
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrocatalytic CO2 reduction: role of the cross-talk at nano-carbon interfaces
Michele
Melchionna
 *a,
Paolo
Fornasiero
ab,
Maurizio
Prato
acd and
Marcella
Bonchio
*e
*a,
Paolo
Fornasiero
ab,
Maurizio
Prato
acd and
Marcella
Bonchio
*e
aDepartment of Chemical and Pharmaceutical Sciences, Center for Energy, Environment and Transport Giacomo Ciamician, University of Trieste and Consortium INSTM, Via L. Giorgieri 1, 34127 Trieste, Italy. E-mail: melchionnam@units.it
bICCOM-CNR, University of Trieste, Via L. Giorgieri 1, 34127 Trieste, Italy
cCenter for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramón 194, Donostia San Sebastián, E-20014, Spain
dIkerbasque, Basque Foundation for Science, Bilbao, Spain
eDepartment of Chemical Science, ITM-CNR, University of Padova and Consortium INSTM, Via F. Marzolo 1, 35131 Padova, Italy. E-mail: marcella.bonchio@unipd.it
First published on 21st October 2021
Abstract
The electrocatalytic CO2 reduction reaction (CO2RR) is an interfacial process, involving a minimum of three phases at the contact point of gaseous CO2 with the electrodic surface and the liquid electrolyte. As a consequence, surface chemistry at composite interfaces plays a central role for CO2RR selectivity and catalysis. Each interface defines a functional boundary, where active sites are exposed to a unique environment, with respect to distal sites in the bulk of organic and inorganic domains. While the individual role of each component-type is hardly predictable “a-solo”, the interface ensemble works via a strategic interplay of individual effects, including: (i) enhanced electrical conductivity, (ii) high surface area and exposure of the interfacial catalytic sites, (iii) favorable diffusion and feeding of reactants, (iv) complementary interactions for the “on/off” stabilization of cascade intermediates, (v) a secondary sphere assistance to lower the activation energy of bottleneck steps, (vi) a reinforced robustness and long-term operation stability. Selected CO2RR case studies are compared and contrasted to highlight how the organic domains of carbon nanostructures merge with metal and metal-oxide active sites to separate tasks but also to turn them into a cooperative asset of mutual interactions, thus going beyond the classic “Divide et Impera” rule.
Broader contextInspired by Nature, conversion of CO2 into added-value chemicals needs a complex synthetic machinery, regulated by confinement of reagents, task-separation, orchestration of rates and functions by making extensive use of specialized bio-interfaces. The expectation for the next generation electrocatalysts is to rival the natural asset, through a creative design of functional interfaces and new contributions across scientific disciplines. The broader vision is to merge materials science and tailored electrocatalytic interfaces with biological routines. Taking the best of the two worlds, by coupling artificial CO2RR with biological CO2 fixation. |
Introduction
Under the Paris Agreement, the United Nations took responsibility for the control of global warming thus counteracting the risks of climate change. This priority action calls into play any possible strategy for CO2 abatement, to “achieve a balance between anthropogenic emissions by sources and removals by sinks of greenhouse gases” (Paris Agreement 5th October 2016, COP21).The current strategic plan for CO2 mitigation contemplates several approaches, among which two are expected to be highly promising (Fig. 1). The “CO2 capture and storage” approach (CCS) is based on sequestration of gaseous CO2 by absorbing materials, that can mineralize CO2 to carbonates.1 However, CCS presents the main issue of CO2 long-term storage safety and stability.2 On the other hand, the “CO2 chemical fixation” approach (CCF), recycling of CO2 into valuable carbon-containing products, offers bright horizons considering: (i) the most convenient storage of liquid CO2-derived products at ambient conditions; (ii) their potential as renewable combustion fuels, powering an overall carbon-neutral energy cycle;3,4 (iii) the added value of a circular atom economy scheme, where key commodity chemicals can be produced from CO2 as the C1-building block via its selective reduction into HCO2H, CH3OH, CH4, and/or C–C coupling products. The CO2 reduction reaction (generally referred to as “CO2RR”) can be performed with different methods including photo-, electro-, thermal and enzymatic catalysis.5 In particular, any fundamental progress on the electrochemical CO2 processing is central to the development of new electro-enzymatic and photo-electrocatalytic schemes, which are gaining increasing attention both from a mechanistic and a synthetic perspective.
 | ||
| Fig. 1 CO2 mitigation approaches highlighting the conversion of CO2 into valuable carbon-containing products, via chemical fixation through reaction pathways carved on tailored potential energy surfaces. | ||
Electrochemical CO2RR, especially if carried out in aqueous phase, is amenable to clean energy schemes and sustainable “green-chemistry” methods, when renewable sources (i.e. solar photovoltaics) are used to generate the required electrical potential, and considering mild catalytic electrohydrogenation conditions that can avoid a direct H2 supply, at high pressure and high temperature conditions. Compared to direct photo-reduction protocols operating upon a photo-induced charge separation and dictated by the photophysical properties of the photoactive materials,6 “dark” electrochemical CO2RR offers the great advantage of tuning the applied potentials (Eap), according to the kinetic and thermodynamic requirements of the envisaged products (overpotential), thus avoiding competitive pathways and favoring selectivity. Moreover, the gap between the fundamental progress on electro-catalyst development and the technical hurdles for the photo-assisted device implementation is expected to be bridged in a shorter time frame as compared to other less-mature technologies.7
The grand challenge of electrochemical CO2RR lies in the design of next-generation electrocatalysts that can sustain a high current efficiency at low overpotential (η = Eap − E0, namely the potential to be applied that exceeds the equilibrium potential, E0), while featuring a favorable selectivity towards target products, long term stability and affordable cost associated to any synthetic protocol, scale-up and recycling. The number of proposed functional molecules and materials is incessantly increasing. Among these, the synthesis of multi-phase, hybrid nano-materials is gaining a big momentum with the precise ambition to control the multi-component structure, morphology and hierarchy of the final composite, while addressing the specific functions of the redox-active core, of secondary-sphere interactions and of relevant interfacial phenomena. With this aim, the engineering of functional organic–inorganic nano-hybrids for CO2RR has the potential to set a new paradigm in the field of electrocatalysis for multi-redox transformations and small molecule activation.8–10
The main problem with CO2 reduction lies in its high chemical inertness so that CO2RR generally proceeds through a complex proton coupled multi-electron mechanism, dictated by both thermodynamic restrictions and kinetic hurdles. The purpose in the synthetic design of hybrid nanomaterials is to bring up synergistic effects that can orchestrate CO2RR by favoring a cooperative interplay of effects including absorption and confinement, multi-site/across-boundary reactivity, interfacial diffusion dynamics. These phenomena, which benefit from the co-localization of organic and inorganic domains, are expected to affect the kinetic of the CO2RR elementary steps, giving access to low-energy mechanistic pathways. Inspiration is drawn from the complexity of CO2RR in biological systems occurring at equilibrium potential and performed by specialized enzymatic machineries.4,11 However, artificial analogs of CO2 reduction enzymes are still far from the biological performance when compared under electrocatalytic conditions in terms of η, current density (j, the measured current divided by the geometric surface area of the working electrode), turnover frequency (TOF), faradaic efficiency (FE, the fraction of consumed charge actually used in the conversion to a given product), selectivity and long-term stability. Significant advancement has been made, considering bio-inspired functional guidelines to shape the electrocatalytic machinery, while avoiding a mere replica of the biological structure.12 This implies that man-made building blocks and their functional assembly will be optimized to counteract both the intrinsic fragility of natural proteins and catalytic co-factors and their high-energy processing within the enzyme active sites. Therefore, artificial catalytic packages for efficient CO2 activation will require a creative design of organic–inorganic composites, displaying modular architectures with the aim to regulate: (i) their composition at the atomic level (including structural defects, hetero-dopants, terminal groups, redox manifold, etc.) (ii) the surface/interface engineering of sub-domain boundaries; (iii) the overall morphology and hierarchical phase arrangement, thus regulating: the active sites distribution, their phase-segregation and inner-sphere proximal contacts, any competent second sphere interactions emerging from the surrounding of the active site, inter-phase reagent diffusion and stabilization of emergent intermediates.10
Here, we discuss the critical points connected with the latest progress on composite electrocatalysts for enhanced CO2 reduction, focusing on the unique role of carbon nanostructures (CNS) as the organic matrix to shape the organic–inorganic interface for the electrocatalytic CO2RR. CNS are generally considered to improve the electrical conductivity/electron transduction of the composite materials, while providing a defined nano-scaffold regulating both the morphology and the electronic properties of the metal-based active phase in the initial state and under electrocatalytic regime. As a consequence, the proper design of CNS–inorganic interface is expected to leverage charge transfer dynamics, reagent diffusion and enhanced structural stability, promoting CO2RR by a synergistic cross-talk across the phase boundaries.
Selected examples will be considered to address the added-value of the hybrid electrocatalytic interface by dissecting the role of the CNS and identifying the new functional capacity of the ensemble. To this aim, we initially highlight the CO2RR aspects that requires a multi-faceted approach to catalyst design. The perspective is to respond to the mechanistic complexity of CO2 activation by considering diverse catalytic effectors (with “effector” meaning an ensemble of modifications that regulates the catalytic behavior, including steric or electronic effects, solvation, polarization, binding, charge separation, redox) that can be implemented both by the nanocarbon network and by a tailored choice of the metal-based catalyst, thus finding the optimal synergy at their interfacial junction. This vision takes inspiration and strength from the multi-functional character of biological machineries specifically adapted for CO2 processing. A prerogative of natural systems is the interplay of hydrophobic and hydrophilic domains, as well of phase-confinement coupled with interfacial communication. The same approach can therefore be rendered within artificial architectures based on nanocarbon composites, as it is showcased by prominent examples reviewed in the following sections. Emerging research directions in this field will also be highlighted that represent new valuable opportunities for future progress.
CO2RR electrocatalysis: the selectivity issue
CO2RR poses some unique challenges, compared to other small molecule activation and energy-related reactions including: oxygen reduction (ORR), hydrogen evolution (HER), water oxidation (WOR), or nitrogen reduction (NRR). The complexity of CO2RR is by far related to the great number of possible products that can be generated under electrocatalytic conditions, implying diverse mechanistic steps and/or consecutive transformations, including the competitive HER occurring in protic media.13–15The selectivity issue calls for attention, as any benefit arising from increasing of the electrocatalyst performance might be neutralized by a selectivity loss, due to a poor control over the diverse reaction coordinates leading to multiple products.16,17 The formation of the radical anion CO2˙− by the first electron reduction occurs at very negative potentials, due to the large structural reorganization of the bent radical anion (E0 = −1.90 V, vs. SHE in an aqueous solution, pH 7),17 and stands as the rate-determining step of a multi-step reduction sequence in CO2RR. In this regard, the thermodynamic potential of proton reduction (HER) at pH 7 (E0 = −0.42 V, vs. SHE) occurs at less negative potentials than the CO2˙− radical anion formation. Overall, compared to CO2RR, HER turns out to be favored when operating in protic electrolytic solutions, such as aqueous media. This generally leads to low faradaic FE for the desired CO2 reduction product, as most of the transferred electrons are used to generate H2. Several strategies can be adopted to overcome this problem:
(i) The use of aprotic solvents and electrolytes to suppress HER;
(ii) A tailored engineering of the catalyst package in terms of its atomic-scale structure, surface and interfacial properties that favor CO2 absorption and diffusion while increasing η for water/proton reduction;18
(iii) The fabrication of porous, mesostructured electrodes, to impact wettability and proton diffusion so to inhibit HER while favoring CO2 enrichment at catalytic sites.19
Indeed, all these choices are instrumental to control the CO2RR selectivity and should be considered together. When we focus on the molecular aspects of the electrocatalytic interface, the first two strategies deserve attention.
(1) Concerning non-aqueous electrolytes, ionic liquids (ILs) may represent a greener alternative to organic solvents, for CO2 solubilization and for stabilization of charged reduction intermediates.20–24 However, due to cost issues, the scale-up of electrochemical devices using IL-based electrolytes is not straightforward. In this respect, the recent application of deep eutectic solvents (DESs) for CO2RR offers a promising perspective. DESs are usually binary/ternary mixtures of hydrogen bond donor/acceptor molecules, whose melting points are substantially lower than those of the separated components, thus exhibiting: low vapor pressure, high conductivity, a wide electrochemical potential window, and high CO2 solubility, as conventional ILs. The advantage of DESs is their ability to significantly decrease the onset potential for the CO2RR, by favoring the proton-coupled electron transfer (PCET) mechanism, which is instrumental to enhance selectivity. As in the case of choline-based DESs, these systems are generally nontoxic and less expensive than ILs, while serving as additive phases and/or organo-catalysts for the electrocatalytic CO2RR.25
(2) Besides the solvent/electrolyte nature, one key strategy to target the HER → CO2RR selectivity switch relies on the assembly of composite electrocatalysts,26 where the selectivity of CO2RR can be regulated by the applied potential. Ideally, the design of a tunable catalytic interface would allow to by-pass the first electron injection step (formation of the CO2˙− radical anion) favoring alternative mechanisms via the stabilization of diverse CO2-based intermediates. The outcome is a definite shift of the CO2RR onset potential at earlier potentials. This was shown by Kanan and Min, who used Pd nanoparticles (5 nm diameter) dispersed on carbon particles (100 nm diameter) for the direct electro-hydrogenation of CO2 to formic acid, at near equilibrium potentials, so ruling out the high-energy formation of the radical anion.27 In this case, the carbon scaffold provides a Pd/C interface with high surface area, thus facilitating the evolution to the active Pd-hydride phase. Therefore, the direct electro-hydrogenation of CO2 turns out to be the dominant pathway at quasi-reversible applied potential. The role of CNS interfaces in tuning CO2RR mechanism and the corresponding product profile will be further addressed in the next sections, by showcasing prominent examples of multifunctional electrocatalytic interfaces featuring the active metal cores and their environment (homo-metal, hetero-metal or metal-oxide environment) in close contact with the nano-carbon phase.
Collaborative catalytic interfaces and multi-functionality for selective CO2RR
Electrocatalytic CO2RR is an interfacial process,28–30 driven by a triple phase boundary at the contact point of gaseous CO2 with the electrodic surface and the liquid electrolyte. Indeed, interfacial CO2RR takes place through sequential CO2 adsorption, surface diffusion and activation at catalytic sites, and the ultimate step of product desorption. Because CO2 diffusion and accumulation are dictated by favorable equilibria at the catalytic sites, the reaction performance depends on their density and on proximal cooperative effects emerging from the active site environment. Therefore, the design of CO2RR electrocatalysts is today flourishing in the field of multi-phase materials, where optimized interfaces hold the key for enhanced catalysis, regulating the steric and electronic properties of the active sites, their distribution with a high interfacial-to-bulk ratio.Herein, types of interfaces under the lens are hybrid heterojunctions that result from the intimate contact of carbon nanostructures (CNS) with catalytically active metal (M) and/or metal oxide (MOx) domains, including single site, multinuclear cores, nanoparticles or extended surfaces.31,32 It is of general knowledge that MOx and CNS scaffolds can improve dispersion and stability of metal nanoparticles, and of single atom catalytic sites.33 Indeed, oxide-based materials are commonly used as robust heterogeneous supports for industrial catalytic applications.34 With respect to electrocatalysis, the added-value of a composite M/MOx@CNS interface lies in its multi-functionality, that can respond to the multiple requirements of the CO2RR mechanism towards the target product. This aspect is of particular relevance for selective CO2RR where specific “effectors”, emerging from the diverse hetero-junctions, can regulate competitive electrocatalytic pathways. As a consequence, in the integrated M/MOx@CNS electrocatalytic platform, reactivity and selectivity are tuned at hybrid interfaces.
For CO2 electrocatalysis, the concept of “collaborative catalytic interfaces” was proved at least a decade ago, when Hori et al. while working on pure Sn, a known catalyst for electrogeneration of HCOOH from CO2,35 noted that the formation of a SnOx native layer on the Sn electrode resulted in an 8-fold increase in j and a 4-fold increase in HCOOH production, measured as FE. In contrast, the removal of the SnOx layer reverted the catalysis to HER, revealing the key role of the metal oxide component for CO2RR selectivity. It was assumed that SnOx could either stabilize the incipient negative charge on CO2 or could act as an electron transfer mediator.36
Table 1 collects the key steps for selective CO2RR generally associated to a target product, together with a literature selection of binary (M@CNS or MOx@CNS) and ternary M/MOx@CNS electrocatalysts, where specific functional effectors are provided by the CNS interface that plays a fundamental role for the reported electrocatalytic performance. As a general concept, each interface defines a functional boundary, where active sites are exposed to a unique environment that differs from that of bulk distal sites. With the aim to pinpoint the intimate features of the electrocatalyst structure and of the multi-phase arrangement, a fundamental tool is certainly provided by the continuous advancement of specialized characterization techniques that allow to screen the evolution of the catalyst active sites under electrocatalytic regime (operando spectroscopies aided by computational studies). Recent progress in these methods has shed light on the impact of the CNS interface not only to enhance the electrocatalyst conductivity and surface area but to modulate the intrinsic properties of the active sites (Table 1), by improving interfacial electron transfer (ET) at redox-sites, dynamics of defect formation (low-coordinated M-sites and MOx oxygen vacancies), binding of CO2RR intermediates, the hydrophobicity and Lewis-base behavior and electronic properties of the active site environment (functionalized and N-doped CNS). It turns out that the introduction of the CNS organic hetero-junction opens new opportunities vis-à-vis the modulation of the CO2RR selectivity versus a target product and overarching HER (Table 1).37
| Main product and CO2RR key functional steps | M/MOx@CNS electrocatalysts | Catalytic effector and role of CNS |
|---|---|---|
| CO | AgNPs@CNT;39 | – Template and ET mediator; |
| (i) Binding to form a carboxylic acid intermediate (*COOH) | AuNPs@GNR;40 SnOx@MWCNT41 | – Enhancement of surface area and of stability; |
| (ii) Low binding energy of the *CO intermediate | – Overpotential tuning; | |
| – Increasing electrochemical surface area (ECSA); | ||
| – Tuning the electronic properties by inner sphere coordination effect, and by a second sphere assistance for the stabilization of reactive intermediates. | ||
| HCOOH | SnO2@NCL;42 Bi2O3@NGQDs;43 Bi2S3/Bi2O3@rGO;44 Bi2S3/Bi2O3@MCCM,45 Co3O4/CeO2@LGC;46 (SbNS–G)47 | – Second sphere N-doped Lewis improve CO2 absorption; |
| (i) One electron reduction to CO2˙− radical | – OCHO* stabilization and accelerated ET; | |
| (ii) Protonation to form the *OCHO intermediate | – Synergistic enhancement of oxygen vacancies in CeO2, that boost the electrochemical active area; | |
| – Enhanced catalytic activity of edge sites due to in situ exfoliation and strong electronic interaction. | ||
| HCOOH by electro-hydrogenation | PdNPs@CNP;27 Cu@CNDs;48 CeO2@CNT;49 Pd/TiO2@CNH50 | – Pd–H phase stabilization; |
| (i) Formation of reactive hydrides (M–H) followed by | – Increase proton diffusion and facilitates HCOOH desorption; | |
| (ii) CO2 insertion to form *COOH | – Facilitate Ce4+/Ce3+ reduction, oxygen vacancy formation favoring CO2 binding and charge-transport properties; | |
| – Synergic stabilization of the PdNPs at the TiO2 shell while facilitating ET and Pd-H formation. | ||
| C2H4 | CuNPs@CNPs/PTFE;51 | – Enhanced surface hydrophobicity to increase CO2 capture and coverage of *CO; |
| (i) Moderate/strong *CO binding energy allowing a cascade reduction events | CuPs@BCF;52 | – Pyridinic-N species facilitates hydrogenation and C–C coupling; |
| (ii) Stabilization of *CHxO and dimerization to *OCCO intermediates | CuNPs@OLC or @SWNT or @RGO;53 | – Stabilization of low-coordinated active sites at stepped surfaces (e.g., terraces, edges, facets, and corners); |
| – Filtering and protection effect, increase in local pH. | ||
| CH4 | CuNPs@CNP;54 | – Confinement of Cu agglomeration, maintain Cu low coordination number. |
| (i) Adsorption of H+ to *H | ||
| (ii) Hydrogenation of *CO to *CHO | ||
| CH3OH | SnOx@rGO;55 | – Facilitate the formation of mixed-valence oxides; |
| (i) Stabilization of *CO allowing multiple electron and proton transfer | CoO/CN/Ni;56 | – Enhancement of interfacial electrons transfer; |
| (ii) Electron rich surface and active sites | Co@SL-NG;57 | – The metal sub-layer improves the interfacial electron distribution at the active sites. |
| CH3CH2OH/CH3CO(OH) | CuNPs@NCS;58 Fh-FeOOH@X–C X = O, N;59 | – Multiple reactive sites with weaker binding of the OCCO intermediates; |
| (i) Stabilization of *CHO and dimerization to *OCCO intermediates | – Tuning the redox behavior of Fe(II) active sites for enhanced selectivity. | |
| (ii) Partial hydrogenation of *OCCO intermediates | ||
| Suppression of H2 (HER) | CuNPs@CNDs;48 MoSx@PEI–rGO;60 | – Block of the HER sites; |
| (i) Weak binding energy with *H | – PEI layer suppresses HER and stabilizes CO2− radical intermediate. | |
| (ii) Favorable CO2 adsorption and diffusion |
The role of carbon nanostructures (CNS)
CNS include several different types of nanometric carbon allotropes with specific morphologies and atom arrangements (i.e. C orbital hybridization). In general, their properties are due to the continuous sp2 carbon atom network, although several other structural features (dopants, defects, functional groups) as well as the overall geometry and confinement effects play an important role.61–63 The properties vary across the range of the diverse structures, and this offers the chance to select the CNS most suitable for the purpose (Table 2). In the realm of heterogeneous catalysis, and more specifically in electrocatalysis related to energy, the modern trend has focused on the use of a few well-known CNS such as graphene (G), graphene oxide (GO), reduced graphene oxide (rGO), single-walled and multi-walled carbon nanotubes (SWCNT and MWCNT respectively), carbon nanohorns (CNH). For these type of applications, the extended conjugation of the π-system enables charge flow along one, two or three directions, and this comes in conjunction with an enhanced surface area. Electron mobility and density hinges on the particular nanocarbon morphology and its band structure, with the opportunity to alter the intrinsic conductivity character (metallic, semiconducting) by controlling a number of variables during the CNS synthesis and processing.64–67| CNS | Schematic structure | Typical geometric characteristics | Typical specific surface areas (SSA) |
|---|---|---|---|
| Single-layer G |

|
10–15 nm (it can increase to several μm depending on method of preparation and number of layers) | – Theoretical SSA of 2630 m2 g−1;70 |
| – Measured SSA of exfoliated G in the range 600–700 m2 g−1;71 | |||
| – SSA ranging between 2–1000 m2 g−1 for monolayer of GO.72 | |||
| SWCNT |
|
Diameter: 1–10 nm | – Theoretical SSA of 2630 m2 g−1;70 |
| Length: 50 nm–1 μm | – SSA ∼ 370 m2 g−1 for pristine SWCNT.73 | ||
| MWCNT |

|
Outer diameter: 2.5–30 nm | – Theoretical SSA depending on diameter number of layers ranges between of 40–800 m2 g−1;70 |
| Length: 10 nm–1 μm | – Measured total SSA in the range 140–270 m2 g−1 for pristine MWCNT.74,75 | ||
| CNH (aggregate) |

|
Diameter of the spherical aggregate: 80–100 nm | – SSA ∼ 300 m2 g−1 (accounting for the contribution of interstitial surface area and individual horns);76 |
| Diameter of the tips: 2–5 nm | – SSA ∼ 1400 m2 g−1 if tip-opened;77 | ||
| Tube length: 40–50 nm | – Theoretical internal SSA for tip-opened CNH of 938 (if the adsorptive is N2 at 77 K) or 1353 (if adsorptive is H2 at 20 K).78 |
In electrocatalysis, the fine-tuning of the interfacial CNS/metal domains is required to take full advantage of such properties. In addition, the CNS may act as electron mediators with the working electrodes, thus creating better bridging with the electrolyte solution and maximizing the redox process.68 However, the understanding of the electronic properties at CNS/metal or metal-oxide interface is severely hampered by the CNS heterogeneity, which, in addition to shape and size variance, also bears a large distribution of defects and surface groups, all playing a possible role in CO2RR.69 Notable attempts in correlating CO2RR activity and selectivity with the CNS interfacial properties rely on the combination of advanced characterization techniques and computational analysis. Genovese et al. used electron microscopy, operando X-ray spectroscopy techniques and DFT simulations to unravel the origins of the high performance in C–C coupling by Fe oxy-hydroxide ferrihydrite-like nanostructures supported on O- and N-doped graphitic carbon where acetic acid evolved as a product with a FE as high as 97%59 (Table 1, Fh-FeOOH@X–C X = O, N). In this system, the Fe redox chemistry is influenced by the carbon-based environment, depending on the nature of the heteroatom dopants and on the applied potential, as highlighted by DFT calculations.
It turns out that the selective formation of CH3COOH occurs at nitrogen-coordinated Fe(II) sites at the interface with the nitrogen-doped carbon (N–C).59 Fh-FeOOH@N–C was indeed stabilized against the complete reduction to Fe(0) clusters. This is not occurring at the oxygen doped carbon interface, where extensive reduction to Fe(0) is responsible for the prevalence of the HER competing pathway in the overall range of negative potential investigated. This work is significant to highlight the complexity of the interface dynamics in CNS/metal hybrids, the synergy effect in tuning the CO2RR selectivity and the impact of the carbon phase substructure involving edges, steps, defects, etc., in directing the evolution of electrocatalytic sites.
A remarkable exploitation of “abrupt” interfaces was recently reported by Sargent et al., describing a composite gas diffusion electrode (GDL). The CO2RR catalyst (CuNP) is sandwiched within two layers, consisting on one side of carbon nanoparticles (CNP) and on the other side of a typical gas porous polytetrafluoroethylene polymer (PTFE) facilitating CO2 mass-transfer and diffusion. Herein, the nanocarbon interface plays a role in stabilizing the CuNPs domains (25 nm) while favoring the electrical contact of the redox-active sites (Table 1 CuNPs@CNPs/PTFE). In this asset, high hydroxide (OH−) concentration is maintained at the catalytic sites, where CO2 is reduced continuously at a fast rate, thus preventing the acidification of the solution via bicarbonate formation. High pH conditions are instrumental to enhance ethylene yield at the copper active sites, leading to a remarkable 70% current efficiency and stability for 100 hours operation.51
The use of 1D, 2D or 3D carbon nanostructures can template the final morphology of the hybrid composite material.10,79 In this respect, 2D graphene (G) has been widely employed for electrocatalytic applications due to the very high surface area combined with an unrivalled mobility of the charge carriers, flexibility and film robustness. Moreover, graphene-supports are known to be highly sensitive to doping and interfacial modifications, however the electrochemical response depends strongly on the graphene synthetic protocols, and therefore on the distribution and density of surface defects and on possible contaminants of the resulting materials.80
The fabrication and use of single layer graphene (SLG) supports have demonstrated a great impact for selective CO2RR. In particular, a straightforward synthesis of partially oxidized cobalt nanoparticles dispersed on nitrogen-doped SLG (Table 1, CoNPs@SL-NG) has been realized by processing single-layer graphene-oxide (GO) with Co(acac)3, H2O, n-butylamine, and DMF (Table 1).57 The surface adhesion of the Co3+ precursor is promoted by the GO oxygen sites, while the n-butylamine ligand is instrumental to prevent CoNPs and GO aggregation during the hydrothermal reduction/condensation step (220 °C for 10 h), serving as nitrogen dopant to incorporate N atoms into the graphene lattice. The electrocatalytic performance of the composite CoNPs@SL-NG was compared and contrasted with that arising from separated CoNPs and SL-NG or from their mechanical mixture, confirming the advantage of the collaborative interface in terms of: (i) current enhancement, (ii) low overpotential, (iii) CO2RR selectivity, (iv) long-term stability for 10 h of electrocatalysis. In this case, while the unsupported CoNPs lead to a mixture of formic acid and methanol, the SL-NG interface favors a multi-proton multi-electron transfer mechanism, so that a j of 4 mA cm−2 is obtained with over 70%, CH3OH selectivity, at low η (280 mV).57
The choice of introducing heteroatom dopants onto the CNS is becoming more and more popular, as it is a way to control the local polarity and therefore acidity/basicity of the carbon surface, as well as its hydrophilicity.81 Moreover, N doping atoms, in particular pyridinic N sites, have proven to function as active sites for CO2RR.82 This strategy has been effective in combination with copper-based catalysts for directing the CO2RR selectivity towards the production of C≥2 hydrocarbons. The electrocatalytic production of higher hydrocarbons from CO2 requires a complex multi-electron/multi-proton transfer mechanism leading to the formation of new C–C bonds (Table 1). Therefore, CO2RR at copper sites is highly appealing, on account of Cu natural abundance on Earth and with regard to its privileged selectivity favoring C≥2 hydrocarbons. These are versatile feed-stocks with higher energy density than C1 products, generally obtained by petroleum refining or by Fischer–Tropsch synthesis with H2 under high temperature conditions.
The crucial role of a pyridinic N-doped graphitic interface has been reported in a butterfly-wing-derived carbon frameworks used as support for Cu particles (1–5 μm in diameter). The butterfly-wing-derived scaffolds consist of small overlapped and elongated rectangular scales of two or more layers, giving rise to a quasi-periodic triangular roof-type ridges (spaced by ∼2 μm), thus showing an extended microporous structure, that guarantees a high-surface area while providing a robust anchoring for the Cu active sites (Table 1 CuPs/BCF).52 In fact, from the measured CO2 adsorption isotherms, it is confirmed that the CO2 uptake for CuPs/BCF is considerably higher than the corresponding BCF-free CuNPs, reaching values as high as 0.17 mmol g−1 at P/P0 (P = equilibrium pressure and P0 = saturation pressure) values of 1.
The porous structure and the high content of pyridinic-N in CuPs/BCF increased the CO2 absorption capacity, while facilitating electron transfer at the Cu active sites. As a result, the energy barrier for the *COOH formation is lowered at the BCF interface, resulting in a reduced Tafel slope (104 mV dec−1 for Cu Ps/BCF) with respect to the isolated components (163 mV dec−1 for copper free BCF, and 128 mV dec−1 for unsupported CuPs) (Fig. 2). The favorable electron and proton-transfer events in the near pyridinic N environment facilitate the accumulation of *CHxO intermediates at Cu sites evolving to C2H4via a C–C coupling reaction. It turns out that CO2RR occurs at the CuPs/BCF electrocatalyst with a FE > 60% for C2H4 at −1.0 V versus RHE, and with a C2H4/CH4 selectivity ratio >50 also in long test periods, which is indicative of a maintained operation stability (100% retention after 24 h). Noteworthy the observed pyridinic N–Cu synergy is effective only at the carbon–metal interface, vanishing at distal regions, as proved by control experiment where the mass ratio of the CuPs/BCF samples is varied to optimize/reduce the interfacial contact.52
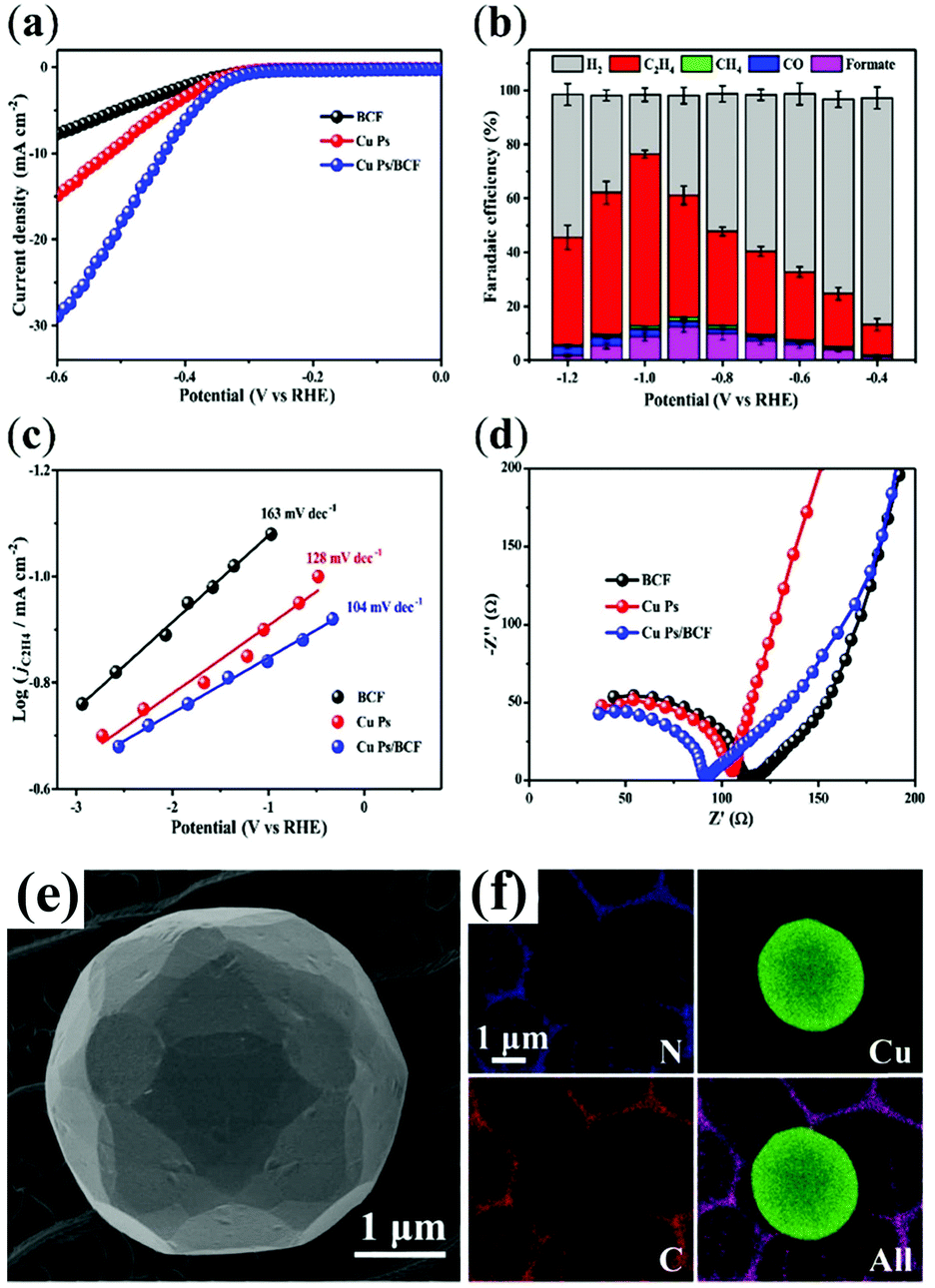 | ||
| Fig. 2 CO2RR by Cu particles supported on butterfly-wing-derived carbon frameworks (CuPs/BCF): (a) LSV curves in CO2-saturated solutions at a scan of 10 mV s−1; (b) FEs for C2H4, CH4, CO, formate, and H2 at various applied potentials; (c) Tafel plots of partial current density of CO2RR for the catalysts, demonstrating the faster kinetics of *COOH formation over the CuPs/BCF; (d) Nyquist plots showing the improved electron transfer ability of the nanohybrid; (e) SEM images of Cu Ps/BCF; (f) EDX elemental mapping of N, Cu, C and superimposed. Adapted with permission from ref. 52. Copyrights 2018, American Chemical Society. | ||
The interplay of Cu nanoparticles (20–40 nm, CuNPs) with different types of CNS interfaces has been investigated by fabricating composite electrocatalysts based on SWCNT, rGO and onion-like carbon (OLC).53 The CO2RR performance of the resulting systems has been compared and contrasted in terms of C2H4/CH4 selectivity, FE, η shift and ECSA, including control experiments using amorphous Vulcan carbon as support (Table 1, Cu/SWCNT, Cu/RGO and Cu/OLC, Fig. 3). In particular, a remarkable 200 mV positive shift of onset potentials was registered for C2H4vs. CH4 production for all CNS-based interfaces, while the Cu/OLC catalyst produced a peak C2H4 faradaic efficiency reaching 60% at −1.8 V vs. Ag/AgCl. Interestingly, the superior activity and selectivity was ascribed to the “core–shell” morphology of the organic–inorganic phases, wherein the OLC porous shell encapsulates the CuNPs. This filters and promotes the CO2 absorption, facilitates its reduction and enhances the concentration of *CO intermediates, while increasing the local pH in proximity of the Cu active sites. All these factors are known to play a key role directing the absorbed CO dimerization thus improving the C2H4 production.
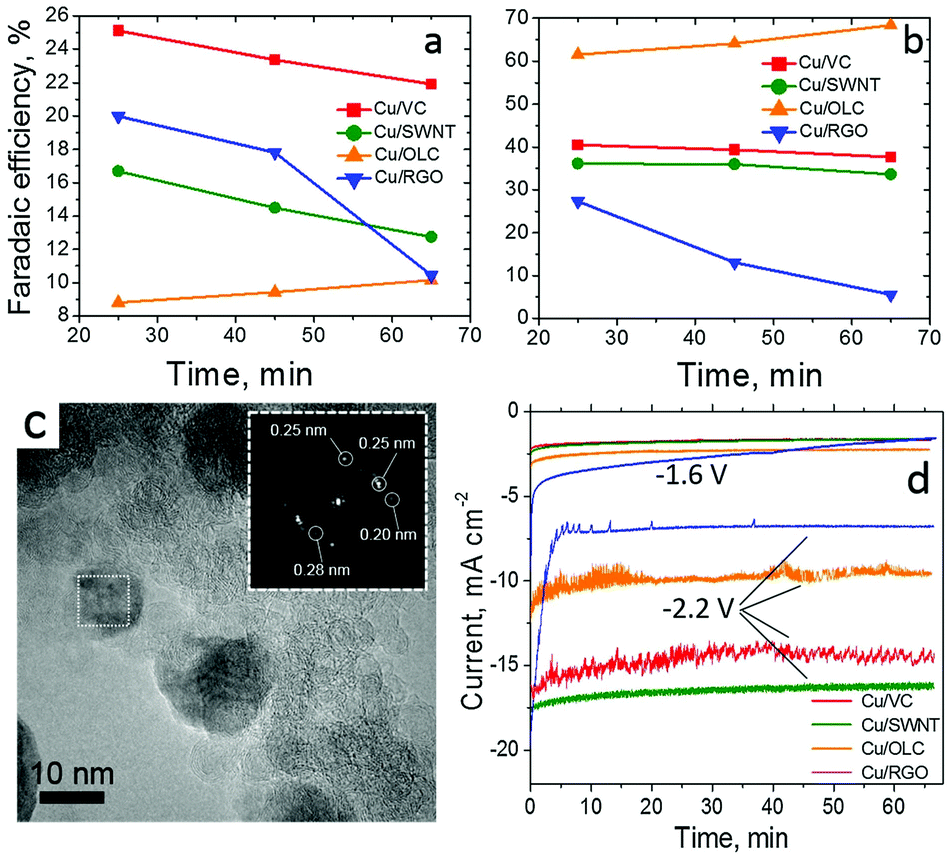 | ||
| Fig. 3 CO2RR by Cu nanoparticles supported on single wall carbon nanotubes (SWNT), reduced graphene oxide (RGO) and onion-like carbon (OLC) and Vulcan Carbon (VC). FE vs. time for (a) CH4, (b) C2H4 on thin films of 20 wt% Cu/VC, 20 wt% Cu/SWNT, 20 wt% Cu/OLC and 20 wt% Cu/RGO catalysts at −1.8 V. Conditions: CO2-saturated 0.1 M KHCO3, 1600 rpm. (c) HRTEM images of 20 wt% Cu/OLC with inset of FFT analysis of CuNPs; (d) current density vs. time measured on thin films of the composite electrocatalysts (20% Cu) at −1.6 V and −2.2 V vs. Ag/AgCl. Reprinted from ref. 53, (2017) with permission from Elsevier. | ||
To further highlight the crucial role of the CNS interface in tuning the copper-sites selectivity, a relevant case study is provided by the use of amine-rich carbon dots (CND)83 in combination with Cu coral-like structures (Table 1, Cu@CNDs).48 In this case, the hybrid interface forms by CND binding at the metal site via the surface amino groups. These latter can also play a major effect directing a preferential CND-mediated interface interaction with CO2. It turns out that CO2RR by Cu@CNDs occurs at a moderate potential of −0.7 V vs. RHE (CO2-saturated 0.5 M KHCO3, pH = 7.2), with HCOOH and CH3OH products accounting for up to 79% FE. Indeed, HCOOH is formed at low η (0.13 V), overarching the H2 evolution reaction (0.5 V) which remains the prevalent one when CND-free copper nanostructures are used as electrocatalysts. A possible explanation for the absence of carbon–carbon coupling products, can be envisaged in the specific effects of the CND amine-rich shell, favoring the desorption of HCOOH that escapes further reduction. The impact of CND terminal amines on the coordination environment of the Cu sites can be crucial to stabilize the redox manifold of the copper sites which is also a key effector to tune selectivity. Combined computational and experimental evidence indicate that the fine tuning of the Cu-redox states can be a promising tool for directing the CO2RR selectivity and that the ratio of oxidized Cu+ to Cu0 active sites can drive the formation of carbon–carbon coupling products, such as C2H4, by promoting the dimerization of CO adsorbed at adjacent sites with complementary electrophilic–nucleophilic affinity.84 Based on these conclusions, we can foresee a key role for heteroatom-doped CNS that can offer an effective environment to regulate the redox properties of Cu active sites at highly conductive electron-rich/electron-poor interfaces.84 In this respect, pulsed electrolysis techniques may be a powerful adjustment knob, e as selection of anodic pulses potential and the pulses time are able to generate in situ the suitable redox states for C≥2 products.85
CNS interfaces applied to CO2RR have been recently designed using a bottom up synthetic approach, from small molecular precursors.40 In particular, the molecular control on the synthesis of graphene nanoribbons (dimens) as support for gold NPs (Table 1, AuNPs@GNR) was found to be essential for the electrocatalytic regulation of Au active sites. The synthesis of the GNR interface can be designed to provide tuneable morphology, dimensions and electronic structure, while enabling the installation of surface functional groups to assist catalysis. The AuNPs@GNR composites (size in the range 0.2 to 1.0 μm) display a high density of AuNPs embedded within a three-dimensional nanocarbon network formed by aggregation of the individual 2D GNR, featuring ultramicroporosity (<0.7 nm) and improved electrochemical surface area (ECSA) compared to bare Au NPs (Fig. 4).40 Noteworthy the GNR composites display an onset potential with significant positive shift in the range −0.36/−0.14 V, compared to the amorphous reference Cblack–AuNP at −0.54 V vs. RHE, producing CO with increased current density (jCO) and FECO values exceeding 80%. This observation provides a direct proof of the intrinsic change of the electronic properties of the active sites as a result of the Mott–Schottky heterojunction formation (Fig. 4).40 Moreover the GNR functionalization with peripheral methyl carboxylate groups leads to a further improvement of the CO2RR performance (Fig. 4C and D). The impact of the GNR functionalization on the CO2RR kinetics was addressed by Tafel analysis, showing slope values for GNRs 1 and 2a (144 and 129 mV dec−1) that are similar to the one of the Cblack–AuNP reference (141 mV dec−1), and consistent in all cases with electron transfer forming the CO2 radical anion in the rate determining step. A lower Tafel slope of just 66 mV dec−1 was found in the case of the 2b derivative, decorated with the methyl carboxylate residues. This observation points to a favorable change in the CO2RR mechanism, which is consistent with a fast electron transfer followed by a rate-limiting chemical step. As a result, the composite AuNP@GNR-2b is the best performing electrocatalyst, indicating that the Au active sites can benefit from a molecular optimization of the GNR terminals, involved in the stabilization of the transition state by a “secondary sphere” effect on the electrocatalytic mechanism.
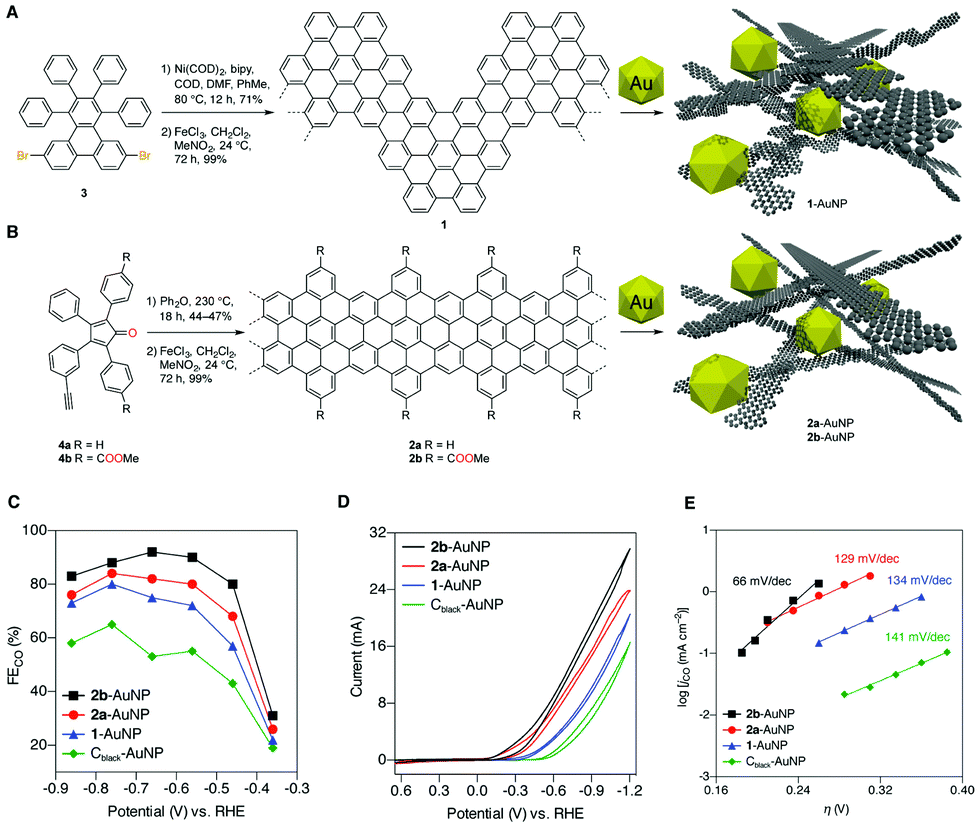 | ||
| Fig. 4 (A) and (B) Design and bottom-up synthesis of GNR–AuNP composite materials and their graphical representation. (C) Faradaic efficiencies for CO production (FECO) by 1-AuNP, 2a-AuNP, 2b-AuNP, and Cblack–AuNP composite electrodes. (D) Electrolysis performed at potentials from −0.37 to −0.87 V vs. RHE in 0.5 M aqueous KHCO3 saturated with CO2 (pH 7.3), with relative cyclic voltammograms for the four composites. The performance of Cblack–AuNP serves as a standard reference. (E) Tafel analysis of CO2RR. Adapted with permission from ref. 40. Copyrights 2017, American Chemical Society. | ||
Along the same lines, a strong electronic interaction has been invoked for a few-layer Sb nanosheet (SbNS) material integrating a graphene interface (G), which is conveniently synthesized in situ by simultaneous cathodic and anodic exfoliation of Sb bulk crystals and graphite respectively.47 The resulting electrocatalyst (SbNS–G, Table 1) outperforms the graphene-free SbNS and the bulk Sb crystal, by converting CO2 to formate at a lower η (0.87 V, CO2-saturated 0.5 M NaHCO3 solution, pH 7.2) and with a FE for formate of 88.5%, while minor products were confirmed to be H2 and CO. The enhanced performance is ascribed to the electrons migration across the Sb-G boundary from graphene to the Sb domains, thus increasing the electron density at the surface of SbNS. Indeed, the electronic distribution at the SbNS–G interface can modify the binding energies of the CO2 reactant and intermediates while tuning the mechanistic envelope of CO2RR. This latter aspect is demonstrated by the consistent decrease of the Tafel slope upon evolution of the electrocatalyst composite, from the pristine Sb bulk (324 mV dec−1) to the exfoliated SbNS (242 mV dec−1) and up to the composite SbNS–G (110 mV dec−1). The remarkable variation of the Tafel slope parameter is associated to a change in the rate-limiting step occurring at the Sb–G interface, facilitating CO2 adsorption so that its one-electron reduction to the *CO2˙− radical anion becomes rate determining.47
The interfacial tuning of CO2RR can benefit from a tailored functionalization of the CNS surface with suitable organic pendants, installed according to optimized synthetic protocols. This was recently demonstrated by preparing SnOx nanosheets/MWCNT hybrids, featuring three different types of pendant groups, namely –COOH, –NH2 or –OH terminals (Table 1, SnOx@MWCNT, Fig. 5).41 The role of the terminal group with diverse proton and electron donor properties can be traced at multiple levels: (i) determining the SnOx loading, as a function of the improved affinity of the nanocarbon surface for the metal oxide phase; (ii) increasing the available ECSA, due to the un-bundling of functionalized MWCNT providing an enhancement of their specific surface areas, and favoring the anchoring of the inorganic phase onto the nanocarbon surface; (iii) tuning the electronic properties of the Sn active sites by a direct inner sphere coordination effect, and by a second sphere assistance for the stabilization of reactive intermediates. Indeed, the experimental results confirmed that both the activity and the selectivity of CO2RR was tuned by the MWCNT functionalization, and that SnOx@MWCNT–NH2 displayed an improved loading of active sites, corresponding also to an enhanced ECSA and a nearly 100% selectivity for CO with maximal j.41 Further analysis on the impact of the organic domains on the CO2RR selectivity needs to be addressed by drawing predictive structure–activity relationships that set the basis for a critical discussion.57 Moreover, several examples in the literature are also showcasing the use of polymeric additives to modify the CNS surface with the primary goal to boost CO2RR while suppressing competitive HER.60,86 In this respect, amorphous molybdenum sulphide (MoSx) was used in combination with graphene oxide (rGO) and polymeric additives including: polyethylenimine (PEI), diallyldimethylammonium chloride (PDDA) or polyethylene glycol (PEG) for the fabrication of MoSx@polymer–rGO modified electrodes (MoSx@PEI–rGO, Table 1) applied to CO2RR in CO2-saturated 0.5 M aqueous NaHCO3 solutions.60 Remarkably, the PEI modified electrocatalyst is capable of reducing CO2 to CO at an overpotential as low as 140 mV, while reaching a maximum FE of 85.1% at an overpotential of 540 mV, and a TOF value of 2.4 s−1 for CO production. This result is unique among the electrocatalysts fabricated with PDDA or PEG polymers, or considering the CO2RR performance and selectivity for the isolated components and for the binary assemblies rGO–PEI, rGO–MoSx, as in all these other cases, HER generating H2 occurs as the dominant process. Mechanistic evidence indicates that the PEI environment slows down the HER kinetics in protic media, as a result of a larger overpotential, ascribed to a specific effect of the polymer amino groups. Vice versa, stabilization of the reduced CO2˙− intermediate through hydrogen bonding and electrostatic interactions with PEI is at the origin of a Tafel slope as low as 74 mV dec−1 obtained with the MoSx@PEI–rGO electrode for CO production, thus explaining the enhancement of the CO2RR selectivity due to the suppression of the HER process (Table 1).
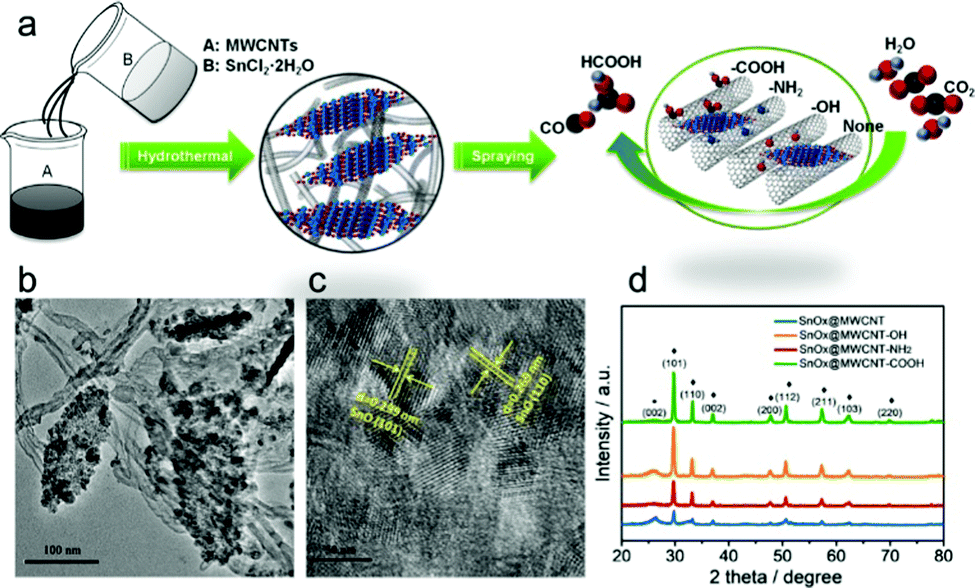 | ||
| Fig. 5 (a) Sketch of the synthetic scheme for SnOx/CNT catalysts, allowing uniform laying of the SnOx nanosheets, (b) and (c) TEM and HRTEM micrographs of the materials, (d) XRD of the nanohybrids with different functional groups on the CNT. Reprinted with permission from ref. 41. Copyright Wiley and sons. | ||
It should be considered that the use of CNS supports in combination with gas-diffusion layers is one key technology to enhance CO2RR, by increasing the local CO2 concentration and its diffusion at the active sites. In this respect a bio-inspired control of hydrophobicity at both the nanoscale and microscale surface structuration, can leverage the so called “plastron effect”, as a result of a gaseous layer trapped at the surface of the electrode.87 The CO2-trapping phenomenon at the electrolyte–electrode interface forms a triple-phase boundary hampering water diffusion and proton reduction while improving the CO2RR selectivity.87
In summary, the case studies reported in Table 1 show that the impact of CNS for CO2RR electrocatalysis can be envisaged at different levels:
(i) CNS with diverse aspect ratios and dimensionality offer a tunable platform to template the morphology of the composite electrocatalyst, tuning the surface area and porous texture (ii) the intimate contact with the metal/metal-oxide phase provides a local modification of the active site properties including the redox state distribution, the density of defects, the hydrophobicity of the environment, with impact on the electron and mass transport phenomena that can modulate the CO2RR selectivity.
(iii) Alterations in crystal packing and in chemical bonding on the CNS surface can be responsible for specific activation/stabilization effects of CO2RR intermediates, thus modulating the kinetics and selectivity of CO2RR.
(iv) Engineering of hydrophobicity at the electrodic surface by a combined use of CNS and polymeric coatings is instrumental to suppress the competing HER.88
Hierarchical metal/metal-oxide@CNS interfaces
The assembly of hierarchical systems offers a multi-level arrangement of catalytic interfaces.89,90 In the realm of hierarchical structures, the core–shell motif emerges as an appealing choice for tailoring the catalyst properties. In particular, the interfacial confinement of metal NPs within a porous metal-oxide environment is expected to be crucial for selective CO2RR. The MOx phase is instrumental considering a combination of beneficial effects to enhance CO2RR, namely: (i) promoting CO2 adsorption at the porous nano-oxide architecture, can increase its concentration at active sites and accelerate its conversion; (ii) facilitated mass transport and gaseous product desorption at tailored metal-oxide surfaces can be a winning strategy to tune kinetics, tandem reactions and the selectivity outcome; (iii) the control of the local pH by buffering the acidity/basicity conditions after the electrocatalytic event can suppress competitive HER; (iv) control of the redox sites dynamics and oxygen vacancies at the interface will impact the CO2 activation modes and the stabilization of reduction intermediates.91 In this light, the fabrication of a triple electrocatalytic interface by integrating the M/MOx motif with the CNS surface is pivotal to mitigate the conductivity-loss ascribed to the insulating oxide phase while mediating interfacial ET to regulate the MOx redox behavior and to shuttle electrons at the active metal sites.38While several examples showcase the prominent appeal of core–shell M/MOx interfaces for CO2RR,92 their combination with CNS surfaces is still underexplored. (Table 1, Pd/TiO2@CNH).
We have recently reported the synthesis of a triple phase interface that is instrumental to boost electrocatalytic CO2RR (Table 1).50,93 Ternary hybrids built on 3D-carbon nanohorn templates (PdNP/TiO2@CNH) with a hierarchical core–shell morphology, exhibited an unprecedented selectivity for formate production, at near equilibrium potential (Fig. 6). Interestingly, Pd-assisted CO2 electro-hydrogenation27 occurred in a broad potential window, and preventing a parallel formation of CO, which is known to poison Pd NPs. As a result, the long term stability of the electrocatalyst was considerably improved.50 Moreover, the conductive and high surface area of CNHs can facilitate electron transfer to the active sites and improve CO2 mass transport versus proton diffusion, thus suppressing HER. Moreover, the presence of TiO2 has the double function of stabilizing the Pd NP, thus avoiding aggregation, and contribute to the fine-tuning of the CO2/H2O binding equilibria. Interestingly, H2 production is associated to the reversible formate decomposition that takes place at near equilibrium potential.50 The hierarchical design of the PdNP/TiO2@CNHcatalyst notably allowed high activity with low loadings of the Pd precious metal, reaching a TOF of 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 h−1 at −0.2 V vs. RHE, which sets a new benchmark in the field.
500 h−1 at −0.2 V vs. RHE, which sets a new benchmark in the field.
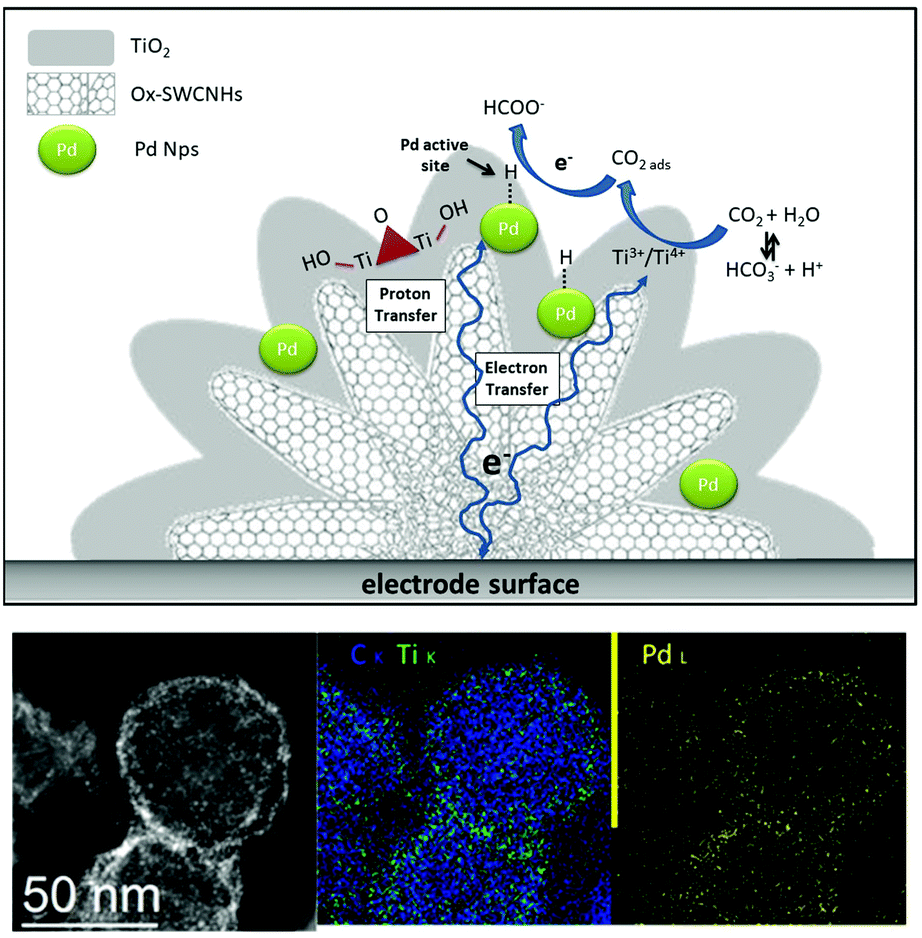 | ||
| Fig. 6 Top: Graphical sketch of the PdNP/TiO2@CNH nanohybrid and of the interfacial synergy towards the reversible HCOOH formation; bottom: HAAFD (black and white) and EDX elemental mapping of C, Ti and Pd confirming the co-location of the three elements. | ||
A considerable step forward was achieved by exploiting a Pd-free CeO2@MWCNT electrocatalytic interfaces for CO2 reduction to formic acid. Operando EXAFS analysis is consistent with the involvement of transient ceria-hydride species being responsible for a direct electro-hydrogenation step. Reduction of ceria and migration of Ce3+ defects appears to be facilitated by the close contact with the conducting MWCNT surface (Fig. 7).49
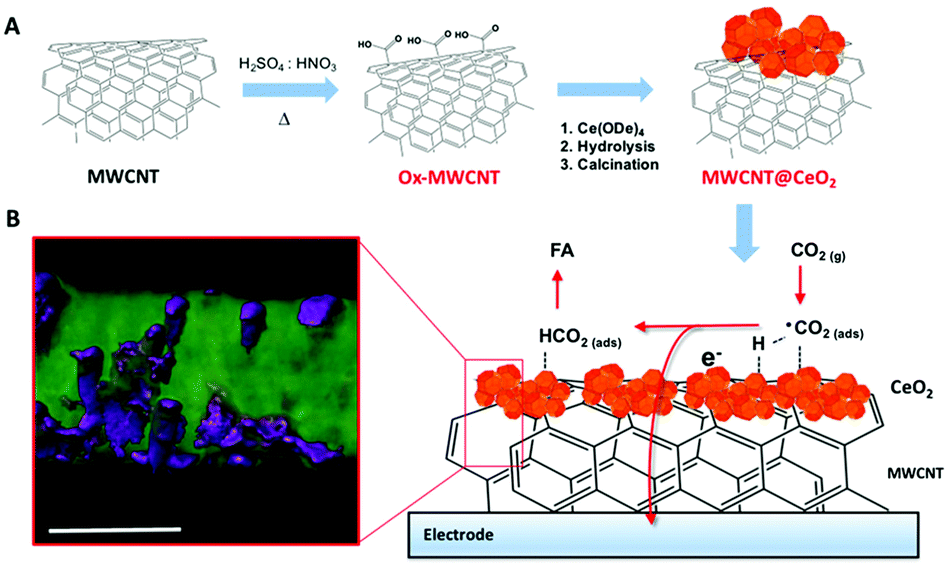 | ||
| Fig. 7 (A) Schematic of MWCNT@CeO2 synthesis involving a first oxidation step of the MWCNT scaffolds, followed by decoration with CeO2-NPs, grown on the MWCNT surface by controlled hydrolysis of Ce4+ tetrakis(decyloxide), Ce(ODe)4, and calcination at 250 °C. (B) STEM tomographic reconstruction of MWCNT@CeO2 (the region of high density corresponding to the CeO2 is rendered with a violet mesh) and sketch of the possible mechanism of CO2 hydrogenation to formic acid. Scale bar, 20 nm. Reprinted with permission from ref. 49. Copyright (2020) American Chemical Society. | ||
Indeed, CeO2 is being considered for CO2RR due to its rich redox chemistry associated with a dynamic evolution of oxygen vacancies under electrocatalytic conditions. These properties are expected to induce additional binding states of key intermediates and direct the CO2RR selectivity.94
Oxygen vacancies have been identified as one crucial feature in various other metal oxide-based electrocatalysts, by virtue of surface charge modulation, which opportunely alters CO2 adsorption and activation, while DFT calculations have confirmed that the CO2RR active sites turn out to be located at the metal–metal oxide interface.95 Moreover, the electrocatalytic activity was found to depend on the percentage of reduced Ce3+ sites, which is facilitated by redistribution of oxygen vacancies from bulk to surface.96,97 A recent example of a three-phase system is based on a CuOy–SnOx/CNT material where the relative composition CuOy/SnOx was systematically varied in order to understand the role of each component in directing the CO2RR process. The product distribution could therefore be changed by altering the atomic % of the metal oxides. The choice of CNT as the model carbon support was not random, as the authors confirm that CNT were ideal carbon platforms guaranteeing both stable anchoring and dispersion of the Cu and Sn species while providing electron conduction pathways during CO2RR catalysis. Therefore, pivotal to the good performance was the efficient contact between the three components.98 An interesting type of hierarchy was obtained by a multi-step synthetic sequence, where the carbon phase, more specifically N-doped carbon (NC), was sandwiched between CoO NPs and Ni NPs (Table 1, Ni/CN/CoO), and its role was to facilitate the electron transfer from the underlying Ni to the catalytically active CoO NP. This ternary hybrid proved to be efficient for the formation of CH3OH as the main product with a FE(CH3OH) of 70.1%, thanks to the electron modulation of the Ni NP, while the reference binary composite without Ni (CN/CoO) led to prevalent formation of HCOOH. Mott–Schottky analysis provided evidence on the alteration of the conduction band (CB) in the presence of the Ni NP, which turns out to be more negative (−0.48 V vs. −0.38 V for the CN/CoO composite), implying a charge redistribution at the CoO/CN interface which drives the selectivity towards methanol (Fig. 8).56
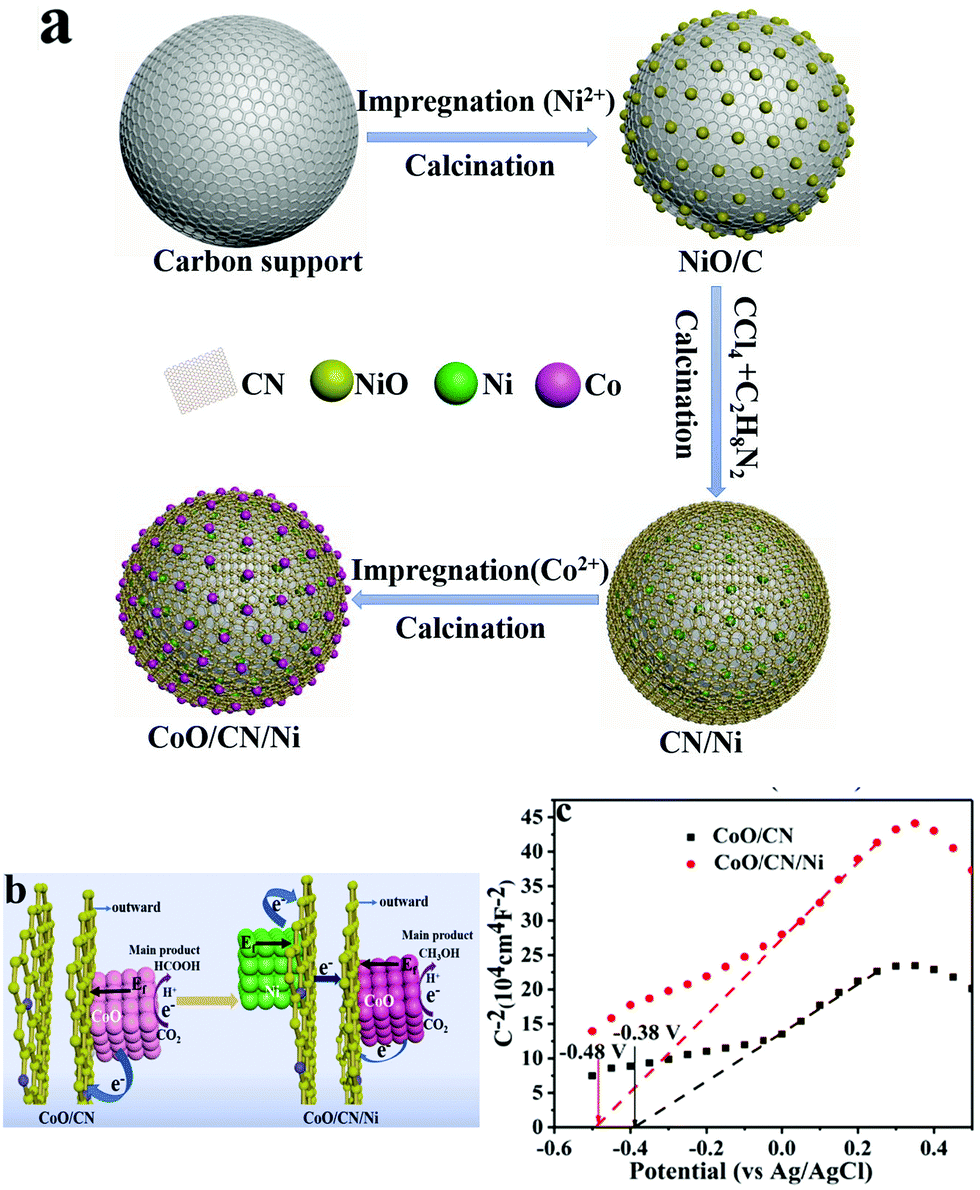 | ||
| Fig. 8 (a) Sketch of the synthetic sequence for the assembly of the ternary CoO/CN/Ni catalyst; (b) graphical representation of the factors controlling product selectivity in CO2RR by the binary and ternary catalysts; (c) Mott–Schottky analysis of the ternary hybrid and of the Ni-free binary catalyst. Reprinted from ref. 56, (2021) with permission from Elsevier. | ||
In the field of molecular metal oxides, polyoxometalates (POMs) deserve attention as this class of molecular oxides have been successfully combined with metal NPs99 and with CNS surfaces.100,101 A remarkable observation was that a highly challenging CO2RR product such as acetate could be formed with FE ca. 49% and a very high j (∼110 mA cm−2) by combining copper nanocubes with a molybdenum-based POM.26 The outstanding performance originates from the interfacial Cu–O–Mo sites (confirmed by XAFS), whereby the Mo modifies electronically the Cu local structure to tune the product selectivity. According to DFT calculations, the key intermediate is *CH3, which can react with CO2 forming acetate with a lower energy profile. Small amounts of other products such as methane, ethylene and ethane, were observed depending on the fractional Cu surface not being covered with the Mo-containing POM, thus highlighting the key role of the Cu–O–Mo interface.26
Conclusions and emerging directions
The engineering of hybrid interfaces can implement CO2RR electrocatalysis by offering the optimal combination of efficiency, selectivity and long-term robustness. Considerable progress has been achieved in the field, guided by an impressive advancement of time-resolved spectroscopies and modelling studies. The fine-tuning of the CO2RR active site properties by a multifaceted interplay of steric and electronic effects can be regulated at the CNS interface, merging the gap between homogeneous and heterogeneous catalysis. Indeed, optimization of metal site stereo-electronics is a well-established concept for molecular catalysis based on the directed evolution of the ligand design (primary sphere of the metal site) and on the role of the local environment (secondary sphere of the metal site). This latter can provide the active site with specific chemical interactions (ionic, acid/base, hydrogen/halogen bonding, etc.) and constrains (geometrical/steric via intermolecular interactions). The same approach can be transferred to heterogeneous electrocatalysts by a proper choice of hybrid organic–inorganic domains where the nanocarbon interface can leverage both primary and secondary sphere effectors installed by surface functionalization methods (Fig. 8). Herein, we will highlight three emerging trends in anticipation of future developments:(1) Surface engineering: controlling facets and defects at multi-phase hybrid interfaces
When a polycrystalline material is considered, the precise identification of the structural features (step, kink, terrace, vacancy, grain boundary) that governs CO2RR is perhaps impossible. Contributions from the different structural elements all sum up, and discerning priorities and synergies is a formidable challenge. One approach to address this complexity is to correlate the binding energies of CO2 and of its reduction intermediates with different structural elements. Typically, single crystals are used as model catalysts to ascribe catalytically relevant structures at specific crystal facets, taking into account that crystallographic hkl indexing is critical for a reliable calculation of their binding mode energetics. However, because of intrinsic limitations of single crystal catalysts in terms of low j, the key step is to synthesize active catalysts with preferential faceting, i.e. controlling the selection of facets of metal nano-particles by suitable effectors. This research opened up a new direction for the engineering of catalytic surfaces with enhanced performance. Single crystal Cu electrodes has served as excellent examples for evaluating the importance of the crystallographic faceting for tuning CO2RR selectivity, particularly in relation to C–C coupling products. Over the years, converging evidence has been collected showing that control on activity and selectivity of Cu surfaces can be attained by determining specific (hkl) directions for crystal growth.102–104 General trends have been drawn for some particular facets of the Cu single crystal, in particular for the (100)-facet, which seems to favor C≥2 products, while CH4 is mainly observed at (111)-facets.104–106 The potential-dependent selectivity of Cu(100), (111), and (751) electrocatalytic thin films prepared by physical vapor deposition (PVD) was investigated by in situ electrochemical scanning tunneling microscopy. This technique revealed that under-coordinated active sites lead to higher selectivity towards C–C coupling products, while with Cu(751) the oxygenated/hydrocarbon product ratio was the highest.107 These results highlight the importance of the crystal growth orientation on suitable extended interfaces, thus paralleling the results obtained with small-dimension single crystals.107 Very recently, Cu single crystals with various morphologies and faceting have been used for the fabrication of GDE, in electrochemical cells where the observed current densities are significantly higher.108 However a game-changer approach would be to direct the growth of active facets by a suitable choice of the hybrid interface contact as demonstrated in the case of graphene-based nanomaterials.109In the realm of structural–activity relationships, one key aspect is the introduction of defects with “ad hoc” distribution and morphology. For CO2RR, the occurrence of surface defects, i.e. atom vacancies, can modulate the CO2 adsorption and the binding energies of emerging intermediates, which results in a change of activity and selectivity.110 It was for example demonstrated that a defect-rich Bi2S3–Bi2O3@rGO nanohybrid interface is determinant for the CO2 adsorption. However, this interface requires to be appropriately tuned as a too high co-localization of vacancies may result in fragility, deterioration and conductivity loss.44 In particular, the ability of oxygen vacancies to lower the activation energy barrier for stabilization of HCOO−* intermediate was recently demonstrated for Co3O4 layered catalysts.97 ZnO nanosheets displayed a CO2 to CO activity that proportionally increased with the content of O vacancies,96 while the presence of oxygen vacancies in Cu/CeO2 was instrumental for accessing catalytic sites for selective reduction to methane.111
The formation of O vacancies in metal oxides can be generated through several pathways such as reduction with H2 or with other chemical reductants (i.e. NaBH4), thermal treatments, plasma-assisted methods, ultrasonication and others, and some of these methods can also be extended to other non-oxide materials such as dichalcogenides or nitrides, where S, Se or N vacancies have been generated.112–114 With this aim, hybrid nanocomposites integrating carbon nanostructures can leverage a highly efficient interfacial charge transfer under electrocatalytic regime, providing a favorable shaping of the metal-oxide phase defects thus boosting CO2RR.49
(2) Bio-inspired catalyst design: shaping cooperative and cascade mechanisms at multi-phase hybrid interfaces
Inspiration from natural born catalysts, i.e. enzymes, has been one priority mission of biomimetic inorganic chemistry, with the twofold aim of (i) providing a better understanding of biosynthetic pathways and (ii) discovering new catalytic manifolds with exceptional selectivity and efficiency rivalling the biological systems within artificial environments.115 Major breakthroughs have been reported in the field of homogeneous catalysis and functional molecular systems, which can be designed to replicate bioinspired mechanistic features. The same vision translated into heterogeneous surfaces, bulk materials and hybrid nano-composites is now considered one emerging research direction with great appeal for electrocatalytic applications.116 Indeed, Nature has adopted a most effective task-separation, modular approach to orchestrate multiple-functions by making extensive use of interfaces and hybrid organic–inorganic domains for biological CO2 processing. Natural enzymes such as carbon monoxide dehydrogenase (CODH) and formate dehydrogenase (FDH) can interconvert CO2, CO, and formate under mild conditions at equilibrium potential. These enzymes exploit a synergy of effects resulting from tailored hydrophobic/hydrophilic protein domains, a “hard–soft” metal coordination environment and multi-site electron and proton transfer pathways, which are tuned by specific second-sphere and long-range stereo-electronic effectors. This strategy can be ideally transferred to the fabrication of organic–inorganic, multi-phase electrocatalytic platforms, shaped along bio-inspired guidelines but using totally synthetic building blocks.50 In particular the combined use of metal/metal oxide domains and carbon nanostructures offers a wide space to explore the impact of the first and second sphere effects on the electrocatalytic active sites. This implies a tailored engineering of the interfacial chemistry at the molecular scale including the positioning of: (i) hetero-metals and/or proximal lattice defects; (ii) localized charges; (iii) proton donor/acceptor groups; (iv) spacers and/or sterically orienting groups (Fig. 9). Moreover, organic additives or surface coatings have been found to enhance the electrocatalyst performance, selectivity and long-term stability by virtue of modifications of surface sites and their binding properties of specific intermediates.117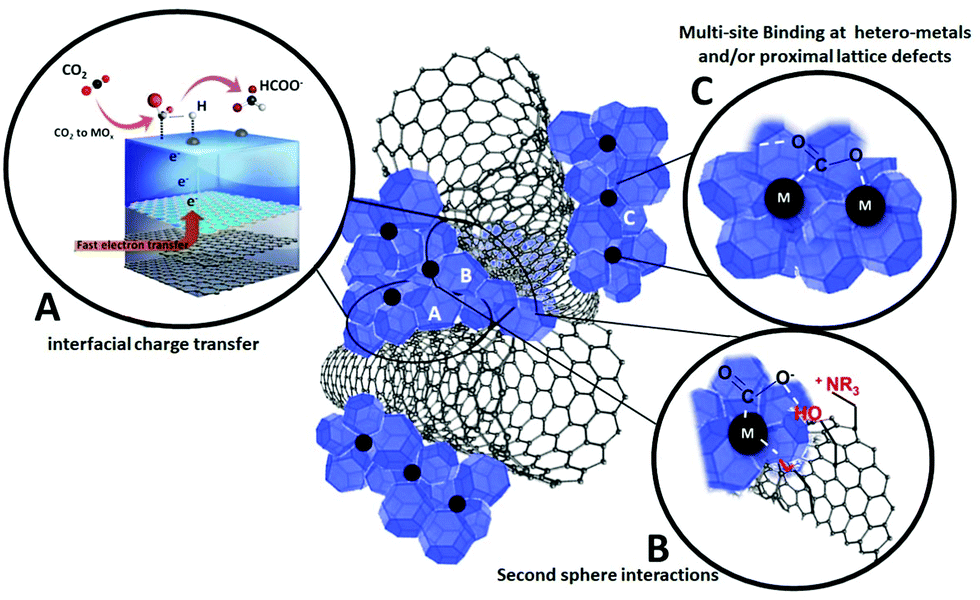 | ||
| Fig. 9 Interfacial effects occurring at multi-phase hybrid electrocatalysts. (A) Improved electron transfer at conductive MOx/CNS interface; (B) second sphere interactions promoted by terminal groups on functionalized CNS; (C) multi-site binding at M/MOx interfaces. | ||
Two main aspects will be instrumental to leverage bio-inspired CO2RR at nanohybrid platforms: (i) low-energy proton-coupled electron transfer (PCET) mechanisms and (ii) sequential catalytic steps that maximize product selectivity (tandem catalysis). In both cases, an opportune interplay of organic and inorganic interfaces can be expected. In PCET, concerted electron and proton transport can be envisaged at metal/metal oxide contacts with conductive carbon nanostructures by installation of proton acceptors/donors with tailored thermodynamic strength (pKa) so to enable multi-site electron and proton transfer events and facilitate CO2RR at low η. This will enhance current efficiency, while broadening the CO2RR selectivity window.118
Tandem catalysis by multiple enzymes, that proceeds in sequential metabolic steps, is the biological way for the continuous fixation of CO2 and its conversion into high-value multicarbon products.119 Along the same concept, a cascade of electrocatalytic steps can be programmed at distal sites on the hybrid multi-phase platform, where CO2RR intermediates are sequentially converted or coupled to increase the complexity of the carbon-based products. The multi-phase composite can differentiate the reactive steps by a stringent confinement of the active sites in the diverse nanodomains, while favoring the interfacial diffusion of reagents and the release of reactive intermediates.120
This synthetic scheme will require a proper choice of the different catalytic subunits, their distribution, coverage density and interfacial connection mediated by the nanocarbon scaffolds. Electrocatalytic analogs of enzymatic cascade reactions pose some formidable challenges with respect to the orchestration of rates and reagent/product diffusion, although retaining great potential for synthetic applications.121 The concept of integrating competitive or cooperative functions emerging from distinct electrocatalytic components in one common platform has been recently proposed for regulating the synthesis gas generation (syngas, CO + H2) by aligning the relative rates of simultaneous CO2RR and HER at the electrode. In this way the needed range of syngas compositions can be achieved by a modular design of the electrocatalytic interface.122
(3) Single atom catalysts
While the idea of using single atom catalysts (SAC) has been in the catalysis landscape for at least a decade,123,124 their role as “frontier catalytic materials” is very recent. The SAC appeal is mainly due to the theoretical maximization of the single metal efficiency, matching the current sustainability trends of enhancing the atom economy process.125 The term SAC does not simply refer to anchoring isolated molecular catalyst on heterogeneous supports. Indeed, SAC is associated to a single-metal atom site highly dispersed on heterogeneous supports and thereby featuring an under-coordinated state. Given their isolated form and lack of ligands, the material environment provides a stabilizing platform to counteract their high surface energy and strong tendency to aggregate.126 CNS, in particularly N-doped, are typical supports for SAC species, in which the metal atom turns out to be stabilized by nitrogen atom coordination,127,128 thus resembling the M–N4 motif of molecular coordination complexes featuring macrocyclic aza-ligands such as porphyrins or tetra-azacyclotetradecane (cyclam-type complexes). However, the heterogeneous SACs display a high degree of structural inhomogeneity, where the M–Nx coordination sites are not well defined, hampering a precise understanding of the metal-substrate or metal-intermediate interaction during CO2RR.129,130 The site-specific CO2RR mechanism in a model Fe-SAC on N-doped carbon supports was recently studied by relying on in situ techniques such as attenuated total reflection infrared (ATR-IR) spectroscopy combined with advanced microscopy and DFT computations. It was found that neither Fe–N4 moieties located in the bulk nor at the edge of a defectless graphitic structure were the competent catalysts, while activity could correlate with the Fe–N4 sites in a defected structure.131 The under-coordinated state of the metal center is proposed as a critical feature for achieving high rates of CO2 reduction over the competing HER in aqueous electrolytes. Representative examples of SAC electrocatalysts for CO2RR are already available. For example, Ni–N4 moieties embedded in graphitic carbon were prepared via a typical synthetic method based on the pyrolysis of Zn/Ni bimetallic zeolitic imidazolate frameworks (ZIF) showing high FE towards CO. The low coordination number for the Ni atom (∼2) was confirmed by EXAFS, and DFT calculations were in agreement with the more favorable energetic profile of the COOH adsorbed intermediate, whereas the fully coordinated Ni–N4 sites were inert. Interestingly, comparison with the energetic profile for the HER process indicated that the optimum for CO2RR activity and selectivity was connected to a Ni–N2V2 moiety (V = coordination vacancy).132 Therefore, the level of control on the coordination number of the competent SAC species is of high relevance for modulating the CO2RR activity.133 To this aim the hierarchical integration of nano-carbon scaffolds is of prominent interest: it increases the surface area and maximizes the exposure of SAC dispersed active sites to the reactant diffusion, while improving conductivity and electron transport at the SAC redox manifold. Indeed, anchoring a monolayer of Ni-based SACs on carbon nanotubes (CNTs) was found to enhance CO2RR TOF values by one order of magnitude than the ZIF-derived SAC.134 Moreover, the use of Ni-imidazolate coordination polymers (Ni-Im) as SAC precursors in combination with CNT, was instrumental to increase the density of Ni-based sites that is mandatory to attain industrial-relevant current densities (Fig. 10).134 Along the same lines of reasoning, metal oxides can be introduced as SAC supports modulating the atomic metal catalytic behavior.135 Therefore, an appealing opportunity arises from composites N-CNS/SAC@MOx to make a three-phase nanohybrid catalyst, where exploration of the triple interface electron transfer dynamics may lead to the emergence of a new paradigm of CO2RR catalysts.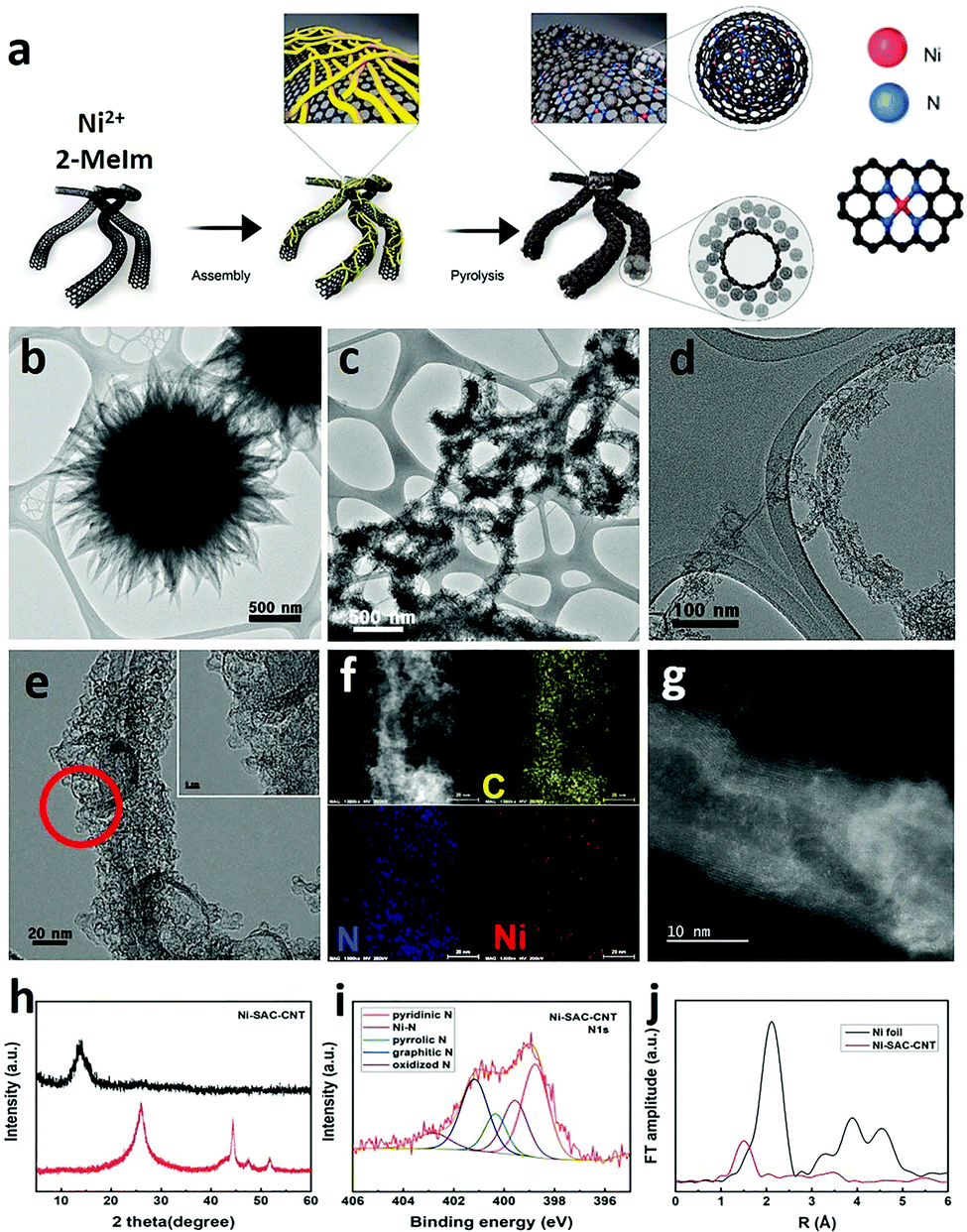 | ||
| Fig. 10 (a) Graphical sketch of the synthesis of the Ni–CNT SAC catalyst; (b–d) TEM of the material at different magnifications; (e) HRTEM and (f) corresponding EDX elemental mapping of C, Ni, N; (g) STEM; (h) XRD patterns before (red) and after acid treatment (black); (i) N 1 s XPS spectra after acid treatments. Atomic contents of N species including pyridinic N, pyrrolic N, graphitic N, and Ni metallic N. (j) Fourier transformations of EXAFS spectra. Reprinted from ref. 134, (2021) with permission from Elsevier. | ||
Techno-economic analysis is expected to guide the evolution of the next-generation CO2RR processes, minimizing the drawbacks and the environmental impact associated to the electrocatalyst fabrication and life cycle analysis (LCA). The selectivity issue and the need to increase performance and stability moving away from alkaline harsh conditions, while minimizing performance losses will be at the heart of expected breakthroughs for CO2 conversion with industrial relevance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. P. is the AXA Chair for Bionanotechnology (2016–2023). This work was supported by the University of Trieste, INSTM, the European Commission (H2020 – RIA-CE-NMBP-25 Program, Grant No. 862030), the European Commission (H2020 – LC-SC3-2019-NZE-RES-CC – Grant no. 884444) and the Italian Ministry of Education MIUR (cofin Prot. 2017PBXPN4). Part of this work was performed under the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency Grant No. MDM-2017-0720.Notes and references
- J. M. Matter, M. Stute, S. Ó. Snæbjörnsdottir, E. H. Oelkers, S. R. Gislason, E. S. Aradottir, B. Sigfusson, I. Gunnarsson, H. Sigurdardottir, E. Gunnlaugsson, G. Axelsson, H. A. Alfredsson, D. Wolff-Boenisch, K. Mesfin, D. F. D. L. R. Taya, J. Hall, K. Dideriksen and W. S. Broecker, Science, 2016, 352, 1312–1314 CrossRef CAS PubMed.
- A. Chen and B.-L. Lin, Joule, 2018, 2, 594–606 CrossRef CAS.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- C. Chen, J. F. Khosrowabadi Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- Y. Liang, Y. Li, H. Wang and H. Dai, J. Am. Chem. Soc., 2013, 135, 2013–2036 CrossRef CAS PubMed.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- H. Mistry, A. S. Varela, S. Kühl, P. Strasser and B. R. Cuenya, Nat. Rev. Mater., 2016, 1, 16009 CrossRef CAS.
- F. A. Armstrong and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14049–14054 CrossRef CAS PubMed.
- A. H. Proppe, Y. C. Li, A. Aspuru-Guzik, C. P. Berlinguette, C. J. Chang, R. Cogdell, A. G. Doyle, J. Flick, N. M. Gabor, R. van Grondelle, S. Hammes-Schiffer, S. A. Jaffer, S. O. Kelley, M. Leclerc, K. Leo, T. E. Mallouk, P. Narang, G. S. Schlau-Cohen, G. D. Scholes, A. Vojvodic, V. W.-W. Yam, J. Y. Yang and E. H. Sargent, Nat. Rev. Mater., 2020, 5, 828–846 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 14834–14837 CrossRef CAS PubMed.
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Nature, 1999, 399, 28–29 CrossRef.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- S. K. Shukla, S. G. Khokarale, T. Q. Bui and J.-P. T. Mikkola, Front. Mater., 2019, 6, 42 CrossRef.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. Salehi-Khojin, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- S. P. Kelley, L. A. Flores, M. S. Shannon, J. E. Bara and R. D. Rogers, Chem. – Eur. J., 2017, 23, 14332–14337 CrossRef CAS PubMed.
- D. V. Vasilyev, A. V. Rudnev, P. Broekmann and P. J. Dyson, ChemSusChem, 2019, 12, 1635–1639 CrossRef CAS PubMed.
- D. Zang, Q. Li, G. Dai, M. Zeng, Y. Huang and Y. Wei, Appl. Catal., B, 2021, 281, 119426 CrossRef CAS.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS PubMed.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- Y. Wang, L. Cao, N. J. Libretto, X. Li, C. Li, Y. Wan, C. He, J. Lee, J. Gregg, H. Zong, D. Su, J. T. Miller, T. Mueller and C. Wang, J. Am. Chem. Soc., 2019, 141, 16635–16642 CrossRef CAS PubMed.
- L. Lv, D. Zha, Y. Ruan, Z. Li, X. Ao, J. Zheng, J. Jiang, H. M. Chen, W.-H. Chiang, J. Chen and C. Wang, ACS Nano, 2018, 12, 3042–3051 CrossRef CAS PubMed.
- Y. Yang, M. Luo, W. Zhang, Y. Sun, X. Chen and S. Guo, Chem, 2018, 4, 2054–2083 CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- A. Bruix, Y. Lykhach, I. Matolínová, A. Neitzel, T. Skála, N. Tsud, M. Vorokhta, V. Stetsovych, K. Ševčíková, J. Mysliveček, R. Fiala, M. Václavů, K. C. Prince, S. Bruyère, V. Potin, F. Illas, V. Matolín, J. Libuda and K. M. Neyman, Angew. Chem., Int. Ed., 2014, 53, 10525–10530 CrossRef CAS PubMed.
- J. L. Dubois, in Metal Oxides in Heterogeneous Catalysis, ed. J. C. Védrine, Elsevier, 2018, pp. 401–549 DOI:10.1016/B978-0-12-811631-9.00008-9.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- Y. Chen and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 1986–1989 CrossRef CAS PubMed.
- M. Melchionna, M. Bonchio, F. Paolucci, M. Prato and P. Fornasiero, Top. Curr. Chem., 2013, 348, 139–180 CrossRef PubMed.
- F. Pan and Y. Yang, Energy Environ. Sci., 2020, 13, 2275–2309 RSC.
- S. Ma, R. Luo, J. I. Gold, A. Z. Yu, B. Kim and P. J. A. Kenis, J. Mater. Chem. A, 2016, 4, 8573–8578 RSC.
- C. Rogers, W. S. Perkins, G. Veber, T. E. Williams, R. R. Cloke and F. R. Fischer, J. Am. Chem. Soc., 2017, 139, 4052–4061 CrossRef CAS PubMed.
- Q. Zhang, Y. Zhang, J. Mao, J. Liu, Y. Zhou, D. Guay and J. Qiao, ChemSusChem, 2019, 12, 1443–1450 CrossRef CAS PubMed.
- B. Zhang, S. Chen, B. Wulan and J. Zhang, Chem. Eng. J., 2021, 421, 130003 CrossRef CAS.
- Z. Chen, K. Mou, X. Wang and L. Liu, Angew. Chem., Int. Ed., 2018, 57, 12790–12794 CrossRef CAS PubMed.
- X. Yang, P. Deng, D. Liu, S. Zhao, D. Li, H. Wu, Y. Ma, B. Y. Xia, M. Li, C. Xiao and S. Ding, J. Mater. Chem. A, 2020, 8, 2472–2480 RSC.
- S. Liu, X. F. Lu, J. Xiao, X. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2019, 58, 13828–13833 CrossRef CAS PubMed.
- Q. Zhang, J. Du, A. He, Z. Liu and C. Tao, J. Solid State Chem., 2019, 279, 120946 CrossRef CAS.
- F. Li, M. Xue, J. Li, X. Ma, L. Chen, X. Zhang, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 14718–14722 CrossRef CAS PubMed.
- S. Guo, S. Zhao, J. Gao, C. Zhu, X. Wu, Y. Fu, H. Huang, Y. Liu and Z. Kang, Nanoscale, 2017, 9, 298–304 RSC.
- G. Valenti, M. Melchionna, T. Montini, A. Boni, L. Nasi, E. Fonda, A. Criado, A. Zitolo, S. Voci, G. Bertoni, M. Bonchio, P. Fornasiero, F. Paolucci and M. Prato, ACS Appl. Energy Mater., 2020, 3, 8509–8518 CrossRef CAS.
- M. Melchionna, M. V. Bracamonte, A. Giuliani, L. Nasi, T. Montini, C. Tavagnacco, M. Bonchio, P. Fornasiero and M. Prato, Energy Environ. Sci., 2018, 11, 1571–1580 RSC.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- Y. Huo, X. Peng, X. Liu, H. Li and J. Luo, ACS Appl. Mater. Interfaces, 2018, 10, 12618–12625 CrossRef CAS PubMed.
- O. Baturina, Q. Lu, F. Xu, A. Purdy, B. Dyatkin, X. Sang, R. Unocic, T. Brintlinger and Y. Gogotsi, Catal. Today, 2017, 288, 2–10 CrossRef CAS.
- Y. Xu, F. Li, A. Xu, J. P. Edwards, S.-F. Hung, C. M. Gabardo, C. P. O’Brien, S. Liu, X. Wang, Y. Li, J. Wicks, R. K. Miao, Y. Liu, J. Li, J. E. Huang, J. Abed, Y. Wang, E. H. Sargent and D. Sinton, Nat. Commun., 2021, 12, 2932 CrossRef CAS PubMed.
- F. C. Romeiro, B. C. Silva, A. S. Martins, M. V. B. Zanoni and M. O. Orlandi, J. CO2 Utiliz., 2021, 46, 101460 CrossRef CAS.
- L. Wang, Y. Xu, T. Chen, D. Wei, X. Guo, L. Peng, N. Xue, Y. Zhu, M. Ding and W. Ding, J. Catal., 2021, 393, 83–91 CrossRef CAS.
- J. Huang, X. Guo, G. Yue, Q. Hu and L. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 44403–44414 CrossRef CAS PubMed.
- Y. Song, R. Peng, D. K. Hensley, P. V. Bonnesen, L. Liang, Z. Wu, H. M. Meyer III, M. Chi, C. Ma, B. G. Sumpter and A. J. Rondinone, ChemistrySelect, 2016, 1, 6055–6061 CrossRef CAS.
- C. Genovese, M. E. Schuster, E. K. Gibson, D. Gianolio, V. Posligua, R. Grau-Crespo, G. Cibin, P. P. Wells, D. Garai, V. Solokha, S. Krick Calderon, J. J. Velasco-Velez, C. Ampelli, S. Perathoner, G. Held, G. Centi and R. Arrigo, Nat. Commun., 2018, 9, 935 CrossRef PubMed.
- F. Li, S.-F. Zhao, L. Chen, A. Khan, D. R. MacFarlane and J. Zhang, Energy Environ. Sci., 2016, 9, 216–223 RSC.
- R. G. Mendes, P. S. Wróbel, A. Bachmatiuk, J. Sun, T. Gemming, Z. Liu and M. H. Rümmeli, Chemosensors, 2018, 6, 60 CrossRef CAS.
- C. Wang, V. Murugadoss, J. Kong, Z. He, X. Mai, Q. Shao, Y. Chen, L. Guo, C. Liu, S. Angaiah and Z. Guo, Carbon, 2018, 140, 696–733 CrossRef CAS.
- M. Melchionna, M. Prato and P. Fornasiero, Catal. Today, 2016, 277, 202–213 CrossRef CAS.
- J. Gao, Y. Ji, Y. Li, J. Zhong and X. Sun, RSC Adv., 2017, 7, 21079–21084 RSC.
- O. Okhay, G. Gonçalves, C. Dias, J. Ventura, E. M. F. Vieira, L. M. V. Gonçalves and A. Tkach, J. Alloys Compd., 2019, 781, 196–200 CrossRef CAS.
- F. I. L. Passos, J. G. da Silva Filho, A. Saraiva-Souza, A. G. Souza Filho, V. Meunier and E. C. Girão, Phys. Rev. B, 2017, 95, 195124 CrossRef.
- D. Janas, K. Z. Milowska, P. D. Bristowe and K. K. K. Koziol, Nanoscale, 2017, 9, 3212–3221 RSC.
- S. Marchesan, M. Melchionna and M. Prato, ACS Nano, 2015, 9, 9441–9450 CrossRef CAS PubMed.
- P. P. Sharma, J. Wu, R. M. Yadav, M. Liu, C. J. Wright, C. S. Tiwary, B. I. Yakobson, J. Lou, P. M. Ajayan and X.-D. Zhou, Angew. Chem., Int. Ed., 2015, 54, 13701–13705 CrossRef CAS PubMed.
- A. Peigney, C. Laurent, E. Flahaut, R. R. Bacsa and A. Rousset, Carbon, 2001, 39, 507–514 CrossRef CAS.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- S. Zhang, H. Wang, J. Liu and C. Bao, Mater. Lett., 2020, 261, 127098 CrossRef CAS.
- M. Eswaramoorthy, R. Sen and C. N. R. Rao, Chem. Phys. Lett., 1999, 304, 207–210 CrossRef CAS.
- A. Beltram, M. Melchionna, T. Montini, L. Nasi, P. Fornasiero and M. Prato, Green Chem., 2017, 19, 2379–2389 RSC.
- S. Inoue, N. Ichikuni, T. Suzuki, T. Uematsu and K. Kaneko, J. Phys. Chem. B, 1998, 102, 4689–4692 CrossRef CAS.
- K. Murata, K. Kaneko, F. Kokai, K. Takahashi, M. Yudasaka and S. Iijima, Chem. Phys. Lett., 2000, 331, 14–20 CrossRef CAS.
- K. Murata, K. Kaneko, W. A. Steele, F. Kokai, K. Takahashi, D. Kasuya, K. Hirahara, M. Yudasaka and S. Iijima, J. Phys. Chem. B, 2001, 105, 10210–10216 CrossRef CAS.
- H. Tanaka, H. Kanoh, M. El-Merraoui, W. A. Steele, M. Yudasaka, S. Iijima and K. Kaneko, J. Phys. Chem. B, 2004, 108, 17457–17465 CrossRef CAS.
- F. Li, D. R. MacFarlane and J. Zhang, Nanoscale, 2018, 10, 6235–6260 RSC.
- L. Wang, A. Ambrosi and M. Pumera, Angew. Chem., Int. Ed., 2013, 52, 13818–13821 CrossRef CAS PubMed.
- R. Arrigo, M. Hävecker, S. Wrabetz, R. Blume, M. Lerch, J. McGregor, E. P. J. Parrott, J. A. Zeitler, L. F. Gladden, A. Knop-Gericke, R. Schlögl and D. S. Su, J. Am. Chem. Soc., 2010, 132, 9616–9630 CrossRef CAS PubMed.
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh, R. Vajtai, A. Z. Yu, R. Luo, J. Lou, G. Ding, P. J. A. Kenis and P. M. Ajayan, Nat. Commun., 2016, 7, 13869 CrossRef CAS PubMed.
- G. Filippini, F. Amato, C. Rosso, G. Ragazzon, A. Vega-Peñaloza, X. Companyó, L. Dell’Amico, M. Bonchio and M. Prato, Chem, 2020, 6, 3022–3037 CAS.
- Z.-Z. Wu, F.-Y. Gao and M.-R. Gao, Energy Environ. Sci., 2021, 14, 1121–1139 RSC.
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845–7848 CrossRef CAS PubMed.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed.
- W. Zhang, Q. Qin, L. Dai, R. Qin, X. Zhao, X. Chen, D. Ou, J. Chen, T. T. Chuong, B. Wu and N. Zheng, Angew. Chem., Int. Ed., 2018, 57, 9475–9479 CrossRef CAS PubMed.
- W. Luc, C. Collins, S. Wang, H. Xin, K. He, Y. Kang and F. Jiao, J. Am. Chem. Soc., 2017, 139, 1885–1893 CrossRef CAS PubMed.
- Z. Deng, J. Ji, M. Xing and J. Zhang, Nanoscale Adv., 2020, 2, 4986–4995 RSC.
- Q. Li, J. Fu, W. Zhu, Z. Chen, B. Shen, L. Wu, Z. Xi, T. Wang, G. Lu, J.-J. Zhu and S. Sun, J. Am. Chem. Soc., 2017, 139, 4290–4293 CrossRef CAS PubMed.
- G. Valenti, A. Boni, M. Melchionna, M. Cargnello, L. Nasi, G. Bertoni, R. J. Gorte, M. Marcaccio, S. Rapino, M. Bonchio, P. Fornasiero, M. Prato and F. Paolucci, Nat. Commun., 2016, 7, 13549 CrossRef CAS PubMed.
- M. Melchionna, M. Bevilacqua and P. Fornasiero, Mater. Today Adv., 2020, 6, 100050 CrossRef.
- D. Gao, Y. Zhang, Z. Zhou, F. Cai, X. Zhao, W. Huang, Y. Li, J. Zhu, P. Liu, F. Yang, G. Wang and X. Bao, J. Am. Chem. Soc., 2017, 139, 5652–5655 CrossRef CAS PubMed.
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS PubMed.
- S. Gao, Z. Sun, W. Liu, X. Jiao, X. Zu, Q. Hu, Y. Sun, T. Yao, W. Zhang, S. Wei and Y. Xie, Nat. Commun., 2017, 8, 14503 CrossRef CAS PubMed.
- S. Huo, Z. Weng, Z. Wu, Y. Zhong, Y. Wu, J. Fang and H. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 28519–28526 CrossRef CAS PubMed.
- Y. Wang and I. A. Weinstock, Chem. Soc. Rev., 2012, 41, 7479–7496 RSC.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. R. Gonzalez, H. Amenitsch, T. Da Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Chem., 2010, 2, 826–831 CrossRef CAS PubMed.
- M. Quintana, A. M. López, S. Rapino, F. M. Toma, M. Iurlo, M. Carraro, A. Sartorel, C. Maccato, X. Ke, C. Bittencourt, T. Da Ros, G. Van Tendeloo, M. Marcaccio, F. Paolucci, M. Prato and M. Bonchio, ACS Nano, 2013, 7, 811–817 CrossRef CAS PubMed.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, ACS Catal., 2017, 7, 1749–1756 CrossRef CAS.
- K. J. P. Schouten, Z. Qin, E. Pérez Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS.
- C. Hahn, T. Hatsukade, Y.-G. Kim, A. Vailionis, J. H. Baricuatro, D. C. Higgins, S. A. Nitopi, M. P. Soriaga and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5918–5923 CrossRef CAS PubMed.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS PubMed.
- S.-H. Liu, J.-S. Lu, Y.-C. Pu and H.-C. Fan, J. CO2 Utiliz., 2019, 33, 171–178 CrossRef CAS.
- L. Zhang, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed.
- Y. Wang, Z. Chen, P. Han, Y. Du, Z. Gu, X. Xu and G. Zheng, ACS Catal., 2018, 8, 7113–7119 CrossRef CAS.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Nørskov and X. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- B. Liu, B. He, H. Q. Peng, Y. Zhao, J. Cheng, J. Xia, J. Shen, T. W. Ng, X. Meng, C. S. Lee and W. Zhang, Adv. Sci., 2018, 5, 1800406 CrossRef PubMed.
- C. Zhang, Y. Shi, Y. Yu, Y. Du and B. Zhang, ACS Catal., 2018, 8, 8077–8083 CrossRef CAS.
- R. H. Holm and E. I. Solomon, Chem. Rev., 2004, 104, 347–348 CrossRef CAS PubMed.
- E. D. Goodman, C. Zhou and M. Cargnello, ACS Cent. Sci., 2020, 6, 1916–1937 CrossRef CAS PubMed.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- J. N. Schrauben, R. Hayoun, C. N. Valdez, M. Braten, L. Fridley and J. M. Mayer, Science, 2012, 336, 1298–1301 CrossRef CAS PubMed.
- T. Schwander, L. Schada von Borzyskowski, S. Burgener, N. S. Cortina and T. J. Erb, Science, 2016, 354, 900–904 CrossRef CAS PubMed.
- C. Xie, C. Chen, Y. Yu, J. Su, Y. Li, G. A. Somorjai and P. Yang, Nano Lett., 2017, 17, 3798–3802 CrossRef CAS PubMed.
- Y. Lum and J. W. Ager, Energy Environ. Sci., 2018, 11, 2935–2944 RSC.
- M. B. Ross, Y. Li, P. De Luna, D. Kim, E. H. Sargent and P. Yang, Joule, 2019, 3, 257–264 CrossRef CAS.
- B. T. Qiao, A. Q. Wang, X. F. Yang, L. F. Allard, Z. Jiang, Y. T. Cui, J. Y. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- J. M. Thomas, Z. Saghi and P. L. Gai, Top. Catal., 2011, 54, 588–594 CrossRef CAS.
- M. Monai, M. Melchionna and P. Fornasiero, in Adv. Catal., ed. C. Song, Academic Press, 2018, vol. 63, pp. 1–73 Search PubMed.
- C. T. Campbell, Nat. Chem., 2012, 4, 597–598 CrossRef CAS PubMed.
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878–1889 CrossRef CAS PubMed.
- C. Xia, Y. Qiu, Y. Xia, P. Zhu, G. King, X. Zhang, Z. Wu, J. Y. Kim, D. A. Cullen, D. Zheng, P. Li, M. Shakouri, E. Heredia, P. Cui, H. N. Alshareef, Y. Hu and H. Wang, Nat. Chem., 2021, 13, 887–894 CrossRef CAS PubMed.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, ACS Catal., 2018, 8, 3116–3122 CrossRef CAS.
- X. Qin, S. Zhu, F. Xiao, L. Zhang and M. Shao, ACS Energy Lett., 2019, 4, 1778–1783 CrossRef CAS.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H.-L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 7607–7611 CrossRef CAS PubMed.
- G. Hwa Jeong, Y. Chuan Tan, J. Tae Song, G.-Y. Lee, H. Jin Lee, J. Lim, H. Young Jeong, S. Won, J. Oh and S. Ouk Kim, Chem. Eng. J., 2021, 426, 131063 CrossRef CAS.
- B. Lu, Q. Liu and S. Chen, ACS Catal., 2020, 10, 7584–7618 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |