
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Specific carbon/iodide interactions in electrochemical capacitors monitored by EQCM technique†
Anetta
Platek-Mielczarek
 ,
Elzbieta
Frackowiak
* and
Krzysztof
Fic
*
,
Elzbieta
Frackowiak
* and
Krzysztof
Fic
*
Institute of Chemistry and Technical Electrochemistry, Poznan University of Technology, Berdychowo 4, Poznan 60-965, Poland. E-mail: elzbieta.frackowiak@put.poznan.pl; krzysztof.fic@put.poznan.pl
First published on 5th March 2021
Abstract
This paper reports on the ion fluxes at the interfaces of various porous carbon electrodes/aqueous solutions of alkali metal cations (Na+, K+ and Rb+) and iodide anions, monitored by an electrochemical quartz crystal microbalance (EQCM). Different electrode material compositions as well as various electrolyte concentrations have also been considered. By tracking the ions during electrochemical polarization, we aimed to identify the reasons for the fading capacitor performance that occurs during long-term operation. The mass change profile suggests that hydroxide anions are responsible for counterbalancing the charge stored on the negative electrode. Furthermore, we found that iodide-based species are physically adsorbed on the carbon surface immediately after the electrodes come into contact with the electrolyte, regardless of the textural properties of the activated carbon used or electrode composition. Apart from the qualitative description, the mass change profiles allow the sequence of pore occupation to be determined. Additionally, the solvation numbers for alkali metals (Na+, K+ and Rb+) and hydroxide-based species have been determined. It is claimed that the solvation number is strongly affected by the electrolyte composition. Apparently, the concentration of water molecules available in the electrolyte cannot be neglected. The outcome of this research has fundamental and application context.
Broader contextProviding a reasonable response to the ever-growing demand from society for devices to store increasingly more energy in the same or a smaller volume, to be available as fast as possible, is a stimulating challenge for researchers in the energy storage field. Thus, to date, various types of materials, electrolytes and technological concepts have been proposed in order to cover all application niches. Stretching the limits of energy storage materials is currently realized at the nanoscale, and a deep understanding of these processes seems to be a crucial step towards the further development of energy storage devices. Our report elucidates the behavior of iodide-based species as the redox-active components of the electrolytic solution in electrochemical capacitors. It has been assumed that the peculiar performance of iodides seriously impacts the lifetime of devices that exploit the pseudocapacitive effect of the iodide-based redox couple. We confirmed that iodide anions demonstrate a specific affinity for the activated carbon surface; furthermore, it appears that they do not actively contribute to the ion flux on the positive electrode and tend to be instantly adsorbed on the carbon surface, as indicated by in situ EQCM measurements. Solvent–ion interactions, ion mixing, and the transport of ions confined in the pores have been considered. |
1. Introduction
An increase in the energy output of electrochemical capacitors is still of very high interest;1,2 hence, the introduction of redox-active electrolytes has been proposed.3–9 Application of an electrolytic solution demonstrating redox activity combines the advantages of electrostatic attraction (short response time) with the beneficial effect of the charge transfer process at the interface (high capacity). Nevertheless, the main drawback for redox-based storage is quite often a limited lifetime. In addition, remarkable self-discharge originating from the shuttling effect and the ‘privileged’ oxidation of the carbon material can be included.10–12Even if the capacitance of redox based systems is improved by incorporating faradaic processes, electrochemical capacitors with aqueous electrolytes cannot replace their organic-based counterparts, regardless of their competitive energy density, but they are highly concurrent due to their power, cost and ‘green’ character. However, limited cyclability, affected by faradaic storage, is still an issue.13,14 For that reason, apart from technological development, there is plenty of room for fundamental research aiming at understanding the processes in energy storage devices.15
It is well known that in situ techniques, despite being powerful tools, display some technical limitations, such as the need for operation in a three-electrode setup; however, with some efforts, these results might be successfully translated to full-device performance.16–20 In this context, the adsorption/desorption of ions monitored by an electrochemical quartz crystal microbalance (EQCM) for a single electrode appears to be advantageous21–24 and has already been applied to monitor capacitive24–29 and faradaic charge storage processes.30 Though, it is widely accepted that for highly porous activated carbon (AC) materials, the ion adsorption process is complex and could encounter several barriers preventing the full material capacity or capacitance from being exploited.17,31,32 In fact, partial or total solvation, the ion mixing effect and ion-pore size mismatching have a strong impact on the process efficiency and thus the energy outcome of the device.33 Thus, it becomes a crucial factor to evaluate and describe the ion behavior in electrochemical capacitors, at least in order to provide a set of features that are necessarily addressed in novel materials and electrolytes.34 Certainly, the correlation of the electrode and electrolyte properties with the electrochemical performance has been reported in numerous reports, presenting various approaches to the capacitance of the electrical double-layer.35–43
In the context of primary reports for specific interactions between cations and redox-active anions and their impact on the capacitor performance, an initial study was performed on the halide ion coupled with a series of alkali metals, such as lithium, sodium, potassium and cesium.4,44 It has been stated that the solvation number of the ion and the ion–solvent interactions play a crucial role in the electrochemical activity of the species. Apart from fundamental studies on iodide-based electrolytes and their performance, extensive studies have been performed to understand the performance decay of capacitors during long-term operation.7,10 Initially, it was reported that iodide anions are confined only on the positive electrode surface after long-term use.6 Since the redox activity of the anion is normally expected at the positive electrode, this result might suggest that the positive electrode is ‘exclusively’ responsible for the aging of the system. Such a statement is in accordance with the literature, where such a phenomenon occurs for EC systems.45–48 Moreover, one can conclude that iodides are only attracted to the positive electrode surface. Such a mechanism seems to be reasonable but does not explain accelerated aging at elevated capacitor voltages since the iodides should also play a role as overcharge protectors.10
Measurements conducted with an EQCM confirmed that specific adsorption of cations or anions results in a difference between the potential of zero charge (pzc) and the potential of zero mass (pzm);49 such a difference is also foreseen for iodide-based electrolytes and may indirectly confirm their confinement in carbon pores. This finding also implies that the presence of certain ions in the system might demonstrate a set of particular interactions. It is well known that iodides are soft (chaotropic) anions. They are presumed to be adsorbed on the electrode/electrolyte interface. However, due to high redox activity, the iodide-based system has not been studied by EQCM during positive polarization.44 As a consequence, the dual charge storage mechanism in ECs has not yet been elucidated in detail.
Thus, this paper offers a comprehensive report on the electrochemical performance of iodide anions at porous carbon/aqueous solution interfaces and their multifunctional role in energy storage mechanisms. Confirmation and description of I2/I−/carbon specific interactions allow these advantageous species to be fully exploited.
2. Experimental
All salts used for electrolyte preparation (NaI, KI, RbI and RbOH) were provided by Sigma-Aldrich (≥99.5%, ACS grade) and prepared at a concentration of 0.1 mol L−1. A concentration of 0.01 mol L−1 has been also applied, demonstrating the effect of salt concentration on ionic fluxes. The activated carbon used in this study was a commercially available YP-50F powder; for comparison purposes YP-80F powder, characterized by different textural properties was used. Both carbons were provided by Kuraray (Japan).Electrodes (coated on the quartz crystal resonator) were prepared as follows: AC powder and a 5 wt% solution of poly(vinylidenedifluoride) (PVDF) binder (Sigma-Aldrich) in N-methyl-2-pyrrolidone (NMP) solvent (Sigma-Aldrich) were mixed together; the adjusted AC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :binder mass ratio in the final electrode material was either 60:40 (w/w) or 80:20 (w/w). The latter one allowed the activated carbon to be a major component in a coating composition. The wet slurry was then uniformly placed by the drop-casting procedure onto a quartz crystal resonator (standard frequency 9 MHz, SEIKO EG&G, Japan) with a stainless steel (SUS304) current collector. Then, the resonators were dried at 50 °C for 12 h. The mass loading of the carbon coating did not exceed 85 μg to follow the rule of a thin and rigid coating layer. Additionally, various mass loadings have also been examined (in the range of 40–300 μg), to verify and exclude experimental errors that might originate from samples of small, non-representative mass. Moreover, the resistance was controlled during the entire experiments to verify the application of the Sauerbrey equation for mass change recalculation.
:binder mass ratio in the final electrode material was either 60:40 (w/w) or 80:20 (w/w). The latter one allowed the activated carbon to be a major component in a coating composition. The wet slurry was then uniformly placed by the drop-casting procedure onto a quartz crystal resonator (standard frequency 9 MHz, SEIKO EG&G, Japan) with a stainless steel (SUS304) current collector. Then, the resonators were dried at 50 °C for 12 h. The mass loading of the carbon coating did not exceed 85 μg to follow the rule of a thin and rigid coating layer. Additionally, various mass loadings have also been examined (in the range of 40–300 μg), to verify and exclude experimental errors that might originate from samples of small, non-representative mass. Moreover, the resistance was controlled during the entire experiments to verify the application of the Sauerbrey equation for mass change recalculation.
The electrochemical cell consists of a polypropylene body with construction adapted for electrochemical quartz crystal microbalance measurements. A resonator with an AC coating (geometric surface area equal to 0.1590 cm2) was placed on the bottom of the cell and served as the working electrode. Platinum wire or stainless-steel foil were used as a counter electrode, respectively to the carbon loading on the working electrode. The reference electrode was the saturated calomel electrode (SCE, +0.241 V vs. SHE), positioned close to the resonator. The 400 μL electrolyte solution (excess) was carefully placed in the cell, avoiding any presence of bubbles.
A supplementary electrochemical study was performed in a 3-electrode Swagelok® cell. For this purpose, a YP-50F-based electrode film was prepared in a mass ratio of 90:5:5 with a polymer binder (PTFE) and conductive additive (C65). The components were mixed in ethanol at 70 °C; after subsequent evaporation of the solvent, the mixture became a dense and plastic mass. The material was rolled into a thin film (approx. 150 μm) and cut into disk-shaped electrodes with a diameter of 10 mm. Then, the electrodes were separated by a glass fiber membrane soaked in the electrolyte solution. Three-electrode measurements were performed with an excess of electrolyte to ensure good contact with the reference electrode.
Additionally, aging performance of the systems operating with 1.0 mol L−1 KI in a Swagelok® cell at two operational voltages, i.e., 1.5 V and 1.2 V has been also demonstrated. Carbon electrode used for this purpose was a self-standing cloth Kynol® 507-20, allowing several post-mortem analyses to be conducted. It has to be noted here that this carbon exhibits textural properties different from YP-50F and YP-80F carbons.
All electrochemical measurements were carried out on a multichannel potentiostat/galvanostat VMP3 (BioLogic, France) connected by an analog input to the quartz crystal analyzer QCA 922 (SEIKO EG&G, Japan). The electrochemical techniques included cyclic voltammetry (CV), constant current charge/discharge and large-amplitude sinusoidal voltammetry (LASV).
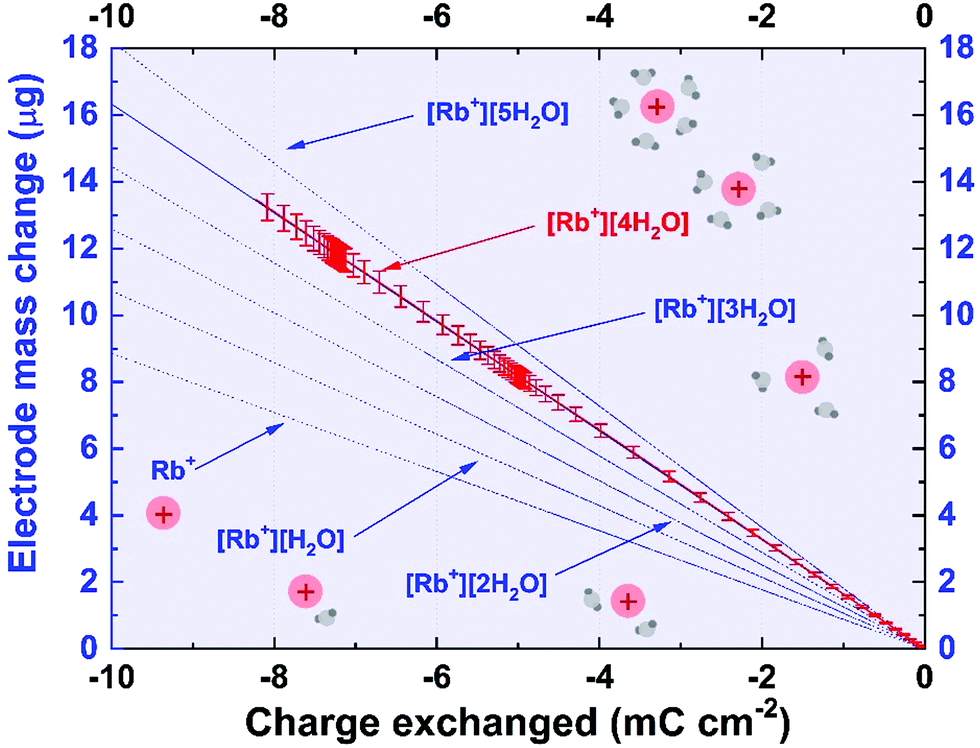
Theory and calculations
Data recorded during cyclic voltammetry were recalculated with respect to Faraday's law (eqn (1)). This approach allowed the results to be compared with the theoretical mass change as a function of the charge in the so-called mass to charge ratio plot: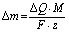 | (1) |
485 C mol−1), and z is the number of electrons exchanged, which equals the valence number of the adsorbed/desorbed ions.
The mass change was calculated directly from the resonator frequency change recorded by the EQCM with respect to the Sauerbrey equation (eqn (2)) for thin and rigid film layers.
| Δf = −f02·Δm/N·ρ | (2) |
It is worth highlighting that the change in the electrolyte viscosity (caused by the high resonance frequency) is not considered to play a significant role in ion transport; it has been verified that the viscosity of the solutions investigated here is constant even at high shear rates (Brookfield viscometer, cone-plate setup); aqueous iodide-based electrolytic solutions are Newtonian fluids.
Calculations have been performed for alkali metals in the form of neat cations as well as taking into account the hydration shell, composed of 1 up to 25 molecules of water; the hydration number depends on the concentration of iodide-based salt.
For the iodine-based species, apart from the single iodide anion (I−), a number of various poly(iodide) forms (I3− and I5−) and iodates (IO3−) have been considered, as their presence at the interface has been previously reported.6
3. Results and discussion
In the first step of the investigation, the corresponding potential range for cation and iodine anion adsorption/desorption was designated using LASV, as presented in Fig. S1 (ESI†). In this case, the capacitive character of the curve is attributed to the electrical double-layer (EDL) charge/discharge process. In contrast, for systems based on faradaic storage (redox reactions), the current and potential are in the same phase. The phase shift is assumed to be a more sensitive factor than the change in the potential or current profiles in typical dc methods. Therefore, LASV profiles allowed the potential range of electrostatic attractions (cation adsorption/desorption) and redox processes (charge transfer reaction) to be determined and confirmed. Furthermore, it was initially assumed that the open circuit potential (labeled EOC) corresponds to the point of zero charge (pzc), as it might be related to the equilibrium state in the system. Taking into account the lowest capacitance response of the system in the 3-electrode system in the case of iodide-based electrolytes, the pzc does not have the same position as the EOC (Fig. S2, ESI†). The point of zero charge is equal to 0.150 V vs. SHE, whereas the EOC is 0.350 V vs. SHE. Moreover, the point of zero mass or charge does not reflect the absolute zero value, but rather the lowest value found in the system. However, one should be aware that the EQCM response is measured together with the potential change; this, for transport-related reasons (e.g., diffusion), might result in a response shifted with respect to the current change. This remark is of high importance for further considerations.The microtextural properties of the YP-50F activated carbon, denoted as AC, are presented in Fig. 1. Nitrogen adsorption/desorption isotherms were recorded at 77 K in order to evaluate the porous texture of activated carbon and its composite with a polymer binder. Besides the application of the Brunauer–Emmett–Teller (BET) equation for highly microporous carbons,50 the surface area has also been calculated by DFT.51,52 Textural characterization of YP-80F activated carbon powder and Kynol® 507-20 tissue are presented in ESI† (Table S1 and Fig. S3, ESI†). One could remark that both activated carbon powders demonstrate slightly different textural properties. YP-50F is characterized by limited pore volume, resulting in a lower specific surface area as compared to YP-80F (0.75 cm3 g−1vs. 1.06 cm3 g−1). Different textural properties of these carbons are maintained for powder and composite used as electrode coating. Such a characteristic allows for a versatile discussion of iodide behavior in the vicinity of carbon materials with different pore structure (Fig. 1A and Fig. S3, ESI†).
 | ||
| Fig. 1 Nitrogen adsorption data for the Kuraray YP-50F materials: (A) pore size distribution with the presumed dimensions of the investigated ions; (B) adsorption/desorption isotherms for the pristine carbon and with the addition of binder. | ||
Both isotherms for activated carbon powder (YP-50F) and its composite are classified as type I (Fig. 1B), according to the IUPAC classification.26,27 On both curves, a small hysteresis loop can be observed in the relatively high pressure range. This hysteresis confirms that the Kuraray YP-50F contains a limited (but still not negligible) fraction of small mesopores that are essential for ensuring ion transport to and from the interface. YP-80F displays higher mesopore fraction while Kynol® 507-20 demonstrates typical microporous character (Table S1 and Fig. S3, ESI†). For all carbons, the micropore range is wide, so it was assumed that the material is capable of storing all kinds of ions from the electrolyte. It must be mentioned that smaller species certainly penetrate pores of larger diameter, and the presence of ultramicropores cannot be neglected. Furthermore, the cations Na+, K+, and Rb+ and anions I−, I3−, and I5− could penetrate the pore volume easily, especially with partially or fully desolvated shells. To conceptualize the ion-pore matching, the presumed ion dimensions are presented in Fig. 1A. Given that data, one can calculate the theoretical capacity of the material and the number of cations and anions that can possibly be stored in the entire volume of pores, with the pore diameter as a discriminant. This is a rough estimation of activated carbon capacity when in contact with an aqueous electrolyte. Assuming that the ions (with and without a solvation shell) are perfectly spherical, the maximum mass uptake (assuming complete filling of the micropore volume) for a 0.1 mol L−1 RbI solution during negative polarization should be in the range of 400–1500 μg (depending on the solvation shell of rubidium) with 400 μL of electrolyte used. Theoretical calculations are depicted in the ESI† and Table S2.
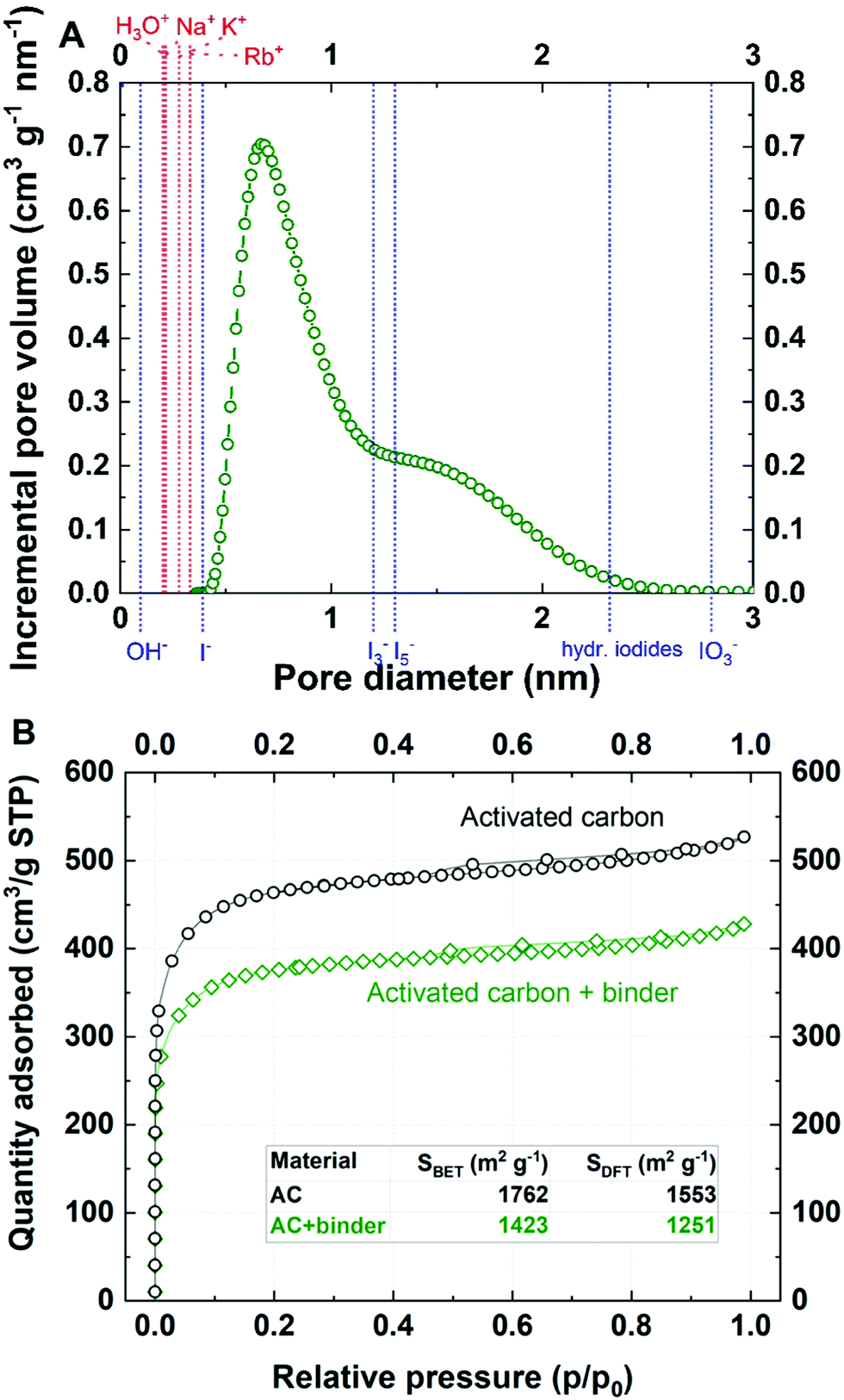
The mass change profile and current response recorded for the activated YP-50F electrode operating with a 0.1 mol L−1 aqueous solution of RbI are presented in Fig. 2.
 | ||
| Fig. 2 Mass change recorded (electrode material: YP-50F; electrolyte: 0.1 mol L−1 RbI) during a cyclic voltammetry scan at 5 mV s−1 presented with: (A) the current measured; (B) the charge exchanged. | ||
Plateaus on the mass change profile (red line) within a potential range of 0.1 and −0.2 V vs. SHE, as well as −0.6 and −0.4 V vs. SHE, are observed due to the difference in the phase of the current, the potential and the resonance frequency. The plateau at the beginning of the CV scan might be explained by the fact that either any ion flux can be detected or the balance of the mass transfer at the interface is equal to zero representing that co-ions and counterions movement is controlled by ion mass. However, the hysteresis loop suggests deep pore penetration, so-called confinement, by the Rb+ species. The ‘discharge’ profile indicates that the cations leave the interface in a two-step process, as two slopes can be distinguished. Thus, it can be stated that during discharge, the cations confined deeply in micropores (ultramicropores) undergo a charge relocation process towards wider pores before release.53 This effect is observed as a slight descent in the mass change profile. Moreover, it suggests that all pores (micro- and mesopores) are filled by the electrolyte; given that, their contribution to the electric double-layer formation is comparable.
As the electrode surface is being repolarized, a negligible mass difference is initially detected. The same conclusion was made with the YP-80F carbon electrode (Fig. S4, ESI†) indicating that ion fluxes vary (in terms of quantity) in the presence of various electrode materials, but the Rb+ (cation) adsorption in iodide solutions follows the same mechanism regardless of ACs. This observation leads to the hypothesis that even if the outer-pore volume should be released faster than the deep pore volume, the more likely charge carriers are relocated in the void space and then subsequently removed.54 However, what might also explain a slight mass change while repolarization occurs is that the carbon surface is not neutral in charge and might cause cation trapping by local negative surface charge carriers. This phenomenon might cause an initial delay in Rb+ release from the micropore volume and seems to be reasonable for our further considerations. Full ion release from the pores is observed as a remarkable mass decrease, while the potential is directed towards the ion-mixing region when co-ions (Rb+ and H3O+) are desorbed and replaced by counterions (I− and OH−).1 Moreover, one should be aware that the small mesopore fraction is not fully involved in the EDL charge/discharge process; it only plays the role of a pore entrance that allows fast charge carrier transport.55 This observation appears to be in accordance with other literature reports concerning discharge profiles where the ions are claimed to be subsequently released. However, we do believe that on the microscopic scale, charge relocation more likely occurs before ion release from the pore volume; a particular interaction between Rb+ and I− might also be considered. Moreover, this process appears to be dominant even in narrow pores (ultramicropores), where steric effects play a major role in ion–ion interactions.56 Hence, deeply confined ions are likely to move towards wider void space (outer surface of micropores) in the repulsion/relocation process and then follow a potential-driven desorption process.57 Therefore, while the potential shifts towards the cation desorption process, the mass change recorded remains nearly unchanged. Strict separation of ongoing phenomena, including the ion-mixing effect, charge carrier relocation/repulsion, crowding effect and others, is unlikely to occur for carbons with wide pore distributions, as these effects may overlap and affect each other.56,58,59
Fig. 2B presents the recalculation of the current measured compared to the charge exchanged during the adsorption/desorption process might be quite insightful. First, as it might be confusing, the point of absolute zero charge is correlated neither with the potential where charge accumulated on the surface equals zero nor with the pzc. One needs to bear in mind that the carbon surface may contain several surface functionalities or defects. Thus, its chemical nature does not necessarily need to be considered neutral; the presence of I2/I−/carbon specific interactions might cause local changes in the properties of the electrode/electrolyte interface. As a consequence, the surface might induce a specific ion affinity that influences the pzc. Therefore, the point of zero charge (pzc) has been determined (Fig. S2, ESI†) by the traditional 3-electrode technique, which allows the lowest capacitance point to be detected over a wide potential range. The point of zero charge is shifted 0.2 V towards negative values with respect to the open circuit potential (EOC) for 0.1 mol L−1 RbI. This finding proves that the EOC does not always correspond to the pzc. At the beginning of the adsorption process, a plateau is observed in the mass change profile. From the charge profile, one can conclude that near 0 V vs. SHE (close to the pzc), the ions approach the surface without any interactions. Therefore, the ions pass each other in order to approach the polarized surface, acting via the so-called ion mixing effect. It is worth highlighting again that the mass change recorded by the EQCM is a result of all ion fluxes at the interface.
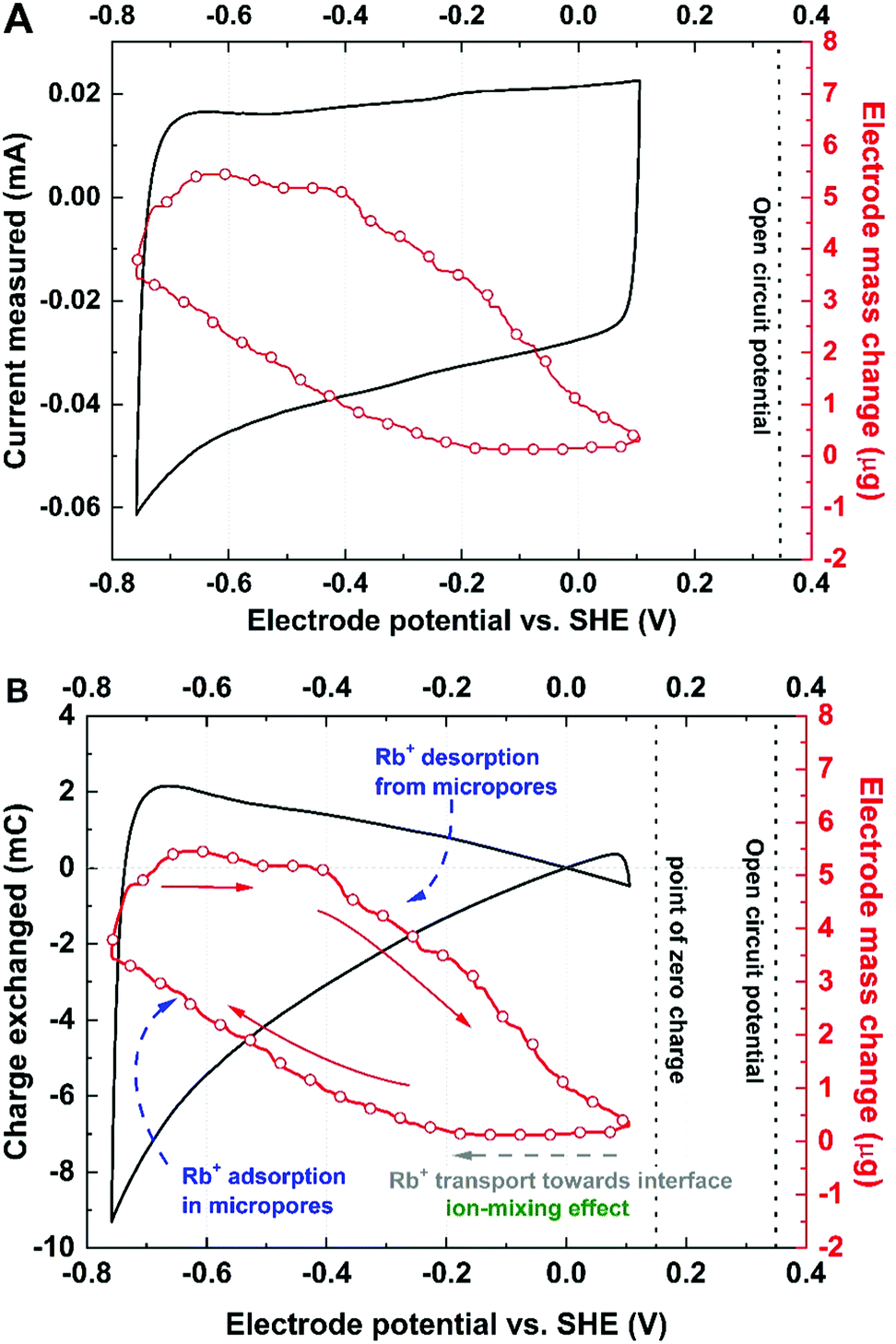
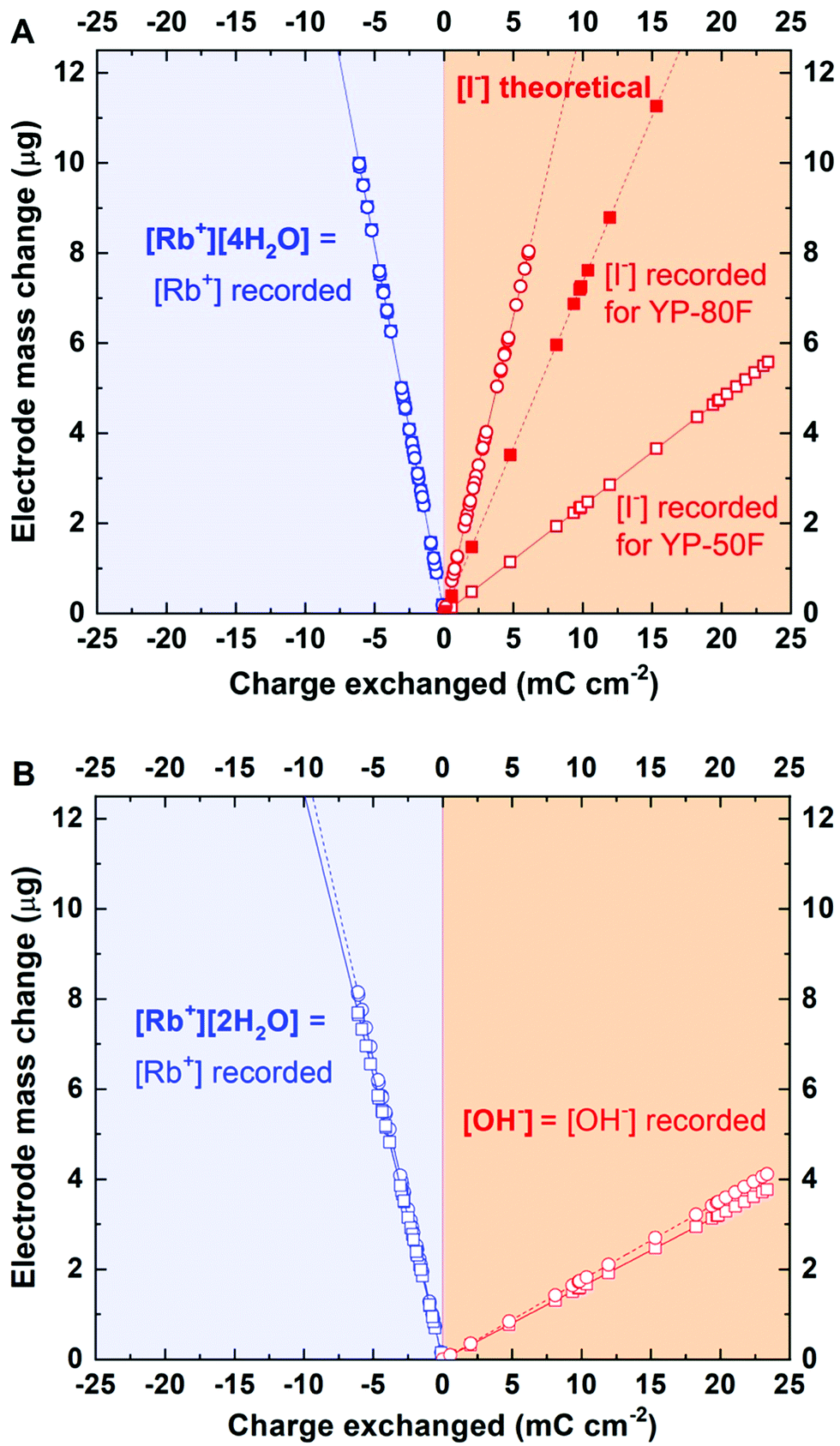
Furthermore, presenting the mass change vs. charge exchanged allows the experimental data to be compared with theoretical calculations for hydration numbers of various species. Based on that approach, it has been found that in the vicinity of YP-50F-based electrode, the solvation number for Rb+ in 0.1 mol L−1 RbI is equal to 4 (Fig. 3).
 | ||
| Fig. 3 Mass vs. charge changes for negative polarization, i.e., cation adsorption. Blue lines indicate theoretical profiles, and the red line indicates experimental data corresponding to the [Rb+]·[4H2O] complex. | ||
As shown in Fig. 3, the hydration shell remarkably influences the mass change profile; for instance, the molar mass for Rb+ hydrated by 5 molecules of water, denoted as [Rb+]·[5H2O], is 175.5 g mol−1, i.e., twice as high as that of the neat Rb+ cation, at 85.5 g mol−1. Such observations lead to the conclusion that the hydration shell, even if lost by the ion during micropore penetration, is ‘left’ and ‘confined’ in the vicinity of the interface or in the mesopore volume and influences the total mass change recorded by the EQCM. Therefore, the solvent molecules significantly impact the transport of ions by means of solvent–ion interactions and cannot be excluded from the ion adsorption mechanisms. Moreover, the ion desolvation process prior to pore penetration cannot be observed when an electrode with a wide pore size distribution is used. As a consequence, it is difficult to determine whether the ions in the observed flux are already partially desolvated or not. Interestingly, for YP-80F carbon, Rb+ cation adsorption occurs together with simultaneous movement of 15 molecules of water (Fig. S4, ESI†). This activated carbon is characterized by significantly higher mesopore fraction in the total pore volume than YP-50F. This might suggest that no steric (textural) hindrance is present in the electrode material; thus, the solvent molecules move easily towards polarized surface. Observations made for ion fluxes in electrolytes of various ion concentrations are spectacular; these data clearly show that the obstacle on the ion flux trajectory impacts the adsorption process (Fig. S5, ESI†). These impediments could result either from textural properties of carbon (e.g., narrow pore size) or electrolyte concentration (too populous ion concentration in the electrolyte volume, thus, strong ion–ion interactions).
Ion fluxes were studied in several stages: first, by applying only negative polarization from the EOC to observe cation adsorption/desorption (Fig. 2 and Fig. S4, ESI†), and second, by applying positive polarization from the EOC to observe ‘anion’ fluxes. Both negative and positive electrode studies with 0.1 mol L−1 RbI electrolyte are presented in Fig. S6A (ESI†) (recorded in Swagelok® cell) and Fig. S7A (ESI†) (recorded in EQCM, 3-electrode set-up). Subsequently, wide-range potential cycling has been implemented to observe all interactions among ions. The cyclic voltammetry profile (recorded at 5 mV s−1) for a wide potential range (Fig. 4 and Fig. S4, ESI†) clearly indicates the region of EDL formation and the region of I−/I2 redox couple activity; these regions are noticeable for both, YP-50F and YP-80F, carbons.
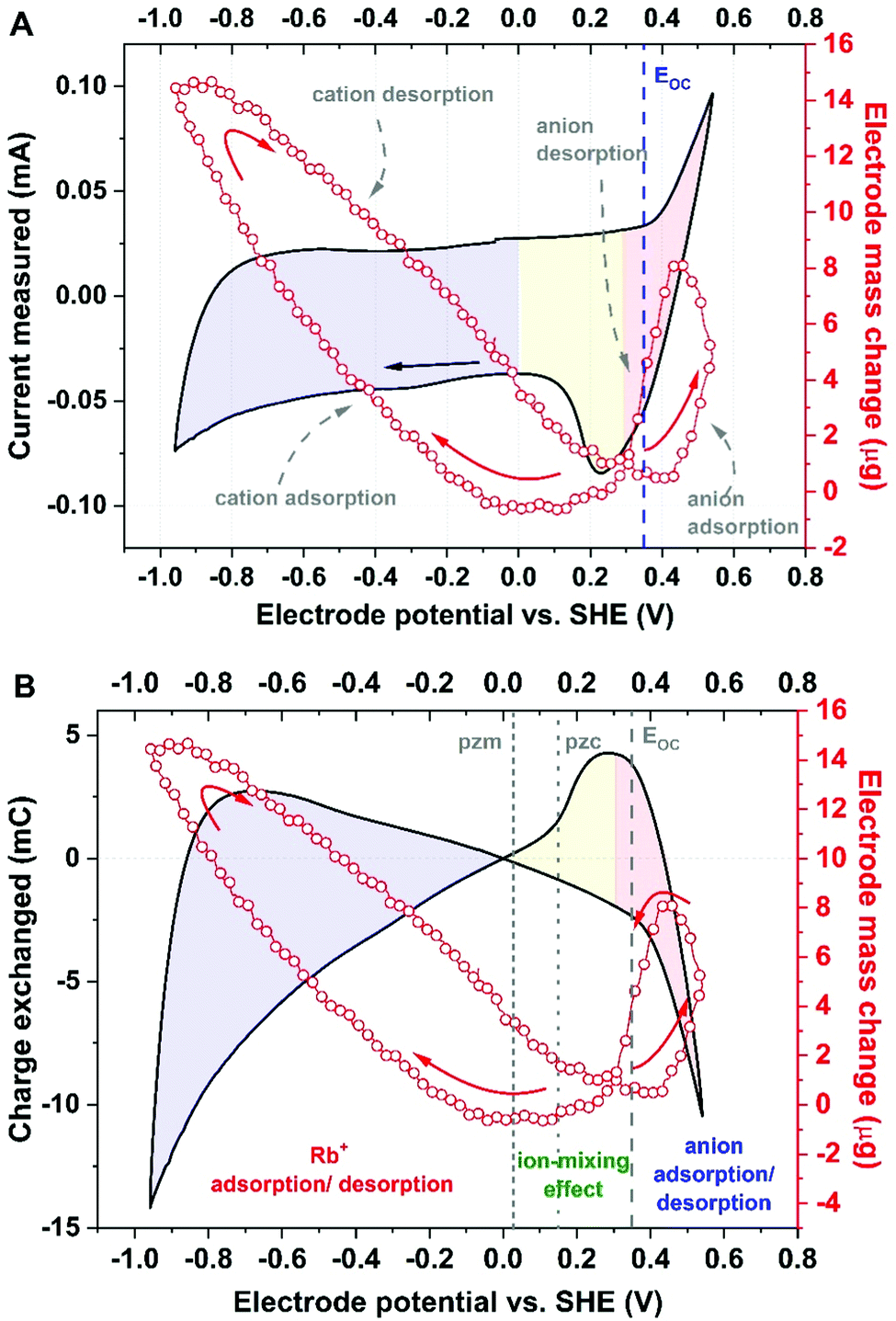 | ||
| Fig. 4 Mass change recorded during a cyclic voltammetry scan at 5 mV s−1 for a wide potential range (electrode: YP-50F; electrolyte: 0.1 mol L−1 RbI) presented with: (A) the current measured; (B) the charge exchanged. | ||
Such a wide scan is recorded in order to capture all nonseparated ion fluxes in the system. One might discuss the poor redox response in terms of the current value recorded for I2/2I− redox activity. It should be noted that the 3-electrode Swagelok® cell (Fig. S6, ESI†) with iodides trapped inside the electrolyte volume ensures a higher current response than the EQCM cell (Fig. S7, ESI†). The generated iodine might evolve from an improperly sealed or open 3-electrode cell and affect the redox efficiency. However, Fig. S8 (ESI†) indicates that both experimental cells display similar electrochemical responses when the same concentration of iodide species is studied. This result shows that 3-electrode studies might be adapted to the full-cell performance.
Surprisingly, the mass change recorded during positive polarization is remarkably smaller than that recorded during negative polarization, despite noticeable iodide/iodine activity (the mass change for cation adsorption/desorption ≫ the mass change for anion adsorption/desorption). It was initially assumed that iodide adsorption (M = 126.9 g mol−1) results in a higher mass change than that for Rb+ (M = 85.5 g mol−1) or is at least similar to that of the already detected during negative polarization [Rb+]·[4H2O] (157.5 g mol−1). In fact, the total mass change in this region is one-half that for cation adsorption/desorption; thus, it has been stated that the simple I− adsorption/desorption process is not directly involved in ion fluxes at the interface.
Self-adjustment of the electrode operating potentials is clearly observed in iodide-based systems, where a negative electrode, of purely capacitive character, operates over a wide potential range to counterbalance the redox capacity of the positive electrode, provided over a very narrow potential range.4,7,10,60–69 It is clearly seen in Fig. 4A that the mass change of anion adsorption/desorption correlates with the iodide/iodine activity. To clarify the rigid electrode response in the redox-active potential range, resistance was plotted as a function of experiment time (for all recorded cycles), as depicted in Fig. S9 (ESI†). Even if reduction–oxidation processes are ongoing, the viscoelastic properties of the carbon coating should remain constant to allow application of the Sauerbrey equation. The resistance change over time is ±1.5%, which meets the rigidity requirements.
However, the profiles of the charge exchange and mass change vs. the electrode potential (Fig. 4B) suggest that the pzc is shifted versus the point of zero mass (pzm) and that the open circuit potential (EOC) is located near the pzc; this finding indicates that the pzm might reflect an equilibrium state of ions at the electrode surface under open circuit conditions close to 0 V vs. SHE. Furthermore, this observation (separation of the pzc, pzm and EOC) confirms the strong affinity of iodide anions for the carbon surface. It has been thus assumed that the potential difference between the pzc and pzm values refers to the overtaking of ions during organized movement in the bulk of the electrolyte. This region has been assigned as the ion mixing domain. Such an explanation might justify the plateau observed in Fig. 2B, most likely representing Rb+ approaching the electrode surface and passing by the anions desorbed from the interface, as mentioned previously for [Rb+]·[4H2O] with a molar mass of 157 g mol−1. Considering mathematical recalculation to find counterions balancing this mass change, one needs to consider either a solvated iodide anion with a mixed solvation shell (either 1 or 2 water molecules), which might be designated [I−]·[H2O]:[I−]·[2H2O] 50%:50% (with an average molar mass 154 g mol−1), or a hydroxide anion complex with water molecules [OH−]·[8H2O] (molar mass 161 g mol−1). To verify these hypotheses and to determine which species counterbalance rubidium cations, the Faraday law has been implemented.
Obviously, during positive polarization, the anions deeply penetrate the volume of micropores. It was initially assumed that the mass change in this region corresponds to certain iodide-based species, as their redox activity generates several forms according to the Pourbaix diagram (Scheme 1):
| 2I− ↔ I2 + 2e− | (3) |
| 3I− ↔ I3− + 2e− | (4) |
| 5I− ↔ I5− + 4e− | (5) |
| 3I2 + 6OH− ↔ 5I− + IO3− + 3H2O | (6) |
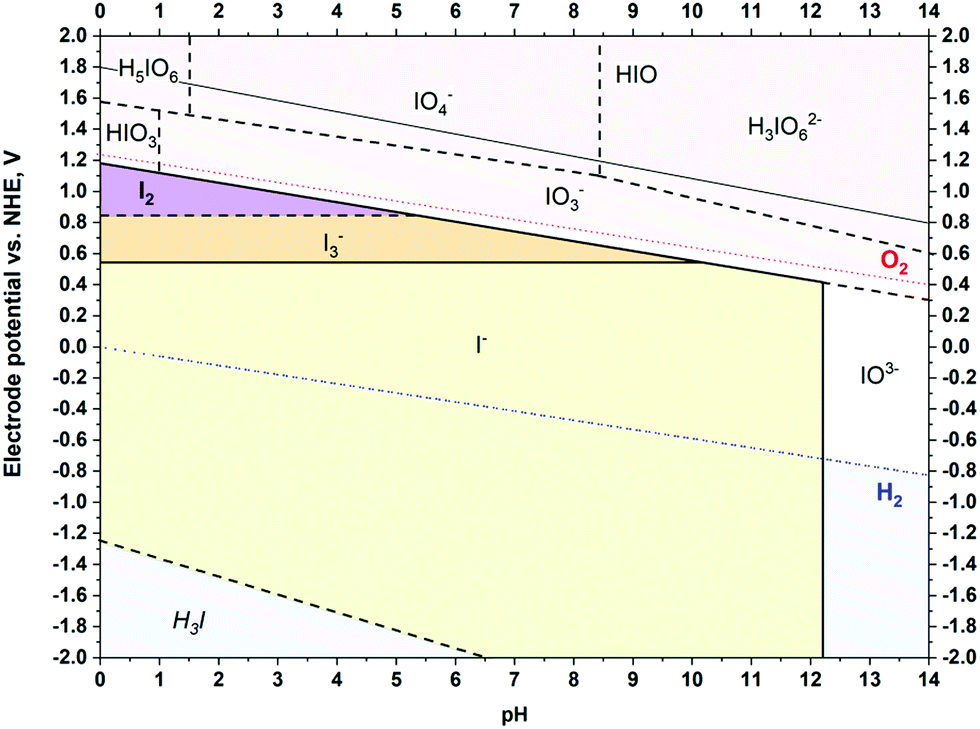 | ||
| Scheme 1 Pourbaix diagram for iodide species calculated for 0.1 mol L−1 solution at 25 °C, based on ref. 70 and 71. | ||
Surprisingly, the mass change recorded is much lower than that predicted from Faraday's law for the I− ions (Fig. 5A) and all polyiodide forms (I3− and I5−) for both studied carbons, YP-50F and YP-80F. Notably, if polyiodides are formed, as described by eqn (4) and (5), we assume that the mass change of the electrode might not be detected, as these polyiodides are formed from the species already adsorbed at the electrode surface, and that the process does not involve any species from the electrolyte bulk (in contrast to the case of IO3−).
 | ||
| Fig. 5 Mass change recorded for a CV scan at 5 mV s−1 (electrode: YP-50F; electrolytes: 0.1 mol L−1): (A) RbI; (B) RbOH. | ||
This peculiar observation prompted a more detailed consideration of the processes possibly occurring at the interface. To identify the species adsorbed/desorbed during the positive polarization, the mass change profiles were recalculated in order to find the molar mass of the species. The resulting mass indicated that certain hydroxide complexes, such as OH−, H3O2−, H7O4−, H9O5−, and OH−·n[H2O], where n = 4, 8, 16, or 20, are responsible for the mass change recorded. Since the mass of these complexes is relatively small, the reference test verifying whether OH−-based species might be detected by EQCM was performed with a 0.1 mol L−1 RbOH solution (Fig. 5B). The mass change profile recorded during positive polarization clearly demonstrates that OH− can be detected by EQCM and that these ions are responsible for charge balancing in iodide-containing solutions. In the alkaline medium, the hydration number for Rb+ changed from 4 to 2. Moreover, for alkaline solution (0.1 mol L−1 RbOH), only OH− ion fluxes have been confirmed, whereas for the 0.1 mol L−1 RbI electrolyte, the movement of [OH−]·2[H2O] has been detected. Thus, cations reveal several solvation numbers, as observed for anions. The same conclusions are valid for YP-80F. Interestingly, the anion adsorption follows similar rules as described for cations; [OH−]·3[H2O] has been found in the ion flux during positive polarization for YP-80F with 0.1 mol L−1 RbI (Fig. 5A). Thus, higher solvation number for anion is observed when carbon with more developed surface area is applied. However, activity of OH− is not that high as for Rb+ cation.
This in situ experiment demystifies the reason for the local pH increase during the long-term performance of iodide-based systems and suggests the tentative origin of the deterioration. From an experimental point of view, the alkalization of the electrolyte after electrochemical operation has already been described. However, in situ studies confirm that the presence of OH− in the electric double-layer is a cause of this local pH change, not a result of ongoing redox reactions.
As indicated by Pourbaix diagram (Scheme 1), for iodide-based systems, the pH increase shifts the redox activity from iodine (I2) and polyiodides (e.g., I3−) towards iodate (IO3−) formation and further salt precipitation caused by limited solubility (at least one order of magnitude lower than that for iodides). Thus, the cycle life might be improved either by maintaining the pH at values <8 or by preventing the positive electrode potential from reaching the value for IO3− formation. For this reason, the nominal voltage of the full cell needs to be carefully selected. Generally, the higher the voltage applied, the shorter the operational time is.45 For iodide-based systems, it has been found that application of elevated voltage (1.5 V) allows the initial capacitance (up to 80%) to be maintained for 120 h at a constant polarization. Furthermore, increasing the voltage dramatically decreases the energetic efficiency of the charge/discharge process (60% at 1.5 V). It is worth noting that up to 1.5 V, the coulombic efficiency reaches 90%. In a cell operating at 1.2 V, both the coulombic and energetic efficiencies of the charge/discharge process are higher than 90%, showing an equilibrated redox process. By limiting the harmful potential of the positive electrode and selecting a lower cell voltage (i.e., 1.2 V), the system is capable of operating for over 2000 h, maintaining the same end-of-life criterion (C/C0 = 80%), as presented in Fig. S10 (ESI†). In addition, Fig. S10C (ESI†) should be considered as an analytical proof of I2 presence in the iodide-based EC after long-term operation. Definitely, the iodide/iodine redox couple is highly active and affects both electrodes. However, this process results in various electrode/electrolyte interface composition; interestingly, more iodine has been found near the counter electrode than near (but not in the bulk of) the working one.
All abovementioned results and discussions indicate that iodide-based solutions might become a very useful component for ECs; if the right mass-balance, separator and optimal potential range are chosen.72 Thus, our findings have a real impact on a full cell performance, explain and complete many observations from iodide implementation in ECs. Moreover, to date, iodides have been successfully employed not only in electrochemical capacitors but also in desalination systems.73–75
Fig. 6 presents a tentative sequence of events at the porous carbon electrode/aqueous iodide solution with respect to the CV profile presented in Fig. 4A. For better clarity, 10 points (steps) of the current response have been distinguished.
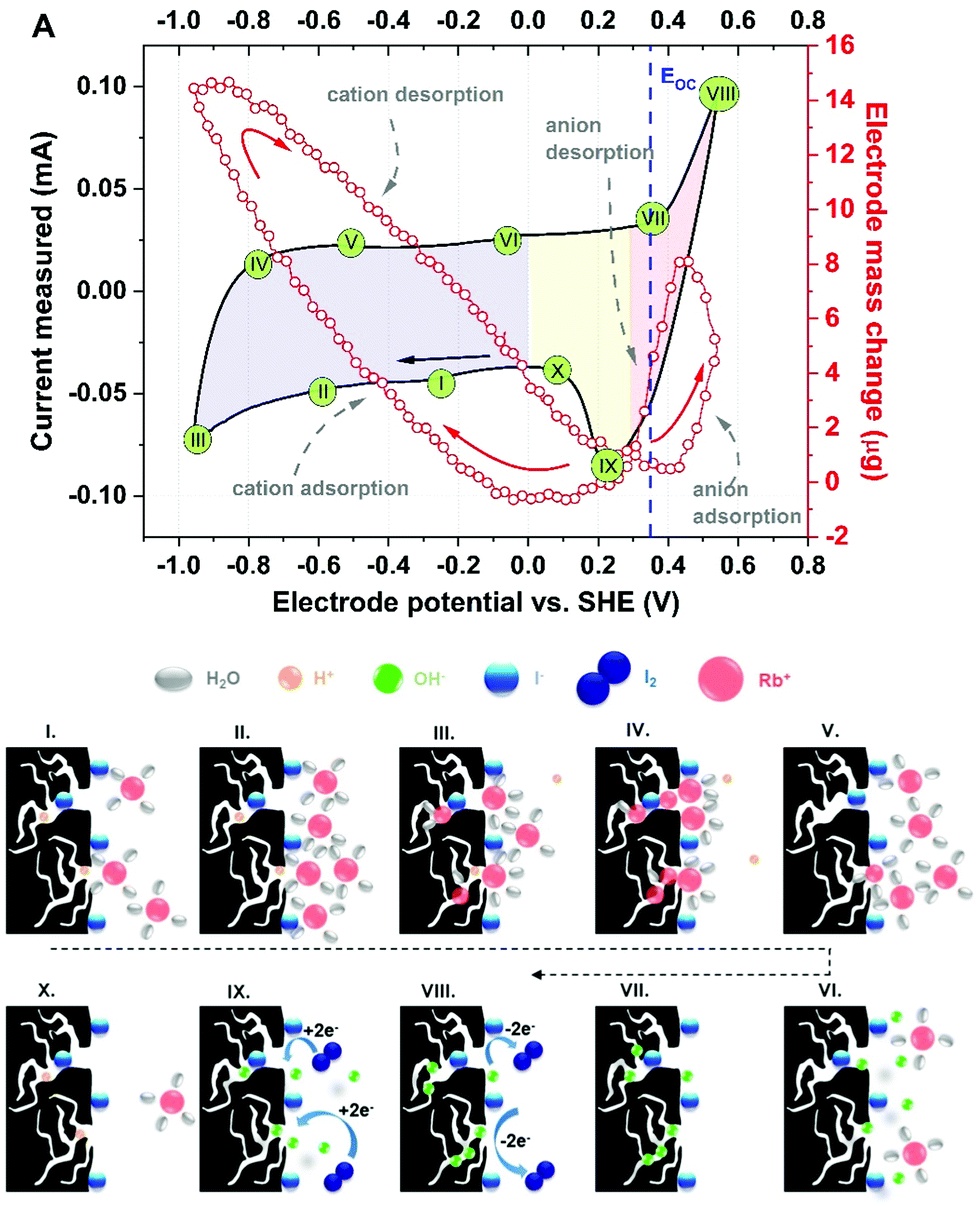 | ||
| Fig. 6 Scheme of the charge/discharge process occurring in the 0.1 mol L−1 RbI solution in the bulk and direct vicinity of YP-50F electrode. | ||
Step I denotes the mass change corresponding to the presence of protons (either H+ or H3O+) inside the pores and three [Rb+]·[4H2O] complexes approaching the electrode/electrolyte interface. Iodide ions most likely remain specifically adsorbed at the electrode surface, even if negative polarization is applied. Step II reflects the situation in which polarization is increasingly more cathodic and rubidium cations are moving towards the electrode. At the maximum cathodic potential (step III, with the highest current response), protons are expelled to the bulk of the electrolyte and replaced by cations penetrating the porous structure. It can be seen that water molecules from the hydration shell are gathered either in the volume of the larger pores (mesopores) or at the entrance to the micropores. When the polarization is reversed (step IV), the mass change initially continues to increase (as it is shifted in phase with the current response), demonstrating the increasing number of cations ‘confined’ in the pores; interestingly, the initial step of the discharge process is observed in the cyclic voltammetry profile. Moreover, the pores are free of protons, as they have moved to the bulk of the electrolyte (in step III). Step V reflects the movement of rubidium cations together with protons and their departure from the electrode/electrolyte interface. On the current plateau, denoted as step VI, the last rubidium cations are exchanged with hydroxide anions that approach the electrode surface. Subsequently, the pores are penetrated by OH− complexes (step VII) in the presence of the adsorbed iodide species. Then, the current increases and reflects the redox activity of iodides, but no mass change is observed (step VIII). This description suggests that the ongoing reaction involves the species present at the interface, as the mass change is not detected. Accordingly, one can conclude that it is possible to observe the redox activity of these species only by the current response, as the in situ gravimetric detection does not reflect any remarkable change. The situation is similar for step IX during the reduction process. Following step X is cation adsorption; in fact, due to their smaller size, protons tend to fill the pores first; simultaneously, rubidium cations approach the surface, and the loop is closed. This description suggests that ion transport at the porous electrode interface and the corresponding energy storage mechanism constitute a multistep process and that various aspects must be considered regarding potential bottlenecks for further development. Moreover, the results show an equilibrium between adsorbed and desorbed species from the electrode/electrolyte interface. These findings also confirm the stability of such a system and encourage the benefit of redox activity in the electrolyte towards improving the overall EC performance. In this particular case, iodides do not affect the movement of co-ions (OH−) and counterions (Rb+ or H+).
The quite surprising behavior of iodides at the porous carbon interface compelled further verification of whether the interactions are cation specific. As it has been already confirmed, I2/2I−/carbon specific interactions are independent of carbon coating composition ratio (60–40 and 80–20), type of AC (YP-50F and YP-80F) and electrolyte concentration (0.01 and 0.1 mol L−1 RbI). In this case, the only factor that left for exploration is the electrolyte composition. Thus, to confirm the assumption of general affinity of iodides towards carbon surface, additional tests with potassium iodide and sodium iodide solutions were carried out. For the 0.1 mol L−1 KI solution, the same experimental schedule was applied (cyclic voltammetry at 5 mV s−1), whereas for the 0.1 mol L−1 NaI solution, another scanning rate, i.e., 2 mV s−1, was applied after confirmation of the same performance at 5 mV s−1. Such a change has been made in order to study the tentative limitation of mass change detection.
Fig. 7A presents the mass change profile recorded in a 0.1 mol L−1 KI solution. It can be observed that the solvation number for K+ is equal to 6. During positive polarization, the mass change profile corresponds to the profile recorded for 0.1 mol L−1 RbI (although a smaller mass change than that for I− might be observed). This finding confirms the iodide affinity for the activated carbon surface, independent of the cation. However, as potassium is characterized by a higher hydration number than rubidium, the same trend is not observed for OH− adsorbed during positive polarization. The mass change recorded indicates adsorption/desorption of [OH−]·[1H2O].
For the 0.1 mol L−1 NaI solution, different scan rates were applied in order to verify whether the conditions of the measurement affect the detection of ion fluxes. The correlation coefficients (R2) (Fig. 5 and 7) calculated from the linear fit are high (>0.7410) and suggest that the scan rate (within applied values) does not discriminate the ions for the electrical double-layer formation; the same ions (with hydration shell) are involved in the ion fluxes.
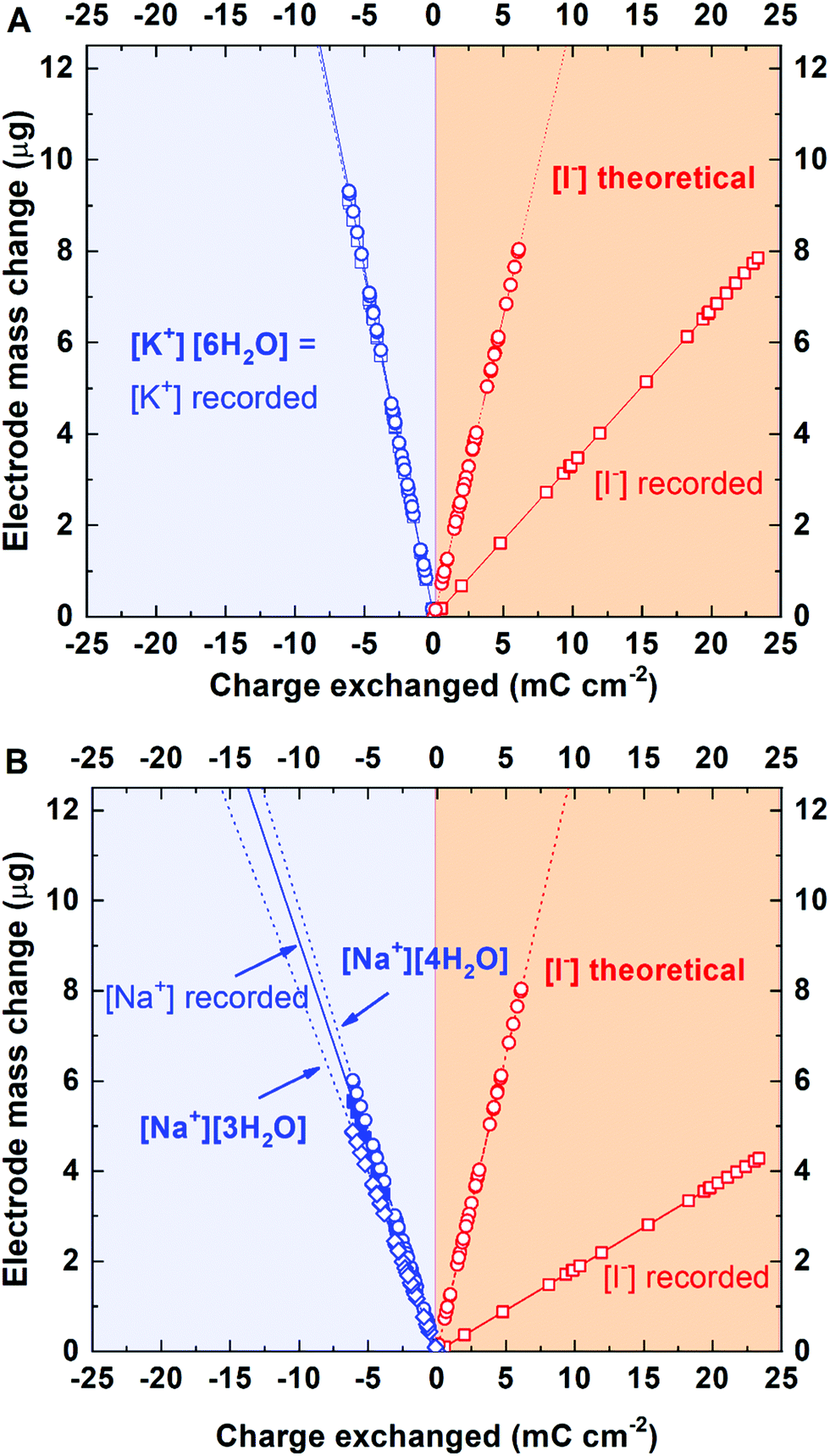 | ||
| Fig. 7 Mass change recorded in various electrolytes during CV scans (electrode: YP-50F; electrolytes: 0.1 mol L−1): (A) KI at 5 mV s−1; (B) NaI at 2 mV s−1. | ||
For Na+, the hydration number is determined to be 4. However, as the fitting line is not straightforward, the mixed composition of the hydration shells (including 3 and 4 water molecules, with preference for [4H2O]) is considered. Interestingly, while cations (Na+) are strongly solvated by several water molecules, the hydroxide anion has no solvation shell. Thus, the OH− species (in 0.1 mol L−1 NaI solution) behaves similarly as that in a hydroxide solution of 0.1 mol L−1 RbOH containing only one anion type.
A summary of the experimental potential ranges and cyclic voltammetry results for all studied electrolytes with YP-50F electrode material, i.e., NaI, KI, RbI, and RbOH, is presented in Fig. S6, S7 and Table S3 (ESI†) recorded either in Swagelok® cell or 3-electrode EQCM set-up. Such information is necessary to determine the correct experimental conditions for the studied systems. Verification of the electrochemical response in the EQCM 3-electrode system is presented in Fig. S8 (ESI†) where cyclic voltammetry with a wide potential range has been presented for Swagelok® and EQCM cell with 0.1 mol L−1 RbI electrolytic solution.
In Table 1, all the results obtained, together with the physicochemical data for the discussed electrolytes, are listed and compared with the results from other literature reports.
Table 1 suggests that the iodide anion/carbon affinity is independent of the cation type present in the solution. Moreover, the affinity is not affected by pH (7.2 for KI and 5.8 for NaI), by the solvation number of cations or by the conductivity. The iodide behavior uncommon for other anions (SO42− and NO3−) used in ECs is a result of partial iodide–carbon bond formation. However, it needs to be considered that only the I2 form might bind to the carbon double bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), as happens during iodine number determination. Due to the amphoteric character of the activated carbon surface, we cannot exclude partial transfer of iodide to iodine even when no polarization is applied to the electrochemical system. Furthermore, this transfer was proved after long term performance (Fig. S10C, ESI†), when I2 has been found in the n-hexane eluate from electrode washing. Besides this method, another one has been used to verify iodide|iodine|carbon interaction. Thermo-programmed desorption coupled with mass spectrometry spectra for pristine YP-50F carbon and one after contact with KI are depicted in Fig. S11 (ESI†). These plots clearly prove evolution of iodide/iodine related species (m/z = 127, but moreover, m/z = 45, m/z = 63, m/z = 64, m/z = 128 and m/z = 129) around 240 °C for sample after being in contact with KI in comparison to pristine one. Furthermore, the energy of the carbon–iodide bond has been estimated as 139 kJ mol−1, suggesting strong interaction between carbon and iodine. Interestingly, considering overall mass loss during thermal decomposition for all studied carbons, i.e., YP-50F, YP-80F and Kynol® 507-20 the mass loss was found higher for iodinated samples than for pristine ones.
C), as happens during iodine number determination. Due to the amphoteric character of the activated carbon surface, we cannot exclude partial transfer of iodide to iodine even when no polarization is applied to the electrochemical system. Furthermore, this transfer was proved after long term performance (Fig. S10C, ESI†), when I2 has been found in the n-hexane eluate from electrode washing. Besides this method, another one has been used to verify iodide|iodine|carbon interaction. Thermo-programmed desorption coupled with mass spectrometry spectra for pristine YP-50F carbon and one after contact with KI are depicted in Fig. S11 (ESI†). These plots clearly prove evolution of iodide/iodine related species (m/z = 127, but moreover, m/z = 45, m/z = 63, m/z = 64, m/z = 128 and m/z = 129) around 240 °C for sample after being in contact with KI in comparison to pristine one. Furthermore, the energy of the carbon–iodide bond has been estimated as 139 kJ mol−1, suggesting strong interaction between carbon and iodine. Interestingly, considering overall mass loss during thermal decomposition for all studied carbons, i.e., YP-50F, YP-80F and Kynol® 507-20 the mass loss was found higher for iodinated samples than for pristine ones.
Finally, the results provide new insight into the performance of iodides at the carbon electrode/aqueous electrolyte interface and give a complementary picture of ion fluxes in such a system. It appears that the change in the electrolyte pH in the vicinity of the positive electrode towards alkaline values4,7,10,62–64,66,83 is most likely caused by OH− transport towards the interface. Initially, it was assumed that hydrogen electrosorption on the negative side might generate such a pH change. Certainly, this effect cannot be excluded, but it appears that the local pH in the positive electrode microporous region is governed by aberrant ion transfer. The pH change has a detrimental impact on the electrode and capacitor performance. At higher pH values, the iodide–iodine redox couple shifts towards IO3−, which is less soluble than I− or I2 (I3−) and exacerbates pore clogging.10,62,63,84 This effect can also be observed by a gradual shift in the rest potential of the positive electrode towards negative values during long term operation.6,77
To date, the formation of polyiodides has been confirmed in several publications.4,6,79 One may wonder why no iodide movement has been found to date. For a carbon nanotube electrode material operating in a 1 mol L−1 NaI solution, I2 has been detected.82 Considering our findings in a set of alkali metal iodide solutions, for various electrolyte concentrations and in the presence of various electrode materials, it is not straightforward to detect the presence of I2 in the electrochemical system. First, the iodide/iodine redox reaction is equilibrated by mass; thus, as long as there is no I2 evacuation from the cell, one will not see the mass change. Moreover, while I2 might evolve from the system, instead of dissolution in iodide solution (as in the case of Lugol's solution), one would observe a decrease in the mass due to I− consumption as a substrate in the reaction (3). The only explanation that might be found in this situation is that I2 species are entrapped in the carbon and cannot be easily released. Thus, activated carbon electrodes appear to be suitable for iodide-based systems, as undesirable obstacles in the form of gaseous I2 are not present in the pore volume.
Furthermore, the higher pH at the positive electrode privileges the oxygen evolution reaction (OER) and aggravates the maximum voltage of the system.13,84
In addition, the presence of adsorbed iodides and formed iodine on both electrodes might (in certain cases) provoke a fast self-discharge if the mass/charge balancing is not made and the optimal potential ranges for the electrodes are not ensured.6,65 Moreover, a study to take advantage of iodate formation by alkalization of an iodide electrolyte has been reported.74 This work confirms the formation of iodate species from iodide while the environment is strongly alkaline. Nevertheless, the iodine presence in the electrode bulk after long term performance proves that iodine–carbon bond is strong enough to remain there despite washing with distilled water, pre-drying and even drying in a high vacuum.
Therefore, the next step should verify whether an electrolyte composed of another species for adsorption at a positive electrode (followed by micropore penetration) improves the cycle life of the device. The results obtained to date6,66,77 suggest that this direction is well chosen. It might be concluded that the presence of iodide and iodine on the negative electrode deteriorates the electrochemical capacitor performance. This hypothesis has been proven in the application of hybrid electrolyte systems, as reported elsewhere.6,77 The application of K2SO4 aqueous electrolyte on the negative side and KI on the positive side allows prolongation of the operational time at elevated voltages.10 Other studies have focused on the immobilization of iodide species on the positive electrode in order to prevent their migration towards the negative electrode.80,81 Such a procedure requires additional steps before the final assembly of the cell; however, it still provides very satisfactory results concerning the stability of the system. Therefore, the full device study shows that adjustment of the electrode potential ranges in line with the operation of the redox-active species is a reasonable approach, as iodide anions have mostly been immobilized on the positive side.
In this manuscript, in situ proof of strong iodide/iodine affinity (∼139 kJ mol−1) towards activated carbon surfaces affecting electrochemical performance has been described in detail. Type of activated carbon used as electrode material, its content in the electrode composite (AC:PVdF), as well as electrolyte concentration and formation (selected ions: Na+, K+, Rb+, I−, OH−) have been studied and discussed. Therefore, we claim that iodide/iodine species are adsorbed onto the carbon surface, and the following consequences might be expected:
(1) Great enhancement of capacity from redox reaction contribution,
(2) Inaccessibility of the surface for effective charge storage in electric double-layer,
(3) High change in the pH towards alkaline values, as OH− species are responsible for counterbalancing cations,
(4) Complex ion behavior during charging/discharging causing spatial hindrance, lowering the coulombic and energetic efficiency of the device.
4. Conclusions
The electrochemical quartz crystal microbalance allows the ion fluxes at the porous carbon electrode/aqueous electrolyte interface to be established.It has been demonstrated that not all of the ions present in the electrolyte take an active part in ion fluxes. It has been shown that I− anions are not responsible for the electrode mass change during positive polarization; hence, it is assumed and confirmed that the charge is equilibrated mostly by hydroxide anions. Iodide/iodine movement was not detected neither during anion adsorption nor anion repulsion. This observation is in agreement for various carbons. It has to be pointed out that I2/2I−/carbon specific interactions do not depend on the iodide salt (at least for the alkali salts, Na+, K+ or Rb+) and conditions of electrochemical test.
Therefore, the lifetime of the electrochemical capacitor based on the iodide/iodine redox couple is limited due to the presence of iodine on the negative electrode.
It has also been stated that in aqueous solutions, even if the ions are partially/fully desolvated, the solvent molecules remain in the vicinity of the interface and can be detected by EQCM. When the electrode material displays more accessible porous structure, the solvent molecule flux accompanies the ion adsorption. Moreover, solvation shell strongly depends on the concentration of solvent molecules in the electrolyte. The higher the salt concentration, the lower the number of water molecules surrounding particular ion.
Hydration numbers were determined for Na+, K+, Rb+, OH− in iodide and alkaline solution with AC based electrode. For each inorganic salt, the size of molecules should be determined by either molecular dynamic simulations or applications of in situ approaches, combining various advanced techniques.
A multistep model of ion transport has been proposed for iodide-based electrochemical capacitors.
This study also has a very sound applied context; namely, the pH change towards alkaline usually observed for capacitors operating with iodide-based electrolytes after aging procedures (regardless of long-term cycling or floating) is explained. This phenomenon has a substantial impact on the cycle life, as the redox activity of iodides is strongly dependent on the pH. For pH values higher than 6, one should not expect the presence of free I2. Furthermore, for pH values higher than 9, one should not expect even the presence of I3−. Thus, the redox system shifts towards IO3− of poor solubility; the precipitation products will block the pores of the carbon electrode and result in a sudden performance fade (quite often observed for systems subjected to aging tests). Finally, competition between the ions (I− and OH−) also appears reasonable, as their dimensions and mobilities are different. However, the presence of both species cannot be excluded, as they need to occupy the electrode surface to balance the charge stored on the counter electrode.
Finally, it has been demonstrated that replacement of organic media (expensive and hazardous) by aqueous iodide-based electrolytes (cheap and environmentally friendly) can be considered a feasible alternative.
In this context, one definitely cannot directly translate the peculiar performance of iodine-based species to any other redox-based system. However, for all of them, several directions for development can be chosen:
(1) Adjustment of the pore distribution that ensures efficient mass transport (including co-ion adsorption) and will not discriminate the ions from being adsorbed at the interface,
(2) Mass- and charge-balanced system that ensures the optimal potential window for redox-operating electrode,
(3) Good separation of the redox-based species for positive and negative electrode that prevents shuttling effect and diminishes self-discharge. The general charge storage mechanism (including pH-dependent processes) should be considered at a first glance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
APM would like to acknowledge the National Science Centre, Poland, for financial support in the framework of the project 2017/25/N/ST4/01738 and scholarship 2019/32/T/ST4/00276. All authors acknowledge the European Commission and the European Research Council for financial support by the Starting Grant project (GA 759603) under the European Union's Horizon 2020 Research and Innovation Programme. For contributing to the aging tests, the authors would like to thank Justyna Piwek. Dr Camelia Matei-Ghimbeu and Eng. Benedicte Rety are gratefully acknowledged for their substantial help in TPD studies.References
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P. L. Taberna, C. P. Grey, B. Dunn and P. Simon, Nat. Energy, 2016, 1, 16070 CrossRef CAS.
- Z. Lin, E. Goikolea, A. Balducci, K. Naoi, P. L. Taberna, M. Salanne, G. Yushin and P. Simon, Mater. Today, 2018, 21, 419–436 CrossRef CAS.
- S. Yamazaki, T. Ito, M. Yamagata and M. Ishikawa, Electrochim. Acta, 2012, 86, 294–297 CrossRef CAS.
- G. Lota, K. Fic and E. Frackowiak, Electrochem. Commun., 2011, 13, 38–41 CrossRef CAS.
- S. Yamazaki, T. Ito, M. Yamagata and M. Ishikawa, ECS Trans., 2012, 41, 15 CrossRef CAS.
- P. Przygocki, Q. Abbas, P. Babuchowska and F. Beguin, Carbon, 2017, 125, 391–400 CrossRef CAS.
- J. Menzel, K. Fic, M. Meller and E. Frackowiak, J. Appl. Electrochem., 2014, 44, 439–445 CrossRef CAS.
- B. Gorska, P. Bujewska and K. Fic, Phys. Chem. Chem. Phys., 2017, 19, 7923–7935 RSC.
- S. E. Chun, B. Evanko, X. Wang, D. Vonlanthen, X. Ji, G. D. Stucky and S. W. Boettcher, Nat. Commun., 2015, 6, 7818 CrossRef CAS PubMed.
- A. Platek, J. Piwek, K. Fic and E. Frackowiak, Electrochim. Acta, 2019, 311, 211–220 CrossRef CAS.
- P. Ratajczak, K. Jurewicz, P. Skowron, Q. Abbas and F. Beguin, Electrochim. Acta, 2014, 130, 344–350 CrossRef CAS.
- K. Fic, M. He, E. J. Berg, P. Novák and E. Frackowiak, Carbon, 2017, 120, 281–293 CrossRef CAS.
- K. Fic, M. Meller, J. Menzel and E. Frackowiak, Electrochim. Acta, 2016, 206, 496–503 CrossRef CAS.
- B. Gorska, E. Frackowiak and F. Beguin, Curr. Opin. Electrochem., 2018, 9, 95–105 CrossRef CAS.
- Z. F. Lin, P. L. Taberna and P. Simon, Curr. Opin. Electrochem., 2018, 9, 18–25 CrossRef CAS.
- M. Hahn, O. Barbieri, R. Gallay and R. Kotz, Carbon, 2006, 44, 2523–2533 CrossRef CAS.
- J. M. Griffin, A. C. Forse, W. Y. Tsai, P. L. Taberna, P. Simon and C. P. Grey, Nat. Mater., 2015, 14, 812–819 CrossRef CAS PubMed.
- J. M. Griffin, A. C. Forse, H. Wang, N. M. Trease, P. L. Taberna, P. Simon and C. P. Grey, Faraday Discuss., 2014, 176, 49–68 RSC.
- Z. F. Lin, P. Rozier, B. Duployer, P. L. Taberna, B. Anasori, Y. Gogotsi and P. Simon, Electrochem. Commun., 2016, 72, 50–53 CrossRef CAS.
- E. Hark, A. Petzold, G. Goerigk, S. Risse, I. Tallo, R. Harmas, E. Lust and M. Ballauff, Carbon, 2019, 146, 284–292 CrossRef.
- J. Agrisuelas, C. Delgado, C. Gabrielli, J. J. Garcia-Jareno, H. Perrot, O. Sel and F. Vicente, J. Solid State Electrochem., 2015, 19, 2555–2564 CrossRef CAS.
- W. L. Gao, H. Perrot and O. Sel, Phys. Chem. Chem. Phys., 2018, 20, 27140–27148 RSC.
- T. Le, D. Aradilla, G. Bidan, F. Billon, M. Delaunay, J. M. Gerard, H. Perrot and O. Sel, Electrochem. Commun., 2018, 93, 5–9 CrossRef CAS.
- W. L. Gao, C. Debiemme-Chouvy, M. Lahcini, H. Perrot and O. Sel, Anal. Chem., 2019, 91, 2885–2893 CrossRef CAS PubMed.
- M. D. Levi, N. Levy, S. Sigalov, G. Salitra, D. Aurbach and J. Maier, J. Am. Chem. Soc., 2010, 132, 13220–13222 CrossRef CAS PubMed.
- M. D. Levi, G. Salitra, N. Levy, D. Aurbach and J. Maier, Nat. Mater., 2009, 8, 872–875 CrossRef CAS PubMed.
- M. D. Levi, M. R. Lukatskaya, S. Sigalov, M. Beidaghi, N. Shpigel, L. Daikhin, D. Aurbach, M. W. Barsoum and Y. Gogotsi, Adv. Energy Mater., 2015, 5, 1400815 CrossRef.
- C. Prehal, C. Koczwara, H. Amenitsch, V. Presser and O. Paris, Nat. Commun., 2018, 9, 4145 CrossRef CAS PubMed.
- Y. C. Wu, P. L. Taberna and P. Simon, Electrochem. Commun., 2018, 93, 119–122 CrossRef CAS.
- P. G. Kitz, M. J. Lacey, P. Novák and E. J. Berg, Anal. Chem., 2019, 91, 2296–2303 CrossRef CAS PubMed.
- Y. Gogotsi, A. Nikitin, H. Ye, W. Zhou, J. E. Fischer, B. Yi, H. C. Foley and M. W. Barsoum, Nat. Mater., 2003, 2, 591–594 CrossRef CAS PubMed.
- X. B. Zhao, B. Xiao, A. J. Fletcher and K. M. Thomas, J. Phys. Chem. B, 2005, 109, 8880–8888 CrossRef CAS PubMed.
- M. Kunze, S. Jeong, E. Paillard, M. Schonhoff, M. Winter and S. Passerini, Adv. Energy Mater., 2011, 1, 274–281 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- T. A. Centeno, M. Sevilla, A. B. Fuertes and F. Stoeckli, Carbon, 2005, 43, 3012–3015 CrossRef CAS.
- T. A. Centeno, O. Sereda and F. Stoeckli, Phys. Chem. Chem. Phys., 2011, 13, 12403–12406 RSC.
- F. Stoeckli and T. A. Centeno, Phys. Chem. Chem. Phys., 2012, 14, 11589–11591 RSC.
- B. Lobato, L. Suarez, L. Guardia and T. A. Centeno, Carbon, 2017, 122, 434–445 CrossRef CAS.
- J. Chmiola, G. Yushin, R. K. Dash, E. N. Hoffman, J. E. Fischer, M. W. Barsoum and Y. Gogotsi, Electrochem. Solid-State Lett., 2005, 8, A357–A360 CrossRef CAS.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- J. Chmiola, G. Yushin, R. Dash and Y. Gogotsi, J. Power Sources, 2006, 158, 765–772 CrossRef CAS.
- C. Largeot, C. Portet, J. Chmiola, P. L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed.
- J. Chmiola, C. Largeot, P. L. Taberna, P. Simon and Y. Gogotsi, Angew. Chem., Int. Ed., 2008, 47, 3392–3395 CrossRef CAS PubMed.
- M. D. Levi, S. Sigalov, G. Salitra, R. Elazari and D. Aurbach, J. Phys. Chem. Lett., 2011, 2, 120–124 CrossRef CAS.
- J. Piwek, A. Platek, E. Frackowiak and K. Fic, J. Power Sources, 2019, 438, 227029 CrossRef CAS.
- M. Zhu, C. J. Weber, Y. Yang, M. Konuma, U. Starke, K. Kern and A. M. Bittner, Carbon, 2008, 46, 1829–1840 CrossRef CAS.
- P. Kurzweil and M. Chwistek, J. Power Sources, 2008, 176, 555–567 CrossRef CAS.
- P. W. Ruch, D. Cericola, A. Foelske-Schmitz, R. Kotz and A. Wokaun, Electrochim. Acta, 2010, 55, 4412–4420 CrossRef CAS.
- M. D. Levi, S. Sigalov, G. Salitra, D. Aurbach and J. Maier, ChemPhysChem, 2011, 12, 854–862 CrossRef CAS PubMed.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- J. Jagiello and J. P. Olivier, J. Phys. Chem. C, 2009, 113, 19382–19385 CrossRef CAS.
- J. Jagiello and J. P. Olivier, Carbon, 2013, 55, 70–80 CrossRef CAS.
- M. V. Fedorov and A. A. Kornyshev, J. Phys. Chem. B, 2008, 112, 11868–11872 CrossRef CAS PubMed.
- J. W. Graydon, M. Panjehshahi and D. W. Kirk, J. Power Sources, 2014, 245, 822–829 CrossRef CAS.
- A. Laheaar, A. Arenillas and F. Beguin, J. Power Sources, 2018, 396, 220–229 CrossRef CAS.
- S. Kondrat and A. Kornyshev, J. Phys.: Condens. Matter, 2010, 23, 022201 CrossRef PubMed.
- H. Yang and Y. Zhang, J. Power Sources, 2015, 273, 223–236 CrossRef CAS.
- S. Kondrat, C. R. Perez, V. Presser, Y. Gogotsi and A. A. Kornyshev, Energy Environ. Sci., 2012, 5, 6474–6479 RSC.
- M. Kaus, J. Kowal and D. U. Sauer, Electrochim. Acta, 2010, 55, 7516–7523 CrossRef CAS.
- G. Lota and E. Frackowiak, Electrochem. Commun., 2009, 11, 87–90 CrossRef CAS.
- P. Skowron, E. Frackowiak and F. Beguin, ECS Trans., 2014, 61, 21–30 CrossRef CAS.
- M. Meller, J. Menzel, K. Fic, D. Gastol and E. Frackowiak, Solid State Ionics, 2014, 265, 61–67 CrossRef CAS.
- E. Frackowiak, M. Meller, J. Menzel, D. Gastol and K. Fic, Faraday Discuss., 2014, 172, 179–198 RSC.
- J. Menzel, K. Fic and E. Frackowiak, Prog. Nat. Sci.: Mater. Int., 2015, 25, 642–649 CrossRef CAS.
- Q. Abbas and F. Beguin, Prog. Nat. Sci.: Mater. Int., 2015, 25, 622–630 CrossRef CAS.
- Q. Abbas, P. Babuchowska, E. Frackowiak and F. Beguin, J. Power Sources, 2016, 326, 652–659 CrossRef CAS.
- Z. Y. Gao, L. C. Zhang, J. L. Chang, Z. Wang, D. P. Wu, F. Xu, Y. M. Guo and K. Jiang, Chem. Eng. J., 2018, 335, 590–599 CrossRef CAS.
- D. Jain, J. Kanungo and S. K. Tripathi, Appl. Phys. A, 2018, 124, 397 CrossRef.
- X. Gao, L. Zu, X. Cai, C. Li, H. Lian, Y. Liu, X. Wang and X. Cui, Nanomaterials, 2018, 8, 335 CrossRef PubMed.
- M. Pourbaix, Atlas of electrochemical equilibria in aqueous solutions, 1974 Search PubMed.
- P. Delahay, M. Pourbaix and P. Van Rysselberghe, J. Chem. Educ., 1950, 27, 683–688 CrossRef CAS.
- E. Frackowiak, K. Fic, M. Meller and G. Lota, ChemSusChem, 2012, 5, 1181–1185 CrossRef CAS PubMed.
- J. Lee, P. Srimuk, R. L. Zornitta, M. Aslan, B. L. Mehdi and V. Presser, ACS Sustainable Chem. Eng., 2019, 7, 10132–10142 CrossRef CAS.
- X. Wang, R. S. Chandrabose, S. E. Chun, T. Zhang, B. Evanko, Z. Jian, S. W. Boettcher, G. D. Stucky and X. Ji, ACS Appl. Mater. Interfaces, 2015, 7, 19978–19985 CrossRef CAS PubMed.
- J. Lee, P. Srimuk, S. Fleischmann, X. Su, T. A. Hatton and V. Presser, Prog. Mater. Sci., 2019, 101, 46–89 CrossRef CAS.
- K. Jayaramulu, D. P. Dubal, B. Nagar, V. Ranc, O. Tomanec, M. Petr, K. K. R. Datta, R. Zboril, P. Gómez-Romero and R. A. Fischer, Adv. Mater., 2018, 30, 1705789 CrossRef PubMed.
- P. Przygocki, Q. Abbas and F. Beguin, Electrochim. Acta, 2018, 269, 640–648 CrossRef CAS.
- S. T. Senthilkumar, R. K. Selvan, Y. S. Lee and J. S. Melo, J. Mater. Chem. A, 2013, 1, 1086–1095 RSC.
- Q. Abbas, H. Fitzek, V. Pavlenko and B. Gollas, Electrochim. Acta, 2020, 337, 135785 CrossRef CAS.
- Q. Abbas, S. Fitzek, S. Dsoke and B. Gollas, Nanomaterials, 2019, 9, 1413 CrossRef CAS PubMed.
- S. Yamazaki, T. Ito, Y. Murakumo, M. Naitou, T. Shimooka, M. Yamagata and M. Ishikawa, J. Power Sources, 2016, 326, 580–586 CrossRef CAS.
- Y. Taniguchi, Y. Ishii, A. Al-Zubaidi and S. Kawasaki, J. Nanosci. Nanotechnol., 2017, 17, 1901–1907 CrossRef CAS.
- M. Meller, K. Fic, J. Menzel and E. Frackowiak, ECS Trans., 2014, 61, 1–8 CrossRef CAS.
- K. Fic, M. Meller and E. Frackowiak, J. Electrochem. Soc., 2015, 162, A5140–A5147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee03867a |
| This journal is © The Royal Society of Chemistry 2021 |