
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From the Birkeland–Eyde process towards energy-efficient plasma-based NOX synthesis: a techno-economic analysis
Kevin H. R.
Rouwenhorst
 *a,
Fatme
Jardali
*b,
Annemie
Bogaerts
*b and
Leon
Lefferts
*a
*a,
Fatme
Jardali
*b,
Annemie
Bogaerts
*b and
Leon
Lefferts
*a
aCatalytic Processes & Materials, MESA+ Institute for Nanotechnology, University of Twente, P.O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: k.h.r.rouwenhorst@utwente.nl; l.lefferts@utwente.nl
bResearch Group PLASMANT, Department of Chemistry, University of Antwerp, Universiteitsplein 1, B-2610 Wilrijk-Antwerp, Belgium. E-mail: fatme.jardali@uantwerpen.be; annemie.bogaerts@uantwerpen.be
First published on 31st March 2021
Abstract
Plasma-based NOX synthesis via the Birkeland–Eyde process was one of the first industrial nitrogen fixation methods. However, this technology never played a dominant role for nitrogen fixation, due to the invention of the Haber–Bosch process. Recently, nitrogen fixation by plasma technology has gained significant interest again, due to the emergence of low cost, renewable electricity. We first present a short historical background of plasma-based NOX synthesis. Thereafter, we discuss the reported performance for plasma-based NOX synthesis in various types of plasma reactors, along with the current understanding regarding the reaction mechanisms in the plasma phase, as well as on a catalytic surface. Finally, we benchmark the plasma-based NOX synthesis process with the electrolysis-based Haber–Bosch process combined with the Ostwald process, in terms of the investment cost and energy consumption. This analysis shows that the energy consumption for NOX synthesis with plasma technology is almost competitive with the commercial process with its current best value of 2.4 MJ mol N−1, which is required to decrease further to about 0.7 MJ mol N−1 in order to become fully competitive. This may be accomplished through further plasma reactor optimization and effective plasma–catalyst coupling.
Broader contextIndustrial nitrogen fixation was first commercialized as the plasma-based Birkeland–Eyde process about a century ago, although this process was eventually outcompeted by the Haber–Bosch process due to the lower energy consumption for nitrogen fixation of the Haber–Bosch process. Nitrogen fixation is currently highly centralized, due to the high temperature and high pressure synthesis of ammonia via the Haber–Bosch process. Due to the emergence of low cost renewable electricity from solar and wind, there is renewed interest in decentralized opportunities for electricity-driven nitrogen fixation. In recent years, computational studies have greatly enhanced the understanding of plasma-based nitrogen fixation. This has allowed for optimized plasma reactors with reduced energy consumption for plasma-based NOX synthesis. This has spurred renewed interest in the plasma-based nitrogen fixation process for decentralized and on-demand fertilizer production. The recent developments are discussed in the current analysis paper, as well as energy consumption targets for renewed commercialization of plasma-based nitrogen fixation. |
Introduction
For over a century, nitrogen (N2) has been industrially fixed into reactive nitrogen (Nr) compounds to increase agricultural yields.1 In order to artificially fix atmospheric N2, different attempts have been made throughout the years, including the Birkeland–Eyde (B–E) process that produces NOX,2 the Frank–Caro (F–C) process that produces calcium cyanamide,3 and the Haber–Bosch (H–B) process that produces ammonia (NH3),4 among others. Nowadays, nitrogen is almost exclusively fixed via the Haber–Bosch process.4 An overview of the annual consumption of fixed nitrogen from various natural sources and from industrial nitrogen fixation technologies is shown in Fig. 1. Guano and Chile saltpetre are natural sources of fixed nitrogen, mostly derived from Chile and Peru.4 Ammonium sulphate is a by-product of coke ovens and of caprolactam production.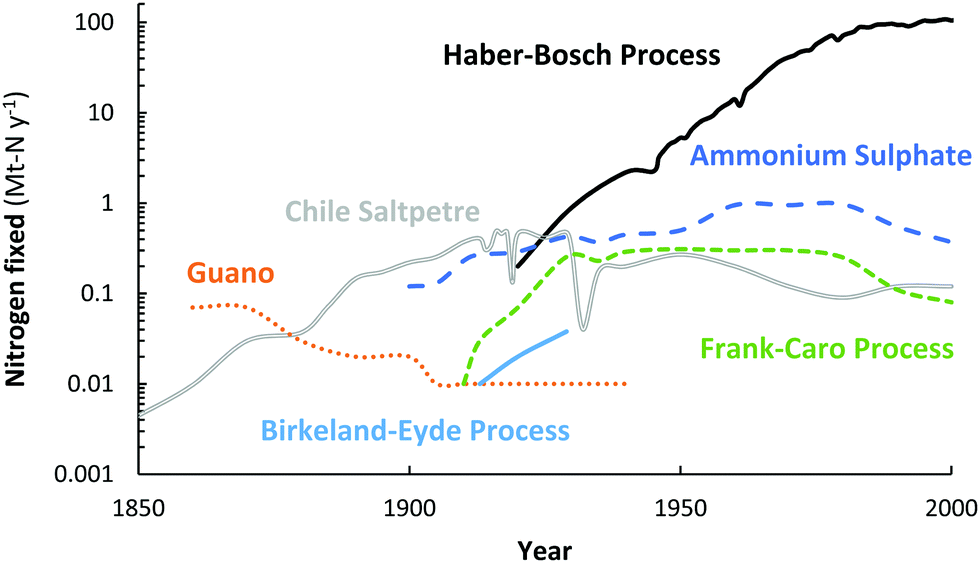 | ||
| Fig. 1 Annual consumption of fixed nitrogen from various natural sources and from industrial nitrogen fixation technologies. Original sources.2,4,5 | ||
In 1903, the first synthetic plasma-based NOX synthesis process was developed and tested in Christiania University (nowadays known as the University of Oslo) by Kristian Birkeland and Samuel Eyde. In the B–E process, air was passed through an electric arc, i.e., a thermal plasma, thereby producing nitrogen oxide (NO) and nitrogen dioxide (NO2) (eqn (1) and (2)). Thereafter, NO2 was concentrated and absorbed in water to form nitric acid (HNO3) (eqn (3)).
Nitric acid can also be produced via the combined Haber–Bosch (H–B) and Ostwald process. In the H–B process, ammonia (NH3) is synthesized from hydrogen (H2) and atmospheric nitrogen (N2) (eqn (4)). The NH3 produced by the H–B process is then oxidized in the Ostwald process to form NO and NO2 (eqn (2) and (5)). Subsequently, the NO2 is absorbed in water to from HNO3. In both processes, the resulting product is HNO3, which can be neutralized with NH3 to form ammonium nitrate (NH4NO3) (eqn (6)). NH4NO3 is primarily used as a fertilizer for agricultural activity and as an explosive for the mining industry. NH4NO3 production accounts for about 75–80% of the HNO3 produced.6 Further uses of HNO3 include nitration reactions, its usage as oxidant and as rocket propellant.
| N2+ O2 ⇌ 2NO. with ΔHor = 180 kJ mol−1. | (1) |
| 2NO + O2 → 2NO2. with ΔHor = −114 kJ mol−1. | (2) |
| 3NO2 + H2O → 2HNO3 + NO. with ΔHor = −117 kJ mol−1. | (3) |
| 3H2 + N2 → 2NH3. with ΔHor = −92 kJ mol−1. | (4) |
| 4NH3 + 5O2 → 4NO + 6H2O. with ΔHor = −905 kJ mol−1. | (5) |
| NH3 + HNO3 → NH4NO3. | (6) |
Throughout the years, different factors played a role in the abandonment of the plasma-based B–E process in favour of the fossil-fuel powered H–B technology, including (i) emergence of low-cost fossil fuels such as coal and natural gas, (ii) the substantially lower energy cost for nitrogen fixation via the thermochemical H–B process (about 0.5–0.6 MJ mol N−1) as compared to the plasma-based B–E process (about 2.4–3.1 MJ mol N−1),7–10 (iii) the higher capital investment for the B–E compared to the combined H–B and Ostwald process,2 and (iv) the higher maintenance cost of the B–E reactor.2,11 Therefore, NOX production via NH3 produced in the H–B process is more cost effective despite the fact that this is actually a detour. Nitrogen in N2 (oxidation state 0) is first reduced to ammonia (oxidation state −3), where after it is oxidized again to NO (oxidation state +2); in fact H2 is burnt in this sequence to drive the overall reaction. Instead, a direct route from N2 (oxidation state 0) to NO (oxidation state +2) in eqn (1) would be an elegant shortcut, which has the potential to be more efficient.
The H–B technology substantially increased the agricultural productivity and it succeeds in sustaining about 50% of the world population.12 Nevertheless, the H–B process suffers from its poor scalability for decentralized production. Thus, industrial plants typically produce at least 100 t-NH3 per day.5 Furthermore, the H–B process operates at high temperatures and high pressures (350–500 °C and 100–300 bar), implying operation with varying load from intermittent renewables is difficult. Therefore, current research focuses on enabling load variation,13 and on NH3 synthesis under milder conditions.14 Eventually, the H–B process may be replaced by a single-pass thermo-catalytic NH3 synthesis process or electrochemical NH3 synthesis.15,16
The emergence of low cost and intermittent renewable electricity may change the preferred choice of technology. Plasma technology offers potential benefits, such as fast turning on and off, and scalability for small communities.9,17 The aim of this paper is to evaluate whether plasma-activated NOX synthesis can become a feasible alternative for nitrogen fixation again in the 21st century, just like it was at the start of the 20th century. We identify how the state-of-the-art plasma nitrogen fixation process compares to the benchmark thermo-catalytic H–B process with the subsequent thermochemical Ostwald process. For this purpose, we will first explain the principles and state-of-the-art of the B–E process, the H–B process and the Ostwald process.
The Birkeland–Eyde process
The B–E process was the first nitrogen fixation process to operate commercially with hydropower in Niagara Falls (Canada). The power supplied to the B–E plant increased from 2.24 kW in 1903 to 238.6 MW in 1928. This commercial plant succeeded in fixing 38 kt-N year−12. About 175 t-air was required to fix 1 t-N via the B–E process.9 The B–E process consumed about 2.4–3.1 MJ mol N−1 and produced 1–2 mol% NO.9,17 A process scheme for the B–E process is shown in Fig. 2. Air was converted to NO in an electric arc formed between two co-axial, water-cooled copper electrodes placed between the poles of a strong electromagnet inside a furnace, for which various alternative configurations were considered.9 Rapid quenching of the dilute nitrogen oxides to 800 – 1000 °C was applied at the reactor outlet to prevent reverse reactions (i.e., converting NO back to N2 and O2).2 The heat of the reaction was recovered in waste heat boilers. Afterwards, oxidation of NO to NO2 took place at a slow rate in a large oxidation chamber. Since the absorption capacity increases with decreasing temperature, the mixture of NO and NO2 leaving the economizer at about 200 °C was further cooled to 50 °C in cooling towers before entering the absorption towers. Finally, NO2 gas was absorbed in water to produce a solution of HNO3. The final stream contained about 30% HNO3 in water.2 The unabsorbed NOX was passed through alkaline absorption columns for further absorption. Despite this second absorption step, about 3% of the produced NOX was purged to the atmosphere.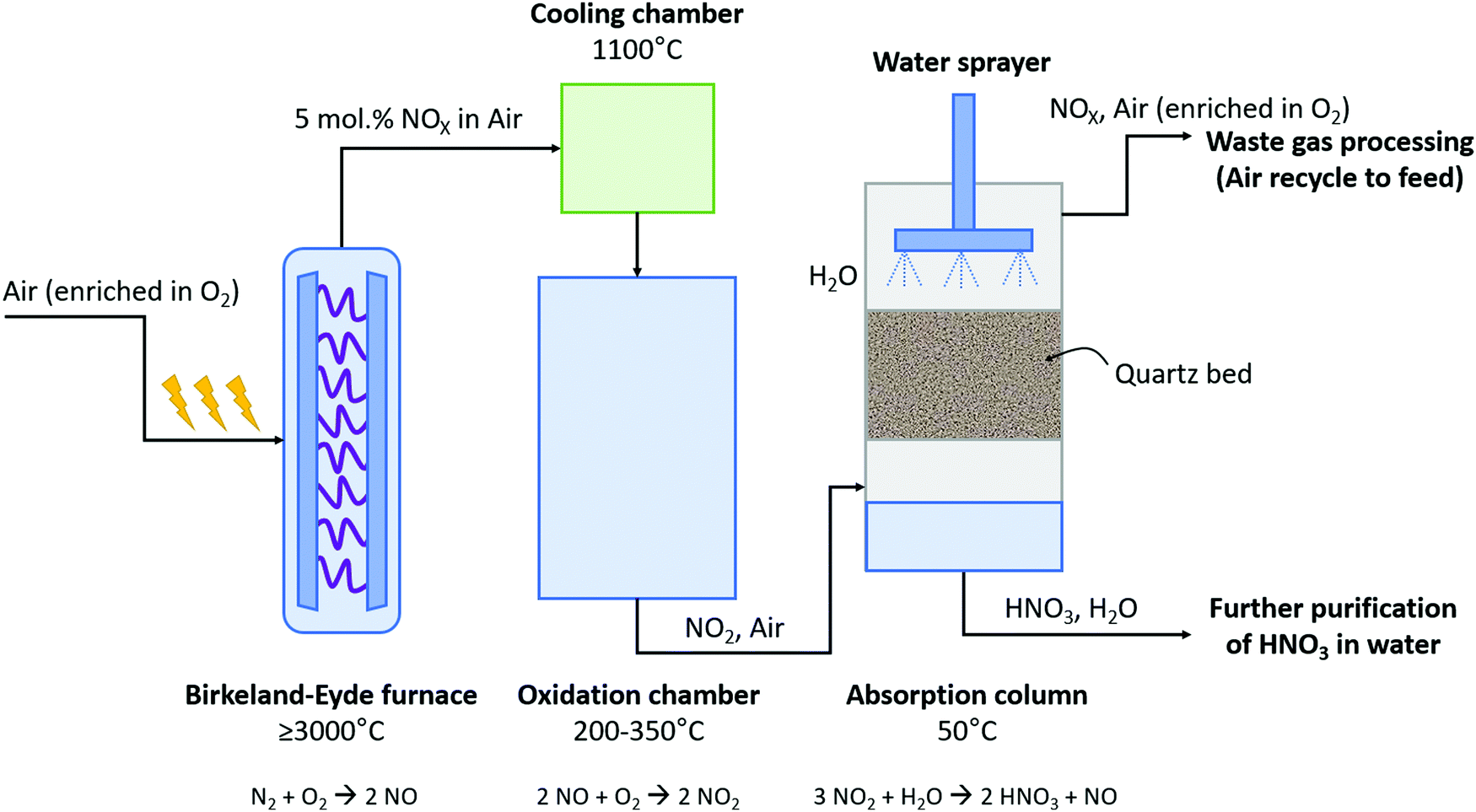 | ||
| Fig. 2 Process scheme for the Birkeland–Eyde industrial nitrogen-fixation process. Inspired by Patil et al.9 | ||
Many ideas have been suggested to reduce the energy consumption of NOX production and improve the performance of the B–E process, as for example the use of a 50–50% mixture of N2 and O2, preheating the inlet gas, applying heat recovery from process gas and operating the furnace at elevated pressures.9 However, not only the plasma reactor is a major contributor to the investment and the energy cost of the B–E process, as also the absorption towers, especially the acid absorption towers, contribute significantly to the CapEx and OpEx.18 According to a 1922 report on nitrogen fixation, the absorption columns compose over 40% of the CapEx and about 30% of the OpEx.18 These absorbers were costly due to the low concentration of NOX at the outlet of the plasma reactor. However, this technology has been optimized for the Ostwald process in previous decades, which could be used in combination with the B–E process as well. More recently, adsorbents, such as BaO, have been used to concentrate NOX for car exhaust catalysts.19 Through temperature swing adsorption (TSA) or pressure swing adsorption (PSA), the concentration of NOX can be increased by using such solid sorbents. Possibly, such solid sorbents can replace or minimize the use of the costly absorption columns in the B–E process.
The Haber–Bosch process combined with the Ostwald process
In 1908, Haber and Le Rossignol demonstrated the feasibility of direct synthesis of 2 kg-NH3 day−1 from N2 and H2 with a table top system operating at 500–550 °C and 100–200 atm, in the presence of an osmium catalyst.1 In the following years, Mittasch and co-workers developed the multicomponent iron catalyst, a less poisonous and more abundant material, as an alternative to osmium for NH3 synthesis,20,21 while Bosch and co-workers solved engineering challenges regarding the operation with H2 at high pressures.22 In 1913, the first ammonia synthesis plant started operating according to the H–B process at BASF in Oppau, Ludwigshafen.20 Nowadays, the H–B process starting from methane consumes about 0.5–0.6 MJ mol N−1. This is the total energy content of the feed methane, of which about two third is transformed into hydrogen, while the remainder is used for heating during the steam methane reforming section for H2 production, as discussed below.23 The energy content of the ammonia product is only 0.32 MJ mol N−1, implying significant heat generation during ammonia synthesis from methane. On the other hand, the H–B process starting from H2O and N2 also consumes about 0.5–0.6 MJ mol N−1 nowadays. The theoretical minimum energy consumption for NH3 synthesis from H2O and N2 is 0.35 MJ mol N−1. The overall yield of the H–B process is typically 97–99%, depending on the source of H2 used.15Schematic diagrams for a natural gas-based H–B process and an electrolysis-based H–B process are shown in Fig. 3. In the former method, H2 is produced from methane (CH4) via steam methane reforming (SMR), in which a mixture of CO, CO2, and H2 is produced. Typically, CH4 is first converted with H2O to CO and H2 in a tubular reformer at 850–900 °C and 25–35 bar, after which the last portion of CH4 conversion is performed by partial oxidation with air at 900–1000 °C, thereby introducing N2 in the gas mixture. The CO is then converted with H2O to CO2 and H2 in a two-stage water–gas-shift reactor, after which CO2 is removed. Traces of CO are converted to CH4 in a methanation step just before the synthesis loop, preventing deactivation of the ammonia synthesis catalyst. The feed gas, mainly composed of H2 and N2, is then compressed and fed to the ammonia synthesis loop operating at typically 100–300 bar, in which the reactants are fed to the ammonia synthesis reactor with iron-based catalysts operating at 350–500 °C. About 15–20% of the feed gas is converted to NH3. The reactor effluent is then cooled down to ambient temperature to condense the NH3 out. The remaining gas is recycled to the NH3 synthesis reactor. This process scheme of NH3 synthesis would be similar to the electrically-driven system. Here, H2 is produced by electrolysis. Purified N2 in the electrolysis-based process is produced in a separate unit by pressure swing adsorption (PSA) or cryogenic distillation.14,24 Due to the different feedstocks for the SMR-based Haber–Bosch process and the electrolysis-based Haber–Bosch process, the heat integration between the process components changes substantially.
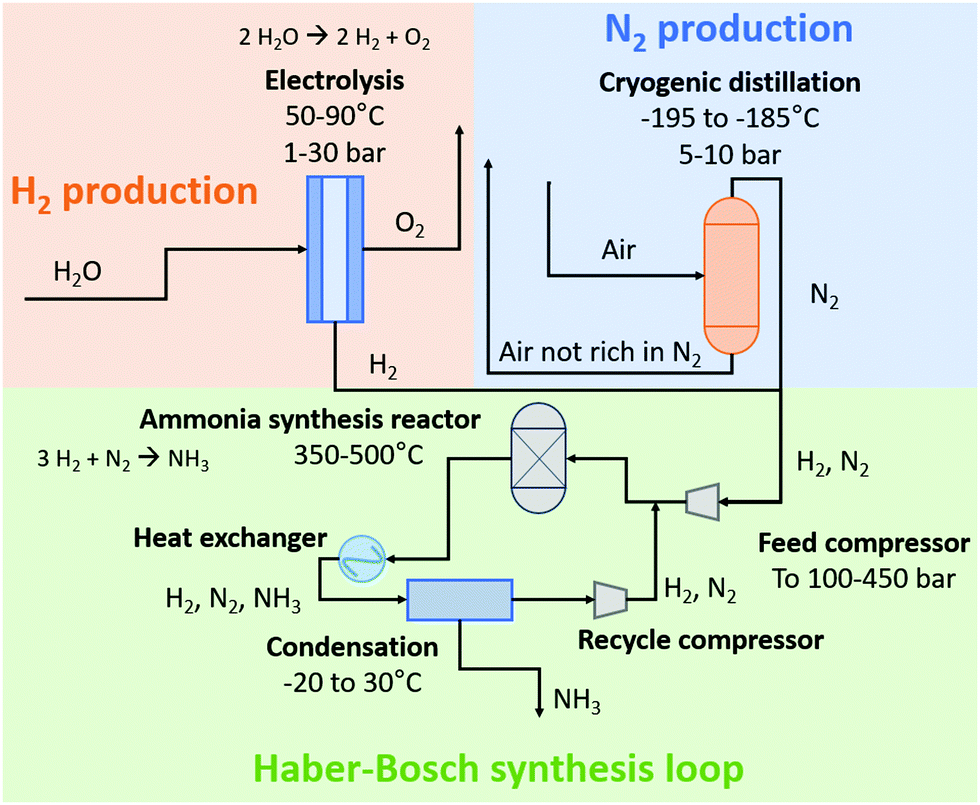 | ||
| Fig. 3 Schematic diagram of the electrolysis-based Haber–Bosch process. For details, see text. Inspired by ref. 15. | ||
The subsequent oxidation process was developed by Wilhelm Ostwald, who patented the ammonia oxidation process in 1902.6 In this process, ammonia is oxidized in the presence of a rhodium–platinum gauze to form NO and H2O at 600–800 °C and 4–10 atm. Afterwards, NO is cooled to about 50 °C and subsequently oxidized to NO2 and absorbed in H2O, producing dilute HNO3. The untreated NO is recycled, while the HNO3 is concentrated by distillation. The overall yield of the Ostwald process is typically 98%. A process scheme of the Ostwald process is shown in Fig. 4, which is similar to the B–E process (see Fig. 2), although less absorption steps are required due to the higher NO2 concentration after the oxidation reactor.
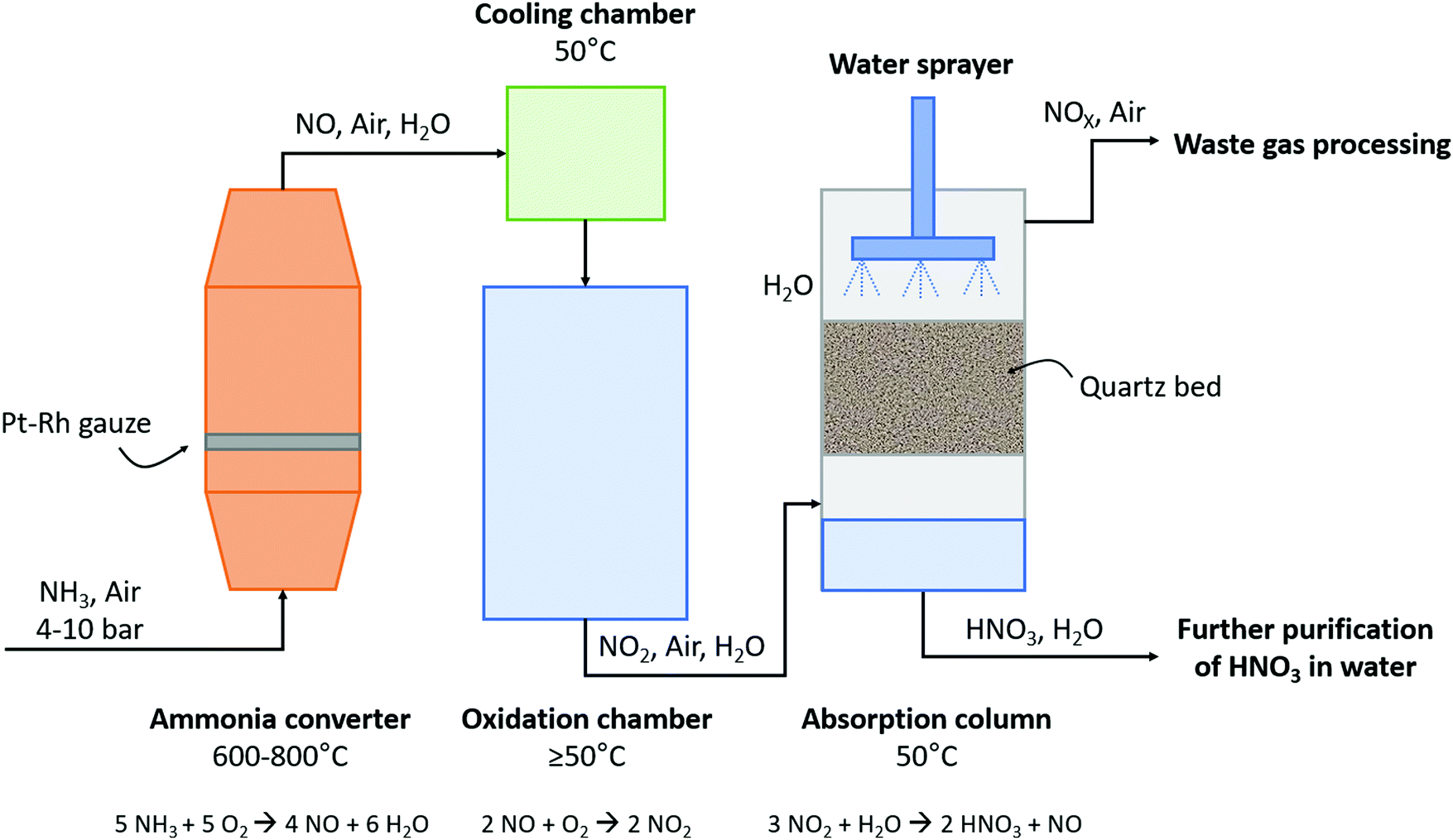 | ||
| Fig. 4 Schematic diagram of the Ostwald process. For details, see text. Reproduced from ref. 25. | ||
State-of-the-art of plasma-based NOX synthesis
As discussed above, plasma-based NOX synthesis was commercialized by Birkeland and Eyde in 1903,26,27 and the energy consumed by the electric arc to generate a thermal plasma for NO synthesis is 2.4–3.1 MJ mol N−17–9. Hereafter we will discuss the state-of-the-art of plasma-based NOX synthesis, as well as potential avenues for improvements.Plasma types and comparison of energy consumption
Various plasma types can be distinguished, namely thermal plasmas, warm plasmas, and non-thermal plasmas. In a thermal plasma, the electrons and the heavier plasma species (molecules, radicals, and ions) are in thermal equilibrium, forming a quasi-neutral plasma bulk. The temperature in a thermal plasma is typically high (order of 104 K). The highest NO equilibrium concentration (about 5 mol%) can be achieved at a gas temperature near 3500 K and at 1 atm.8 The NO formed is also prone to decomposition after the plasma, forming N2 and O2 again. Therefore, rapid quenching of the gas is required at a rate of several millions of Kelvins per second.28,29 However, even if thermal plasma reactors are optimized, the theoretical minimum energy consumption for thermal plasmas is 0.72 MJ mol N−1, which means that the energy efficiency of thermal plasma cannot compete with nitric acid produced from an electrolysis-based Haber–Bosch process (about 0.6 MJ mol N−1). Here, the energy consumption refers to the electricity input for nitrogen fixation. The theoretical minimum energy consumption for thermal plasmas is based on the assumption that both N2 and O2 dissociate completely in the plasma, considering the bond-dissociation energies of N2 (945 kJ mol−1) and O2 (498 kJ mol−1).In a non-thermal plasma, on the other hand, the electrons are not in equilibrium with the heavier plasma species, resulting in a substantially higher electron temperature as compared to the gas temperature, which is typically near room temperature. This potentially allows for selectively activating molecules with a strong chemical bond, such as N2 (about 9.79 eV).30 This is relevant for NO formation, as breaking the triple N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond is rate-limiting for the formation of NO. The O2 dissociation step takes place more easily, because of the somewhat weaker O
N bond is rate-limiting for the formation of NO. The O2 dissociation step takes place more easily, because of the somewhat weaker O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond (about 5.12 eV). Depending on the actual electron temperature, electrons can excite the molecules to various vibrational and electronic states. In typical non-thermal plasmas, such as dielectric barrier discharges (DBDs), the electron temperature is typically several eV, which mainly gives rise to electronic excitation.17
O double bond (about 5.12 eV). Depending on the actual electron temperature, electrons can excite the molecules to various vibrational and electronic states. In typical non-thermal plasmas, such as dielectric barrier discharges (DBDs), the electron temperature is typically several eV, which mainly gives rise to electronic excitation.17
In between thermal and non-thermal plasmas, we can identify so-called warm plasmas, such as gliding arc (GA) and microwave (MW) plasmas, in which the electron temperature is still higher than the gas temperature, but the latter can be several 1000 K.17 The electron temperature is typically 1–2 eV,17 which is more beneficial for vibrational excitation of the molecules than in non-thermal plasmas (see eqn (7) for vibrational excitation). This gives rise to more efficient NOX formation in warm plasmas.
Indeed, the NO formation rate via the reaction of atomic oxygen with N2 by the so-called vibrationally-promoted Zeldovich mechanisms (eqn (8)) is enhanced upon increasing the population of N2 vibrational levels in the plasma. The chain mechanism of NO synthesis is closed by the exergonic reaction given by Equation 9.28,31 The sum of eqn (8) and (9) then gives a net energy consumption of 0.2 MJ mol N−1 for NOX synthesis (cf.Table 1), i.e., lower than NOX synthesis via the electrolysis-based Haber–Bosch process combined with the Ostwald process. Therefore, exploiting the non-equilibrium phenomena in a plasma is a promising approach to increase the energy efficiency of plasma-based processes for nitrogen fixation.
| Technology | Best available technology (MJ mol N−1) | Minimum energy consumption* (MJ mol N-1) |
|---|---|---|
| Thermochemical process (electrolysis-based Haber–Bosch + Ostwald) | 0.615 | 0.3532 |
| Thermal plasma (Birkeland–Eyde process) | 2.4–3.17–9 | 0.72 |
| Warm plasma (Gliding arc reactor) | 2.433 | 0.534 |
| Plasma with only vibrationally-promoted Zeldovich mechanism (only vibrational excitations in N2) | — | 0.28 |
It should also be noted that unproductive electronic excitation and ionization channels in real plasma reactors lead to a higher minimum energy consumption than for an hypothetical plasma reactor operating exclusively via the vibrationally-promoted Zeldovich mechanism (eqn (8)). The distribution of productive and unproductive N2 activation channels leads to a theoretical minimum energy consumption of about 0.5 MJ mol N−1 (see Table 1) for a gliding arc plasma reactor, which is a warm plasma type.28,33,34 The different plasma activation channels for N2 and O2 in various plasma reactors are shown in Fig. 6.
In practice, the energy consumption is even higher, which is due to vibrational–translational relaxation (hence depopulating the N2 vibrational levels), and NOX decomposition after the plasma if the temperature does not drop fast enough. Plasma radicals may also recombine to form O2 and N2 again, implying all energy is lost as heat. Lastly, decomposition of NOX products in the plasma will further limit the energy efficiency. With increasing NOX concentration, the probability of plasma-activation of NOX increases, thereby promoting the reaction back to N2 and O2.
| e− + N2 → e− + N2(v). | (7) |
| O + N2(v) → NO + N. | (8) |
| N + O2 → NO + O. | (9) |
The enthalpy of formation for NO is 90 kJ mol N−1 and any addition of energy input above that level leads to the formation of heat. Thus, even in case of the Zeldovich mechanism with an energy consumption of 0.2 MJ mol N−1, 55% of the energy in the reactor is lost as heat. In case of thermal dissociation of the triple NN bond (945 kJ mol−1) and double OO bond (498 kJ mol−1), only 12% of the energy is stored in the NO bond whereas 88% in converted to heat.
Plasma catalysis
A potential avenue to improve the energy efficiency of the process beyond optimizing the plasma is the introduction of a catalyst. Catalysts are used in most chemical processes to decrease the reactor size, as well as to operate at milder operating conditions and to lower t energy requirement. Various authors have attempted the use of metal and metal oxide catalysts for plasma-based NOX synthesis.35,36 However, up till now, results are inconclusive on whether there is an actual catalytic effect rather than a change in the physiochemical plasma properties d to the introduction of a packing material into the reactor.8,36,37 A change of packing material is known to modify the plasma properties, and thereby the conversion.38 Some synergistic effects between plasma and catalyst have however been proposed. Rapakoulias et al.35 investigated NO synthesis in the presence of transition metal oxides, such as molybdenum trioxide (MoO3) and tungsten trioxide (WO3) catalysts (e.g. n-type semiconductors). The authors proposed that the vibrationally excited N2 molecules undergo dissociative adsorption on the catalytic surface (eqn (10)). This may occur because n-type semiconductors donate electrons because of their easy ionization. Therefore, the adsorbed molecule can accept electrons to its anti-bonding π* orbital, leading to its pre-dissociation.39 Then, the atomic nitrogen may react with surface oxygen, forming NO upon desorption (eqn (11)). The oxygen vacancy can then be replenished by oxygen from the gas phase (eqn (12)), thereby oxidizing the transition metal surface, according to a Mars–van Krevelen redox mechanism.40 | (10) |
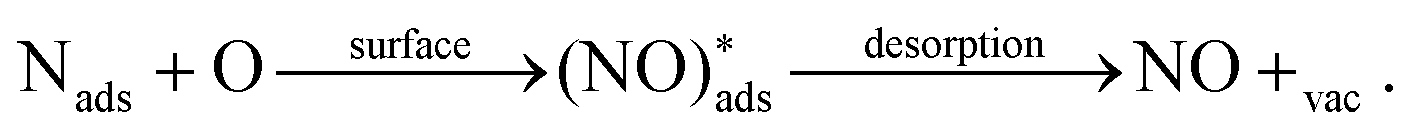 | (11) |
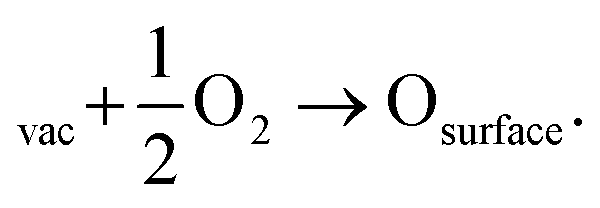 | (12) |
A limitation of a thermally-active catalyst is that it always catalyses not only the NOX synthesis reaction but also the reverse decomposition reaction.45 As the equilibrium at mild conditions is completely towards the formation of N2 and O2, a metal catalyst with thermal catalytic activity will in principle mainly form N2 and O2 under mild conditions.46 The presence of a surface could improve the performance only if it would enhance an irreversible reaction step, e.g. a quenching reaction of a highly activated species, leading to the formation of NOX.47 This can potentially be achieved with metal oxide catalysts, or metals inactive for NOX decomposition such as Ag and Au. However, at ambient temperatures, a catalytic effect was not observed for NOX synthesis on alumina-supported W-, Co- and Pb-oxides in a dielectric barrier discharge (DBD) reactor,36 and any change in activity must be attributed to modifications in the physiochemical plasma properties due to the introduction of a packing material into the reactor. On the other hand, metal oxides become active for NO decomposition at elevated temperatures.45,48–50
Performance of various plasma reactors
Various plasma types and plasma reactors have been investigated for NOX production after the earlier research on thermal plasma (i.e., the electric arc).26,27,33,51 This includes spark discharges,52–55 radio-frequency crossed discharge,56 laser-produced discharge,57 corona discharges,52,58 glow discharges,53,59 (packed bed) dielectric barrier discharges (PB) DBD,36,53 (pulsed) (gliding) arc discharges,34,53,60–62 microwave (MW) discharges,63–65 and plasma jets in contact with water.66–76A summary of the reported energy consumption and the product concentration in various plasma reactors is listed in Table 2. Additionally, the reported NOX concentration and energy consumption are shown in Fig. 5. A distinction is made between various types of plasma reactors.
| Plasma type | Product (concentration) | Energy cost (MJ mol N−1) | Ref. |
|---|---|---|---|
| Electric arc (Birkeland–Eyde) | NO (2%) | 2.4–3.1 | 26,27,51 |
| Spark discharge | NO and NO2 | 20.27, 40 | 52,55 |
| Transient spark discharge | NO and NO2 | 8.6 | 54 |
| Pin-to-plane ns-pulsed spark discharge | NO and NO2 | 5.0–7.7 | 53 |
| Radio-frequency crossed discharge | HNO3 | 24–108 | 56 |
| Laser-produced discharge | NO and NO2 | 8.9 | 57 |
| (Positive/negative) DC corona discharge | NO and NO2 | 1057/1673 | 52 |
| Pulsed corona discharge | HNO3 | 186 | 58 |
| Pin-to-plane DC glow discharge | NO and NO2 | 7 | 53 |
| Pin-to-pin DC glow discharge | NO and NO2 (0.7%) | 2.8 | 59 |
| Dielectric barrier discharge | NO and NO2 (0.6%) | 56–140 | 53 |
| Packed dielectric barrier discharge | NO and NO2 (0.5%) | 18 | 36 |
| DC plasma arc jet | NO (6.5%) | 3.6 | 60 |
| Propeller arc | NO and NO2 (0.4%) | 4.2 | 53 |
| Pulsed milli-scale gliding arc | NO and NO2 (1–2%) | 2.8–4.8 | 61,62 |
| Gliding arc plasmatron | NO and NO2 (1.5%) | 3.6 | 34 |
| Rotating gliding arc | NO and NO2 (5.4%) | 2.5 | 33 |
| Microwave plasma | NO and NO2 (0.6%) | 3.76 | 63 |
| Microwave plasma with catalyst | NO (6%) | 0.84 | 64 |
| Microwave plasma with magnetic field | NO (14%) | 0.28 | 65 |
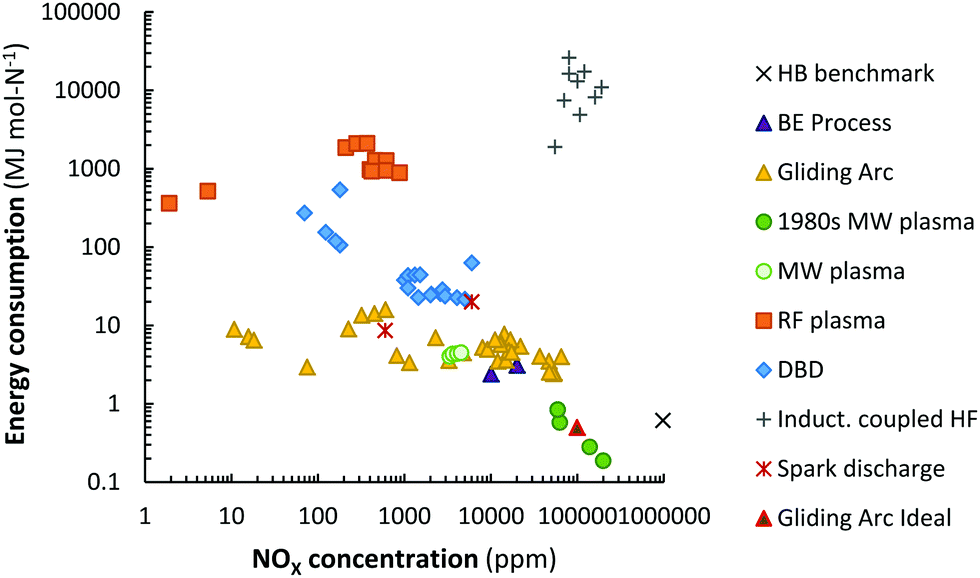 | ||
| Fig. 5 Comparison of energy consumption for NO production in various plasma reactors. Original references: 1980s low pressure MW plasma,64,65,77,78 MW plasma,63 gliding arc,33,34,53,60,62,79–82 RF plasma,83,84 DBD,36,53,85,86 inductively coupled HF,35 spark discharge.54,55 | ||
Among the different plasma types, warm plasmas, such as gliding arcs (GA), atmospheric pressure glow discharges (APGD) and microwave plasmas (MW), have been explored extensively for gas conversion applications.17 As explained above, warm plasmas are a special type of plasma that include both thermal and non-thermal plasma characteristics. The gas temperature is typically a few 1000 K, while the electron temperature is still higher (1–2 eV), thus, providing warm plasmas with non-equilibrium (or non-thermal) characteristics. However, the vibrational temperature is (nearly) equal to the gas temperature, resulting in vibrational–translational (VT) equilibrium.87,88 Therefore, warm plasmas are also known as quasi-thermal plasmas.
Different GA reactor configurations have shown promise for gas conversion applications.17,34,61,62,89,90 GA plasmas are characterized by reduced electric fields below 100 Td, resulting in electron energies around 1 eV. Such electron energies are most beneficial for vibrational excitation of the gas molecules (see Fig. 6a).17 Wang et al.62 investigated NOX formation mechanisms in a pulsed-power milliscale GA reactor, while Vervloessem et al.34 studied NOX formation in a reverse-vortex flow gliding arc plasmatron (GAP). The chemical kinetics modelling results showed that the vibrationally excited N2 molecules can reduce the energy barrier of the non-thermal Zeldovich mechanism O + N2(v) → NO + N, providing an energy-efficient way for NO production.
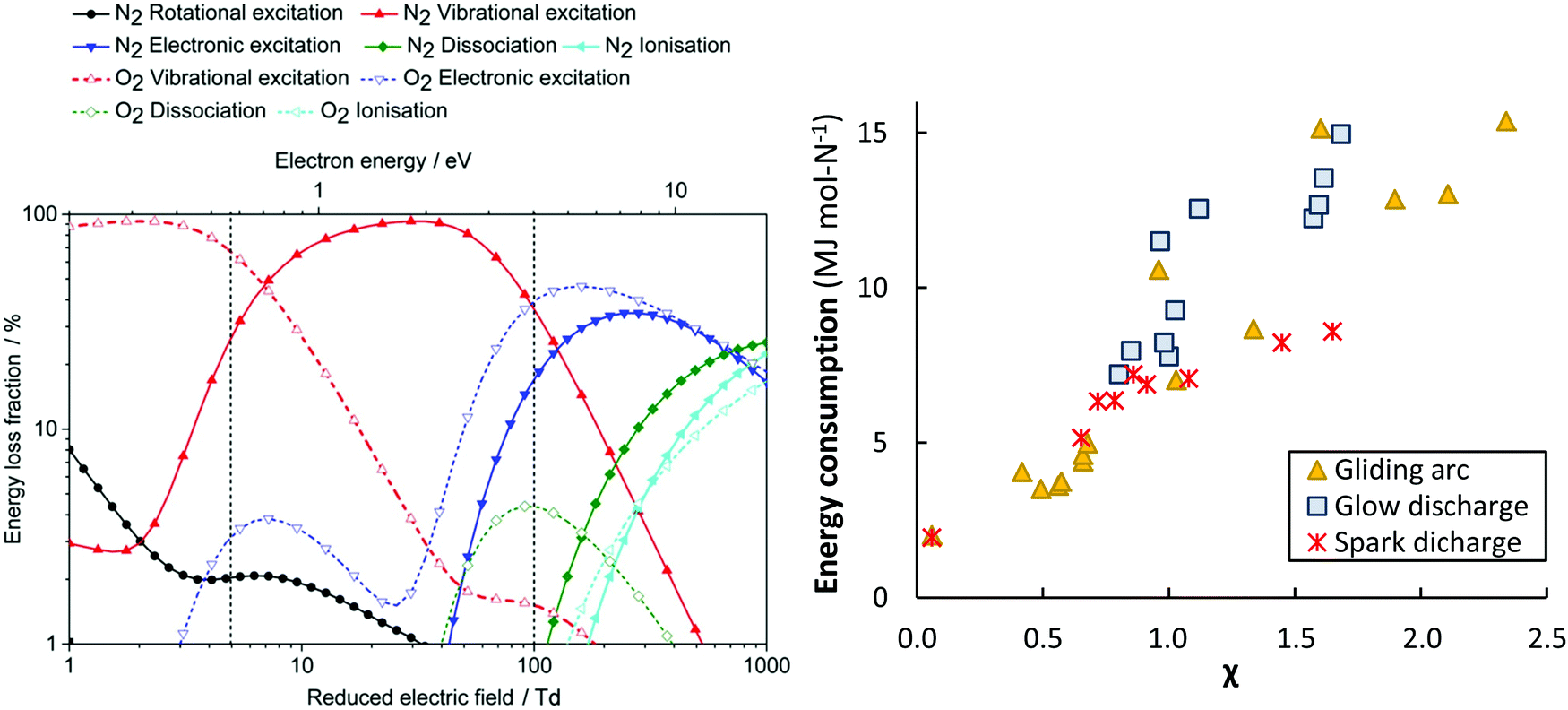 | ||
Fig. 6 Left: The dominant plasma-activation channels in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 N2:O2 stream. Reproduced from ref. 62. Reduced electric fields of 5–100 Td correspond to GA and MW plasmas, while the region above 100 Td corresponds to a DBD reactor.17 Right: The apparent energy cost as function of the χ factor, as proposed by Pei et al.53 Original reference: gliding arc,34,53,62,80,81,91–95 glow discharge,53 spark discharge.53,54 :50 N2:O2 stream. Reproduced from ref. 62. Reduced electric fields of 5–100 Td correspond to GA and MW plasmas, while the region above 100 Td corresponds to a DBD reactor.17 Right: The apparent energy cost as function of the χ factor, as proposed by Pei et al.53 Original reference: gliding arc,34,53,62,80,81,91–95 glow discharge,53 spark discharge.53,54 | ||
Moreover, the high gas temperature (>3000 K) leads to significant thermal dissociation of the lower N2 vibrational levels, whose vibrational distribution function exhibits a Boltzmann shape. In fact, thermal reactions are quite efficient at the high temperatures reached in GA reactors. The limitation in the overall N2 conversion is rather the fraction of gas treated by the GA plasma. For instance, only 15% of the gas is estimated to pass through the plasma arc in the GAP and the rest of the gas by-passes through the reactor without contacting the plasma.90,96 Vervloessem et al.34 reported a NOX yield of 1.5% at an energy consumption of 3.6 MJ mol N−1. Through reactor optimization and by preventing the transfer of vibrational energy from N2 to O2, the authors showed that the energy consumption can potentially decrease to 0.5 MJ mol N−1.34
Janda et al.54 studied NOX production in a transient spark discharge. This type of spark discharge starts from a streamer phase, i.e. a non-thermal plasma, and is subsequently transformed into short spark current pulses which generate a thermal plasma. The self-pulsing feature of the discharge avoids thermalization of the plasma.97,98 The spark phase is characterized by a high chemical activity due to the high electron density achieved (about 1017 cm−3). The excited nitrogen molecules (N2*) were observed in both the streamer and the spark phases and the energy consumption for NOX production was 8.6 MJ mol N−154. Pavlovich et al.55 developed a spark-glow discharge reactor, where the generated plasma discharge had a spark phase (thermal plasma) and glow phase (non-thermal plasma) in one cycle. The authors were able to control the percentage of glow phase by fine-tuning the voltage waveforms. The spark phase, which had a very high electron density and energy, generated more NO, while the glow phase promoted the oxidation of NO to NO2. However, the energy consumption of NOX production was as high as 40 MJ mol N−1. In general, such plasma types have a limited volume, resulting in a limited fraction of the N2 gas being exposed to the plasma, and thus a limited amount of NOX produced.
Packed bed DBD reactors have also been studied, because of the possibility to enhance the product selectivity and the energy efficiency by combining the plasma with a catalyst. Patil et al.36 studied NOX production in a DBD packed with different catalyst support materials (α-Al2O3, γ-Al2O3, TiO2, MgO, TaTiO3, and quartz wool). The authors obtained the best results with a γ-Al2O3 catalyst with the smallest particle size of 250–160 μm. However, the obtained energy cost was high (18 MJ mol N−1) and the product yield low (0.5 mol%), compared to other atmospheric pressure plasma reactors.36 These poor results obtained in a DBD could be explained by the high reduced electric field, i.e. above 100–200 Td, which creates highly energetic electrons, resulting mainly in electronic excitation, ionization, and dissociation, instead of vibrational excitation (see Fig. 6a), and thus not exploiting the most energy-efficient NOX formation pathway through the vibrationally-induced Zeldovich mechanism.17
The best results in terms of product yield and energy consumption were obtained in low-pressure MW plasmas. The energy consumption obtained in a MW plasma with catalyst was stated to be 0.84 MJ mol N−1 for an NO concentration of 6 mol%.64 The highest NO concentration of 14% and lowest energy cost of 0.28 MJ mol N−1 were reported for a MW plasma with magnetic field (so-called electron cyclotron resonance).65 However, these values were reported in the 1980s and have not been reproduced in recent years. A similar situation exists for plasma-based CO2 splitting, where results from the 1980s could not be reproduced with similar reactors in recent years.99 Therefore, the reported energy yield calculations for plasma-based NOX synthesis in a MW plasma from the 1980s should be assessed critically.
These MW plasmas operated at reduced pressures (down to 66 mbar), which indeed promote vibrational–translational non-equilibrium, and thus the vibrational-induced Zeldovich mechanisms. Hence, this partially explains their high product yields and low energy consumption. However, the low reported energy consumptions only account for the plasma power and do not include the energy consumed by both the vacuum equipment and the reactor cooling system. Therefore, the overall energy cost of NOX production in a MW plasma would be higher. Operation of MW reactors at higher pressures is also possible, but heat losses increase due to increased collision frequency.100
In 2010, Kim et al.63 reported a performance of 3.76 MJ mol mol N−1 and 0.6% NOX, similar to that of GA reactors, but for a MW plasma at a pressure slightly below atmospheric and for an input power between 60 and 90 W and at a fixed flow rate of 6 L min−1 (see Fig. 5). Power pulsing in a MW reactor may suppress unfavourable vibrational–translational relaxation, hence increasing the vibrational temperature, and thus the vibrational–translational non-equilibrium, needed for (the most energy-efficient) vibration-induced dissociation of N2.101
Pei et al.53 investigated four different plasma types, i.e. DBD, glow, spark and arc-type, and identified a key parameter (so-called χ factor, eqn (13)) that appeared to correlate the energy cost of NOX production with a range of different discharges (see Fig. 6b). The authors showed that NOX production efficiency can mainly be controlled by the average electric field and the average gas temperature of the discharge.
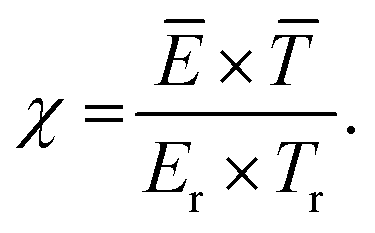 | (13) |
![[T with combining macron]](https://www.rsc.org/images/entities/i_char_0054_0304.gif) (K) are the average electric field and average gas temperature of the discharge under study, respectively, while Er. (i.e. 1.43 kV cm−1) and Tr (i.e. 1800 K) are chosen to normalize the parameter to a reference condition. The authors chose a DC glow discharge with a gap of 5 mm and a discharge current of 45 mA as a reference condition because of its simplicity and stability, i.e. the discharge conditions can be easily reproduced for reference. By decreasing the χ factor, e.g. by decreasing the electric field and/or the average gas temperature of the discharge, the energy cost can be reduced. The two important mechanisms that control the energy efficiency of NOX production in any type of discharge are (i) efficient electron-impact activation of N2 molecules to facilitate NOX formation, which is influenced by the electric field, and (ii) rapid thermal quenching of NO to prevent its conversion back to N2 and O2 molecules when the gas temperature drops more slowly. N atoms formed at high electric fields are an important pathway for NOX decomposition.62 The authors suggested various methods to decrease the average gas temperature, such as cooling the reactor walls with water, using short duration high current pulses, and extending t discharge length.33
(K) are the average electric field and average gas temperature of the discharge under study, respectively, while Er. (i.e. 1.43 kV cm−1) and Tr (i.e. 1800 K) are chosen to normalize the parameter to a reference condition. The authors chose a DC glow discharge with a gap of 5 mm and a discharge current of 45 mA as a reference condition because of its simplicity and stability, i.e. the discharge conditions can be easily reproduced for reference. By decreasing the χ factor, e.g. by decreasing the electric field and/or the average gas temperature of the discharge, the energy cost can be reduced. The two important mechanisms that control the energy efficiency of NOX production in any type of discharge are (i) efficient electron-impact activation of N2 molecules to facilitate NOX formation, which is influenced by the electric field, and (ii) rapid thermal quenching of NO to prevent its conversion back to N2 and O2 molecules when the gas temperature drops more slowly. N atoms formed at high electric fields are an important pathway for NOX decomposition.62 The authors suggested various methods to decrease the average gas temperature, such as cooling the reactor walls with water, using short duration high current pulses, and extending t discharge length.33
Finally, NOX production has also been reported by plasma jets flowing in (ambient) air (or N2 atmosphere), and interacting with water.66–76 Generally, the focus of this research was on NH3/NH4+ formation, but NO2− and NO3− formation was also reported, due to the presence of oxygen. The combination of plasma jets and water potentially allows for removal of the product NOX, thereby preventing its decomposition by the plasma.
Comparison of direct plasma-based NOX synthesis and the Haber–Bosch process combined with the Ostwald process
In this section, we assess the techno-economic feasibility of a direct plasma-based NOX synthesis process with subsequent conversion to HNO3, in comparison to an electrolysis-based Haber–Bosch process combined with the Ostwald process for HNO3 production. Both processes produce nitric acid from water, air, and electrical power exclusively. To the best of our knowledge, direct cost analyses comparing the direct plasma-based NOX synthesis process and the H–B process combined with the Ostwald process have not been reported yet.2,102The production capacity considered is 100 t-HNO3 day−1, i.e. a factor 1000 smaller than world-scale Haber–Bosch plants, at an electricity cost of 20 € MW h−1. The cases considered are (1) the electrolysis-based Haber–Bosch process combined with the Ostwald process (EHB + O base-case), (2) the plasma-based NOX synthesis process at an energy cost of 2.4 MJ mol N−1 (PL base-case, based on the recent results of Jardali et al.33 for gliding arc plasmas), and (3) the potential plasma-based NOX synthesis process at an energy cost of 0.5 MJ mol N−1 (PL potential). The energy consumption of 0.5 MJ mol N−1 is based on the theoretically minimum attainable energy consumption in a gliding arc reactor,34 as listed in Table 1.
Capital expenditure
The capital expenditure for the electrolysis-based Haber–Bosch process and the Ostwald process (e.g., the EHB + O base-case) is estimated from cost-scaling relations.103,104 The capital expenditure for the plasma-based NOX synthesis process (PL) is estimated from the cost-scaling relations for the Ostwald process, and from reported costs of plasma reactors. The current estimated cost for the plasma-reactor is 0.90 € W-1, based on a recent estimate of Van Rooij et al.105 for microwave reactors, as well as the cost of power supplies for DBD reactors (about 1.00–2.00 € W−1 for a few hundreds of W). The estimated cost for plasma generators is expected to decrease to 0.05 € W−1 for large-scale application.105A comparison of the capital expenditure for the electrolysis-based Haber–Bosch process, combined with the Ostwald process, and the plasma-based NOX synthesis process is shown in Fig. 7. The ‘high’ case and ‘low’ case refer to a plasma generator cost of 0.90 € W−1 and 0.05 € W−1, respectively. As shown in Fig. 7, the cost of a PL base-case is nearly on par with the EHB + O base-case. Upon improving the energy consumption from 2.4 MJ mol N−1 to 0.5 MJ mol N−1 or upon decreasing the cost of the plasma generator, the capital expenditure of the plasma-based process is about half to one third that of the EHB + O base-case. Thus, the plasma-based NOX synthesis process has potentially a highly competitive capital expenditure, especially when the cost of the plasma generator becomes as low as 0.05 € W−1.
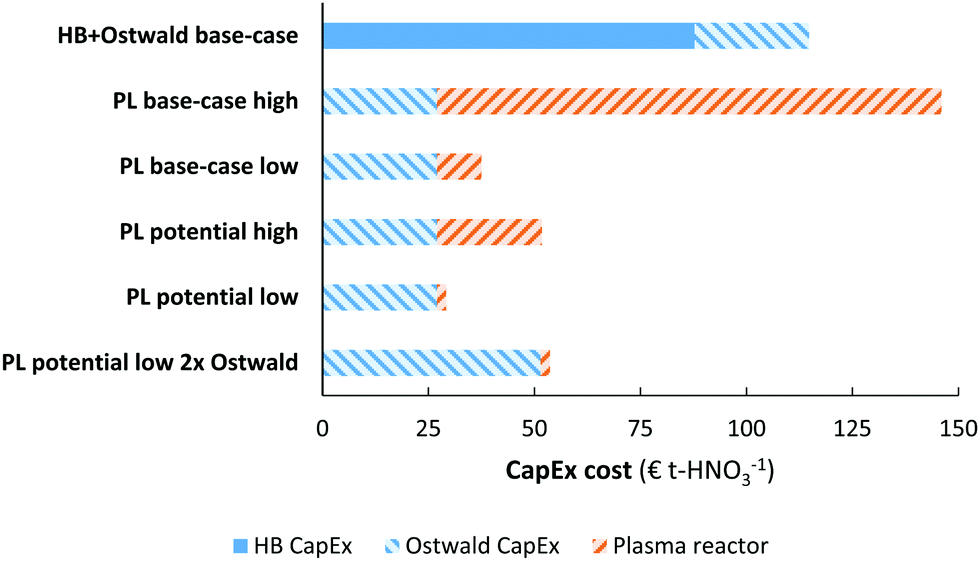 | ||
| Fig. 7 Comparison of the capital expenditure for various HNO3 synthesis methods. Cost-scaling numbers from ref. 103 for the electrolysis-based Haber–Bosch process, from ref. 104 for the Ostwald process, and ref. 105 for the plasma reactor. See text for more information. The annuity is assumed to be 10%. | ||
We assumed that the CapEx for the plasma process is similar to that of the Ostwald process (apart from the plasma reactor), due to the similarity in the downstream NOX absorption steps. However, the NOX concentration may be lower in case of plasma-based NOX synthesis (see Fig. 5). Therefore, an additional unit operation may be required to concentrate the produced NOX for the plasma-based NOX synthesis process. Therefore, we also show the CapEx for the plasma-based NOX process (PL) with double the equipment required for downstream NOX absorption and conversion to HNO3. As shown in Fig. 7, the CapEx of the PL process is lower, even if twice the equipment capacity is required for the NOX absorption in the PL process as compared to the EHB + O base-case process.
Effect of energy consumption
The energy consumption is another important descriptor for the operational cost of a process (see Fig. 8). The cases presented in Fig. 7 are also shown in Fig. 8a. It is clear that the energy consumption has a major impact on the total cost of HNO3 production, and a minor increase in the capital expenditure has little effect on the overall economics on the process. Thus, it is reasonable to focus on the energy consumption of the process. | ||
| Fig. 8 (a) Cost breakdown of the total cost of nitric acid production, for the cases considered in Fig. 7. The ‘high’ case and ‘low’ case refer to a plasma generator cost of 0.90 € W−1 and 0.05 € W−1, respectively. Process capacity 100 t-HNO3 day−1, electricity cost 20 € MW h−1. Oxygen is added to account for the lower oxygen content in air, as compared to the nitrogen content in air. At the process scale of 100 t-HNO3 day−1, about 1300 m3-O2 h−1 is required, which costs about 14–28 € t-HNO3−1.106 The operational costs apart from the electricity cost is assumed to be 2% of the CapEx. (b) Effect of the energy consumption of the plasma-based NOX synthesis process on the total cost of nitric acid production. The solid and dotted line represent the plasma process with a plasma reactor cost of 0.90 € W−1 and 0.05 € W−1, respectively. The orange square represents the total cost of nitric acid for a reference electrolysis-based Haber–Bosch process combined with an Ostwald process. Process capacity 100 t-HNO3 day−1, electricity cost 20 € MW h−1. | ||
The effect of the energy consumption on the nitric acid cost in the plasma-based NOX synthesis process is shown by the solid and dotted lines in Fig. 8b, from which it follows that the plasma-based NOX synthesis process becomes competitive with the electrolysis-based Haber–Bosch process combined with the Ostwald process at an energy consumption of 0.7 MJ mol N−1. As listed in Table 1, this is not attainable for thermal plasmas, as these plasmas have a minimum energy consumption of 0.72 MJ mol N−1. However, warm plasmas may attain the required energy consumption below 0.7 MJ mol N−1 (see Table 1).
Effect of electricity cost & process capacity
It should be noted that the current market value of HNO3 is about 250–350 € t-HNO3−1, while the predicted cost of HNO3 production for the EHB + O base-case and the PL potential low cases is as high as 890 € t-HNO3−1 and 655 € t-HNO3−1 for an electricity cost of 20 € MW h−1. The relatively low market value of HNO3 is mainly due to the low cost of fossil-based feedstocks, such as natural gas and coal.107 As shown in Fig. 8b, the CapEx only has a minor effect on the total cost of HNO3 production at the process scale considered (100 t-HNO3 day−1). Thus, the cost of electricity is a common descriptor for sustainable HNO3 production from the electrolysis-based Haber–Bosch process combined with the Ostwald process and the plasma-based NOX synthesis process, as compared to fossil-based HNO3 production.The cost of nitric acid production as function of the electricity cost is shown in Fig. 9. It is immediately clear that chemicals produced with electricity require low electricity cost (<5–10 € MW h−1) in order to become cost-competitive with fossil-based HNO3 production. The lowest solar auction prices in recent years are in the range 15–20 € MW h−1, implying the electricity-driven processes may become competitive with fossil-based processes in the upcoming decades.
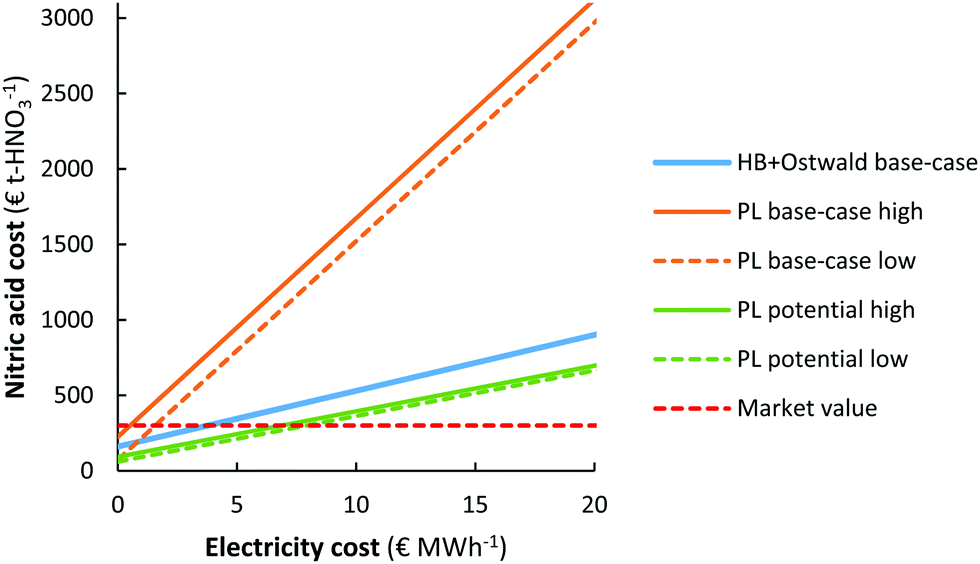 | ||
| Fig. 9 Effect of the electricity cost on the cost of nitric acid production. Process capacity 100 t-HNO3 day−1. The same cases are considered as in Fig. 7. | ||
It should be noted, however, that the cost of HNO3 depends on the geographic location. While the market value is as low as 250–350 € t-HNO3−1 in some locations where the cost of transportation is minimal, the cost at remote locations (e.g., the interior of sub-Saharan Africa) can be multiple times that of the production cost108,109 so that electricity driven processes may become favourable at higher electricity cost.
Effect of process capacity
As shown in Fig. 10, the plasma-based NOX synthesis process has the benefit over the Haber–Bosch process combined with the Ostwald process that the capital expenditure for ammonia synthesis is not required. This means there is potential for decentralized HNO3 synthesis, instead of importing HNO3 to remote locations.109 While the Haber–Bosch process suffers from a high CapEx upon scale-down to capacities below 50 t-HNO3 day−1, the plasma-based NOX synthesis process may be scaled down more effectively (see Fig. 10). Hence, plasma-based NOX synthesis may be used for decentralized nitrogen fixation. It should be noted, however, that scale-down below 1 t-HNO3 day−1 also becomes less economical for the plasma-based NOX synthesis process, due to an increase in oxygen purification cost upon scale-down.106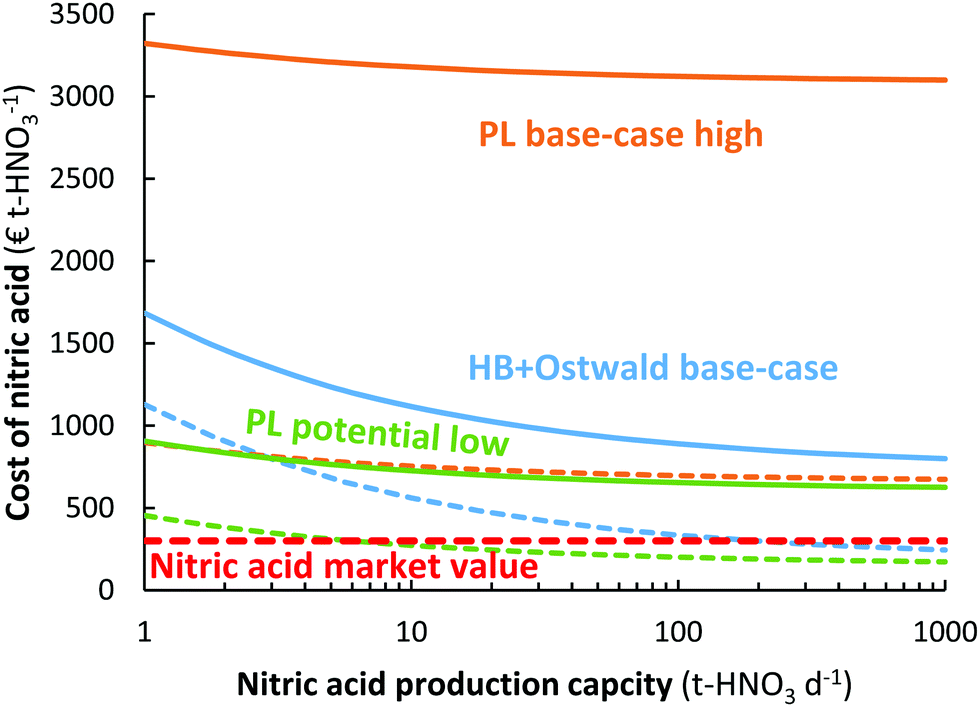 | ||
| Fig. 10 Effect of nitric acid production capacity on the cost of nitric acid for the electrolysis-based Haber–Bosch process combined with the Ostwald process, as well as for the plasma-based NOX synthesis process. The full and dotted lines represent an electricity cost of 20 € MW h−1 and 5 € MW h−1, respectively. The high pressure Haber–Bosch process becomes less energy-efficient upon scale down below 10 t-HNO3 day−1.14,110 The HB + Ostwald base-case, PL base-case, and PL potential case are the same as in Fig. 8. | ||
Further improving the performance of plasma-based NOX synthesis
In recent years, various studies have reported on combination of experimental and modelling work for plasma-based NOX synthesis.33,34,62,91 This has improved the understanding of the dominant reaction pathways in real plasma reactors under relevant reaction conditions. However, the energy cost of plasma-based NOX synthesis remains higher than for the benchmark electrolysis-based Haber–Bosch process combined with the Ostwald process (see Fig. 5). Thus, further performance improvement is required, beyond optimizing experimental conditions, e.g. inspired by modelling.Modelling can, however, also help to improve the reactor design to improve the contacting of gas with plasma so that a larger fraction of gas actually passes through the plasma. This is now often a limitation in for instance gliding arc plasma reactors,34,99 thus limiting the overall gas conversion. Such modelling can describe gas flow dynamics, arc plasma behaviour and plasma chemistry, tracing the gas molecules through the reactor. This allows evaluation of the exact plasma conditions to which molecules are exposed, resulting on optimal conversion by the plasma, as recently demonstrated.33,111
Besides enhancing the gas fraction passing through the plasma, attention should also be paid to fast quenching, i.e. cooling, of the gas downstream of the plasma, avoiding the backward reaction, i.e. decomposition of NOX to N2 and O2. The major beneficial effects of fast quenching were recently studied in detail for CO2 conversion in plasma,112 but the same principle also applies to NOX synthesis. In addition, heat integration is required, using the heat released during gas cooling for pre-heating the gas before entering the plasma reactor,82.
Finally, as discussed in Section 2.2, catalytic enhancement of plasma-based NOX synthesis is an option to increase the NOX yield at the same energy input. Such materials should not catalyse the decomposition of NOX molecules, as this would even decrease the NOX yield as compared to pure plasma-based NOX synthesis. Secondly, the use of NOX sorbents may be beneficial. Removal of NOX species from the plasma environment may prevent the subsequent decomposition of the product by the plasma. Catalyst particles or sorbent particles may be introduced in or after the plasma reactor as a fixed bed, a trickle bed, or a fluidized bed.
Conclusion
We have evaluated the state-of-the-art for plasma-based NOX synthesis. From a techno-economic analysis, it follows that plasma-based NOX synthesis is potentially viable for electricity-based HNO3 production. As compared to the electrolysis-based Haber–Bosch process combined with the Ostwald process, the plasma-based NOX synthesis process benefits from a lower capital expenditure. The current energy cost of ≥2.4 MJ mol N−191 is however still too high to be competitive with the electrolysis-based Haber–Bosch process combined with the Ostwald process, which consumes about 0.6 MJ mol N−115. Plasma-based NOX synthesis will become a highly-competitive alternative to the Haber–Bosch process combined with the Ostwald process, if the energy consumption can be decreased to 0.7 MJ mol−1via smart reactor design, tuning the chemistry and vibrational kinetics, avoiding back-reactions, or combination with catalysts. Thus, plasma technology may become an effective turnkey technology compatible with intermittent electricity.113Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the TKI-Energie from Toeslag voor Topconsortia voor Kennis en Innovatie (TKI) from the Ministry of Economic Affairs and Climate Policy, the Excellence of Science FWO-FNRS project (FWO grant ID GoF9618n, EOS ID 30505023), and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No 810182 – SCOPE ERC Synergy project).References
- A. S. Travis, Nitrogen Capture: The Growth of an International Industry (1900–1940), Springer International Publishing, 2018 DOI:10.1007/978-3-319-68963-0.
- F. A. Ernst, Industrial Chemical Monographs: Fixation of Atmospheric Nitrogen, London, UK: Chapman & Hall, Ltd, 1928 Search PubMed.
- A. S. Travis, The Synthetic Nitrogen Industry in World War I: Its Emergence and Expansion. The Synthetic Nitrogen Industry in World War I, 2015 DOI:10.1007/978-3-319-19357-1.
- V. Smil, Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production, Cambridge, MA, 2004 Search PubMed.
- J. R. Brightling, Ammonia and the fertiliser industry: The development of ammonia at Billingham, Johnson Matthey Technol. Rev., 2018, 62(1), 32–47, DOI:10.1595/205651318X696341.
- M. Thiemann, E. Scheibler and K. W. Wiegand, Nitric acid, nitrous acid, and nitrogen oxides, in Ullmann's Encyclopedia of Industrial Chemistry, 2000 DOI:10.1002/14356007.a17_293.
- K. H. R. Rouwenhorst, P. M. Krzywda, N. E. Benes, G. Mul and L. Lefferts, Ammonia, 4. Green Ammonia Production, in Ullmann's Encyclopedia of Industrial Chemistry, 2020 DOI:10.1002/14356007.w02_w02.
- N. Cherkasov, A. O. Ibhadon and P. Fitzpatrick, A review of the existing and alternative methods for greener nitrogen fixation, Chem. Eng. Proc.: Proc. Intens., 2015, 90, 24–33, DOI:10.1016/j.cep.2015.02.004.
- B. S. Patil, Q. Wang, V. Hessel and J. Lang, Plasma N2-fixation: 1900–2014, Catal. Today, 2015, 256, 49–66, DOI:10.1016/j.cattod.2015.05.005.
- G. Wang, A. Mitsos and W. Marquardt, Renewable production of ammonia and nitric acid, AIChE J., 2020, 66(6), 1–9, DOI:10.1002/aic.16947.
- V. Smil, Global population and the nitrogen cycle, Sci. Am., 1997, 277(1), 76–81, DOI:10.1038/scientificamerican0797-76.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, How a century of ammonia synthesis changed the world, Nat. Geosci., 2008, 1(10), 636–639, DOI:10.1038/ngeo325.
- J. Armijo and C. Philibert, Flexible production of green hydrogen and ammonia from variable solar and wind energy: Case study of Chile and Argentina, Int. J. Hydrogen Energy, 2020, 45(3), 1541–1558, DOI:10.1016/j.ijhydene.2019.11.028.
- K. H. R. Rouwenhorst, A. G. J. Van Der Ham, G. Mul and S. R. A. Kersten, Islanded ammonia power systems: Technology review & conceptual process design., Renewable Sustainable Energy Rev., 2019, 114, 109339, DOI:10.1016/j.rser.2019.109339.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Current and future role of Haber–Bosch ammonia in a carbon-free energy landscape, Energy Environ. Sci., 2020, 13(2), 331–344, 10.1039/C9EE02873K.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker and A. N. Simonov, A roadmap to the ammonia economy, Joule, 2020, 4(6), 1186–1205, DOI:10.1016/j.joule.2020.04.004.
- A. Bogaerts and E. C. Neyts, Plasma technology: an emerging technology for energy storage, ACS Energy Lett., 2018, 3(4), 1013–1027, DOI:10.1021/acsenergylett.8b00184.
- Nitrate Division, O. O. W. D. and Fixed Nitrogen Research Laboratory Department of Agriculture, Report on the Fixation and Utilization of Nitrogen, 1922 Search PubMed.
- E. Fridell, M. Skoglundh, B. Westerberg, S. Johansson and G. Smedler, NOx storage in barium-containing catalysts, J. Catal., 1999, 183(2), 196–209, DOI:10.1006/jcat.1999.2415.
- E. Farber, From chemistry to philosophy: The way of Alwin Mittasch (1869–1953), Chymia, 1966, 11, 157–178, DOI:10.2307/27757266.
- A. Mittasch and W. Frankenburg, Early studies of multicomponent catalysts, Adv. Catal., 1950, 2(C), 81–104, DOI:10.1016/S0360-0564(08)60375-2.
- G. Prieto and F. Schüth, The Yin and Yang in the development of catalytic processes: Catalysis research and reaction engineering, Angew. Chem., Int. Ed., 2015, 54(11), 3222–3239, DOI:10.1002/anie.201409885.
- K. H. R. Rouwenhorst, P. M. Krzywda, N. E. Benes, G. Mul and L. Lefferts ( 2020). Ammonia production technologies, in Techno-Economic Challenges of Green Ammonia as Energy Vector, ed. R. Bañares-Alcántara and A. Valera-Medina, Elsevier Science Publishing Co Inc., pp. 41–84. DOI:10.1016/B978-0-12-820560-0.00004-7.
- A. Sánchez and M. Martín, Scale up and scale down issues of renewable ammonia plants: Towards modular design, Sustainable Prod. Consumption, 2018, 16, 176–192, DOI:10.1016/j.spc.2018.08.001.
- askIITians. (2020). Nitric Acid, Retrieved September 1, 2020, from https://www.askiitians.com/iit-jee-s-and-p-block-elements/nitric-acid/.
- K. Birkeland, On the oxidation of atmospheric nitrogen in electric arcs, Trans. Faraday Soc., 1906, 2, 98–116 RSC.
- S. Eyde, Oxidation of atmospheric nitrogen and development of resulting industries in Norway, J. Ind. Eng. Chem., 1912, 4, 771–774 CrossRef CAS.
- V. D. Rusanov, A. A. Fridman and G. V. Sholin, The physics of a chemically active plasma with nonequilibrium vibrational excitation of molecules, Phys.-Usp., 1981, 24(6), 447–474 CrossRef.
- P. R. Ammann and R. S. Timmins, Chemical reactions during rapid quenching of oxygen–nitrogen mixtures from very high temperatures, AIChE J., 1966, 12, 956–963 CrossRef CAS.
- P. Mehta, P. Barboun, D. B. Go, J. C. Hicks and W. F. Schneider, Catalysis enabled by plasma activation of strong chemical bonds: A review, ACS Energy Lett., 2019, 4(5), 1115–1133, DOI:10.1021/acsenergylett.9b00263.
- L. R. Winter and J. G. Chen, N2 fixation by plasma-activated processes, Joule, 2020, 1–16, DOI:10.1016/j.joule.2020.11.009.
- H. Liu, Ammonia Synthesis Catalysts: Innovation and Practice, World Scientific, 2013 DOI:10.1142/8199.
- F. Jardali, S. van Alphen, J. Creel, H. A. Eshtehardi, M. Axelsson, R. Ingels and A. Bogaerts, NOx production in a rotating gliding arc plasma: potential avenue for sustainable nitrogen fixation, Green Chem., 2021, 23(4), 1748–1757, 10.1039/D0GC03521A.
- E. Vervloessem, M. Aghaei, F. Jardali, N. Hafezkhiabani and A. Bogaerts, Plasma-based N2 fixation into NOx: Insights from modeling toward optimum yields and energy costs in a gliding arc plasmatron, ACS Sustainable Chem. Eng., 2020, 8(26), 9711–9720, DOI:10.1021/acssuschemeng.0c01815.
- D. Rapakoulias, S. Cavadias and J. Amouroux, Processus catalytiques dans un réacteur à plasma hors d’équilibre II. Fixation de l’azote dans le système N2–O2, Rev. Phys. Appl., 1980, 15(7), 1261–1265, DOI:10.1051/rphysap:019800015070126100.
- B. S. Patil, N. Cherkasov, J. Lang, A. O. Ibhadon, V. Hessel and Q. Wang, Low temperature plasma-catalytic NOx synthesis in a packed DBD reactor: Effect of support materials and supported active metal oxides, Appl. Catal., B, 2016, 194, 123–133, DOI:10.1016/j.apcatb.2016.04.055.
- V. Hessel, A. Anastasopoulou, Q. Wang, G. Kolb and J. Lang, Energy, catalyst and reactor considerations for (near)-industrial plasma processing and learning for nitrogen-fixation reactions, Catal. Today, 2013, 211, 9–28, DOI:10.1016/j.cattod.2013.04.005.
- I. Michielsen, Y. Uytdenhouwen, J. Pype, B. Michielsen, J. Mertens, F. Reiniers and A. Bogaerts, CO2 dissociation in a packed bed DBD reactor: First steps towards a better understanding of plasma catalysis, Chem. Eng. J., 2017, 326, 477–488, DOI:10.1016/j.cej.2017.05.177.
- A. Gicquel, S. Cavadias and J. Amouroux, Heterogeneous catalysis in low-pressure plasmas, J. Phys. D: Appl. Phys., 1986, 19, 2013–2042 CrossRef CAS.
- P. Mars and D. W. van Krevelen, Oxidations carried out by means of vanadium oxide catalysts, Chem. Eng. Sci., 1954, 3, 41–59, DOI:10.1016/S0009-2509(54)80005-4.
- L. Diekhöner, H. Mortensen and A. Baurichter, N2 dissociative adsorption on Ru(0001): The role of energy loss, J. Chem. Phys., 2001, 115(19), 9028–9035, DOI:10.1063/1.1413746.
- L. Romm, G. Katz, R. Kosloff and M. Asscher, Dissociative chemisorption of N2 on Ru(001) enhanced by vibrational and kinetic energy: Molecular beam experiments and quantum mechanical calculations, J. Phys. Chem. B, 1997, 101(12), 2213–2217, DOI:10.1021/jp962599o.
- H. E. Pfnür, C. T. Rettner, J. Lee, R. J. Madix and D. J. Auerbach, Dynamics of the activated dissociative chemisorption of N2 on W(110): A molecular beam study, J. Chem. Phys., 1986, 85(12), 7452–7466, DOI:10.1063/1.451334.
- A. Bogaerts, X. Tu, J. C. Whitehead, G. Centi, L. Lefferts, O. Guaitella and M. Carreon, The 2020 plasma catalysis roadmap, J. Phys. D: Appl. Phys., 2020, 53, 1–51, DOI:10.1088/1361-6463/ab9048.
- C. Shi, X. F. Yang, A. M. Zhu and C. T. Au, Catalytic activities of tungsten nitride for NO dissociation and reduction with hydrogen, Catal. Today, 2004, 93–95, 819–826, DOI:10.1016/j.cattod.2004.06.102.
- H. Ma and W. F. Schneider, DFT and microkinetic comparison of Pt, Pd and Rh-catalyzed ammonia oxidation, J. Catal., 2020, 383, 322–330, DOI:10.1016/j.jcat.2020.01.029.
- K. H. R. Rouwenhorst, Y. Engelmann, K. Van’t Veer, R. S. Postma, A. Bogaerts and L. Lefferts, Plasma-driven catalysis: Green ammonia synthesis with intermittent electricity, Green Chem., 2020, 22(19), 6258–6287, 10.1039/D0GC02058C.
- Q. Sun, Z. Wang, D. Wang, Z. Hong, M. Zhou and X. Li, A review on the catalytic decomposition of NO to N2 and O2: Catalysts and processes, Catal. Sci. Technol., 2018, 8(18), 4563–4575, 10.1039/c8cy01114a.
- M. Haneda and H. Hamada, Recent progress in catalytic NO decomposition, Compt. Rend. Chim., 2016, 19(10), 1254–1265, DOI:10.1016/j.crci.2015.07.016.
- N. Imanaka and T. Masui, Advances in direct NOx decomposition catalysts, Appl. Catal., A, 2012, 431–432, 1–8, DOI:10.1016/j.apcata.2012.02.047.
- T. Taugbøl, E. M. Andersen, U. Grønn and B. F. Moen, Rjukan – Notodden Industrial Heritage Site, 2015, retrieved from https://www.visitrjukan.com/en/content/download/6296/35783/file/Verdensarv_engelsk_industriarv_Nomination.pdf.
- N. Rehbein and V. Cooray, NOx production in spark and corona discharges, J. Electrostat., 2001, 51–52(1–4), 333–339, DOI:10.1016/S0304-3886(01)00115-2.
- X. Pei, D. Gidon, Y. J. Yang, Z. Xiong and D. B. Graves, Reducing energy cost of NOx production in air plasmas, Chem. Eng. J., 2019, 362, 217–228, DOI:10.1016/j.cej.2019.01.011.
- M. Janda, V. Martišovitš, K. Hensel and Z. Machala, Generation of antimicrobial NOx by atmospheric air transient spark discharge, Plasma Chem. Plasma Proc., 2016, 36, 767–781, DOI:10.1007/s11090-016-9694-5.
- M. J. Pavlovich, T. Ono, C. Galleher, B. Curtis, D. S. Clark, Z. Machala and D. B. Graves, Air spark-like plasma source for antimicrobial NOx generation, J. Phys. D: Appl. Phys., 2014, 47(50), 505202, DOI:10.1088/0022-3727/47/50/505202.
- W. S. Partridge, R. B. Parlin and B. J. Zwolinski, Fixation of nitrogen in a crossed discharge, Ind. Eng. Chem., 1954, 46(7), 1468–1471 CrossRef CAS.
- M. Rahman and V. Cooray, NOx generation in laser-produced plasma in air as a function of dissipated energy, Opt. Laser Technol., 2003, 35(7), 543–546, DOI:10.1016/S0030-3992(03)00077-X.
- W. Bian, J. Shi and X. Yin, Nitrogen fixation into water by pulsed high-voltage discharge, IEEE Trans. Plasma Sci., 2009, 37(1), 211–218, DOI:10.1109/TPS.2008.2007585.
- X. Pei, D. Gidon and D. B. Graves, Specific energy cost for nitrogen fixation as NOxusing DC glow discharge in air, J. Phys. D: Appl. Phys., 2020, 53, 044002, DOI:10.1088/1361-6463/ab5095.
- J. F. Coudert, J. M. Baronnet, J. Rakowitz and P. Fauchais, Synthesis of nitrogen oxides in a plasma produced by a jet arc generator, in Symp. Int. Chim. Plasmas, 1977 Search PubMed.
- B. S. Patil, F. J. J. Peeters, G. J. van Rooij, J. A. Medrano, F. Gallucci, J. Lang and V. Hessel, Plasma assisted nitrogen oxide production from air: Using pulsed powered gliding arc reactor for a containerized plant, AIChE J., 2018, 64(2), 526–537, DOI:10.1002/aic.15922.
- W. Wang, B. Patil, S. Heijkers, V. Hessel and A. Bogaerts, Nitrogen fixation by gliding arc plasma: Better insight by chemical kinetics modelling, ChemSusChem, 2017, 10(10), 2145–2157, DOI:10.1002/cssc.201700095.
- T. Kim, S. Song, J. Kim and R. Iwasaki, Formation of NOx from air and N2/O2 mixtures using a nonthermal microwave plasma system, Jpn. J. Appl. Phys., 2010, 49, 126201, DOI:10.1143/JJAP.49.126201.
- B. Mutel, O. Dessaux and P. Goudmand, Energy cost improvement of the nitrogen oxides synthesis in a low pressure plasma, Rev. Phys. Appl., 1984, 19(6), 461–464, DOI:10.1051/rphysap:01984001906046100.
- R. I. Asisov, V. K. Givotov, V. D. Rusanov and A. Fridman, High energy chemistry, Sov. Phys., 1980, 14, 366 Search PubMed.
- P. Peng, P. Chen, M. Addy, Y. Cheng, Y. Zhang, E. Anderson and R. Ruan, In situ plasma-assisted atmospheric nitrogen fixation using water and spray-type jet plasma, Chem. Commun., 2018, 54(23), 2886–2889, 10.1039/c8cc00697k.
- Y. Gorbanev, E. Vervloessem, A. Nikiforov and A. Bogaerts, Nitrogen fixation with water vapor by non-equilibrium plasma: Towards sustainable ammonia production, ACS Sustainable Chem. Eng., 2020, 8(7), 2996–3004, DOI:10.1021/acssuschemeng.9b07849.
- J. R. Toth, N. H. Abuyazid, D. J. Lacks, J. N. Renner and R. M. Sankaran, A plasma-water droplet reactor for process-intensified continuous nitrogen fixation at atmospheric pressure, ACS Sustainable Chem. Eng., 2020, 8(39), 14845–14854, DOI:10.1021/acssuschemeng.0c04432.
- P. Peng, C. Schiappacasse, N. Zhou, M. Addy, Y. Cheng, Y. Zhang, E. Anderson, D. Chen, Y. Wang, Y. Liu, P. Chen and R. Ruan, Plasma in situ gas–liquid nitrogen fixation using concentrated high-intensity electric field, J. Phys. D: Appl. Phys., 2019, 52(49), 494001, DOI:10.1088/1361-6463/ab3ea6.
- Y. Kubota, K. Koga, M. Ohno and T. Hara, Synthesis of ammonia through direct chemical reactions between an atmospheric nitrogen plasma jet and a liquid, Plasma Fus. Res., 2010, 5, 042, DOI:10.1585/pfr.5.042.
- S. Kumari, S. Pishgar, M. E. Schwarting, W. F. Paxton and J. M. Spurgeon, Synergistic plasma-assisted electrochemical reduction of nitrogen to ammonia, Chem. Commun., 2018, 54(95), 13347–13350, 10.1039/c8cc07869f.
- R. Hawtof, S. Ghosh, E. Guarr, C. Xu, R. M. Sankaran and J. N. Renner, Catalyst-free, highly selective synthesis of ammonia from nitrogen and water by a plasma electrolytic system, Asian J. Chem., 2019, 31(2), 1–10, DOI:10.1126/sciadv.aat5778.
- T. Haruyama, T. Namise, N. Shimoshimizu, S. Uemura, Y. Takatsuji, M. Hino and M. Kohno, Non-catalyzed one-step synthesis of ammonia from atmospheric air and water, Green Chem., 2016, 18(16), 4536–4541, 10.1039/c6gc01560c.
- T. Sakakura, S. Uemura, M. Hino, S. Kiyomatsu, Y. Takatsuji, R. Yamasaki and T. Haruyama, Excitation of H2O at the plasma/water interface by UV irradiation for the elevation of ammonia production, Green Chem., 2018, 20(3), 627–633, 10.1039/c7gc03007j.
- T. Sakakura, N. Murakami, Y. Takatsuji, M. Morimoto and T. Haruyama, Contribution of discharge excited atomic N, N2*, and N2+ to a plasma/liquid interfacial reaction as suggested by quantitative analysis, ChemPhysChem, 2019, 20(11), 1467–1474, DOI:10.1002/cphc.201900212.
- T. Sakakura, N. Murakami, Y. Takatsuji and T. Haruyama, Nitrogen fixation in a plasma/liquid interfacial reaction and its switching between reduction and oxidation, J. Phys. Chem. C, 2020, 124(17), 9401–9408, DOI:10.1021/acs.jpcc.0c02392.
- L. S. Polak, A. A. Ovsiannikov, D. I. Slovetsky and F. B. Vurzel, Theoretical and applied plasma chemistry, Nauka (Science), 1975 Search PubMed.
- A. A. Ivanov, Plasma physics, Sov. Phys., 1975, 1, 147 Search PubMed.
- J. Krop, E. Krop and I. Pollo, Calculated amounts of nitric oxide in a nitrogen–oxygen plasma jet, Chem. Plazmy, 1979, 242–249 Search PubMed.
- X. Hao, A. M. Mattson, C. M. Edelblute, M. A. Malik, L. C. Heller and J. F. Kolb, Nitric oxide generation with an air operated non-thermal plasma jet and associated microbial inactivation mechanisms, Plasma Proc. Polym., 2014, 11(11), 1044–1056, DOI:10.1002/ppap.201300187.
- Y. D. Korolev, O. B. Frants, N. V. Landl and A. I. Suslov, Low-current plasmatron as a source of nitrogen oxide molecules, IEEE Trans. Plasma Sci., 2012, 40(11), 2837–2842, DOI:10.1109/TPS.2012.2201755.
- R. Ingels, Energy efficient process for producing nitrogen oxide, Norway, 2012 Search PubMed.
- H. Patel, R. K. Sharma, V. Kyriakou, A. Pandiyan, S. Welzel and M. N. Tsampas, Plasma-activated electrolysis for cogeneration of nitric oxide and hydrogen from water and nitrogen, ACS Energy Lett., 2019, 4(9), 2091–2095, DOI:10.1021/acsenergylett.9b01517 rapid-communication.
- A. V. Pipa, T. Bindemann, R. Foest, E. Kindel, J. Röpcke and K.-D. Weltmann, Absolute production rate measurements of nitric oxide by an atmospheric pressure plasma jet (APPJ), J. Phys. D: Appl. Phys., 2008, 41, 194011, DOI:10.1088/0022-3727/41/19/194011.
- Q. Sun, A. Zhu, X. Yang, J. Niu and Y. Xu, Formation of NOx from N2 and O2 in catalyst-pellet filled dielectric barrier discharges at atmospheric pressure, Chem. Commun., 2003, 1418–1419, 10.1039/B303046F.
- A. A. Abdelaziz and H.-H. Kim, Temperature-dependent behavior of nitrogen fixation in nanopulsed dielectric barrier discharge operated at different humidity levels and oxygen contents, J. Phys. D: Appl. Phys., 2020, 53, 114001, DOI:10.1088/1361-6463/ab5c78.
- S. Heijkers, R. Snoeckx, T. Kozák, T. Silva, T. Godfroid, N. Britun and A. Bogaerts, CO2 conversion in a microwave plasma reactor in the presence of N2: Elucidating the role of vibrational levels, J. Phys. Chem. C, 2015, 119(23), 12815–12828, DOI:10.1021/acs.jpcc.5b01466.
- A. Berthelot and A. Bogaerts, Modeling of CO2 splitting in a microwave plasma: How to improve the conversion and energy efficiency, J. Phys. Chem. C, 2017, 121(15), 8236–8251, DOI:10.1021/acs.jpcc.6b12840.
- A. Czernichowski, Gliding arc. Applications to engineering and environment control, Pure Appl. Chem., 1994, 66(6), 1301–1310, DOI:10.1351/pac199466061301.
- M. Ramakers, G. Trenchev, S. Heijkers, W. Wang and A. Bogaerts, Gliding arc plasmatron: Providing an alternative method for carbon dioxide conversion, ChemSusChem, 2017, 10(12), 2642–2652, DOI:10.1002/cssc.201700589.
- B. S. Patil, J. Rovira Palau, V. Hessel, J. Lang and Q. Wang, Plasma nitrogen oxides synthesis in a milli-scale gliding arc reactor: Investigating the electrical and process parameters, Plasma Chem. Plasma Proc., 2016, 36(1), 241–257, DOI:10.1007/s11090-015-9671-4.
- Y. Wang, A. W. DeSilva, G. C. Goldenbaum and R. R. Dickerson, Nitric oxide production by simulated lightning: Dependence on current, energy, and pressure, J. Geophys. Res. Atmos., 1998, 103(D15), 19149–19159, DOI:10.1029/98JD01356.
- I. Adamovich, W. Rich, P. Chernukho and S. Zhdanok, Analysis of the power budget and stability of high-pressure nonequilibrium air plasmas, in 31st Plasmadynamics and Laser Conference, Denver (CO), 2000 Search PubMed.
- T. Namihira, S. Katsuki, R. Hackam, H. Akiyama and K. Okamoto, Production of nitric oxide using a pulsed arc discharge, IEEE Trans. Plasma Sci., 2002, 30(5), 1993–1998, DOI:10.1109/TPS.2002.807502.
- T. Namihira, S. Sakai, M. Matsuda, D. Wang, T. Kiyan, H. Akiyama and K. Toda, Temperature and nitric oxide generation in a pulsed arc discharge plasma, Plasma Sci. Technol., 2007, 9(6), 747–751, DOI:10.1088/1009-0630/9/6/26.
- E. Cleiren, S. Heijkers, M. Ramakers and A. Bogaerts, Dry reforming of methane in a gliding arc plasmatron: Towards a better understanding of the plasma chemistry, ChemSusChem, 2017, 10(20), 4025–4036, DOI:10.1002/cssc.201701274.
- M. Janda, V. Martišovitš, K. Hensel, L. Dvonč and Z. Machala, Measurement of the electron density in transient spark discharge, Plasma Sources Sci. Technol., 2014, 23(6), 065016, DOI:10.1088/0963-0252/23/6/065016.
- M. Janda, T. Hoder, A. Sarani, R. Brandenburg and Z. Machala, Cross-correlation spectroscopy study of the transient spark discharge in atmospheric pressure air, Plasma Sources Sci. Technol., 2017, 26, 055010, DOI:10.1088/1361-6595/aa642a.
- A. Bogaerts and G. Centi, Plasma technology for CO2 conversion: A personal perspective on prospects and gaps, Front. Energy Res., 2020, 8, 1–23, DOI:10.3389/fenrg.2020.00111.
- P. J. Bruggeman, F. Iza and R. Brandenburg, Foundations of atmospheric pressure non-equilibrium plasmas, Plasma Sources Sci. Technol., 2017, 26, 123002, DOI:10.1088/1361-6595/aa97af.
- S. Van Alphen, V. Vermeiren, T. Butterworth, D. C. M. van den Bekerom, G. J. Van Rooij and A. Bogaerts, Power pulsing to maximize vibrational excitation efficiency in N2 microwave plasma: A combined experimental and computational study, J. Phys. Chem. C, 2019, 124(3), 1765–1779, DOI:10.1021/acs.jpcc.9b06053.
- A. Anastasopoulou, R. Keijzer, S. Butala, J. Lang, G. Van Rooij and V. Hessel, Eco-efficiency analysis of plasma-assisted nitrogen fixation, J. Phys. D: Appl. Phys., 2020, 53, 234001, DOI:10.1088/1361-6463/ab71a8.
- E. R. Morgan, J. F. Manwell and J. G. McGowan, Sustainable ammonia production from U.S. offshore wind farms: A techno-economic review, ACS Sustainable Chem. Eng., 2017, 5(11), 9554–9567, DOI:10.1021/acssuschemeng.7b02070.
- M. S. Peters, K. D. Timmerhaus and R. E. West, Plant Design and Economics for Chemical Engineers, Mac Graw and Hill, 2003 Search PubMed.
- G. J. van Rooij, H. N. Akse, W. A. Bongers and M. C. M. van de Sanden, Plasma for electrification of chemical industry: A case study on CO2 reduction, Plasma Phys. Controlled Fus., 2018, 60, 014019, DOI:10.1088/1361-6587/aa8f7d.
- M. J. Kirschner, A. Alekseev, S. Dowy, M. Grahl, L. Jansson, P. Keil and C. Windmeier, Oxygen, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2017 DOI:10.1002/14356007.a18_329.pub2.
- M. Appl, in Ammonia: Principles and Industrial Practice, 1st edn, ed. M. Appl, Wiley-VCH Verlag GmbH, Weinheim (Germany), 1999 DOI:10.1002/9783527613885.
- R. M. Nayak-Luke and R. Bañares-Alcántara, Techno-economic viability of islanded green ammonia as a carbon-free energy vector and as a substitute for conventional production, Energy Environ. Sci., 2020, 13(9), 2957–2966, 10.1039/d0ee01707h.
- A. Anastasopoulou, S. Butala, B. Patil, J. Suberu, M. Fregene, J. Lang and V. Hessel, Techno-economic feasibility study of renewable power systems for a small-scale plasma-assisted nitric acid plant in Africa, Processes, 2016, 4(4), 54, DOI:10.3390/pr4040054.
- B. Lin, T. Wiesne and M. Malmali, Performance of a small-scale Haber process: A techno-economic analysis, ACS Sustainable Chem. Eng., 2020, 8(41), 15517–15531, DOI:10.1021/acssuschemeng.0c04313.
- S. Van Alphen, F. Jardali, J. Creel, G. Trenchev, R. Snyders and A. Bogaerts, Sustainable gas conversion by gliding arc plasmas: A new modelling approach for reactor design improvement, Sustainable Energy Fuels, 2021, 5(6), 1786–1800, 10.1039/D0SE01782E.
- V. Vermeiren and A. Bogaerts, Plasma-based CO2 conversion: To quench or not to quench?, J. Phys. Chem. C, 2020, 124(34), 18401–18415, DOI:10.1021/acs.jpcc.0c04257.
- R. Brandenburg, A. Bogaerts, W. Bongers, A. Fridman, G. Fridman, B. R. Locke and K. K. Ostrikov, White paper on the future of plasma science in environment, for gas conversion and agriculture, Plasma Proc. Polym., 2018, 1–18, DOI:10.1002/ppap.201700238.
| This journal is © The Royal Society of Chemistry 2021 |