
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous electrical pumping membrane process for seawater lithium mining†
Zhen
Li
,
Chunyang
Li
,
Xiaowei
Liu
,
Li
Cao
,
Peipei
Li
,
Ruicong
Wei
,
Xiang
Li
,
Dong
Guo
 ,
Kuo-Wei
Huang
and
Zhiping
Lai
*
,
Kuo-Wei
Huang
and
Zhiping
Lai
*
Division of Physicals Science and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: Zhiping.lai@kaust.edu.sa
First published on 30th March 2021
Abstract
Seawater contains significantly larger quantities of lithium than is found on land, thereby providing an almost unlimited resource of lithium for meeting the rapid growth in demand for lithium batteries. However, lithium extraction from seawater is exceptionally challenging because of its low concentration (∼0.1–0.2 ppm) and an abundance of interfering ions. Herein, we creatively employed a solid-state electrolyte membrane, and design a continuous electrically-driven membrane process, which successfully enriches lithium from seawater samples of the Red Sea by 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times (i.e., from 0.21 to 9013.43 ppm) with a nominal Li/Mg selectivity >45 million. Lithium phosphate with a purity of 99.94% was precipitated directly from the enriched solution, thereby meeting the purity requirements for application in the lithium battery industry. Furthermore, a preliminary economic analysis shows that the process can be made profitable when coupled with the Chlor-alkali industry.
000 times (i.e., from 0.21 to 9013.43 ppm) with a nominal Li/Mg selectivity >45 million. Lithium phosphate with a purity of 99.94% was precipitated directly from the enriched solution, thereby meeting the purity requirements for application in the lithium battery industry. Furthermore, a preliminary economic analysis shows that the process can be made profitable when coupled with the Chlor-alkali industry.
Broader contextOcean is a vast reservoir of resources that may provide a solution to a widespread concern on the future supply of lithium due to the rapid growth in demand for lithium batteries in electrical devices and vehicles. However, the extraction of lithium from seawater is one of the grand challenges among others such as direct caption of CO2 from atmosphere, removal of refractory pollutes from wastewater to name a few in separation science, because of their extremely low concentrations that generally incur formidable energy consumptions. In this report, we sieved the lithium ion by a novel glass-type ceramic electrolyte membrane and designed a continuous electrical pumping membrane process that has successfully enriched lithium by 43000 times from a real seawater sample with high separation efficiencies to all other interference ions. It was further demonstrated that the cost of energy was affordable. Hence, our method may serve as a feasible approach to secure the lithium supply for future energy usage.
|
Introduction
Lithium is quickly emerging as a strategically important commodity due to the inevitable transition from internal combustion engine vehicles to electric cars, the development of intermittent renewable energies, and the widespread use of portable electrical devices. Commercial lithium is mainly produced from land-based resources such as salt-lake brines and high-grade ores using a chemical precipitation process that is technically and economically feasible only when the lithium concentration is at a level of hundreds of ppm.1–3 However, the lithium reserve on land is limited, and is geographically uneven. In 2018, the global lithium demand reached 0.28 Mtons (Li2CO3 equivalent),1 and this will be expected to be boosted to 1.4–1.7 Mtons (Li2CO3 equivalent) by 2030, while the lithium land reserves are expected to be exhausted by 2080.3As a possible unlimited and location-independent lithium supply, the ocean contains approximately 5000 times more lithium than is found on land.3–5 However, the extraction of lithium from seawater is extremely challenging because of its low concentration (∼0.2 ppm) and the high concentration of competing ions (i.e., >13000 ppm of sodium, magnesium, calcium, and potassium ions, among others). Thus, in recent years, some innovative ideas have been proposed for the extraction of lithium from seawater, including systems based on adsorption, electrodialysis, and electrolysis, but none have shown promise for practical application. In the context of adsorption, absorbents such as FePO4,4,6 HMnO2,7–10 and crown ethers11–14 have been found to exhibit a moderate Li/Na selectivity. In addition, Cui et al.4 recently reported a pulse electrochemical adsorption process and used a TiO2 coating to improve the Li/Na selectivity of the LiFePO4 electrode. However, the adsorption process still suffers from slow kinetics, and regeneration is required. Furthermore, the electrodialysis system developed by Hoshino15–17 used a lithium-selective membrane to achieve Li/Na separation, but the lithium concentration in the recovery stream was further reduced, thereby rendering the recovery process more challenging. Moreover, Zhou et al.3 developed an electrolysis process to produce metallic lithium from seawater, but this process required concentrated LiClO4-based organic electrolytes and high voltages (>4.5 V). Mixing of the highly oxidative perchlorate with reductive metallic lithium and flammable organic electrolytes in a confined space may also cause serious safety issues.
The presence of monovalent ions, such as sodium and potassium, is not a significant issue in the conventional precipitation method since their salts are highly soluble. Instead, the lithium concentration and the ratio of lithium to other multivalent ions, such as Mg2+ and Ca2+, are the key factors to consider. In terms of separation, the membrane process is one of the most energy-efficient methods, with a potential to save up to 90% energy in many industrially important separation processes.18 In addition, this process runs continuously, and is easy to scale up.19–22 Unlike conventional membrane processes where the transport proceeds down the concentration gradient, the electrically-driven membrane process can up-grade the concentration; this system has been commercialised for use in the purification of hydrogen.21,22 As lithium possesses one of the smallest ionic sizes, we considered that it could be technically feasible to use a molecular sieving membrane to enrich lithium and to remove multivalent ions at an affordable energy cost. After enrichment, the lithium can be readily extracted using the conventional precipitation method. Thus, we herein report the design of a continuous electrically driven membrane process to enrich lithium from seawater samples of the Red Sea by 43000 times with a high Li/Mg selectivity. Lithium phosphate is then precipitated directly from the enriched solution, and a preliminary economic analysis is carried out to demonstrate the profitability of the process.
Results and discussion
Lithium extraction setup
The electrical pumping membrane process employed for the purpose of this study is illustrated in Fig. 1a and b. The electrical cell was divided into three compartments, namely the cathode compartment, the feed compartment, and the anode compartment. The cathode and the feed compartments were separated by a dense glass-type Li0.33La0.56TiO3 (LLTO) membrane with a diameter of ∼20 mm and a thickness of ∼55 μm (Fig. 1e). The LLTO membrane was prepared by sintering LLTO powder (Fig. S1a and b, ESI†) to a molten state to form a dense glass-type membrane. The high magnification SEM image (Fig. S1c, ESI†) showed that the membrane surface was smooth with no grainboundaries. The mechanical test showed that the membrane had a stress of 110 MPa and a ductility of 0.066%, which is a typical ceramic-type of mechanical property: hard but brittle (Fig. S2, ESI†). The membrane thickness is important. The membrane preparation process was optimized to yield a thickness ∼10 times thinner than those reported in literature,3,23,24 which is one of the factors critical to achieving a high Li+ permeance (Fig. S3, ESI†). From the other hand, the membrane is strong enough to form a stable membrane during the test (Fig. S4, ESI†).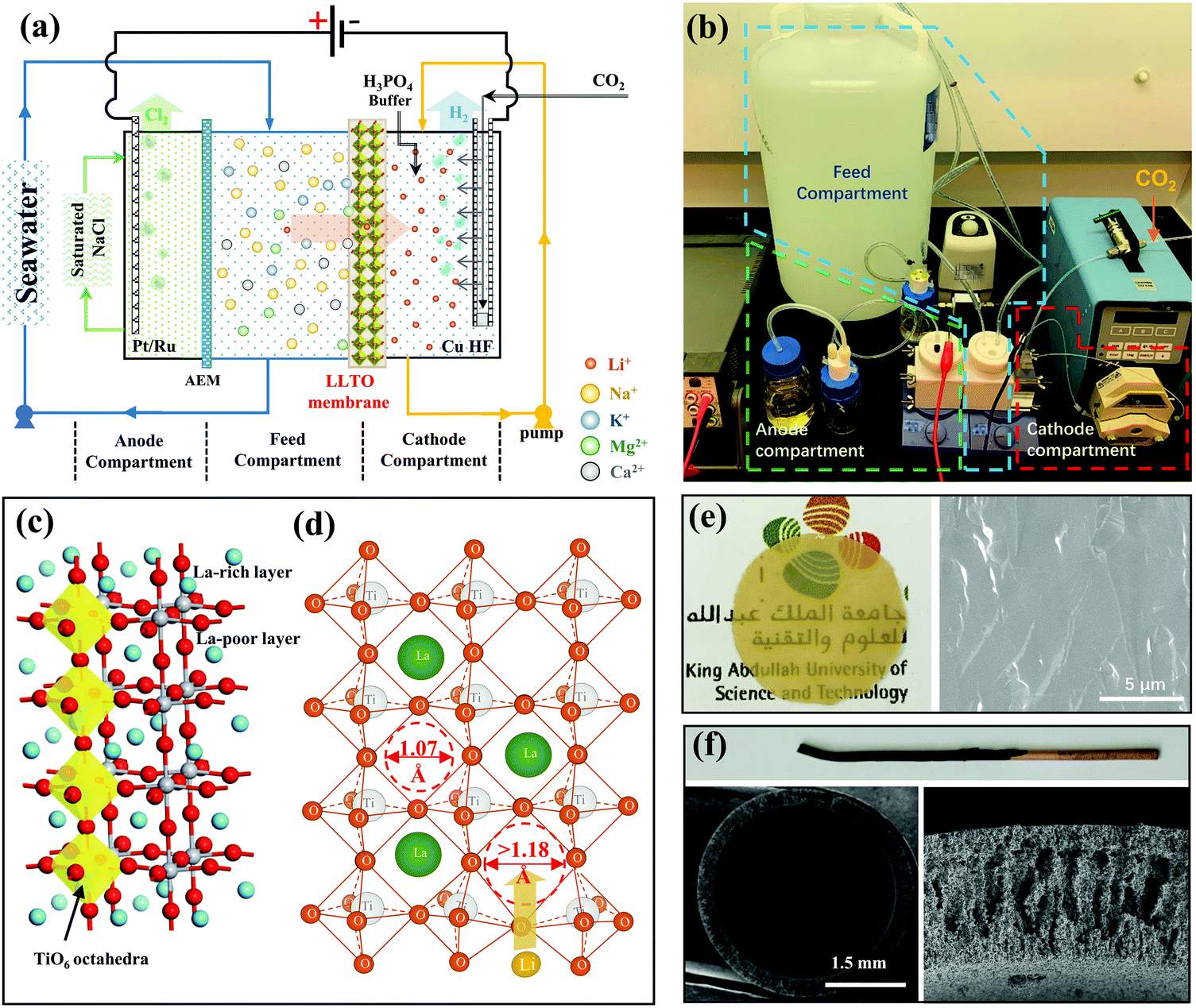 | ||
| Fig. 1 Setup of the continuous electrical pumping membrane process. (a) Schematic illustration of the three-compartment electrical cell to continuously enrich lithium from the feed solution to the cathode compartment and simultaneously generate H2 and Cl2 at the cathode and anode, respectively; (b) photographic image showing the enrichment setup; (c) the crystal structure of LLTO in ball-and-stick mode; (d) illustration of the percolation of lithium ions in the LLTO lattice; (e) images showing the glass-type LLTO membrane (∼20 mm in diameter); (f) images showing the copper hollow fibre cathode, which is coated by catalytic Pt/Ru (dark colour) at one end. | ||
LLTO is one of the superior solid-state lithium ion superconductors. Its high lithium ion conductivity and high selectivity to other ions can be explained from its crystal structure. LLTO has a perovskite-type crystal structure as illustrated in Fig. 1c.25–28 The crystal structure of the LLTO membrane was confirmed by XRD (Fig. S4, ESI†). The lattice framework of LLTO consists of interconnected TiO6 octahedra forming cubic cages that accommodate Li+ and La3+. The large La3+ ions act as support pillars to stabilize the crystal structure. The high valency of La3+ causes an alternative arrangement of La-rich layers and La-poor layers along the c-axis, and generates abundant vacancies in the structure that allow intercalation of Li+. The transport of Li+ from one cage to the others needs to pass through a square window of 1.07 Å that is defined by four neighboring TiO6 tetrahedra. The size of Li+ (1.18 Å) is slightly bigger,29–31 which requires a slight distoration in framework to enlarge the windows (Fig. 1d) and this is possible due to thermal vibrations of the TiO6 octahedra. Other ions present in seawater (i.e., Na+, K+, Mg2+, Ca2+, etc.) are much larger than lithium ion, which requires a substantial larger distortion and thus a much higher energy barrier to transport.3,15,23,24 Hence, from the property of LLTO we expect that the LLTO membrane will allow fast transport of Li+ but blocks all other major ions present in seawater.
The feed compartment and the anode compartment were separated by an anion exchange membrane (AEM) that allows the transport of anions only. The anode was a standard Pt–Ru electrode, but a metallic copper hollow fibre (Cu HF) was used as the cathode (Fig. 1f), which was further coated with 2.0 mg cm−2 Pt–Ru (Fig. S5, ESI†) to facilitate the hydrogen-evolution reaction. The copper hollow fibre had a standard finger-like porous structure,32 allowing CO2 to be introduced from the inner channel and be blown out through the porous wall, to ultimately be released uniformly into the cathode compartment. The released CO2 created an acidic environment near the cathode, which, as previously reported,33 enhances the faradaic efficiency at high current densities. Concentrated H3PO4 was further used as an auxiliary solution to control the pH, whereby CO2 and H3PO4 form a buffer solution to maintain the pH of the cathode compartment between 4.5 and 5.5 to protect the LLTO membrane from alkaline corrosion.
The feed stream was circulated between the feed compartment and a large volume feed tank (Fig. 1b). The cathode stream was also circulated through a micropump, but the volume was much smaller than that of the feed stream to ensure a stage-cut <5%, and thus maintain an almost constant feed concentration (within 5% deviation based on mass balance) during the entire process. The anode compartment was filled with a saturated NaCl solution. A voltage of 3.25 V was applied, which triggered the following electrochemical reactions at the cathode and anode.
Cathode
| CO2 + H2O + e− → HCO3− + 1/2H2↑ | (R1) |
| H3PO4 + HCO3− → xH2PO4− + 2(1–x) HPO42− + H2CO3 | (R2a) |
| H2PO4− + e− → HPO42− + 1/2H2↑ | (R2b) |
Anode
| Cl− − e− → 1/2Cl2↑ | (R3) |
During the electrical pumping membrane process, hydrogen was continuously generated from the cathode through reactions (R1) and (R2b), thereby driving the transport of lithium from the feed compartment through the LLTO membrane to be enriched in the cathode compartment. Simultaneously, chlorine gas was released from the anode compartment through reaction (R3) (Fig. S6, ESI†), since it does not dissolve in a saturated NaCl solution, thereby driving the transport of Cl− and/or other anions (i.e., HCO3−, H2PO4−, HPO42−) from the feed to the anode compartment through the AEM membrane.
Lithium extraction test
Subsequently, we demonstrated the enrichment of lithium from real seawater over 5 stages to give a level of ∼9000 ppm. In the first stage, Red Sea water (fetched from location 22° 18.38′ N, 038° 53.12′ E) was used as the feed solution and deionised water was used as the initial cathode solution. In the 2nd to 5th stages, the enriched lithium solution from the previous stage was used as the feed solution and the initial cathode stream. The operation time of each stage was fixed at 20 h. Such an arrangement allows the design of a membrane cascade as illustrated in Fig. S7 (ESI†) to achieve the lithium enrichment in a compact membrane assembly. Table 1 lists the concentrations of the major ions in seawater after each stage. With the exception of lithium, which was continuously enriched from the seawater level (0.21 ppm) to ∼9000 ppm, all other ions exhibited significantly reduced concentrations and remained almost constant after the 2nd stage. The membrane had a Li/Mg selectivity of 45916 and a Li/Na selectivity of 16277 in the first stage. While after the 5th stage, a Li/Mg ratio of 6090 and a nominal Li/Mg selectivity of more than 45 million were achieved.
| Li/ppm | Na/ppm | K/ppm | Mg/ppm | Ca/ppm | |
|---|---|---|---|---|---|
| Seawater | 0.21 ± 0.01 | 12356.40 ± 96.99 | 746.56 ± 28.42 | 1565.16 ± 22.60 | 483.59 ± 7.00 |
| 1st step | 75.17 ± 0.84 | 271.12 ± 6.30 | 5.82 ± 0.21 | 1.37 ± 0.025 | 0.54 ± 0.024 |
| 2nd step | 976.32 ± 12.03 | 299.10 ± 6.98 | 6.82 ± 0.21 | 1.41 ± 0.025 | 0.54 ± 0.025 |
| 3rd step | 2444.17 ± 24.24 | 301.53 ± 6.69 | 7.35 ± 0.22 | 1.42 ± 0.030 | 0.55 ± 0.025 |
| 4th step | 4432.51 ± 39.49 | 303.14 ± 6.41 | 7.53 ± 0.23 | 1.46 ± 0.025 | 0.55 ± 0.025 |
| 5th step | 9013.43 ± 149.83 | 305.25 ± 7.04 | 7.71 ± 0.22 | 1.48 ± 0.037 | 0.56 ± 0.025 |
Fig. 2a shows the current recorded at each stage over time, whereby it is apparent that the current remains relatively stable after a sharp surge in the initial stage, which is due to the adsorption of ions onto the electrode and the membrane. Only in stage 5 did the current decrease slightly over time. As mentioned above, the feed concentration was maintained relatively constant during the entire process, but the concentration at the cathode stream was increased continuously. Hence, the stable current in the first four stages indicates that at low concentrations, the ion transport rate is mainly determined by the feed concentration rather than the concentration difference across the membrane. From the data presented in Fig. 2a, the steady-state current was further plotted vs. the lithium feed concentration (Fig. 2b). As shown, the current increased with the feed concentration in an approximately proportional manner during the 2nd to 5th stages, but was exceptionally low in the 1st stage, thereby indicating that transport through the membrane was limited by the available lithium in the feed. The first stage is thus the rate-determining stage of the entire process, but its extraction rate, that is determined to be 13.43 mg (ppm dm−2 h−1)−1, still far surpasses the rate of traditional absorption process and electrodialysis process, as shown in Table S1 (ESI).†Fig. 2c shows the number of ions passing through the membrane at each stage. The amount of Li+ increases from the 1st to the 5th stage, which confirms the increasing transport rate upon increasing the feed concentration. In terms of the other ions, only in the first stage was there a substantial amount of Na+ passing through the membrane (i.e., ∼300 ppm). This is inevitable due to the fact that the ratio of Li/Na in seawater is so low that some Na+ can compete with Li+ to enter the LLTO lattice.4,23 However, in the remainder of the stages, all interference ions were almost completely blocked. Moreover, the total faradaic efficiencies of all stages were close to 100% (Fig. 2d). In the first stage, ∼47.06% of electrical energy was used to transport lithium, while in the remainder of the stages, ∼100% of electrical energy was used for lithium migration. Based on these data, we estimated the total electricity required to enrich 1 kg lithium from seawater to 9000 ppm in five stages to be 76.34 kW h. Simultaneously, 0.87 kg H2 and 31.12 kg Cl2 were collected from the cathode and the anode, respectively. Taking the US electricity price of US$ 0.065 per kW h into consideration, the total electricity cost for this process is approximately US$ 5.0. In addition, based on the 2020 prices of hydrogen and Cl2 (i.e., US$ 2.5–8.0 per kg and US$ 0.15 per kg, respectively),34 the side-product value is approximately US$ 6.9–11.7, which can well compensate for the total energy cost. It should also be noted that the current Cl2 utilisation capacity in the chlor-alkali industry is ∼80 Mtons y−1. Even in the case where all the world lithium capacity is produced from our extraction process, the amount of Cl2 produced will be <3 Mtons, and so will have very little effect on the total market. It is also noted that the total concentration of other salts after the first stage is less than 500 ppm, which implies that after lithium harvest, the remaining water can be treated as freshwater. Hence, the process also has a potential to integrate with seawater desalination to further enhance its economic viability.
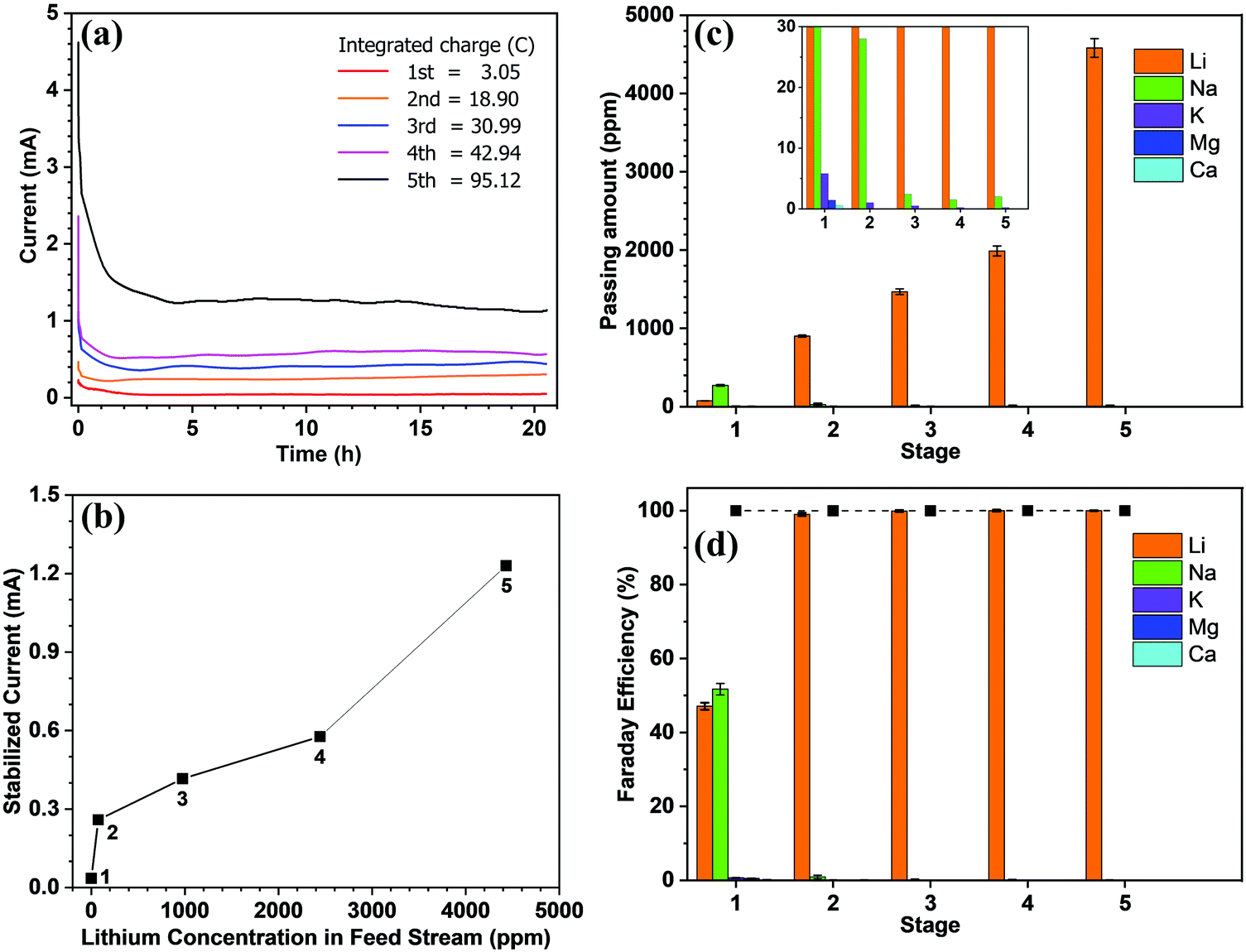 | ||
| Fig. 2 Lithium extraction from seawater using the continuous electrical pumping membrane setup (effective membrane area = 2.01 cm2, voltage = 3.25 V): (a) the chronoamperometric curve at each stage; integrating the area under the curve gives the total charge passing through the membrane in Coulombs for each stage; (b) the steady-state current vs. the lithium feed concentration at different stages; (c) the amount of different ions passing through the membrane during each stage; the inset shows the plots at low range to help view the results of the interfering ions; (d) the contributed faradaic efficiencies of the different ions for each stage. The solid squares indicate the total faradaic efficiencies at the various stages. | ||
It is further noted that the total energy consumption is proportional to the number of stages. However, the stable current curve shown in Fig. 2a implies that extending the processing time at each stage will render it possible to enrich the lithium concentration to a greater extent, and thereby reduce the number of stages (Fig. S8, ESI†). However, this will be conducted at the penalty of a low production rate. The exceptionally slow transport rate in the first stage (Fig. 2b) indicates that the lithium enrichment in the first stage will be a crucial design parameter in optimising the energy-productivity trade-off. In this study, the duration of the first stage was determined based on the product purity, which requires the Mg concentration to be <2.0 ppm.35,36 Hence, the first stage was stopped when the Mg2+ concentration reached ∼1.5 ppm, as shown in Table 1. Under these conditions, the lithium concentration reached ∼75 ppm.
Lithium product
Lithium can be readily precipitated out in the form of Li3PO4 from the 5th stage enrichment stream by adjusting the pH to 12.25 using a 2.0 M NaOH solution (Fig. S9, ESI†). The sediment was separated by centrifugation, rinsed using deionised water, and then dried under vacuum. The collected white powder (Fig. 3a) was characterised by X-ray powder diffraction (XRD, Fig. 3b) spectroscopy, whereby the XRD pattern fit well with the standard pattern of Li3PO4 (PDF#25-1030) without any impurity signals being detected. Further quantitative elemental analyses showed that the purity of Li3PO4 was 99.94 ± 0.03%, and the weight percentages of Na, K, Mg, and Ca in the product were 194.53 ± 7.80, 0.99 ± 0.02, 25.16 ± 0.83, and 17.18 ± 0.57 ppm, respectively, which meet the requirements of lithium battery-grade purity (China standard, YS/T582-2013).35,36 In addition, Fig. S10 (ESI†) showed that the product precipitated from the 4th stage could also meet the purity requirement, but could not be achieved from the 3rd stage.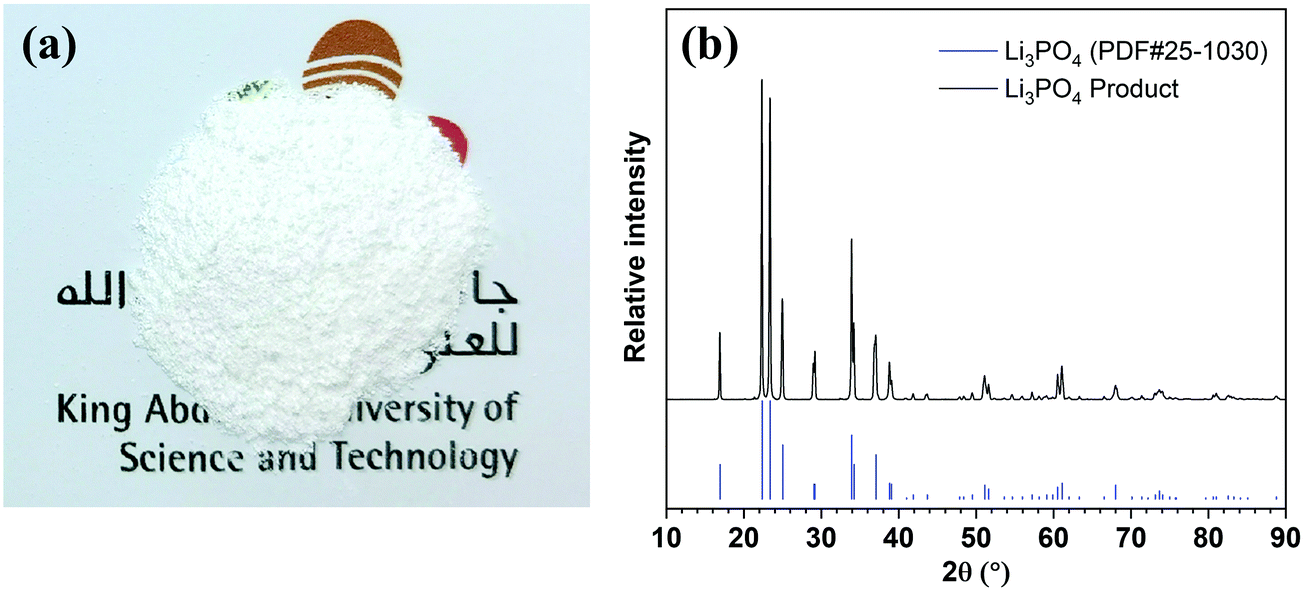 | ||
| Fig. 3 The Li3PO4 product precipitated from the 5th enrichment solution. (a) A photographic image of the collected powder. (b) The XRD pattern of the collected powder. All diffraction peaks of the product matched with the standard Li3PO4 XRD pattern. | ||
Conclusions
We herein reported the design of a continuous electrical pumping membrane process, which successfully enriched lithium from seawater samples of the Red Sea. The success of our process depends on a number of prominent features. Firstly, the thin and dense glass-type Li0.33La0.57TiO3 (LLTO) membrane provides efficient separation between lithium and other interfering ions, in addition to a high lithium permeation rate. Secondly, the separation of the anode compartment from the feed compartment by an anion exchange membrane and the use of a saturated NaCl solution in the anode compartment allow the release of Cl2. This is necessary to prevent the dissolution of the highly soluble Cl2 in the large volume of feed stream. Thirdly, the use of a CO2 and phosphate buffer solution stabilises the pH and prolongs the lifetime of the membrane. Indeed, it was found that the LLTO membrane could be used for >2000 h with a negligible decay in performance. Finally, the use of a metallic copper hollow fibre enhanced the faradaic efficiency to ∼100% in all stages. The combination of enrichment with the conventional precipitation method make the process less sensitive to the interference of soluble ions. The energy consumption is greatly reduced. Cost analysis showed that the value of the by-product could well overcome the energy cost. Although a rigorous economic analysis will be still necessary to include other capital and operating expenses, it is arguable that the energy cost is the major expenditure in this process. Furthermore, the process possesses further potential for optimisation, and for its combination with seawater desalination to create innovative designs under the energy-water nexus scheme, which will further improve the process profitability. Hence, it is expected that our approach will lead to the development of a promising process to secure the supply of lithium for future energy uses.Experimental
Fabrication of LLTO membranes
LLTO nanoparticles were prepared using a sol–gel process. LiNO3 (Sigma-Aldrich, USA, 99.99%) and La(NO3)3 (Sigma-Aldrich, USA, 99.999%) were dissolved in 25% aqueous citric acid (Sigma-Aldrich, USA, 99.0%). Subsequently, titanium(IV) butoxide (Sigma-Aldrich, USA, 97.0%) was added dropwise to the mixture under intense stirring (1000 rpm) and then heated to 100 °C to obtain a homogenous solution. The mole ratio of LiNO3, La(NO3)3, titanium(IV) butoxide and citric acid in the final solution was 0.363:0.57:1.00:6.53. The solution was dried under continuous stirring at 150 °C. The obtained solid was sintered at 600 °C for 4 h and then at 1050 °C for 20 h under air with both heating and cooling rates of 2 °C min−1. The resulting white LLTO powder was ball-milled at 300 rpm for 12 h to obtain nanoparticles of ∼200 nm in diameter (Fig. S1, ESI†). After ball-milling, the LLTO nanoparticles were loaded into a tungsten carbide (WC) pellet mould and pressed into disks to form the green bodies of the membranes with a diameter of 22 mm and a thickness of 70 μm. No binder is needed. The green bodies were sintered in a high temperature furnace first at 1050 °C for 4 h to release CO2 and NOx, and then at 1275 °C for 8 h in order to reach a molten state to form glass-type dense LLTO membranes. The heating and cooling rates of the sintering process were set to 2 °C min−1. During the sintering process, about 10% LiNO3 was vaporized. Hence, the final chemical formula of the LLTO membrane was Li0.33La0.57TiO3 determined from the ICP elemental analysis. The LLTO membrane has a perovskite crystal structure that is confirmed by XRD.
Preparation of the copper hollow fibre electrodes
The copper hollow fibres were prepared through a nonsolvent induced phase separation method followed by a high temperature sintering process.26 Copper powder (99%, ∼1 μm particle size, Shanghai Xianxin New Material Tech. Co., Ltd, China) was mixed with polysulfone (PSE, Ultrason E6020P, BASF, Germany), polyvinylpyrrolidone (MW ∼ 10000, Alfa Aesar, USA), and N-methylpyrrolidone (NMP, 99.5%, Alfa Aesar, USA) at a weight ratio of 64.4:6.2:1.5:27.9 to form a homogenous dope solution, which was then spun through a tube-in-orifice spinneret. The obtained hollow fibres were sintered at 600 °C for 3 h under air and then reduced in an atmosphere of hydrogen/argon (volume ratio = 2:8) at 650 °C for 6 h. The Pt/Ru catalyst (50% on Kejenblack, FuelCellStore, USA) was wetted with deionised water and then mixed with Nafion solution (12.5% in dimethylformamide, Sigma-Aldrich, USA) in weight ratio of 7:3. The Pt/Ru:Nafion mixture was sprayed on the copper hollow fibre surface at a level of 2.0 mg cm−2.
Process of lithium extraction
The Red Sea water sample was fetched from location 22° 18.38′ N, 038° 53.12′ E. The water sample was filtered by fine filter papers (Whatman® acid treated and low metal grade, Sigma-Aldrich, USA) and then used without further treatment. The LLTO membrane and the AEM membrane (Fumasep FAA-3-20, FuelCellStore, USA) were assembled into the electrical cell and sealed using an O-ring. The solution volume circulated in the feed was 25 L for the first stage, and 2.5 L for the remainder of the stages. For all stages, the solution volumes at the cathode and anode compartments were fixed at 1.5 and 25 mL, respectively. A catalytic Pt–Ru carbonic cloth gas diffusion electrode (FuelCellStore, USA) was used as the anode, and the Pt–Ru-coated copper hollow fibre was used as the cathode. The copper hollow fibre was connected to a CO2 gas cylinder at a controlled flow rate of 6.0 mL min−1. Concentrated H3PO4 was further used as an auxiliary solution to control the pH of the cathode compartment between 4.5 and 5.5. The released Cl2 was adsorbed by CH2Cl2 to avoid contamination of the air, while hydrogen was collected using a gas sampling bag. A constant voltage of 3.25 V was applied through a Tektronix© 2450-EC potentiostat.The nominal Li/Mg selectivity (β) was calculated by the following equation,
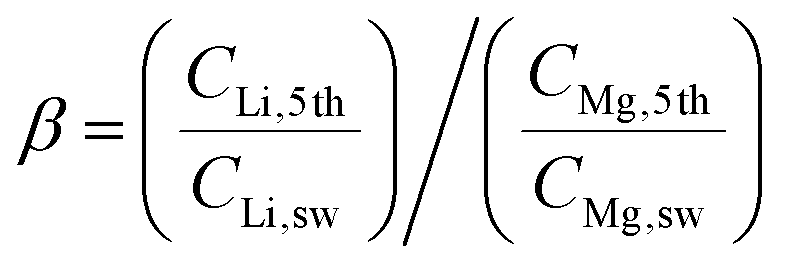
We also calculated the Li/Mg selectivity (SLi/Mg) and Li/Na selectivity (SLi/Na) of the first stage by the following equation,
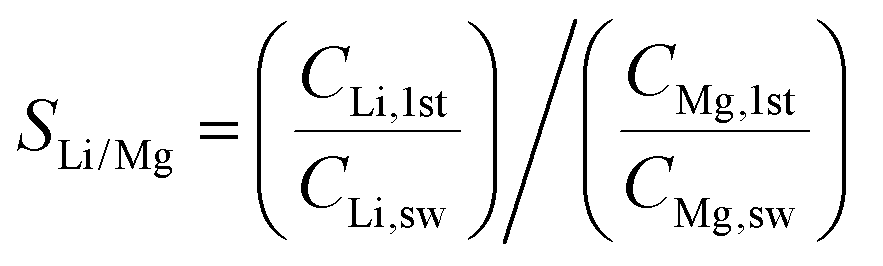
where CLi,1st, CMg,1st, CLi,sw, CMg,sw, CNa,1st, and CNa,sw, are the mole concentrations of Li+, Mg2+ and Na+ in the 1st enriched stream and seawater, respectively.

The first stage is the rate-determining stage of the entire process, and the extraction rate of the first stages was calculated by the following equation:
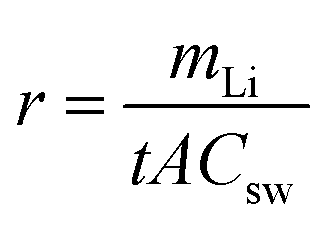
Characterisation
SEM (FEI© Magellan) with a beam energy of 5 kV was used for imaging the samples coated with a 5 nm-thick Ir film. XRD (Bruker© D8 twin) was carried out using Cu Kα radiation at 5.0–90.0° with a scan rate of 0.01° per second. ICP-OES was carried out using a PerkinElmer© Optima 8300 instrument. Liquid samples for ICP-OES were diluted with a 1% aqueous nitric acid where necessary, while solid samples were initially dissolved in a small amount of a 70% aqueous nitric acid solution using a microwave digester and then diluted to the required concentration range using a 1% aqueous nitric acid solution.Calculation of the energy consumption and the faradaic efficiency
The chronoamperometric curves shown in Fig. 2a were integrated over time to obtain the total charge, Qj, for each stage j. Multiplying the total charge with the applied voltage, 3.25 V, gave the total energy consumption, Ej, for each stage.The faradaic efficiency was calculated by the following equation,
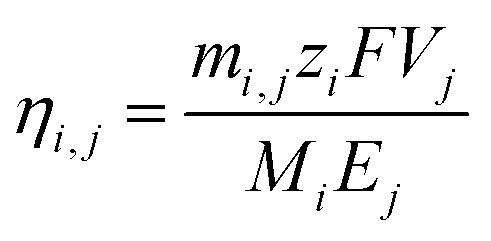
Author contributions
Z. Lai and Z. Li conceived the idea and took the lead in writing the manuscript. Z. Li conducted the lithium enrichment experiments. C. Li synthesized the LLTO membranes. X. Liu performed the ICP measurements. L. Cao and D. Guo carried out the electrochemical testing. P. Li and X. Li acquired the SEM images. R. Wei built the lithium enrichment setup. K.-W. Huang and all other authors provided critical feedback and helped shape the research, analysis, and manuscript. Z. Lai supervised the project.Conflicts of interest
All authors declare no competing interests.Acknowledgements
This work was financially supported by the KAUST baseline fund BAS/1/1375-01 and the KAUST competitive research fund URF/1/3769-01.References
- B. Swain, Sep. Purif. Technol., 2017, 172, 388–403 CrossRef CAS.
- C. Grosjean, P. H. Miranda, M. Perrin and P. Poggi, Renewable Sustainable Energy Rev., 2012, 16, 1735–1744 CrossRef.
- S. Yang, F. Zhang, H. Ding, P. He and H. Zhou, Joule, 2018, 2, 1648–1651 CrossRef.
- C. Liu, Y. Li, D. Lin, P.-C. Hsu, B. Liu, G. Yan, T. Wu, Y. Cui and S. Chu, Joule, 2020, 4, 1459–1469 CrossRef CAS.
- M. Diallo, M. Kotte and M. Cho, Environ. Sci. Technol., 2015, 49, 9390 CrossRef CAS PubMed.
- Z. Zhao, X. Si, X. Liu, L. He and X. Liang, Hydrometallurgy, 2013, 133, 75–83 CrossRef CAS.
- H.-J. Hong, T. Ryu, I.-S. Park, M. Kim, J. Shin, B.-G. Kim and K.-S. Chung, Chem. Eng. J., 2018, 337, 455–461 CrossRef CAS.
- L. Tang, S. Huang, Y. Wang, D. Liang, Y. Li, J. Li, Y. Wang, Y. Xie and W. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 9775–9781 CrossRef CAS PubMed.
- T. Ryu, A. Rengaraj, Y. Haldorai, J. Shin, S. R. Choe, G.-W. Lee, S.-K. Hwang, Y.-K. Han, B.-G. Kim and Y. S. Huh, Solid State Ionics, 2017, 308, 77–83 CrossRef CAS.
- Y. Han, S. Kim, S. Yu, N. V. Myung and H. Kim, J. Ind. Eng. Chem., 2020, 81, 115–123 CrossRef CAS.
- Y. S. Kurniawan, R. R. Sathuluri, K. Ohto, W. Iwasaki, H. Kawakita, S. Morisada and M. Miyazaki, Sep. Purif. Technol., 2019, 211, 925–934 CrossRef.
- M. Yazdanpanah, M. M. Zahedi and M. Ziyaadini, Anal. Methods, 2019, 11, 2720–2725 RSC.
- R. E. C. Torrejos, G. M. Nisola, H. S. Song, J. W. Han, C. P. Lawagon, J. G. Seo, S. Koo, H. Kim and W.-J. Chung, Hydrometallurgy, 2016, 164, 362–371 CrossRef CAS.
- Q. Zhu, X. Ma, H. Pei, J. Li, F. Yan, Z. Cui, H. Wang and J. Li, Sep. Purif. Technol., 2020, 116940 CrossRef CAS.
- T. Hoshino, Desalination, 2015, 359, 59–63 CrossRef CAS.
- T. Hoshino, Desalination, 2013, 317, 11–16 CrossRef CAS.
- T. Hoshino, Fusion Eng. Des., 2013, 88, 2956–2959 CrossRef CAS.
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed.
- P. Ball, Membr. Technol., 2000, 2000, 10–13 CrossRef.
- M. R. I. Amat, PhD thesis, Universitat Politècnica de Catalunya, 2017.
- G. Bernardo, T. Araújo, T. da Silva Lopes, J. Sousa and A. Mendes, Int. J. Hydrogen Energy, 2020, 45, 7313–7338 CrossRef CAS.
- L. Schorer, S. Schmitz and A. Weber, Int. J. Hydrogen Energy, 2019, 44, 12708–12714 CrossRef CAS.
- F. Zhang, S. Yang, Y. Du, C. Li, J. Bao, P. He and H. Zhou, Chem. Commun., 2020, 56, 6396–6399 RSC.
- X. Zhao, H. Zhang, Y. Yuan, Y. Ren and N. Wang, Chem. Commun., 2020, 56, 1577–1580 RSC.
- Y. Inaguma and M. Nakashima, J. Power Sources, 2013, 228, 250–255 CrossRef CAS.
- X. Li, J. Liang, X. Yang, K. R. Adair, C. Wang, F. Zhao and X. Sun, Energy Environ. Sci., 2020, 13, 1429–1461 RSC.
- C. Ma, C. Liang, K. More, C. Nan and M. Chi, Energy Environ. Sci., 2014, 7, 1638–1642 RSC.
- J. Bae, Y. Li, J. Zhang, X. Zhou, F. Zhao, Y. Shi, J. B. Goodenough and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 2096–2100 CrossRef CAS PubMed.
- Y. Sun, P. Guan, Y. Liu, H. Xu, S. Li and D. Chu, Crit. Rev. Solid State Mater. Sci., 2019, 44, 265–282 CrossRef CAS.
- Y. Inaguma, J. Yu, Y. J. Shan, M. Itoh and T. Nakamuraa, J. Electrochem. Soc., 1995, 142, L8–L11 CrossRef CAS.
- H. Kawai and J. Kuwano, J. Electrochem. Soc., 1994, 141, L78–L79 CrossRef CAS.
- B. Wang and Z. Lai, J. Membr. Sci., 2012, 405-406, 275–283 CrossRef CAS.
- R. Kas, K. K. Hummadi, R. Kortlever, P. de Wit, A. Milbrat, M. W. J. Luiten-Olieman, N. E. Benes, M. T. M. Koper and G. Mul, Nat. Commun., 2016, 7, 10748 CrossRef CAS PubMed.
- O. J. Guerra, J. Eichman, J. Kurtz and B.-M. Hodge, Joule, 2019, 3, 2425–2443 CrossRef.
- K. He, Z.-Y. Zhang, L. Alai and F.-S. Zhang, J. Hazard. Mater., 2019, 375, 43–51 CrossRef CAS PubMed.
- W. Liu, G.-W. Chu, S.-C. Li, S. Bai, Y. Luo, B.-C. Sun and J.-F. Chen, Chem. Eng. J., 2019, 377, 119929 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee00354b |
| This journal is © The Royal Society of Chemistry 2021 |