
Activating the I0/I+ redox couple in an aqueous I2–Zn battery to achieve a high voltage plateau†
Xinliang
Li
a,
Mian
Li
*b,
Zhaodong
Huang
a,
Guojin
Liang
a,
Ze
Chen
a,
Qi
Yang
a,
Qing
Huang
 b and
Chunyi
Zhi
*acd
b and
Chunyi
Zhi
*acd
aDepartment of Materials Science and Engineering, City University of Hong Kong, 83 Tat Chee Avenue, Kowloon, Hong Kong 999077, China. E-mail: cy.zhi@cityu.edu.hk
bEngineering Laboratory of Advanced Energy Materials, Ningbo Institute of Materials Technology & Engineering, Chinese Academy of Sciences, Ningbo, Zhejiang 315201, China. E-mail: limian@nimte.ac.cn
cCentre for Functional Photonics, City University of Hong Kong, Kowloon, Hong Kong 999077, China
dCenter for Advanced Nuclear Safety and Sustainable Development, City University of Hong Kong, Kowloon, Hong Kong 999077, China
First published on 8th December 2020
Abstract
Rechargeable iodine conversion batteries possess promising prospects for portable energy storage with complete electron transfer and rich valence supply. However, the reaction is limited to the single I−/I0 redox at a potential of only 0.54 V vs. the standard hydrogen electrode (SHE), leading to a low voltage plateau at 1.30 V when Zn is employed as the anode. Herein, we show how to activate the desired reversible I0/I+ redox behavior at a potential of 0.99 V vs. SHE by electrolyte tailoring via F− and Cl− ion-containing salts. The electronegative F− and Cl− ions can stabilize the I+ during charging. In an aqueous Zn ion battery based on an optimized ZnCl2 + KCl electrolyte with abundant Cl−, the I-terminated halogenated Ti3C2I2 MXene cathode delivered two well-defined discharge plateaus at 1.65 V and 1.30 V, superior to all reported aqueous I2–metal (Zn, Fe, Cu) counterparts. Together with the 108% capacity enhancement, the high voltage output resulted in a significant 231% energy density enhancement. Metallic Ti3C2I2 benefits the redox kinetics and confines the interior I species, leading to exceptional cyclic durability and rate capability. In situ Raman and ex situ multiple spectral characterizations clarify the efficient activation and stabilization effects of Cl− (F−) ions on reversible I0/I+ redox. Our work is believed to provide new insight into designing advanced I2–metal batteries based on the newly discovered I−/I0/I+ chemistry to achieve both high voltage and enhanced capacity.
Broader contextAqueous conversion-type I2–Zn battery has been intensely studied and is regarded as a promising inherent safe energy storage device. To date, the electrochemical activity of reported I2–Zn batteries stops at the reversible I−/I0 conversion, corresponding to two electron transfer or lower. Limited by the unstable nature of positive I+ ions in the electrolytes, reversible multivalent transitions for I species, with high capacity and output voltage, is constrained. Thus, the reported aqueous Zn–I2 batteries can only achieve a low output voltage plateau at ∼1.30 V. In this work, we proposed an efficient inter-halogen strategy for non-negative halogen fixation to fully activate and stabilize the unrealized high I0/I+ redox at 0.99 V vs. SHE via a facile electrolyte tailoring method. With the support of F− or Cl− additives, the resultant multivalent conversion of I−/I0/I+ led to a significantly improved electrochemical performance, including capacity, output voltage and energy density. This discovery is an exemplary effort to bring new insight to the aqueous I2–Zn batteries, and the developed brand-new I0/I+ chemistry can be extended to design other advanced I2–metal batteries such as Li, Mg and Al. |
Introduction
Developing intrinsic safe and advanced energy storage devices is a matter of continuing concern, among which stationary aqueous rechargeable iodine (I2) batteries are promising portable candidates.1–3 Unlike intercalation analogs that require repeated ion shuttle, the I2 batteries, relating to a solid–liquid conversion reaction featuring a stable discharge plateau, fully gain and lose electrons via reversible redox reaction between the iodine element and ion without the problem of electrode collapse.4–6 So far, various metal–I2 battery systems, including Li-, Na-, K-, Mg-, Fe-, Al-, and Zn–I2, have been explored and reported.3,7–11 However, for all of these systems, their common drawback is attributed to the electrochemical activity stopping at the I−/I0 conversion, corresponding to the theoretical redox potential of only 0.54 V vs. SHE or lower (Scheme 1).12,13 The existing monovalent state conversion under low potential is the most obvious bottleneck, restraining further breakthroughs of I2 batteries in terms of output voltage and energy density.14 | ||
| Scheme 1 Illustration of the reactions of iodine batteries. Reversible I−/I0 conversion at 0.54 V vs. SHE can be easily achieved but the desired reversible I0/I+ redox at high 0.99 V vs. SHE is difficult due to the unstable nature of positive I+ ions in the adopted electrolyte. Correspondingly, it is difficult to achieve the high voltage plateau at 1.65 V in an I2//Zn battery. | ||
On the other hand, the redox chemistry of positive I0/I+ located at a potential of 0.99 V vs. SHE was more attractive than the I−/I0 couple.15,16 For example, once the I0/I+ redox reaction can be activated in the aqueous Zn battery, the expected output plateau will exceed the vast majority of reported cathodes of Zn batteries, and an enhanced capacity can also be expected.17,18 In general, the electrochemical performance of a battery depends on the indispensable conductive carrier, whose electron conduction efficiency and affinity to the active substance directly determine the reaction kinetics and cycle durability. However, these factors are not applicable to redox systems.2,19 Unlike the I− ions, positive I+ ions are difficult to stabilize in conventional electrolytes. Previous attempts focusing on positive valence redox of iodine have never been successful due to poor reversibility and toleration.13–15 In principle, the identified effective mutual containment between inter-halogen offers a new viable strategy for non-negative halogen fixation, which is expected to stabilize positive I+ and hence, activate the I0/I+ redox chemistry. Unfortunately, this area has not been explored yet.13,19
Herein, we demonstrated multivalent redox chemistry of iodine (I−/I0/I+) in an aqueous Zn ion battery by F− or Cl−-rich electrolytes, employing I terminated halogenated Ti3C2I2 MXene cathode and Zn foil anode. The resultant Ti3C2I2//Zn battery with an optimized electrolyte of 2 M ZnCl2 + 1 M KCl was equipped with a two-step reaction: in addition to the conventional I−1/I0 conversion at 1.30 V, an unprecedented I0/I+ redox reaction occurred at 1.65 V, resulting in a significantly improved electrochemical performance. As expected, the emerging high voltage plateau strengthened the capacity and energy density to 205% and 330%, respectively. Also, benefiting from the efficient electron conduction and the confinement effect of the Ti3C2I2 MXene interlayer, the reaction kinetics was remarkably enhanced, and the shuttle effect was effectively suppressed. Accordingly, a decent cyclic lifespan of over 2800 with 80% capacity retention and excellent rate capability with 207 mA h g−1 Ti3C2I2 at 0.5 A g−1 and 126 mA h g−1 Ti3C2I2 at 5 A g−1 was achieved. In situ Raman characterization revealed that the strong interaction of free Cl− ions in the electrolyte with I+ ions at high voltage was the decisive factor to activate and stabilize the reversible redox reaction of the I0/I+ couple.
Results and discussions
Activating and stabilizing redox reactions of the I0/I+ couple via F− or Cl− ion additives
Halogenated MXene was produced via a novel molten salt etching process using Ti3AlC2 MAX (JCPDS: 52-0875; Fig. S1, ESI†) ceramic precursor and CuI2 etchant (details are given in the Experimental section of ESI†). The etching reaction follows the equation:| Ti3AlC2 + 5CuI → Ti3C2I2 + 5Cu + AlI3↑ |
The scanning electron microscopy (SEM) image shows the as-produced MXene particles exhibiting the representative accordion-like morphology with a lateral size of around 6 μm (Fig. 1a).20 Also, the laminated feature of Ti3C2I2 sublayers was revealed by the section-view of the transmission electron microscopy (TEM) image (Fig. 1b). The energy-dispersive X-ray spectroscopy (EDX) analysis indicates the internal elemental composition that contains Ti, C and I elements with an atomic ratio of approximately 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2 (Fig. S2, ESI†), which was different from typical Ti3C2 MXene with electrochemically inert –O/OH/F/Cl surficial groups produced using conventional wet chemical etching methods (Fig. 1c).21–23 The departure of Al-layers and formation of I terminations led to the uniform interlayer galleries of about 1.25 nm, as evidenced by the high-resolution TEM (HRTEM) image in Fig. 1d. Further quantitative analysis confirms that the atomic ratio of Ti and I elemental compositions were about 3:2, with negligible Al and O contents. Hence, the obtained MXene was conventionally referred to as Ti3C2I2.24–26 The X-ray diffraction (XRD) patterns in Fig. 1e reveal that the emerging diffraction peak at 7.15° was indexed to the (002) crystal plane of Ti3C2I2 MXene after etching, indicating an expanded interlayer spacing of about 12.5 Å (8.9 Å of Ti3AlC2), matching the HRTEM result well. The other peaks at 14.5°, 21.4°, and 28.8° were indexed to (004), (006), and (008) planes.22,27–29 The smaller strength ratio between (002) and (004) planes can be attributed to the capability difference in scattering X-ray caused by I terminations compared to traditional Ti3C2TX MXene. Besides, the X-ray photoelectron spectroscopy (XPS) spectrum in Fig. 1f shows the distinct I3d signal at 620–630 eV and the absence of the Al2p signal, in addition to the peaks of Ti2p and C1s.
:2:2 (Fig. S2, ESI†), which was different from typical Ti3C2 MXene with electrochemically inert –O/OH/F/Cl surficial groups produced using conventional wet chemical etching methods (Fig. 1c).21–23 The departure of Al-layers and formation of I terminations led to the uniform interlayer galleries of about 1.25 nm, as evidenced by the high-resolution TEM (HRTEM) image in Fig. 1d. Further quantitative analysis confirms that the atomic ratio of Ti and I elemental compositions were about 3:2, with negligible Al and O contents. Hence, the obtained MXene was conventionally referred to as Ti3C2I2.24–26 The X-ray diffraction (XRD) patterns in Fig. 1e reveal that the emerging diffraction peak at 7.15° was indexed to the (002) crystal plane of Ti3C2I2 MXene after etching, indicating an expanded interlayer spacing of about 12.5 Å (8.9 Å of Ti3AlC2), matching the HRTEM result well. The other peaks at 14.5°, 21.4°, and 28.8° were indexed to (004), (006), and (008) planes.22,27–29 The smaller strength ratio between (002) and (004) planes can be attributed to the capability difference in scattering X-ray caused by I terminations compared to traditional Ti3C2TX MXene. Besides, the X-ray photoelectron spectroscopy (XPS) spectrum in Fig. 1f shows the distinct I3d signal at 620–630 eV and the absence of the Al2p signal, in addition to the peaks of Ti2p and C1s.
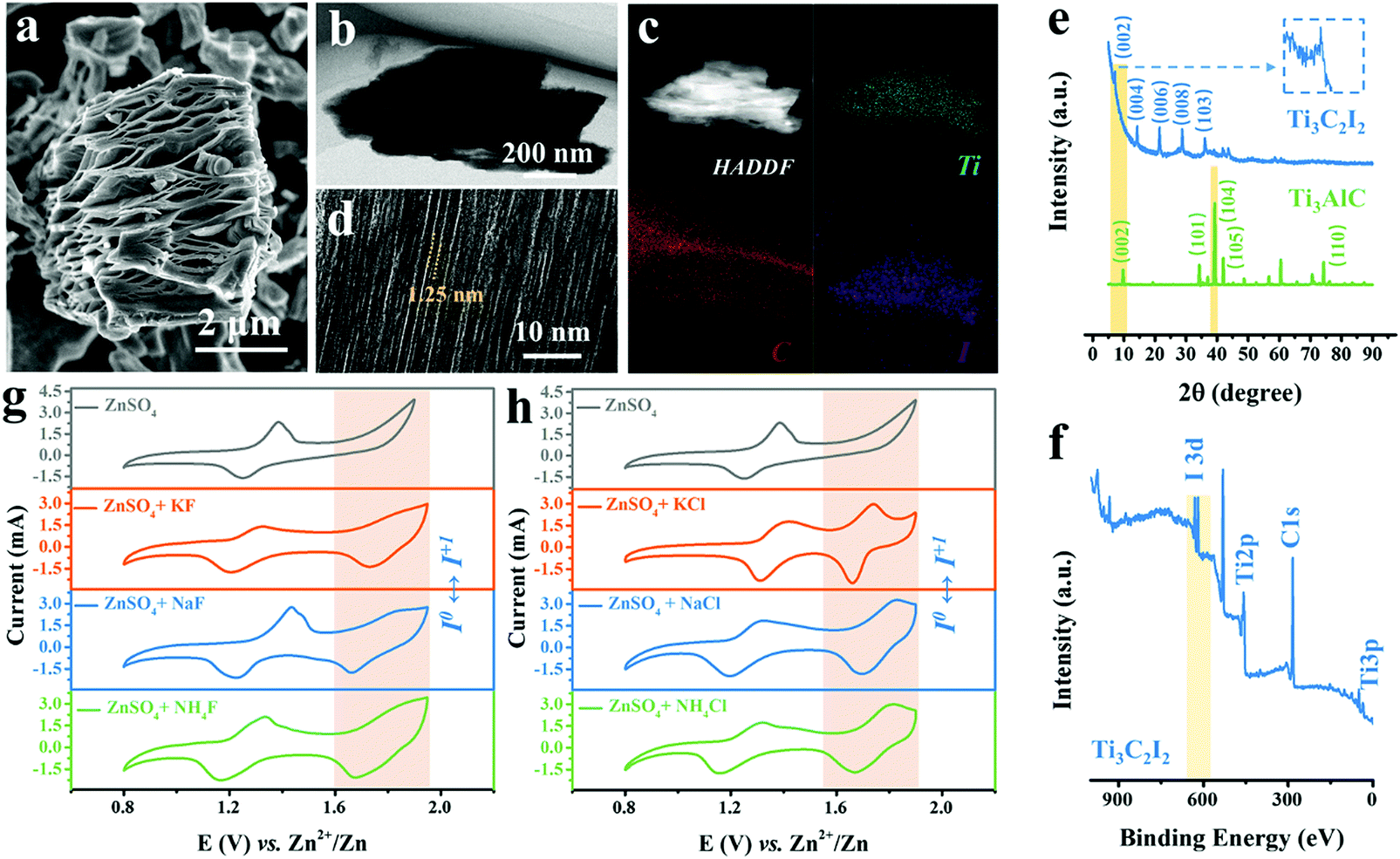 | ||
| Fig. 1 (a) SEM image, (b) TEM image, (c) corresponding EDX mapping of Ti, C, I elements, (d) HRTEM image of Ti3C2I2 MXene. (e) XRD pattern of Ti3C2I2 MXene before and after molten salt etching. (f) Survey XPS spectrum of Ti3C2I2 MXene. (g) CV curves of the assembled Zn ion battery at 10 mV s−1 employing Ti3C2I2 MXene cathode, Zn anode, and aqueous 2 M ZnSO4 electrolyte without/with various F− ion additives, including KF, NaF, and NH4F salts, (h) Cl− ion additives including KCl, NaCl, and NH4Cl salts. | ||
To perform the electrochemical analysis, we assembled the Ti3C2I2 cathode (Fig. S3, ESI†), Zn metal anode, and aqueous electrolytes containing 2 M ZnSO4 electrolyte with/without various F− or Cl− ion additives. KF, NaF, NH4F, or KCl, NaCl, NH4Cl salts were used to provide the F− or Cl− ions in the designed electrolytes. The Ti3C2I2 MXene cathode terminated with I was endowed with natural electrochemical activity, which was different from the conventional pseudocapacitive Ti3C2TX (–O/OH/F/Cl).30–33Fig. 1g depicts a comparison of cyclic voltammogram (CV) profiles at 10 mV s−1 of the full batteries tested in the three aqueous electrolytes with different F−containing additives. Clearly, for all curves, two obvious redox peak pairs located at around 1.32/1.23 V and 1.84/1.66 V vs. Zn2+/Zn were recognized within 0.8–1.9 V. It should be noted that only the redox peak pairs at 1.32/1.23 V were detected in the pure ZnSO4 electrolyte under the same conditions. In addition to the I−/I0 redox (theoretical value: 1.30 V), another I0/I+ conversion peak was activated with the support of F− ions (further evidence will be provided later). It should be noted that different cations will not affect the function of F− ions. A side effect was that the reversibility of the I0/I+ conversion was inferior to that of the I−/I0 redox, manifested by a lower peak current response in the anodic scanning. Furthermore, for all Cl− counterparts, similar electrochemical features with two pairs of redox peaks during cycling were identified (1.42/1.32 V and 1.74/1.66 V), as evidenced in Fig. 1h. Remarkably, the reversibility of the high-potential I0/I+ reaction was significantly enhanced in comparison to that of the F− ion containing electrolytes.34,35 Upon cathodic scanning, the peak potential referred to the conversion from I0 to I+ was blue-shifted in the three electrolytes, indicating the enhanced reaction kinetics. Then, in the subsequent anodic scanning, the matching reduction peak was seen with a higher current response and a narrow half-width, representing the excellent redox efficiency of the I0/I+ couple.2 Moreover, the introduction of K+ cations at this time has been recognized to raise the redox potential of the I−/I0 transition, that is, lower the conversion energy barrier, superior to Na+ and NH4+ ions. Besides, as evidenced by the galvanostatic charge–discharge (GCD) curves of Ti3C2I2//ZnSO4 + KF//Zn and Ti3C2I2//ZnSO4 + KCl//Zn batteries, two discharge plateaus located around 1.62 and 1.30 V were detected, which performed better in the ZnSO4 + KCl electrolyte (Fig. S4, ESI†). The above results demonstrate that both free electronegative F− and Cl− anions are capable of strongly activating and stabilizing the desired I0/I+ redox reaction, with the Cl− ions being much more effective.
Electrochemical behavior of Ti3C2I2//Zn battery with ZnCl2 + KCl electrolyte
To further reveal the advantage of Cl− ions in stabilizing the high-potential I0/I+ redox, 2 M ZnCl2 electrolyte was chosen to replace 2 M ZnSO4 together with 1 M KCl to achieve abundant Cl−1. The resultant cyclic voltammetry (CV) curve of Ti3C2I2//ZnCl2 + KCl//Zn full cell scanned at 10 mV s−1 in the voltage window of 0.8–1.8 V is displayed in Fig. 2a, and a battery with 2 M ZnSO4 electrolyte was also tested for comparison. As expected, two redox couples were observed at 1.36/1.28 V and 1.74/1.66 V, suggesting that a two-step reaction occurs during the cycle, distinct from the ZnSO4 system with only one pair of redox peaks at 1.36/1.22 V. Three points are worth noting here: first, the reduction potential of I−/I0 in the ZnCl2 + KCl electrolyte at 1.28 V upon the anodic process was located above that of ZnSO4 (1.22 V). Second, during the cathodic scanning, the oxidation potential of I0/I+ reached up to 1.74 V, close to the theoretical value. Third, the reduction peak of the I0/I+ couple at 1.66 V was significantly narrowed with a remarkable increase in the current response, indicating a more prominent conversion characteristic.7 Accordingly, in the designed ZnCl2 + KCl electrolyte, the reversibility and stability of both couples were all significantly improved, and hence a more efficient conversion reaction was constructed. Such noticeable changes became more visually distinct in the corresponding GCD curves, in which the two well-defined discharge plateaus at 1.30 V and 1.65 V stand for I−/I0 and I0/I+ couple conversion, respectively, totally differing from the single plateau at 1.26 V with ZnSO4 electrolyte (Fig. 2b). For traditional Ti3C2OF MXene in such electrolyte, only typical pseudocapacitive feature can be detected, as evidenced by Fig. S5 (ESI†).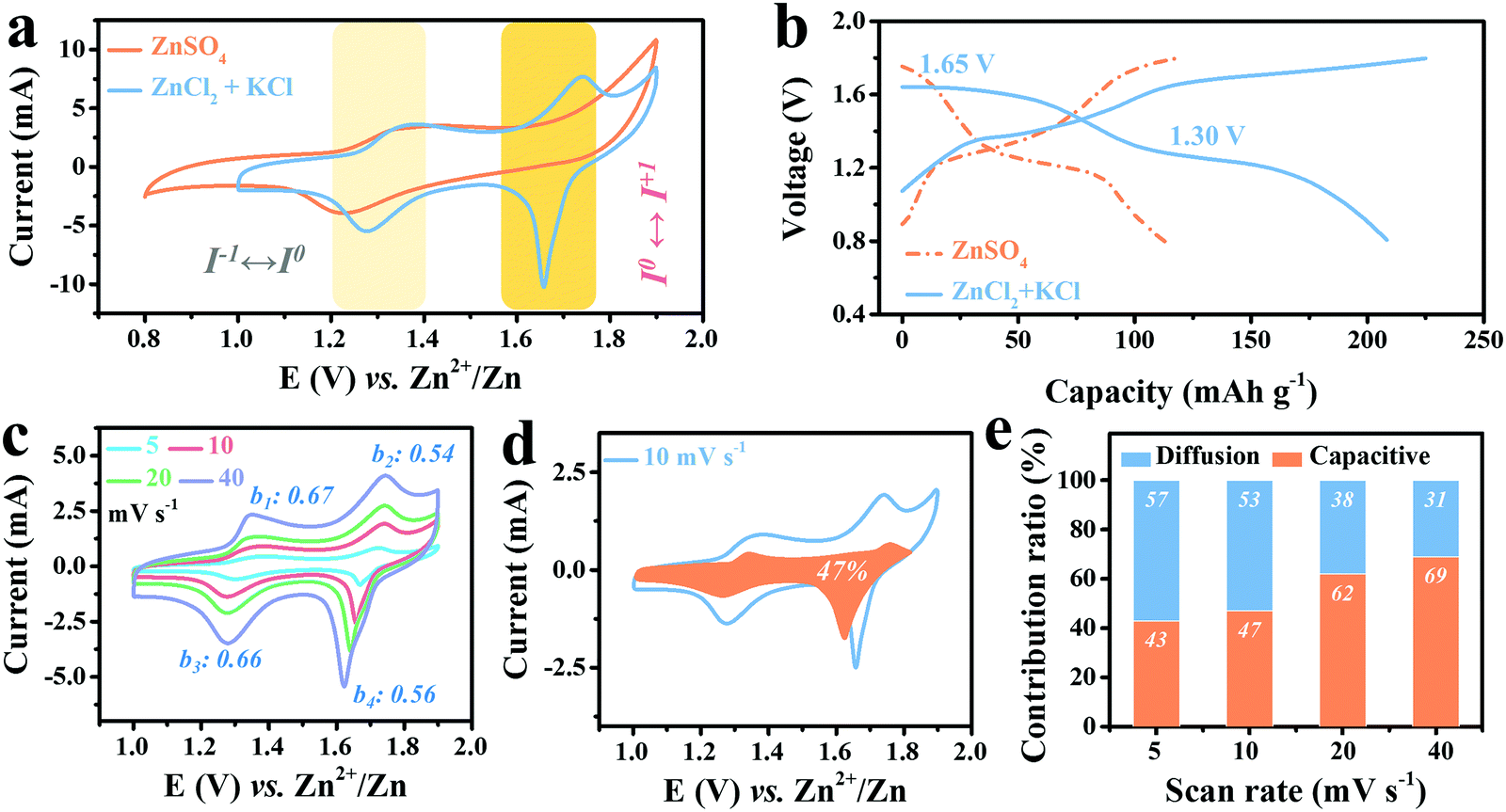 | ||
| Fig. 2 (a) CV curves at 10 mV s−1, (b) GCD curves at 0.5 A g−1 of Ti3C2I2//Zn battery based on optimized ZnCl2 + KCl electrolyte and conventional ZnSO4 electrolytes. (c) CV curves of Ti3C2I2//ZnCl2 + KCl//Zn battery recorded at different scan rates of 5, 10, 20 and 40 mV s−1. The calculated b values are placed near the corresponding redox peaks. (d) The representative CV curve was recorded at 10 mV s−1 with a marked region representing the capacitive-controlled contribution of about 47%. (e) The calculated capacitive and diffusion-controlled contributions at different scan rates of 5, 10, 20 and 40 mV s−1. | ||
Moreover, to better reveal the internal redox kinetics evolution, CV curves scanned at different current rates were collected. As shown in Fig. 2c, in the high sweep range of 4–40 mV s−1, the two pairs of identified redox peaks always remain stable, while the current response of the I0/I+ couple increases more significantly than that of I−/I0 as the rate accelerates. Such efficient kinetics has never been achieved in carbon-support I2 batteries, which undoubtedly benefits from the superior electron conduction of metallic Ti3C2.8,36,37 Moreover, the b values were then calculated to elucidate the charge storage process of the cathode according to the law as follows:8
| i = avb |
| i(V) = k1v + k2v1/2 |
A comprehensive electrochemical evaluation including rate capability and cycling stability of the Ti3C2I2//ZnCl2 + KCl//Zn full cell vs. Zn anode was further conducted. Meanwhile, the stable and reversible Zn anode chemistry in the ZnCl2 + KCl electrolyte was clarified by testing the symmetric Zn//ZnCl2 + KCl//Zn and asymmetric Cu//ZnCl2 + KCl//Zn batteries (Fig. S7–S9, ESI†).39,40Fig. 3a shows the rate performance, in which a capacity of up to 207 mA h g−1 Ti3C2I2 was delivered at 0.5 A g−1 with two well-defined plateaus of 1.65 and 1.30 V.28 When compared with the widely reported aqueous I2–Zn batteries with only redox of the I−/I0 couple, the value was unprecedented even though the heavy Ti3C2 MXene was employed.2 Over 61% of the capacity could be retained when the current density was sharply increased by 10 times to 5 A g−1. Once the current density dropped to 0.5 A g−1, the capacity could be recovered immediately, suggesting an excellent rate capability. On the contrary, with the ZnSO4 electrolyte, the capacity was below 105 mA h g−1 Ti3C2I2 (Fig. S10, ESI†). As shown in the corresponding GCD curves in Fig. 3b, two well-defined discharge voltage plateaus were always detected for all rates, and a small voltage hysteresis (0.08–0.11 V) was believed to benefit from the highly conductive MXene skeleton.37
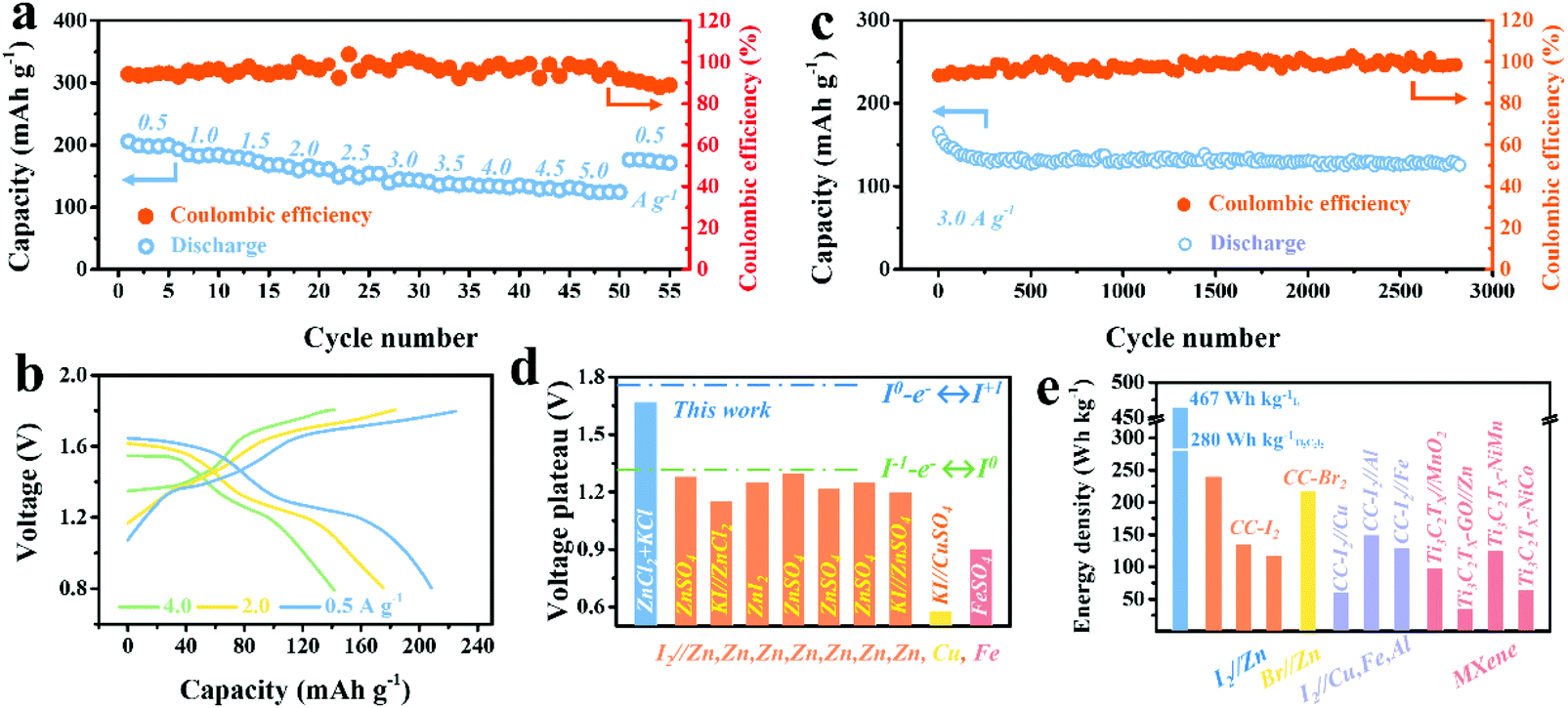 | ||
| Fig. 3 (a) Rate capability of Ti3C2I2//ZnCl2 + KCl//Zn battery cycled at various rates of 0.5–5.0 A g−1. (b) The corresponding GCD curves of 0.5, 2.0, and 4.0 A g−1. (c) The long-term cyclability of Ti3C2I2//ZnCl2 + KCl//Zn battery at 3.0 A g−1. (d) Comparison of discharge voltage plateaus of this work to reported aqueous I2–metal (Cu, Fe, Zn) systems.2,3,10–12,16,42,43 (e) Comparison of the energy density of this work to reported aqueous I2–metal, Br//Zn, MXene-based systems.3,7,10,11,43–48 | ||
The long-term cyclic capability of the battery was further investigated by GCD mode at 3 A g−1, as depicted in Fig. 3c. The Ti3C2I2//ZnCl2 + KCl//Zn battery delivered excellent cyclic durability of over 2800 cycles with a decent CE of over 98%. The capacity retention of about 80% was finally achieved. In addition to stable anode chemistry, the effective binding of the Ti3C2I2 cathode to active iodine species (I−/I0/I+) was reasonably indispensable.36 Owing to the confinement effect, which originated from the nano-sized interlayer galleries, the shuttle effect was impressively suppressed, resulting in the reversible redox of the interior I− ions without undesirable leakage.41 The emerging high plateau contributed to capacity enhancement and improved energy density. The resultant energy density calculated based on the Ti3C2I2 mass reached 280 W h kg−1 Ti3C2I2 (467 W h kg−1 I2 based on only I mass). Quantitative analysis showed that the extended plateau region (above 1.4 V) occupied 52% of the capacity and 69.8% of the energy density (Fig. S11, ESI†). With Cl− ions in the electrolyte, I0/I+ redox chemistry at 1.65 V was fully activated besides I−/I0 conversion at 1.30 V, which was much higher than that achieved in other aqueous I2–metal batteries reported, including I2–Zn, Fe, and Cu systems, as summarized in Fig. 3d.2,3,10–12,16,42,43 Accordingly, the resultant energy density of this work was significantly superior to the reported aqueous I2–Zn, Fe, Al, Cu, Br2//Zn and Ti3C2TX MXene based counterparts, as depicted in Fig. 3e.3,7,10,11,43–48
Reaction mechanism of reversible redox of the I0/I+ couple
To understand how the Cl− ions activate and stabilize the I0/I+ redox, in situ Raman, ex situ SEM, EDX, and ultraviolet-visible absorption spectroscopy (UV-vis) measurements were performed. Fig. 4a exhibits the representative GCD curve at 0.5 A g−1, marked with selected voltage points for the in situ Raman characterization, with a voltage window of 0.8–1.8 V and a voltage interval of 0.1 V (Fig. S12, ESI†). Detailed Raman spectra are plotted in Fig. 4b. The characteristic peak at 149 cm−1 of Ti3C2 and peaks at 1366 and 1627 cm−1 of carbon black remain unchanged, implying no phase transition of Ti3C2I2 MXene.31,41 Meanwhile, the persistent peaks located at 305 cm−1 were assigned to the electrolyte (Fig. S13, ESI†). It should be noted that when the voltage exceeds 1.4 V, a new peak appears around 200 cm−1 and becomes stronger as the charge progresses and disappears when the voltage drops below 1.4 V in the subsequent discharging process. This abrupt change was more evident in the refined Raman map near the 200 cm−1 regions with a voltage interval of 0.05 V, as displayed in Fig. 4c and Fig. S14 (ESI†). The switchable peak only exists in the voltage window of 1.4–1.8 V that corresponds to the I−/I0 conversion range, owing to the formation of I–Cl bonds.49,50 This observation clearly demonstrates that once the I+ ions are produced, the highly electronegative free Cl− ions could bond with them, resulting in the stable existence of I+ and reversible redox, which cannot be achieved by other electrolytes.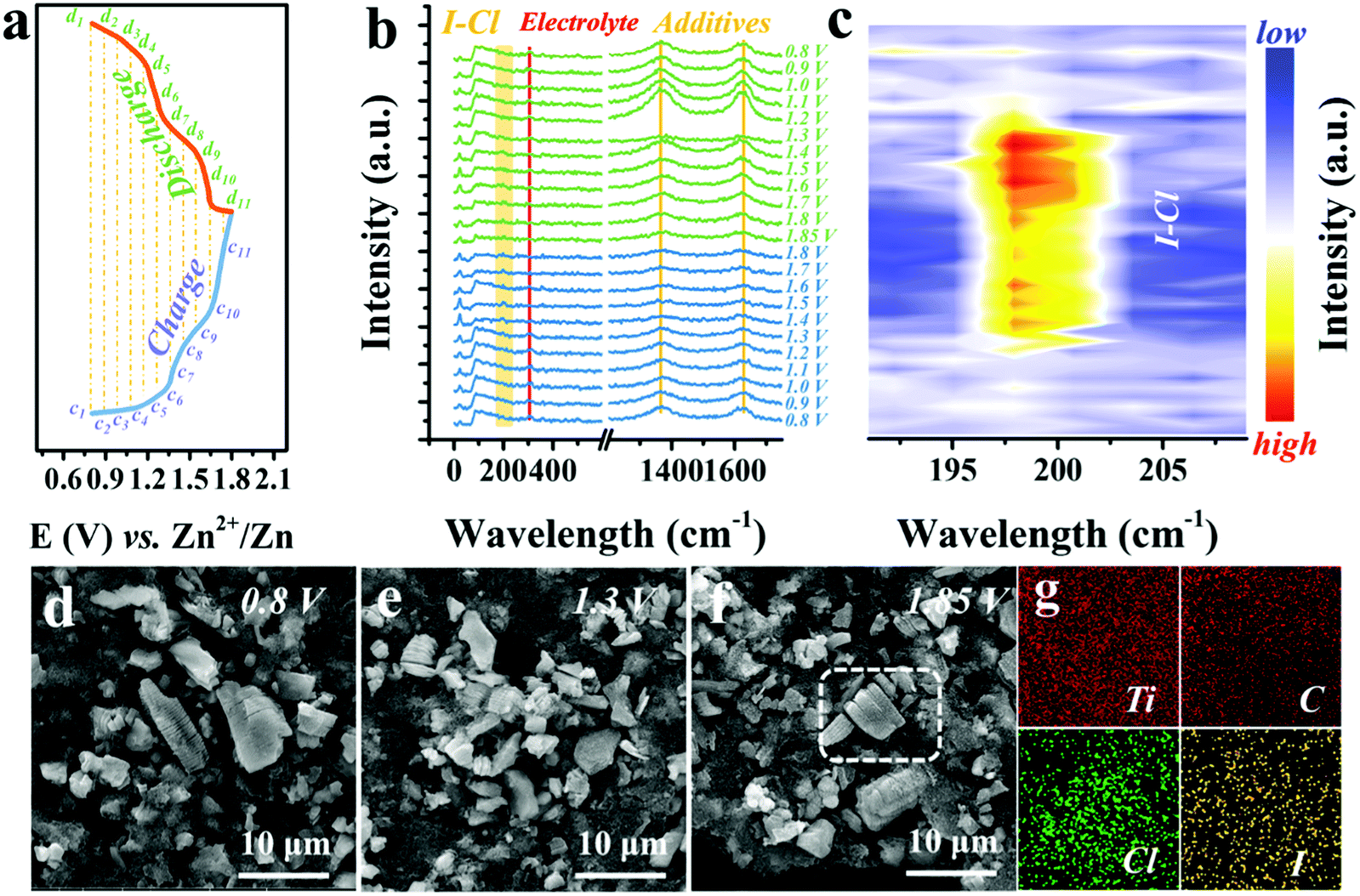 | ||
| Fig. 4 (a) GCD curve with marked voltage points for in situ and ex situ characteristics. (b) In situ Raman spectra of the Ti3C2I2 cathodes at different states with the voltage interval of 0.1 V. (c) Refined Raman map around 200 cm−1 with a voltage interval of 0.05 V. SEM images of the Ti3C2I2 cathodes after 1000 cycles at different voltages, (d) 0.8 V, (e) 1.3 V, and (f) 1.85 V. (g) EDX mapping data of Ti, C, I, Cl elements of the selected region in (f) at fully charged state. | ||
Also, in terms of the conversion reaction within 1.4 V, UV-vis spectra confirm a single and direct redox of the I−/I0 couple with no intermediate present, such as the polyiodides (I3−, 290 cm−1) (Fig. S15, ESI†).2,51 Besides, SEM images of Ti3C2I2 cathode after 1000 cycles at different charged states (0.8 V, 1.3 V and 1.85 V) showed microstructure evolution (Fig. 4d–f). All observed Ti3C2I2 MXene particles were still intact, and the representative layered morphology remained distinct. No visible collapse trace could be noted benefiting from the rigid ceramic nature of Ti3C2I2 MXene.28 As expected, both signals of Cl and I were simultaneously detected in Ti3C2I2 particles with a consistent distribution pattern in the EDX mapping data (Fig. 4g).
Conclusion
Traditional iodine batteries can only utilize the I−/I0 conversion reaction, whose potential was at 0.54 V vs. SHE, leading to a low voltage plateau at ∼1.30 V when Zn was used as the anode. Herein, we have achieved a new aqueous reversible redox of the I0/I+ couple by employing F− or Cl− ion-containing electrolytes. Inspired by this observation, we developed a Ti3C2I2 MXene/Zn battery with Cl− rich ZnCl2 + KCl electrolyte. The battery delivered two well-defined discharge plateaus at 1.65 V and 1.30 V corresponding to the shuttle between I−/I0/I+, distinct from the single plateau at 1.28 V with pure ZnSO4 electrolyte. The unprecedented high plateau significantly improved the capacity and energy density of this system, which reached up to 207 mA h g−1 Ti3C2I2 and 280 W h kg−1 Ti3C2I2 (that is, 467 W h kg−1 I2), far exceeding the upper limit of the reported I2–Zn system equipped with the low potential I−/I0 redox. The high voltage plateau region remarkably contributed to over 52% capacity and 69.8% energy output. Owing to the exceptional electron conduction and confinement effect of the MXene skeleton, excellent cyclic durability (2800 cycles) and rate capability were achieved (207 mA h g−1 Ti3C2I2 at 0.5 A g−1; 126 mA h g−1 Ti3C2I2 at 5 A g−1). In situ Raman, ex situ SEM, EDX, and UV-vis measurements elucidated the stable Ti3C2I2 cathode and the critical role of free Cl− ions in activating and stabilizing reversible I0/I+ redox at high potential. This work demonstrates a facile and scalable strategy via electrolyte tailoring towards developing high-potential I2–metal batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Key R&D Program of China under Project 2019YFA0705104. H. Q. thanks the supports of the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (Grant No. 2019R01003) and Ningbo's top-talent team program for financial support.References
- K. Lu, Z. Hu, J. Ma, H. Ma, L. Dai and J. Zhang, Nat. Commun., 2017, 8, 527 CrossRef.
- C. Bai, F. Cai, L. Wang, S. Guo, X. Liu and Z. Yuan, Nano Res., 2018, 11, 3548–3554 CrossRef CAS.
- H. Pan, B. Li, D. Mei, Z. Nie, Y. Shao, G. Li, X. S. Li, K. S. Han, K. T. Mueller, V. Sprenkle and J. Liu, ACS Energy Lett., 2017, 2, 2674–2680 CrossRef CAS.
- Z. Liu, Y. Huang, Y. Huang, Q. Yang, X. Li, Z. Huang and C. Zhi, Chem. Soc. Rev., 2020, 49, 180–232 RSC.
- L. Suo, O. Borodin, W. Sun, X. Fan, C. Yang, F. Wang, T. Gao, Z. Ma, M. Schroeder, A. von Cresce, S. M. Russell, M. Armand, A. Angell, K. Xu and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 7136–7141 CrossRef CAS.
- C. Yang, J. Chen, T. Qing, X. Fan, W. Sun, A. von Cresce, M. S. Ding, O. Borodin, J. Vatamanu, M. A. Schroeder, N. Eidson, C. Wang and K. Xu, Joule, 2017, 1, 122–132 CrossRef CAS.
- H. Tian, S. Zhang, Z. Meng, W. He and W.-Q. Han, ACS Energy Lett., 2017, 2, 1170–1176 CrossRef CAS.
- F. Wang, Z. Liu, C. Yang, H. Zhong, G. Nam, P. Zhang, R. Dong, Y. Wu, J. Cho, J. Zhang and X. Feng, Adv. Mater., 2020, 32, e1905361 CrossRef.
- F. Bertasi, F. Sepehr, G. Pagot, S. J. Paddison and V. Di Noto, Adv. Funct. Mater., 2016, 26, 4860–4865 CrossRef CAS.
- C. Bai, H. Jin, Z. Gong, X. Liu and Z. Yuan, Energy Storage Mater., 2020, 28, 247–254 CrossRef.
- H. Li, M. Li, X. Zhou and T. Li, J. Power Sources, 2020, 449, 227511 CrossRef CAS.
- J. J. Hong, L. Zhu, C. Chen, L. Tang, H. Jiang, B. Jin, T. C. Gallagher, Q. Guo, C. Fang and X. Ji, Angew. Chem., Int. Ed., 2019, 58, 15910–15915 CrossRef CAS.
- C. Yang, J. Chen, X. Ji, T. P. Pollard, X. Lu, C. J. Sun, S. Hou, Q. Liu, C. Liu, T. Qing, Y. Wang, O. Borodin, Y. Ren, K. Xu and C. Wang, Nature, 2019, 569, 245–250 CrossRef CAS.
- Z. Wang, X. Meng, K. Chen and S. Mitra, ACS Appl. Mater. Interfaces, 2018, 10, 30257–30264 CrossRef CAS.
- Z. Wang, X. Meng, K. Chen and S. Mitra, Energy Storage Mater., 2019, 19, 206–211 CrossRef.
- Z. Wang, J. Huang, Z. Guo, X. Dong, Y. Liu, Y. Wang and Y. Xia, Joule, 2019, 3, 1289–1300 CrossRef CAS.
- Z. Huang, A. Chen, F. Mo, G. Liang, X. Li, Q. Yang, Y. Guo, Z. Chen, Q. Li and B. Dong, Adv. Energy Mater., 2020, 2001024 CrossRef CAS.
- G. Liang, F. Mo, D. Wang, X. Li, Z. Huang, H. Li and C. Zhi, Energy Storage Mater., 2019, 25, 86–92 CrossRef.
- H. Tian, T. Gao, X. Li, X. Wang, C. Luo, X. Fan, C. Yang, L. Suo, Z. Ma, W. Han and C. Wang, Nat. Commun., 2017, 8, 14083 CrossRef CAS.
- X. Li, X. Yin, C. Song, M. Han, H. Xu, W. Duan, L. Cheng and L. Zhang, Adv. Funct. Mater., 2018, 28, 1803938 CrossRef.
- X. Tang, D. Zhou, P. Li, X. Guo, C. Wang, F. Kang, B. Li and G. Wang, ACS Cent. Sci, 2019, 5, 365–373 CrossRef CAS.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall’Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS.
- X. Li, M. Li, Q. Yang, G. Liang, Z. Huang, L. Ma, D. Wang, F. Mo, B. Dong and Q. Huang, Adv. Energy Mater., 2020, 2001791 CrossRef CAS.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS.
- M. Li, J. Lu, K. Luo, Y. Li, K. Chang, K. Chen, J. Zhou, J. Rosen, L. Hultman and P. Eklund, J. Am. Chem. Soc., 2019, 141, 4730–4737 CrossRef CAS.
- Y. Li, H. Shao, Z. Lin, J. Lu, L. Liu, B. Duployer, P. O. Persson, P. Eklund, L. Hultman and M. Li, Nat. Mater., 2020, 1–6 Search PubMed.
- Q. Yang, Z. Huang, X. Li, Z. Liu, H. Li, G. Liang, D. Wang, Q. Huang, S. Zhang and S. Chen, ACS Nano, 2019, 13(7), 8275–8283 CrossRef CAS.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS.
- M. Q. Zhao, X. Xie, C. E. Ren, T. Makaryan, B. Anasori, G. Wang and Y. Gogotsi, Adv. Mater., 2017, 29, 1702410 CrossRef.
- Y. Xia, T. S. Mathis, M. Q. Zhao, B. Anasori, A. Dang, Z. Zhou, H. Cho, Y. Gogotsi and S. Yang, Nature, 2018, 557, 409–412 CrossRef CAS.
- X. Wang, T. S. Mathis, K. Li, Z. Lin, L. Vlcek, T. Torita, N. C. Osti, C. Hatter, P. Urbankowski and A. Sarycheva, Nat. Energy, 2019, 4(3), 241–248 CrossRef CAS.
- X. Li, M. Li, Q. Yang, D. Wang, L. Ma, G. Liang, Z. Huang, B. Dong, Q. Huang and C. Zhi, Adv. Energy Mater., 2020, 2001394 CrossRef CAS.
- G. Liang, Y. Wang, Z. Huang, F. Mo, X. Li, Q. Yang, D. Wang, H. Li, S. Chen and C. Zhi, Adv. Mater., 2020, 1907802 CrossRef CAS.
- L. Ma, S. Chen, C. Long, X. Li, Y. Zhao, Z. Liu, Z. Huang, B. Dong, J. A. Zapien and C. Zhi, Adv. Energy Mater., 2019, 9, 1902446 CrossRef CAS.
- X. Tang, D. Zhou, P. Li, X. Guo, C. Wang, F. Kang, B. Li and G. Wang, ACS Cent. Sci., 2019, 5, 365–373 CrossRef CAS.
- X. Li, M. Li, Q. Yang, H. Li, H. Xu, Z. Chai, K. Chen, Z. Liu, Z. Tang and L. Ma, ACS Nano, 2020, 541–551 CrossRef.
- X. Li, L. Ma, Y. Zhao, Q. Yang, D. Wang, Z. Huang, G. Liang, F. Mo, Z. Liu and C. Zhi, Mater. Today Energy, 2019, 14, 100361 CrossRef.
- X. Xie, S. Liang, J. Gao, S. Guo, J. Guo, C. Wang, G. Xu, X. Wu, G. Chen and J. Zhou, Energy Environ. Sci., 2020, 13, 503–510 RSC.
- Q. Yang, G. Liang, Y. Guo, Z. Liu, B. Yan, D. Wang, Z. Huang, X. Li, J. Fan and C. Zhi, Adv. Mater., 2019, 31, 1903778 CrossRef CAS.
- C. Sun, X. Shi, Y. Zhang, J. Liang, J. Qu and C. Lai, ACS Nano, 2020, 14, 1176–1184 CrossRef CAS.
- Y. Li, L. Liu, H. Li, F. Cheng and J. Chen, Chem. Commun., 2018, 54, 6792–6795 RSC.
- Y. Man, Q. Hao, F. Chen, X. Chen, Y. Wang, T. Liu, F. Liu and N. Li, ChemElectroChem, 2019, 6, 5872–5875 CrossRef CAS.
- D. Zhang, J. Cao, X. Zhang, N. Insin, R. Liu and J. Qin, ACS Appl. Energy Mater., 2020, 3, 5949–5964 CrossRef CAS.
- J. H. Lee, Y. Byun, G. H. Jeong, C. Choi, J. Kwen, R. Kim, I. H. Kim, S. O. Kim and H. T. Kim, Adv. Mater., 2019, 31, e1904524 CrossRef.
- Q. Wang, S. Wang, X. Guo, L. Ruan, N. Wei, Y. Ma, J. Li, M. Wang, W. Li and W. Zeng, Adv. Electron. Mater., 2019, 5, 1900537 CrossRef CAS.
- J. Zheng, X. Pan, X. Huang, D. Xiong, Y. Shang, X. Li, N. Wang, W.-M. Lau and H. Y. Yang, Chem. Eng. J., 2020, 396, 125197 CrossRef CAS.
- S. Wang, Q. Wang, W. Zeng, M. Wang, L. Ruan and Y. Ma, Nano-Micro Lett., 2019, 11, 70 CrossRef.
- W. B. Person, G. R. Anderson, J. N. Fordemwalt, H. Stammreich and R. Forneris, J. Chem. Phys., 1961, 35, 908–914 CrossRef CAS.
- P. Klaeboe, J. Am. Chem. Soc., 1967, 89, 3667–3676 CrossRef CAS.
- Y. Li, L. Liu, H. Li, F. Cheng and J. Chen, Chem. Commun., 2018, 54, 6792–6795 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee03086d |
| This journal is © The Royal Society of Chemistry 2021 |