. Rivas, M. Jaysankar, W. Song, T. Aernouts, J. Poortmans and R. Gehlhaar, ACS Appl. Mater. Interfaces, 2019, 11, 16517–16526 CrossRef CAS PubMed
- S. Akin, N. Arora, S. M. Zakeeruddin, M. Grätzel, R. H. Friend and M. I. Dar, Adv. Energy Mater., 2020, 10, 1903090 CrossRef CAS
- E. Aydin, M. Bastiani and S. Wolf, Adv. Mater., 2019, 31, 1900428 CrossRef PubMed
- F. Gao, Y. Zhao, X. Zhang and J. You, Adv. Energy Mater., 2020, 10, 1902650 CrossRef CAS
- H. Zhang, M. K. Nazeeruddin and W. C. H. Choy, Adv. Mater., 2019, 31, 1805702 CrossRef PubMed
- W. Qi, X. Zhou, J. Li, J. Cheng, Y. Li, M. J. Ko, Y. Zhao and X. Zhang, Sci. Bull., 2020, 65, 2022–2032 CrossRef CAS
- Y. Li, H. Wu, W. Qi, X. Zhou, J. Li, J. Cheng, Y. Zhao, Y. Li and X. Zhang, Nano Energy, 2020, 77, 105237 CrossRef CAS
- S. Akin, B. Dong, L. Pfeifer, Y. Liu, M. Graetzel and A. Hagfeldt, Adv. Sci., 2021, 8, 2004593 CrossRef
- S. Gharibzadeh, B. Abdollahi Nejand, M. Jakoby, T. Abzieher, D. Hauschild, S. Moghadamzadeh, J. A. Schwenzer, P. Brenner, R. Schmager, A. A. Haghighirad, L. Weinhardt, U. Lemmer, B. S. Richards, I. A. Howard and U. W. Paetzold, Adv. Energy Mater., 2019, 9, 1803699 CrossRef
- S. Gharibzadeh, I. M. Hossain, P. Fassl, B. A. Nejand, T. Abzieher, M. Schultes, E. Ahlswede, P. Jackson, M. Powalla, S. Schäfer, M. Rienäcker, T. Wietler, R. Peibst, U. Lemmer, B. S. Richards and U. W. Paetzold, Adv. Funct. Mater., 2020, 30, 1909919 CrossRef CAS
- Y. Liu, S. Akin, L. Pan, R. Uchida, N. Arora, J. V. Milić, A. Hinderhofer, F. Schreiber, A. R. Uhl, S. M. Zakeeruddin, A. Hagfeldt, M. I. Dar and M. Grätzel, Sci. Adv., 2019, 5, eaaw2543 CrossRef CAS PubMed
- Z. Li, J. Zhang, S. Wu, X. Deng, F. Li, D. Liu, C. Lee, F. Lin, D. Lei, C.-C. Chueh, Z. Zhu and A. K. Y. Jen, Nano Energy, 2020, 78, 105377 CrossRef CAS
- M. A. Mahmud, T. Duong, Y. Yin, H. T. Pham, D. Walter, J. Peng, Y. Wu, L. Li, H. Shen, N. Wu, N. Mozaffari, G. Andersson, K. R. Catchpole, K. J. Weber and T. P. White, Adv. Funct. Mater., 2020, 30, 1907962 CrossRef CAS
- T. Duong, H. Pham, T. C. Kho, P. Phang, K. C. Fong, D. Yan, Y. Yin, J. Peng, M. A. Mahmud, S. Gharibzadeh, B. A. Nejand, I. M. Hossain, M. R. Khan, N. Mozaffari, Y. Wu, H. Shen, J. Zheng, H. Mai, W. Liang, C. Samundsett, M. Stocks, K. McIntosh, G. G. Andersson, U. Lemmer, B. S. Richards, U. W. Paetzold, A. Ho-Ballie, Y. Liu, D. Macdonald, A. Blakers, J. Wong-Leung, T. White, K. Weber and K. Catchpole, Adv. Energy Mater., 2020, 10, 1903553 CrossRef CAS
- T. Zhu, D. Zheng, J. Liu, L. Coolen, T. Pauporté, T. Pauporte and T. Pauporté, ACS Appl. Mater. Interfaces, 2020, 12, 37197–37207 CrossRef CAS PubMed
- D. Luo, W. Yang, Z. Wang, A. Sadhanala, Q. Hu, R. Su, R. Shivanna, G. F. Trindade, J. F. Watts, Z. Xu, T. Liu, K. Chen, F. Ye, P. Wu, L. Zhao, J. Wu, Y. Tu, Y. Zhang, X. Yang, W. Zhang, R. H. Friend, Q. Gong, H. J. Snaith and R. Zhu, Science, 2018, 360, 1442–1446 CrossRef CAS PubMed
- K. Lee, J. Kim, H. Yu, J. W. Lee, C. M. Yoon, S. K. Kim and J. Jang, J. Mater. Chem. A, 2018, 6, 24560–24568 RSC
- F. Zhang, Q. Huang, J. Song, Y. Zhang, C. Ding, F. Liu, D. Liu, X. Li, H. Yasuda, K. Yoshida, J. Qu, S. Hayase, T. Toyoda, T. Minemoto and Q. Shen, Sol. RRL, 2020, 4, 1900243 CrossRef CAS
- H. Dong, M. Yue, S. Pang, W. Zhu, D. Chen, H. Xi, Z. Lin, J. Chang, J. Zhang, Y. Hao and C. Zhang, Sol. RRL, 2019, 3, 1900291 CrossRef CAS
- C. Zhang, S. Wu, L. Tao, G. M. Arumugam, C. Liu, Z. Wang, S. Zhu, Y. Yang, J. Lin, X. Liu, R. E. I. Schropp and Y. Mai, Adv. Energy Mater., 2020, 10, 2002004 CrossRef CAS
- S. Tan, I. Yavuz, M. H. Weber, T. Huang, C. Chen, R. Wang, H. Wang, J. H. Ko, S. Nuryyeva, J. Xue, Y. Zhao, K. Wei, J. Lee and Y. Yang, Joule, 2020, 4, 2426–2442 CrossRef CAS
- H. Kim, S. Lee, D. Y. Lee, M. J. Paik, H. Na, J. Lee and S. Il Seok, Adv. Energy Mater., 2019, 9, 1902740 CrossRef CAS
- X. Zheng, B. Chen, J. Dai, Y. Fang, Y. Bai, Y. Lin, H. Wei, X. C. Zeng and J. Huang, Nat. Energy, 2017, 2, 17102 CrossRef CAS
- Q. Zhou, Y. Gao, C. Cai, Z. Zhang, J. Xu, Z. Yuan and P. Gao, Angew. Chem., 2021, 133, 8384–8393 CrossRef
- H. Wang, C. Zhu, L. Liu, S. Ma, P. Liu, J. Wu, C. Shi, Q. Du, Y. Hao, S. Xiang, H. Chen, P. Chen, Y. Bai, H. Zhou, Y. Li and Q. Chen, Adv. Mater., 2019, 31, 1904408 CrossRef CAS PubMed
- T. M. Koh, V. Shanmugam, X. Guo, S. S. Lim, O. Filonik, E. M. Herzig, P. Müller-Buschbaum, V. Swamy, S. T. Chien, S. G. Mhaisalkar and N. Mathews, J. Mater. Chem. A, 2018, 6, 2122–2128 RSC
- K. T. Cho, G. Grancini, Y. Lee, E. Oveisi, J. Ryu, O. Almora, M. Tschumi, P. A. Schouwink, G. Seo, S. Heo, J. Park, J. Jang, S. Paek, G. Garcia-Belmonte and M. K. Nazeeruddin, Energy Environ. Sci., 2018, 11, 952–959 RSC
- Y. Cho, A. M. Soufiani, J. S. Yun, J. Kim, D. S. Lee, J. Seidel, X. Deng, M. A. Green, S. Huang and A. W. Y. Ho-Baillie, Adv. Energy Mater., 2018, 8, 1703392 CrossRef
- P. Chen, Y. Bai, S. Wang, M. Lyu, J.-H. Yun and L. Wang, Adv. Funct. Mater., 2018, 28, 1706923 CrossRef
- Y. Lin, Y. Bai, Y. Fang, Z. Chen, S. Yang, X. Zheng, S. Tang, Y. Liu, J. Zhao and J. Huang, J. Phys. Chem. Lett., 2018, 9, 654–658 CrossRef CAS PubMed
- M. Malekshahi Byranvand, F. Behboodi-Sadabad, A. Alrhman Eliwi, V. Trouillet, A. Welle, S. Ternes, I. M. Hossain, M. R. Khan, J. A. Schwenzer, A. Farooq, B. S. Richards, J. Lahann and U. W. Paetzold, J. Mater. Chem. A, 2020, 8, 20122–20132 RSC
- J. Peng, D. Walter, Y. Ren, M. Tebyetekerwa, Y. Wu, T. Duong, Q. Lin, J. Li, T. Lu, M. A. Mahmud, O. L. C. Lem, S. Zhao, W. Liu, Y. Liu, H. Shen, L. Li, F. Kremer, H. T. Nguyen, D. Choi, K. J. Weber, K. R. Catchpole and T. P. White, Science, 2021, 371, 390–395 CrossRef CAS PubMed
- R. Wang, J. Xue, K.-L. L. Wang, Z.-K. K. Wang, Y. Luo, D. Fenning, G. Xu, S. Nuryyeva, T. Huang, Y. Zhao, J. L. Yang, J. Zhu, M. Wang, S. Tan, I. Yavuz, K. N. Houk and Y. Yang, Science, 2019, 366, 1509–1513 CrossRef CAS PubMed
- X. Liu, Z. Yu, T. Wang, K. L. Chiu, F. Lin, H. Gong, L. Ding and Y. Cheng, Adv. Energy Mater., 2020, 10, 2001958 CrossRef CAS
- S. Yang, J. Dai, Z. Yu, Y. Shao, Y. Zhou, X. Xiao, X. C. Zeng and J. Huang, J. Am. Chem. Soc., 2019, 141, 5781–5787 CrossRef CAS
- C. M. Wolff, L. Canil, C. Rehermann, N. Ngoc Linh, F. Zu, M. Ralaiarisoa, P. Caprioglio, L. Fiedler, M. Stolterfoht, S. Kogikoski, I. Bald, N. Koch, E. L. Unger, T. Dittrich, A. Abate and D. Neher, ACS Nano, 2020, 14, 1445–1456 CrossRef CAS PubMed
- Z. Liu, F. Cao, M. Wang, M. Wang and L. Li, Angew. Chem., 2020, 132, 4190–4196 CrossRef
- J. Xu, C. C. Boyd, Z. J. Yu, A. F. Palmstrom, D. J. Witter, B. W. Larson, R. M. France, J. Werner, S. P. Harvey, E. J. Wolf, W. Weigand, S. Manzoor, M. F. A. M. van Hest, J. J. Berry, J. M. Luther, Z. C. Holman and M. D. McGehee, Science, 2020, 367, 1097–1104 CrossRef CAS PubMed
- F. Peña-Camargo, P. Caprioglio, F. Zu, E. Gutierrez-Partida, C. M. Wolff, K. Brinkmann, S. Albrecht, T. Riedl, N. Koch, D. Neher and M. Stolterfoht, ACS Energy Lett., 2020, 5, 2728–2736 CrossRef
- S. Yang, S. Chen, E. Mosconi, Y. Fang, X. Xiao, C. Wang, Y. Zhou, Z. Yu, J. Zhao, Y. Gao, F. De Angelis and J. Huang, Science, 2019, 365, 473–478 CrossRef CAS PubMed
- F. Wang, Y. Zhang, M. Yang, D. Han, L. Yang, L. Fan, Y. Sui, Y. Sun, X. Liu, X. Meng and J. Yang, Adv. Funct. Mater., 2021, 31, 2008052 CrossRef CAS
- P. Caprioglio, M. Stolterfoht, C. M. Wolff, T. Unold, B. Rech, S. Albrecht and D. Neher, Adv. Energy Mater., 2019, 9, 1901631 CrossRef
- J. Dagar, M. Fenske, A. Al-Ashouri, C. Schultz, B. Li, H. Köbler, R. Munir, G. Parmasivam, J. Li, I. Levine, A. Merdasa, L. Kegelmann, H. Näsström, J. A. Marquez, T. Unold, D. M. Többens, R. Schlatmann, B. Stegemann, A. Abate, S. Albrecht and E. Unger, ACS Appl. Mater. Interfaces, 2021, 13, 13022–13033 CrossRef CAS PubMed
- B. Park, N. Kedem, M. Kulbak, D. Y. Lee, W. S. Yang, N. J. Jeon, J. Seo, G. Kim, K. J. Kim, T. J. Shin, G. Hodes, D. Cahen and S. Il Seok, Nat. Commun., 2018, 9, 3301 CrossRef PubMed
- P. Fassl, V. Lami, A. Bausch, Z. Wang, M. T. Klug, H. J. Snaith and Y. Vaynzof, Energy Environ. Sci., 2018, 11, 3380–3391 RSC
- P. Fassl, Y. Zakharko, L. M. Falk, K. P. Goetz, F. Paulus, A. D. Taylor, J. Zaumseil and Y. Vaynzof, J. Mater. Chem. C, 2019, 7, 5285–5292 RSC
- K. Wang, W. S. Subhani, Y. Wang, X. Zuo, H. Wang, L. Duan and S. (Frank) Liu, Adv. Mater., 2019, 31, 1902037 CrossRef CAS PubMed
- D. Kim, H. J. Jung, I. J. Park, B. W. Larson, S. P. Dunfield, C. Xiao, J. Kim, J. Tong, P. Boonmongkolras, S. G. Ji, F. Zhang, S. R. Pae, M. Kim, S. B. Kang, V. Dravid, J. J. Berry, J. Y. Kim, K. Zhu, D. H. Kim and B. Shin, Science, 2020, 3433, eaba3433 Search PubMed
- D. S. Lee, J. S. Yun, J. Kim, A. M. Soufiani, S. Chen, Y. Cho, X. Deng, J. Seidel, S. Lim, S. Huang and A. W. Y. Ho-Baillie, ACS Energy Lett., 2018, 3, 647–654 CrossRef CAS
- J.-W. Lee, Z. Dai, T.-H. Han, C. Choi, S.-Y. Chang, S.-J. Lee, N. De Marco, H. Zhao, P. Sun, Y. Huang and Y. Yang, Nat. Commun., 2018, 9, 3021 CrossRef PubMed
- M. Jung, T. J. Shin, J. Seo, G. Kim and S. Il Seok, Energy Environ. Sci., 2018, 11, 2188–2197 RSC
- Z. Wang, Q. Lin, F. P. Chmiel, N. Sakai, L. M. Herz and H. J. Snaith, Nat. Energy, 2017, 2, 17135 CrossRef CAS
- D. H. Kim, C. P. Muzzillo, J. Tong, A. F. Palmstrom, B. W. Larson, C. Choi, S. P. Harvey, S. Glynn, J. B. Whitaker, F. Zhang, Z. Li, H. Lu, M. F. A. M. van Hest, J. J. Berry, L. M. Mansfield, Y. Huang, Y. Yan and K. Zhu, Joule, 2019, 3, 1734–1745 CrossRef CAS
- C. Fei, M. Zhou, J. Ogle, D.-M. Smilgies, L. Whittaker-Brooks and H. Wang, J. Mater. Chem. A, 2019, 7, 23739–23746 RSC
- N. Li, Z. Zhu, C. C. Chueh, H. Liu, B. Peng, A. Petrone, X. Li, L. Wang and A. K.-Y. Jen, Adv. Energy Mater., 2017, 7, 1–9 Search PubMed
- S. Xiong, Z. Hou, S. Zou, X. Lu, J. Yang, T. Hao, Z. Zhou, J. Xu, Y. Zeng, W. Xiao, W. Dong, D. Li, X. Wang, Z. Hu, L. Sun, Y. Wu, X. Liu, L. Ding, Z. Sun, M. Fahlman and Q. Bao, Joule, 2021, 5, 467–480 CrossRef CAS
- X. Wang, K. Rakstys, K. Jack, H. Jin, J. Lai, H. Li, C. S. K. Ranasinghe, J. Saghaei, G. Zhang, P. L. Burn, I. R. Gentle and P. E. Shaw, Nat. Commun., 2021, 12, 52 CrossRef CAS PubMed
- F. Zhang and K. Zhu, Adv. Energy Mater., 2020, 10, 1902579 CrossRef CAS
- S. Liu, Y. Guan, Y. Sheng, Y. Hu, Y. Rong, A. Mei and H. Han, Adv. Energy Mater., 2020, 10, 1902492 CrossRef CAS
- J. Wang, G. Jin, Q. Zhen, C. He and Y. Duan, Adv. Mater. Interfaces, 2021, 8, 2002078 CrossRef CAS
- X. Zhou, W. Qi, J. Li, J. Cheng, Y. Li, J. Luo, M. J. Ko, Y. Li, Y. Zhao and X. Zhang, Sol. RRL, 2020, 4, 2000308 CrossRef CAS
- D. Prochowicz, R. Runjhun, M. M. Tavakoli, P. Yadav, M. Saski, A. Q. Alanazi, D. J. Kubicki, Z. Kaszkur, S. M. Zakeeruddin, J. Lewiński and M. Grätzel, Chem. Mater., 2019, 31, 1620–1627 CrossRef CAS
- L. Krückemeier, U. Rau, M. Stolterfoht and T. Kirchartz, Adv. Energy Mater., 2020, 10, 1902573 CrossRef
- L. Krückemeier, B. Krogmeier, Z. Liu, U. Rau and T. Kirchartz, Adv. Energy Mater., 2021, 11, 2003489 CrossRef
- Z. Liu, L. Krückemeier, B. Krogmeier, B. Klingebiel, J. A. Márquez, S. Levcenko, S. Öz, S. Mathur, U. Rau, T. Unold and T. Kirchartz, ACS Energy Lett., 2019, 4, 110–117 CrossRef CAS
- B. Krogmeier, F. Staub, D. Grabowski, U. Rau and T. Kirchartz, Sustainable Energy Fuels, 2018, 2, 1027–1034 RSC
- T. Kirchartz, J. A. Márquez, M. Stolterfoht and T. Unold, Adv. Energy Mater., 2020, 10, 1904134 CrossRef CAS
- P. Fassl, V. Lami, F. J. Berger, L. M. Falk, J. Zaumseil, B. S. Richards, I. A. Howard, Y. Vaynzof and U. W. Paetzold, Matter, 2021, 4, 1391–1412 CrossRef
- S. Zhang, S. M. Hosseini, R. Gunder, A. Petsiuk, P. Caprioglio, C. M. Wolff, S. Shoaee, P. Meredith, S. Schorr, T. Unold, P. L. Burn, D. Neher and M. Stolterfoht, Adv. Mater., 2019, 31, 1901090 CrossRef PubMed
- H. Jin, E. Debroye, M. Keshavarz, I. G. Scheblykin, M. B. J. Roeffaers, J. Hofkens and J. A. Steele, Mater. Horizons, 2020, 7, 397–410 RSC
- W.-Q. Wu, P. N. Rudd, Z. Ni, C. H. Van Brackle, H. Wei, Q. Wang, B. R. Ecker, Y. Gao and J. Huang, J. Am. Chem. Soc., 2020, 142, 3989–3996 CrossRef CAS PubMed
- P. Caprioglio, C. M. Wolff, O. J. Sandberg, A. Armin, B. Rech, S. Albrecht, D. Neher and M. Stolterfoht, Adv. Energy Mater., 2020, 10, 2000502 CrossRef CAS
- N. E. Courtier, Phys. Rev. Appl., 2020, 14, 024031 CrossRef CAS
- M. A. Green and A. W. Y. Ho-Baillie, ACS Energy Lett., 2019, 4, 1639–1644 CrossRef CAS
- Z. Liu, J. Siekmann, B. Klingebiel, U. Rau and T. Kirchartz, Adv. Energy Mater., 2021, 2003386 CrossRef CAS
- H. Guthrey and J. Moseley, Adv. Energy Mater., 2020, 10, 1903840 CrossRef CAS
- J. Hu, I. W. H. Oswald, S. J. Stuard, M. M. Nahid, N. Zhou, O. F. Williams, Z. Guo, L. Yan, H. Hu, Z. Chen, X. Xiao, Y. Lin, Z. Yang, J. Huang, A. M. Moran, H. Ade, J. R. Neilson and W. You, Nat. Commun., 2019, 10, 1276 CrossRef PubMed
- B. G. H. M. Groeneveld, S. Adjokatse, O. Nazarenko, H. Fang, G. R. Blake, G. Portale, H. Duim, G. H. ten Brink, M. V. Kovalenko and M. A. Loi, Energy Technol., 2020, 8, 1901041 CrossRef CAS
- K. Du, Q. Tu, X. Zhang, Q. Han, J. Liu, S. Zauscher and D. B. Mitzi, Inorg. Chem., 2017, 56, 9291–9302 CrossRef CAS PubMed
- K. Thirumal, W. K. Chong, W. Xie, R. Ganguly, S. K. Muduli, M. Sherburne, M. Asta, S. Mhaisalkar, T. C. Sum, H. Sen Soo and N. Mathews, Chem. Mater., 2017, 29, 3947–3953 CrossRef CAS
- P. Cai, X. Wang, H. J. Seo and X. Yan, Appl. Phys. Lett., 2018, 112, 153901 CrossRef
- X. Chen, H. Lu, Z. Li, Y. Zhai, P. F. Ndione, J. J. Berry, K. Zhu, Y. Yang and M. C. Beard, ACS Energy Lett., 2018, 3, 2273–2279 CrossRef CAS
- S. Béchu, M. Ralaiarisoa, A. Etcheberry and P. Schulz, Adv. Energy Mater., 2020, 10, 1904007 CrossRef
- R. D. Chavan, D. Prochowicz, P. Yadav, M. M. Tavakoli, A. Nimbalkar, S. P. Bhoite and C. K. Hong, Sol. RRL, 2019, 3, 1900294 CrossRef CAS
- T. Hellmann, C. Das, T. Abzieher, J. A. Schwenzer, M. Wussler, R. Dachauer, U. W. Paetzold, W. Jaegermann and T. Mayer, Adv. Energy Mater., 2020, 10, 2002129 CrossRef CAS
- N. K. Noel, S. N. Habisreutinger, A. Pellaroque, F. Pulvirenti, B. Wenger, F. Zhang, Y.-H. Lin, O. G. Reid, J. Leisen, Y. Zhang, S. Barlow, S. R. Marder, A. Kahn, H. J. Snaith, C. B. Arnold and B. P. Rand, Energy Environ. Sci., 2019, 12, 3063–3073 RSC
- M. Caputo, N. Cefarin, A. Radivo, N. Demitri, L. Gigli, J. R. Plaisier, M. Panighel, G. Di Santo, S. Moretti, A. Giglia, M. Polentarutti, F. De Angelis, E. Mosconi, P. Umari, M. Tormen and A. Goldoni, Sci. Rep., 2019, 9, 15159 CrossRef PubMed
- B. Philippe, B. Park, R. Lindblad, J. Oscarsson, S. Ahmadi, E. M. J. Johansson and H. Rensmo, Chem. Mater., 2015, 27, 1720–1731 CrossRef CAS
- A. A. Sutanto, P. Caprioglio, N. Drigo, Y. J. Hofstetter, I. Garcia-Benito, V. I. E. Queloz, D. Neher, M. K. Nazeeruddin, M. Stolterfoht, Y. Vaynzof and G. Grancini, Chem, 2021, 7, 1903–1916 CAS
- S. Draguta, J. A. Christians, Y. V. Morozov, A. Mucunzi, J. S. Manser, P. V. Kamat, J. M. Luther and M. Kuno, Energy Environ. Sci., 2018, 11, 960–969 RSC
- J. Emara, T. Schnier, N. Pourdavoud, T. Riedl, K. Meerholz and S. Olthof, Adv. Mater., 2016, 28, 553–559 CrossRef CAS PubMed
- Q. Wang, Q. Dong, T. Li, A. Gruverman and J. Huang, Adv. Mater., 2016, 28, 6734–6739 CrossRef CAS PubMed
- H. Kanda, N. Shibayama, A. J. Huckaba, Y. Lee, S. Paek, N. Klipfel, C. Roldán-Carmona, V. I. E. Queloz, G. Grancini, Y. Zhang, M. Abuhelaiqa, K. T. Cho, M. Li, M. D. Mensi, S. Kinge and M. K. Nazeeruddin, Energy Environ. Sci., 2020, 13, 1222–1230 RSC
- A. Axt, I. M. Hermes, V. W. Bergmann, N. Tausendpfund and S. A. L. Weber, Beilstein J. Nanotechnol., 2018, 9, 1809–1819 CrossRef CAS PubMed
- J. L. Garrett and J. N. Munday, Nanotechnology, 2016, 27, 245705 CrossRef PubMed
- E. M. Lanzoni, T. Gallet, C. Spindler, O. Ramirez, C. K. Boumenou, S. Siebentritt and A. Redinger, Nano Energy, 2021, 88, 106270 CrossRef CAS
- D. H. Cao, C. C. Stoumpos, C. D. Malliakas, M. J. Katz, O. K. Farha, J. T. Hupp and M. G. Kanatzidis, APL Mater., 2014, 2, 091101 CrossRef
- R. A. Street, S. E. Ready, F. Lemmi, K. S. Shah, P. Bennett and Y. Dmitriyev, J. Appl. Phys., 1999, 86, 2660–2667 CrossRef CAS
- Y. Sun, Z. Zhou, Z. Huang, J. Wu, L. Zhou, Y. Cheng, J. Liu, C. Zhu, M. Yu, P. Yu, W. Zhu, Y. Liu, J. Zhou, B. Liu, H. Xie, Y. Cao, H. Li, X. Wang, K. Liu, X. Wang, J. Wang, L. Wang and W. Huang, Adv. Mater., 2019, 31, 1806562 CrossRef PubMed
- Y.-M. Xie, X. Xu, C. Ma, M. Li, Y. Ma, C.-S. Lee and S.-W. Tsang, ACS Appl. Mater. Interfaces, 2019, 11, 25909–25916 CrossRef CAS PubMed
- S. Chen, X. Wen, J. S. Yun, S. Huang, M. Green, N. J. Jeon, W. S. Yang, J. H. Noh, J. Seo, S. Il Seok and A. Ho-Baillie, ACS Appl. Mater. Interfaces, 2017, 9, 6072–6078 CrossRef CAS PubMed
- S. I. Rahman, B. S. Lamsal, A. Gurung, A. H. Chowdhury, K. M. Reza, N. Ghimire, B. Bahrami, W. Luo, R. S. Bobba, J. Pokharel, A. Baniya, A. R. Laskar, K. Emshadi, M. T. Rahman and Q. Qiao, ACS Appl. Mater. Interfaces, 2020, 12, 41312–41322 CrossRef CAS PubMed
- T. Gallet, R. G. Poeira, E. M. Lanzoni, T. Abzieher, U. W. Paetzold and A. Redinger, ACS Appl. Mater. Interfaces, 2021, 13, 2642–2653 CrossRef CAS PubMed
- K. M. Reza, A. Gurung, B. Bahrami, A. H. Chowdhury, N. Ghimire, R. Pathak, S. I. Rahman, M. A. R. Laskar, K. Chen, R. S. Bobba, B. S. Lamsal, L. K. Biswas, Y. Zhou, B. Logue and Q. Qiao, Sol. RRL, 2021, 5, 2000740 CrossRef CAS
- D. Toth, B. Hailegnaw, F. Richheimer, F. A. Castro, F. Kienberger, M. C. Scharber, S. Wood and G. Gramse, ACS Appl. Mater. Interfaces, 2020, 12, 48057–48066 CrossRef CAS PubMed
- D. Kim, J.-H. Yun, M. Lyu, J. Kim, S. Lim, J. S. Yun, L. Wang and J. Seidel, J. Phys. Chem. C, 2019, 123, 14144–14151 CrossRef CAS
- T. Gallet, D. Grabowski, T. Kirchartz and A. Redinger, Nanoscale, 2019, 11, 16828–16836 RSC
- J. Yang, S. Xiong, J. Song, H. Wu, Y. Zeng, L. Lu, K. Shen, T. Hao, Z. Ma, F. Liu, C. Duan, M. Fahlman and Q. Bao, Adv. Energy Mater., 2020, 10, 2000687 CrossRef CAS
- C. Chen, Z. Song, C. Xiao, R. A. Awni, C. Yao, N. Shrestha, C. Li, S. S. Bista, Y. Zhang, L. Chen, R. J. Ellingson, C.-S. Jiang, M. Al-Jassim, G. Fang and Y. Yan, ACS Energy Lett., 2020, 5, 2560–2568 CrossRef CAS
- S. Tan, I. Yavuz, N. De Marco, T. Huang, S. Lee, C. S. Choi, M. Wang, S. Nuryyeva, R. Wang, Y. Zhao, H. Wang, T. Han, B. Dunn, Y. Huang, J. Lee and Y. Yang, Adv. Mater., 2020, 32, 1906995 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee01508g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |