
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-oxidation of pinic acid in the aqueous phase: a mechanistic investigation under acidic and basic pH conditions†
Jéssica Vejdani
Amorim
 a,
Xinyang
Guo
a,
Tania
Gautam
a,
Rongyan
Fang
a,
Christian
Fotang
a,
Florence J.
Williams
b and
Ran
Zhao
*a
a,
Xinyang
Guo
a,
Tania
Gautam
a,
Rongyan
Fang
a,
Christian
Fotang
a,
Florence J.
Williams
b and
Ran
Zhao
*a
aDepartment of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada. E-mail: rz@ualberta.ca
bDepartment of Chemistry, University of Iowa, Iowa City, Iowa 52242-1294, USA
First published on 1st July 2021
Abstract
Atmospheric aqueous phases (cloud and fog droplets, aerosol liquid water) are important reaction media for the processing of water-soluble organic acids (OAs). The photochemistry of these species is known to contribute to the formation of aqueous secondary organic aerosols (aqSOA). OAs arising from the gas-phase oxidation of α-pinene, such as cis-pinonic acid (CPA) or pinic acid (PA), are considered relevant aqSOA precursors, as they are oxygenated compounds and expected to be sufficiently water-soluble to undergo cloudwater processing. Although PA is a major oxidation product of α-pinene, the mechanism of its aqueous-phase photo-oxidation has never been studied previously. In this work, we performed the first in-depth investigation for aqueous-phase OH-oxidation of PA. Given that the pH of atmospheric aqueous phases is highly variable, and it has been increasing over the past 40 years, we were particularly interested in the impact of pH on the reaction mechanisms and product yields. Experiments were performed using a combination of offline and online MS techniques to identify PA and monitor the evolution of PA OH-oxidation products. We identified PA as a novel source of important α-pinene SOA tracers, including 3-methyl-1,2,3-butanetricarboxylic acid (MBTCA), and norpinic acid (NPA). Furthermore, we show a drastic difference in the yield of the oxidation products of PA under acidic and basic pH conditions. Specifically, we observed the yields of NPA and MBTCA at 3.4% and 2.6% under pH 2 but at 10% and 5.0%, respectively, at pH 8. These yield differences are likely due to a charge transfer reaction pathway unique to the aqueous phase. Such impact of pH on the reaction mechanism of OA has never been shown before.
Environmental significanceThis work represents the first in-depth mechanistic investigation for the OH-oxidation of pinic acid (PA) in the aqueous phase. PA is a major product of α-pinene, but its chemistry in the aqueous phase has never been studied. We identified norpinic acid (NPA) and 3-methyl-1,2,3-butanetricarboxylic acid (MBTCA) as major products from PA. We also observed drastically different molar yields of NPA and MBTCA under the acidic and basic conditions, likely due to a charge transfer reaction occurring only to the pinate ion. These results serve as a demonstration that solution pH can alter photochemical mechanisms. The rapidly changing cloud and fog pH in the ambient atmosphere can be affecting photochemistry and the products of organic acids. |
1. Introduction
Water-soluble organic acids (OAs) can undergo photochemistry in atmospheric aqueous phases (cloud, fog droplets, and aerosol liquid water). The chemistry occurring in cloudwater, also referred to as cloudwater processing, results in the decay of OAs, and further formation of secondary organic aerosols (SOA).1–3 The gas-phase oxidation of biogenic volatile organic compounds (BVOC) are known sources of OAs and other multifunctional compounds found in cloud and fog waters.4–7 One of the largest BVOCs emitted annually is α-pinene; thus, numerous studies focused on its gas-phase kinetics and mechanisms leading to SOA formation.4,5,8–10 It has been recognized that the aqueous-phase processing of semi-volatile oxidation products of α-pinene can contribute to SOA formation,1,6,11,12 but these processes are characterized poorly and limited to a few model compounds, such as cis-pinonic acid (CPA) and limononic acid.13–15 Among the multifunctional α-pinene products, CPA and pinic acid (PA) are first-generation OAs.5,16,17 CPA is the most targeted among the OAs mainly due to its commercial availability, while PA is rarely the target of systematic studies, since it is not readily available, and needs to be synthesized.PA was identified first by Holloway et al.9 in 1955 as the product of liquid-phase ozonolysis of α-pinene. Forty years later, Christoffersen et al.18 initiated a series of smog chamber studies that identified PA as one of the main oxidation products of α-pinene gas-phase ozonolysis and as an important α-pinene SOA component. In terms of known physicochemical properties of PA, Bilde et al.19 applied tandem differential mobility analysis to measure the evaporation rate and vapor pressure of PA and other semi-volatile oxidation products of α-pinene. Kołodziejczyk et al.20 used a differential scanning microcalorimetry technique to measure PA's thermal properties, as well as a solid–liquid phase equilibria method for the determination of PA solubility in water. An interesting finding from Kołodziejczyk et al.20 is that PA is the most water-soluble among other key α-pinene SOA oxidation products (PA > cis-norpinic acid > CPA > cis-norpinonic acid). When cloudwater is present in an air mass, PA and other OAs from α-pinene will be present exclusively in the aqueous phase.21 For chemical reactions of PA, a detailed gas-phase formation mechanism of PA has been proposed by Jenkin et al.22 The formation of PA in the gas phase and its particle-phase OH-oxidation has been investigated by many studies,8,16,18,23 but there is a lack of information on PA kinetics and thermodynamics in atmospheric aqueous phases, representing a major missing piece in multiphase atmospheric chemistry models.
The aqueous-phase OH-oxidation mechanism shares many similarities with that in the gas phase; however, mechanisms unique to the aqueous phase may lead to reaction pathways that are absent in the gas phase. Acid–base chemistry24 and rapid formation of carboxylic acids from aldehydes are representative examples.25 In addition, a unique charge transfer can occur to carboxylate but not carboxylic acid, giving rise to pH-dependent reaction mechanisms (Fig. 1). Briefly, the OH radical can react with an OA through H-abstraction and addition to the “R” group. H-abstraction from an O–H bond is slow, hence the OH radical does not react with the carboxyl group (COOH) rapidly. On the other hand, the OH radical is reactive to the carboxylate group (ROO−) by abstracting an electron from it.26 Our recent study24 investigated, for the first time, the pH dependence of the OH-reactivity of PA. The major finding was that the kinetics of PA's aqueous-phase oxidation is not affected significantly by the medium pH, likely because reaction with the “R” group dominates the OH reactivity of PA. Currently, there is no information available related to possible mechanistic differences under acidic and basic conditions. The pH-dependence of OH-oxidation is thought to be less significant compared to the sulfate radical (SO4−) and nitrate radical (NO3), which are more effective one-electron oxidants than the OH radical.26 The overall objective of this investigation was to better understand the fundamental mechanism of PA OH-oxidation in the aqueous phase, given that it has never been studied previously. In particular, we aimed to identify and quantify major products arising from PA OH-oxidation. The impact of solution pH on the reaction mechanism and product formations was also investigated, as it remains a poorly explored research area.
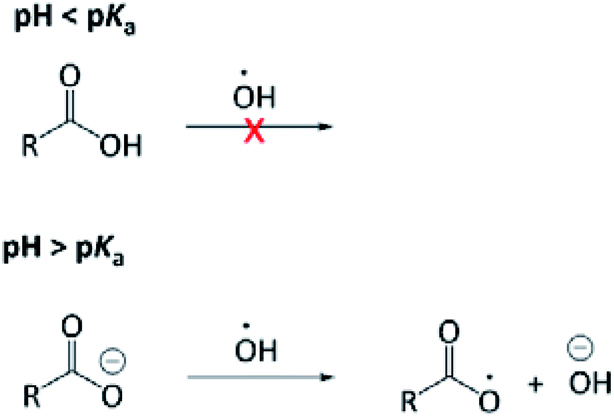 | ||
| Fig. 1 A schematic illustration of the carboxylate charge transfer reaction occurring only when pH of the solution is greater than the pKa of the carboxylic acid. This charge transfer reaction does not proceed with the carboxyl group (i.e., pH < pKa). | ||
2. Experimental
2.1 Materials
The following chemicals were purchased from Sigma Aldrich: CPA (98%), hydrogen peroxide (H2O2, 30% in water), sulfuric acid (50% in water), and potassium hydrogen phthalate (>99.95%). Chemicals were also purchased from Fisher Chemical: tricarballylic acid (TCA, 99%), sodium hydroxide (98%), acetonitrile HPLC grade, and formic acid 99.0+%. All chemicals were used without further purification. PA was custom synthesized and purified as previously described.242.2 Aqueous phase OH-oxidation
The aqueous-phase photo-oxidation of PA was performed using a photoreactor emitting UV-light centered at 310 nm (UVB). H2O2 was added to the solution as the source of OH radical. As shown in previously published data,24 the photoreactor (Rayonet, PRP200) emitting UVB light was able to efficiently photolyze H2O2 into OH radicals at atmospherically relevant concentrations, which have been estimated to be 1.4 × 10−16 to 8.0 × 10−12 M.1,26 We conducted an H2O2 control experiment to confirm that PA does not undergo direct photolysis in the absence of H2O2, and this result has already been published in Amorim et al.24https://www.zotero.org/google-docs/?DVqqW5https://www.zotero.org/google-docs/?DVqqW5https://www.zotero.org/google-docs/?DVqqW5https://www.zotero.org/google-docs/?DVqqW5. The UVB light also does not interact with products detected in this work. However, we note that the UVB light can directly photolyze reaction products, especially those containing carbonyl and peroxidic functional groups. The reaction progress was monitored via offline and online methods, as shown in the schematic of the experimental setup in Fig. 2. Electrospray ionization (ESI) high-resolution liquid chromatography (LC)-mass spectrometry (MS) was employed for the offline analysis, and online particle-into-liquid sampler (PILS) coupled to ESI-MS was used for the online analysis of PA reaction products. The experimental conditions used are summarized in Table 1. PA concentrations in ambient cloudwater are likely very diluted. van Pinxteren et al.27 have reported sub-μM concentrations of PA in cloudwater samples collected at a mountain site in Germany. In this work, the initial concentration of PA was chosen to be 114 or 330 μM for the offline and online experiments, respectively. These concentrations were necessary to provide sufficient product signals for the mechanistic analysis. To ensure the accuracy of the initial concentration of PA, the PA stock solution was standardized using proton nuclear magnetic resonance (1H NMR) spectroscopy (see Fig. S1 for details in the ESI†). | ||
| Fig. 2 Experimental apparatus. | ||
| Offline LC-MS | Online MS | |
|---|---|---|
| Quartz vessel | 15 mL | 150 mL |
| Volume of PA solution | 5 mL | 100 mL |
| PA concentration | 114 μM | 330 μM |
| H2O2 concentration | 5 mM | 20 mM |
| Solution pH | 2 and 8 | Uncontrolled |
| Sample collection time | 0, 5, 7.5 and 15 min | Continuous |
| Volume of sample collected | 300 μL | — |
| Total length of reaction | 15 min | 60 min |
For the offline reaction monitoring, the effect of pH on the reaction mechanism of PA was evaluated, and the pH of the solution was adjusted by adding H2SO4 50% wt (to pH 2) or NaOH 0.5 M (to pH 8). From the decay rate of PA, it was estimated that the steady-state concentration of OH ([OH]ss) was at 1.4 × 10−12 M at pH 2 and 7.4 × 10−13 M at pH 8. This concentration is within the range of the estimated concentrations in ambient cloudwater.26,28 The contribution of direct photolysis to the overall PA decay has been previously investigated and presented by Amorim et al.24 All offline experiments were conducted in triplicates to ensure reproducibility and to enable error analysis for quantitative information.
For the online analysis (details in Section 2.3.2), the pH of the solutions was kept uncontrolled, i.e., with no addition of acid or base. These salts can cause the suppression of the analyte signal in ESI; thus, no base or acid was added for the online analysis. To overcome the detection limit of the online MS, a higher initial PA concentration (330 μM) was chosen. To maintain a similar [OH]ss, H2O2 concentration was increased accordingly to 20 mM, which resulted in an estimated [OH]ss of 6.69 × 10−13 M. A duplicated online analysis was conducted to ensure the reproducibility. Given that this experiment required a higher concentration and a larger volume of PA solution, we could not afford more replicates with a limited amount of PA synthesized.
2.3 Offline and online sampling and analysis
For the online PILS-ESI-MS experiments, signals from the PA + H2O2 solution were recorded until a stable signal was obtained using the MS. The OH-oxidation is then initiated by turning the UV lamps on. The MS was operated in the continuous selected-ion monitoring (SIM) acquisition mode to track the evolution of selected species. A list of targeted species was generated by consulting the literature,13,34 and our own offline measurements. To assist product identification, the MS was occasionally operated in flow injection mode, with the experimental solution injected with a syringe pump. During this time, the MS was operated under the MS/MS mode (MS2). A list of parameters for the triple quadrupole MS is summarized in the ESI (Table S1†).
2.4 Determination of the acid dissociation constant (pKa) of PA
To better understand the acid dissociation equilibria of PA, PA's pKa was experimentally determined via titration with NaOH. The pKa value can be determined fairly accurately from the titration curve using the “half-volume” method.35 Therefore, 25.0 mL of 5.0 mM PA solution was titrated manually with 5.0 mM NaOH. The NaOH solution was standardized previously by titration with potassium hydrogen phthalate. The pH of the PA solution was measured continuously with a calibrated pH meter (Sartorius).3. Results and discussion
3.1 Determination of pKa
PA contains two ionizable carboxyl functional groups sharing a chemically similar environment; thus, under acidic conditions, the dissociation of both acidic protons will happen independently. Although PA is a di-acid, having two dissociation constants (Ka,1 and Ka,2), only one equivalence point was noticeable on the titration curve shown in Fig. 3. This indicates that the difference in PA's first and second dissociation constants is small, and the equivalence point observed is from the merged neutralization of both acidic protons.36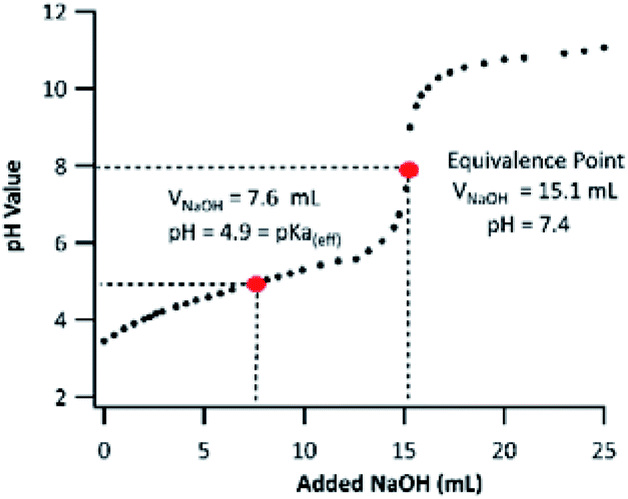 | ||
| Fig. 3 Titration curve of 5.0 mM PA using a 5.0 mM NaOH titrant. The horizontal axis presents the volume of titrant added (VNaOH). | ||
Although it is not possible to determine PA's first and second pKa values experimentally via acid–base titration, one can use the “half-volume” method to determine its “effective” pKa (pKa(eff)). The pKa(eff) is useful to estimate the fraction of neutral PA and pinate anion (PAn) in a solution. By applying the method described in Section 2.4, the PA's pKa(eff) was measured to be 4.9. To the best of our knowledge, the pKa value of PA has never been determined experimentally. A computationally determined value of 4.48 was used in the literature.37 Based on our observation, subsequent experiments in this work were conducted under pH values of 2 and 8. The PA's pKa(eff) indicates that the neutral form of PA dominates at pH 2 (99.9%), while at pH 8, the majority of PA will be dissociated fully and be present as PAn. The pH values of ambient cloudwater and fogs fall between 2 and 7.38,39 Therefore, in atmospheric aqueous phases, PA can exist as a mixture of its neutral form and as a PAn anion. The ratio of which varies drastically with changing cloudwater compositions.38 For this reason, it is important to fundamentally study PA oxidation mechanisms under both acidic and basic conditions, in which PA exists only in one of the two forms.
3.2 Offline (−)ESI-LC-MS for elemental analysis
The products arising from the aqueous-phase PA OH-oxidation were detected and identified using (−)ESI-LC-MS. Many peaks were detected at retention times shorter than that of PA, which indicates the formation of more oxygenated compounds. Table 2 shows the major oxidation products detected, with their elemental compositions and potential identities. We included all detected peaks with a signal intensity larger than 5.2 × 104 cps, and the m/z values correspond to their deprotonated forms ([M–H]−). We have also performed CPA OH-oxidation and detected all the oxidation products previously identified by Witkowski et al.13 (Table S2†). The results of the CPA OH-oxidation experiment provide both confidence for our experimental approach and a means of comparing mechanistic insights of CPA and PA, two structurally related compounds.| MW (g mol−1) | [M–H]− | Elemental composition | Identitya | Ref.b |
|---|---|---|---|---|
| a If missing, the compound has not been identified previously. b References. | ||||
| 144 | 143 | C7H12O3 | ||
| 156 | 155 | C8H12O3 | ||
| 158 | 157 | C7H10O4 | ||
| 172 | 171 | C8H12O4 | Terpenylic acid or norpinic acid (NPA) | 40 |
| 188 | 187 | C9H16O4 | Hydroxynorpinic acid | 41 |
| 190 | 189 | C8H14O5 | ||
| 200 | 199 | C9H12O5 | ||
| 202 | 201 | C9H14O5 | ||
| 204 | 203 | C8H12O6 | 3-Methyl-1,2,3-butanetricarboxylic acid (MBTCA) and/or isomers | 42 |
| 216 | 215 | C9H12O6 | ||
| 218 | 217 | C9H14O6 | ||
3.3 Online PILS-MS for formation and evolution of oxidation products
The evolution of PA OH-oxidation followed by the formation of its oxidation products can be observed in Fig. 4. The UV lamps were turned on to initiate OH-oxidation at time 0, which is indicated by a vertical dashed line. To avoid signal saturation, the signal of PA with a naturally existing 13C isotope (m/z 186 instead of m/z 185) was monitored and plotted. Immediately following the onset of OH-oxidation, the PA signal decayed rapidly, and numerous product signals appeared and subsequently reacted away. The online PILS-MS method was applied to monitor some of the major products detected using offline (−)ESI-LC-MS (Table 2). At the beginning of the experiment, a background signal was obtained by sampling the solution containing PA and H2O2 but without light. The background signal for the PA solution shows that some products were already present before the start of the experiment, which is likely attributed to reaction during the storage time. Control experiments (Section 2.2) showed that PA does not react with H2O2 under dark conditions, therefore, the products observed are not due to rapid oxidation as H2O2 is added to PA solution. Furthermore, these impurities are present in trace amounts, as their signal is smaller than that of the PA 13C isotope (1.1% abundance). The 1H NMR technique used for PA standardization did not detect any impurities, except for formic acid. We are confident that impurities at these concentrations have minimal impact on our mechanistic analysis.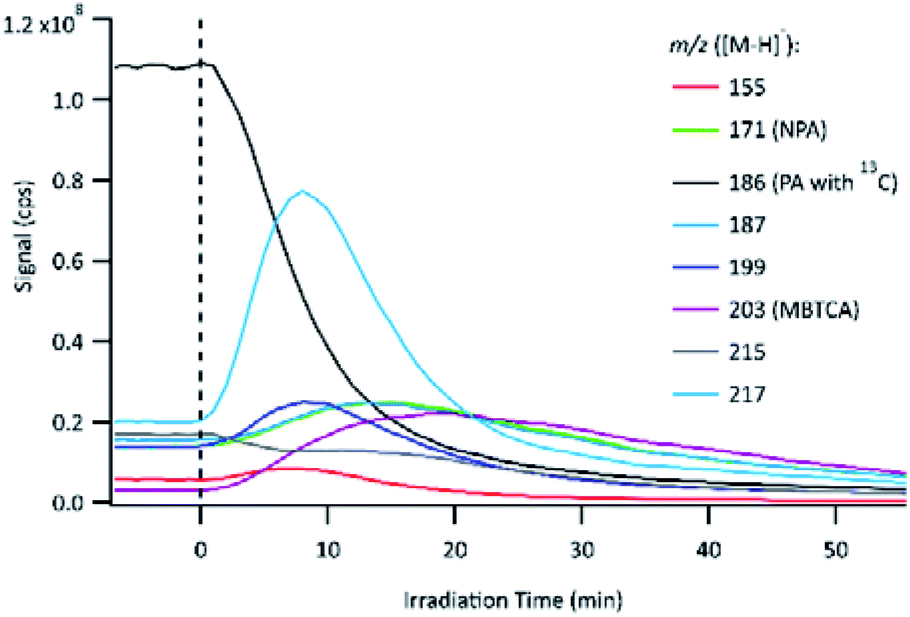 | ||
| Fig. 4 Temporal evolution of PA (monitored as a 13C isomer) and its major products during OH-oxidation, monitored using the online PILS-ESI-MS technique. The vertical dashed line indicates the onset of OH-oxidation. | ||
3.4 MS2 for further structural confirmation
The structural information of selected PA OH-oxidation products was confirmed using the fragmentation patterns from the triple-quadrupole MS instrument. Fig. 5 shows the fragmentation results for three major oxidation products with m/z 171, m/z 203, and m/z 217. The fragmentation spectra of a few other compounds monitored with MS2 are available in the ESI Fig. S2.† The MS2 spectra were collected by direct infusion of the reaction solution without separation; therefore, the fragmentation patterns represent those of mixed isobaric compounds.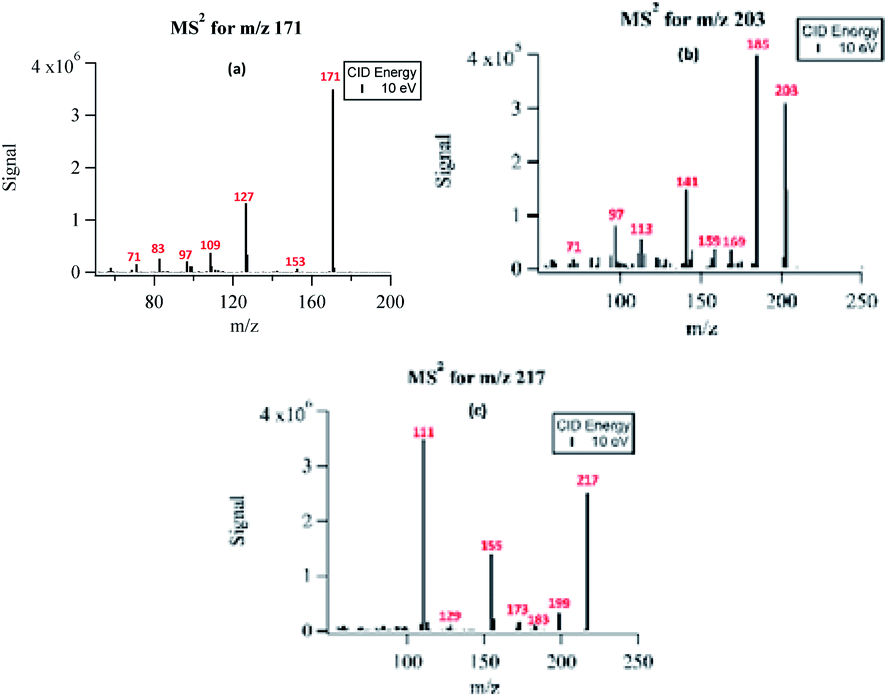 | ||
| Fig. 5 Fragmentation spectra of (a) NPA (m/z 171), (b) MBTCA (m/z 203), and (c) m/z 217 under uncontrolled pH conditions. | ||
The compound detected as m/z 171 ion produced fragment ions of m/z 153 (−18 au) and m/z 83 (−88 au), indicating the loss of H2O, and the presence of up to two carboxylic acids, respectively. The ion detected at m/z 171 was identified as the major PA oxidation product. Previous studies have shown that both gas-phase ozonolysis and aqueous-phase OH-oxidation of α-pinene SOA leads to the formation of compounds of m/z 171.24,41 These compounds have been assigned to be terpenylic acid (a mono-acid) and/or NPA (a diacid).40 We propose that the peak at m/z 171 in our study is mainly attributed to NPA, based on the observed loss of 88 au, which is indicative of the presence of two carboxylic groups. The formation mechanisms for NPA are shown in Sections 3.6.1 and 3.6.2.
For the compound with m/z 203, fragment ions include m/z 185 (−18 au; neutral loss of H2O), m/z 173 (−44 au; neutral loss of CO2), m/z 141 (−62 au; neutral loss of CO2 and H2O), and m/z 97 (−106 au; neutral loss of H2O and two CO2). Although small, the presence of a product ion of m/z 71 represents a combined loss of three CO2, which implies the presence of three carboxyl groups in its structure. The fragmentation pattern of the product detected as m/z 203 was similar to that of MBTCA and its isomers.42,43 Offline analysis with (−)ESI-LC-MS detected a dominant isomer of m/z 203 at Rt = 0.67 min and a few minor ones. To confirm that the dominant isomer is attributable to MBTCA, we compare the extracted ion chromatogram (EIC) of m/z 203 to that of TCA, which exhibits a peak at Rt = 0.59 min (Fig. 6). Given that MBTCA and TCA are structurally similar triacids (Fig. S3†), we are confident that the major isomer of m/z 203 at Rt = 0.67 min is MBTCA. Although the formation of MBTCA from PA has never been reported before, its formation from OH-oxidation of CPA has been previously confirmed.13,43 For comparison, we have also added the EIC of m/z 203 from CPA OH-oxidation to Fig. 6. Interestingly, as opposed to PA, our results show that m/z 203 arising from CPA is attributed to multiple isomers, and the contribution of MBTCA (peak at Rt = 0.63 min) may not be significant.
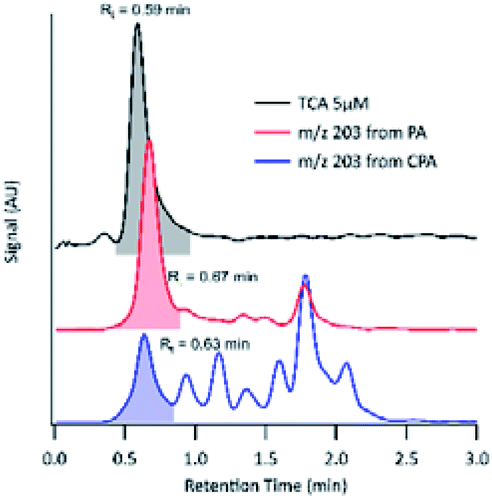 | ||
| Fig. 6 EICs of tricarballylic acid (TCA) (black), m/z 203 arising from PA OH-oxidation (red), and m/z 203 arising from the CPA OH-oxidation (blue). The y-axis has been offset for clarity. EICs for m/z 203 are those from the pH 8 experiments. The EIC for MBTCA at pH 2 is not shown here, as it differs from pH 8 only in terms of products signal intensity. | ||
The product detected as m/z 217 is also a major product of PA OH-oxidation, and it produced fragmentation ions at m/z 199 (−18 au; neutral loss of H2O), m/z 183 (−34 au, neutral loss of H2O2), m/z 173 (−44 au; neutral loss of CO2), m/z 155 (neutral loss of CO2 and H2O), m/z 129 (−88 au, presence of up to two carboxylic acids). The loss of 34 au is a fragmentation pattern unique to the products detected at m/z 217. Previous studies have attributed this pattern to the presence of a hydroperoxy group (ROOH);44 as such, m/z 217 is most likely to be a pinic hydroperoxide, forming as a first-generation product of PA as we propose in Section 3.6.1. This result is in agreement with CPA's oxidation product of m/z 215, which is a 2 au difference from PA's product. Witkowski et al.13 proposed that m/z 215 is a pinonic hydroperoxide. To confirm that m/z 217 from PA photo-oxidation and m/z 215 from CPA photo-oxidation are indeed organic peroxides, we performed iodometry-assisted LC-MS.29 This technique takes advantage of the selectivity of iodometry towards organic peroxides and serves as a powerful method for qualitative confirmation of peroxides. We applied the same technique described by Zhao et al.29 on a PA OH-oxidation sample collected at 5 min of the reaction progress (Section 2.2) to ensure that m/z 217 has not been further oxidized. Fig. 7 shows a comparison of the product detected as m/z 217 and its isomers when treated with and without iodometry. We note that all peaks were depleted by iodometry, which is an indication that the products with m/z 217 appear to be peroxides (Fig. 7(a)). A very similar observation was made for m/z 215 from CPA photo-oxidation (Fig. 7(b)), indicating that some of its isomers are organic peroxides that form via a mechanism equivalent to that of m/z 217 in PA photo-oxidation.
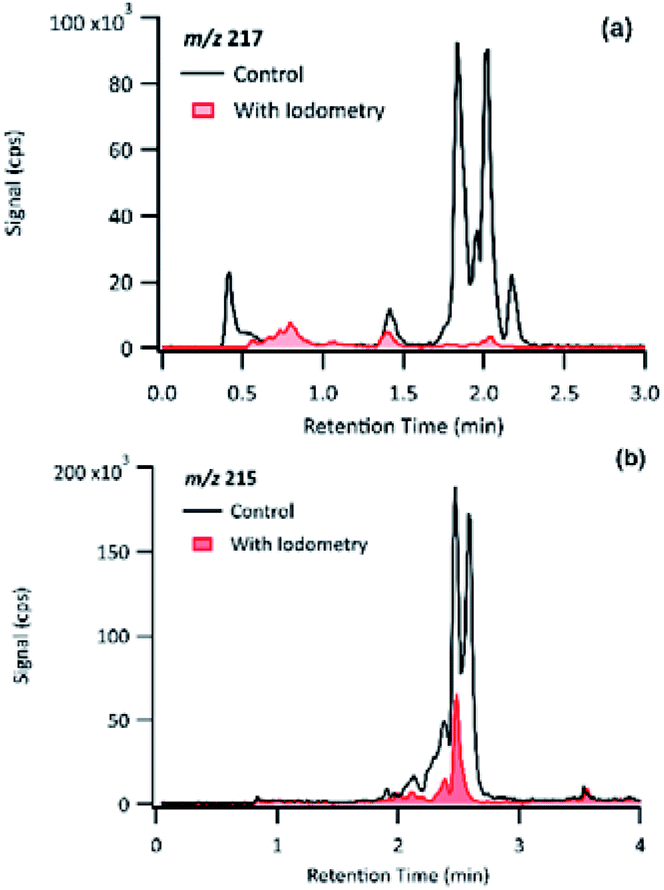 | ||
| Fig. 7 Comparison of the EICs of (a) m/z 217 from PA OH-oxidation and (b) m/z 215 from CPA OH-oxidation treated with and without iodometry and measured by (−)ESI-LC-MS. | ||
3.5 NPA and MBTCA yield at pH 2 and pH 8
As shown in previous sections, the photo-oxidation of PA leads to the formation of many products; among which NPA and MBTCA have been of interest as known tracers of α-pinene SOA, and involved in the formation of SOA.6 The EICs of NPA and MBTCA formed via PA OH-oxidation at pH 2 and 8 are shown in Fig. 8(a) and (b), respectively. The most noteworthy observation is a significant difference in the signal intensities for NPA and MBTCA, with a greater amount detected under the basic conditions. Therefore, it is evident that the pH of the aqueous phase affects PA's reaction mechanism. | ||
| Fig. 8 EICs for peaks of (a) m/z 171 (NPA), and (b) m/z 203 (MBTCA) arising from PA OH-oxidation, and (c) m/z 203 (MBTCA) arising from CPA OH-oxidation. EICs were detected with offline (−)ESI-LC-MS. The shaded area presents 1σ from the triplicates. | ||
To provide a quantitative comparison between pH 2 and 8, we determine the aqueous-phase molar yield (Yaq) based on the ratio of concentration of the product formed ([Product]), to change in PA concentration (Δ[PA]) over reaction time (t), as expressed in eqn (1):
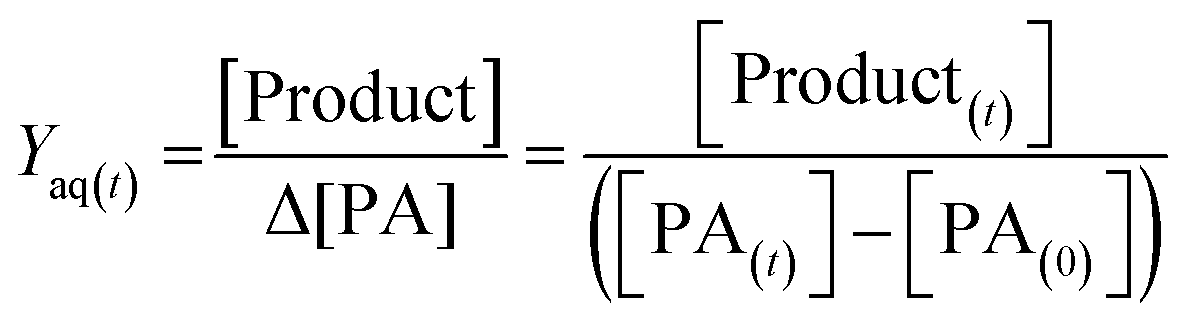 | (1) |
[PA(t)] represents the concentration of PA remaining, while [Product(t)] represents the concentration of either NPA or MBTCA formed at time t. It is important to note that Yaq varies as a function of time, as PA is depleted in time, and the products also undergo further OH-oxidation. In this work, we found that the Yaq values were relatively constant until 60% of PA was consumed; therefore, we have decided to report Yaq by averaging those from all the samples with at least 40% of PA remaining. There are a few advantages in this method. First, this amount of decay ensures an accurate determination of Δ[PA]. Second, multi-generational OH-oxidation significantly complicates the product distribution, and focusing on the unset of the experiment allows more confident identification and quantification of NPA and MBTCA. Third, NPA and MBTCA have not undergone extensive photo-oxidation themselves by this time; therefore, Yaq obtained this way likely represents the maximum yield of NPA and MBTCA.
As there are no commercial standards available for NPA and MBTCA, these two products were quantified assuming their ionization efficiencies are similar to those of PA and TCA, respectively. These surrogates were chosen due to their structural similarities to NPA and MBTCA (Fig. S3†). Note that TCA has been previously used as a surrogate to study reaction mechanisms of MBTCA.6 In this work, a PA and TCA calibration curve was prepared and analysed using (−)ESI-LC-MS as described in Section 2.3.1. The signal of MBTCA was related to that of TCA for quantification, while the signal of NPA was related to that of PA.
The Yaq of NPA and MBTCA calculated with this method are summarized in Table 3. The mean values were obtained from the triplicates, while the uncertainty range indicates 1σ after propagating uncertainties related to all the parameters in eqn (1). The Yaq values of both compounds are higher by more than a factor of 2 under pH 8. Such an impact of pH on the photochemical products of OAs has been suspected but never been reported in the literature.45 In Section 3.6.2, we propose a mechanistic explanation for this observation.
For comparison, we have also calculated MBTCA's Yaq arising from CPA OH-oxidation. Although a peak attributable to MBTCA has been observed from CPA OH-oxidation, as shown in Fig. 8(c), this peak was below the limit of detection of our (−)ESI-LC-MS technique. As discussed in Section 3.4, numerous isomers of m/z 203 are detected, and we consider the peak at Rt = 0.63 min to be MBTCA. We focus on Yaq at t = 5 min, at which 40% of CPA remains. For this calculation, the contribution of the direct photolysis of CPA must be considered. CPA contains a carbonyl group that absorbs the UVB light equipped in our photoreactor. Judging from our previous study,24 in which the same lamp setting was employed, 30% of CPA decay was due to direct photolysis, while the remaining 30% was due to OH-oxidation. In this work, we assumed that MBTCA from CPA is solely from the 30% of CPA reacted via OH-oxidation. Based on the detection limit of MBTCA (0.21 μM), the upper-limit estimate for MBTCA's Yaq from CPA is 0.1%. MBTCA's Yaq from CPA was found to be significantly smaller than its Yaq from PA. While Yaq of MBTCA has never been reported in the literature, the gas-phase yield is described to be <1%.13,42 The Yaq determined by our study is low due to our finding that m/z 203 from CPA is attributable to numerous isomers, and the relative contribution of MBTCA is minor. Overall, our results show that MBTCA's Yaq from PA OH-oxidation is much greater than that of CPA, which is the currently known precursor of MBTCA.
3.6 Proposed reaction mechanisms of PA OH-oxidation
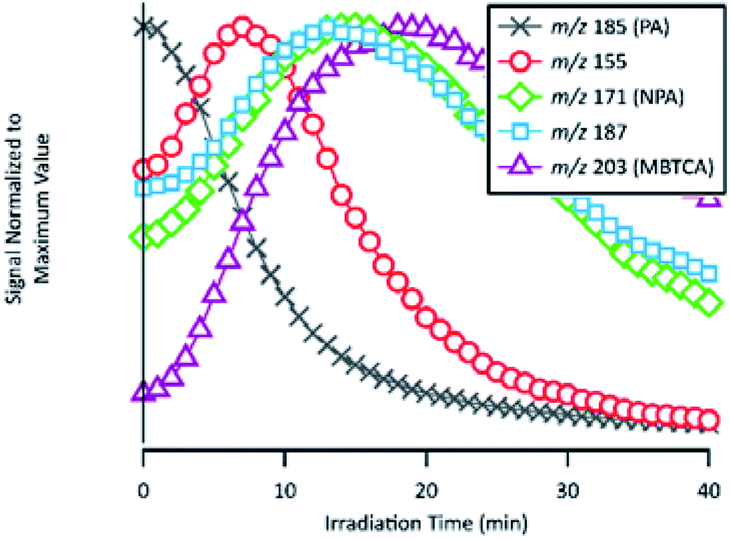 | ||
| Fig. 9 Temporal evolutions of compounds related to the formation of NPA and MBTCA: PA, m/z 155, NPA, m/z 187 and MBTCA. Signals are normalized to their maximum values. | ||
As already discussed in Section 3.4, the major products of m/z of 171 and m/z 203 are NPA and MBTCA, respectively. By observing the formation and decay profile of each compound, we propose a series of mechanisms illustrated in Fig. 10. As the initial step of OH-oxidation, H-abstraction by OH radicals can occur at multiple positions on a PA molecule (1–6). For any of these positions, the resulting peroxy radical (RO2) can react with a hydroperoxy radical (HO2), giving rise to an organic hydroperoxide (ROOH) with m/z 217. Fig. 10 shows the case in which H-abstraction occurs at position C-3. H-abstraction at different positions of PA results in different structural isomers of this compound, likely explaining the presence of multiple peaks on its EIC (Fig. 7(a)). Fig. 10 further illustrates the cases in which the initiation step occurs at the 1 and 4 positions, followed by O2 addition and formation of RO2. Reactions of NO are not expected to be significant in atmospheric aqueous phases due to the low solubility of NO.47 The peroxy radicals formed mainly react with another RO2 in solution or with an HO2, forming the corresponding alkoxy radical (RO).48 The ROs further undergo β-scission to form a variety of possible products, including an aldehyde and a ketone of m/z 155. From Table 2, the accurate mass indicated the elemental composition of C8H12O3, in agreement with those proposed in Fig. 10. We considered that the ion at m/z 155 is an important intermediate product leading to the formation of NPA (m/z 171), and MBTCA (m/z 203).
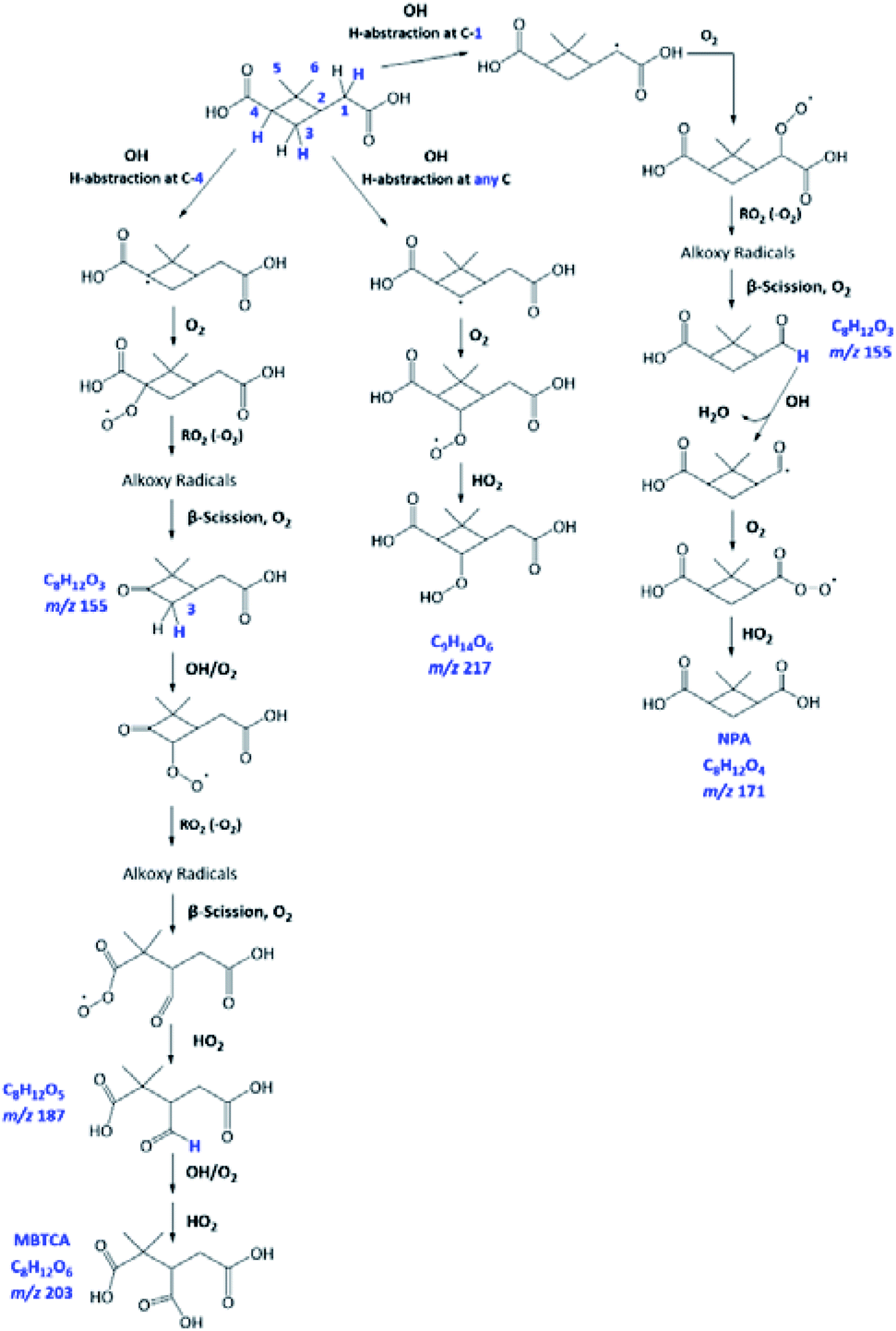 | ||
| Fig. 10 Proposed mechanism of NPA, MBTCA, and one of the m/z 217 isomers formed from PA OH-oxidation at pH 2. Note that multiple isomers of m/z 217 can form, and the structure shown here is an example. | ||
The m/z 155 intermediates can undergo multiple reaction pathways, and we herein focus on those leading to NPA and MBTCA. In an aqueous solution, aldehydes can be hydrated, forming their geminal diols. Thus, for the aldehyde product of m/z 155 (right), the OH radicals can abstract a hydrogen atom from both aldehydic and geminal diol forms.25 The respective alkyl radicals formed are also in hydration equilibrium. This geminal–diol pathway is unique to the atmospheric aqueous phase and it does not occur via gas-phase oxidation. The addition of dissolved O2 to the alkyl radicals formed leads to two types of peroxy radicals. A peroxy radical (not shown here), which dissociates rapidly to form NPA. In parallel, the acylperoxy radical shown in Fig. 10 can either hydrate or react with a HO2 to form NPA. The observation that NPA is a major product of PA explains a previous finding by Amorim et al.24 The OA with m/z 171 exhibited a completely different decay profile during OH-oxidation of α-pinene SOA, indicating that it was forming from other species. The results obtained here can, therefore, confirm that the OA at m/z 171 was likely NPA and was formed from PA present in the SOA extract.
In the case of the ketone product of m/z 155 (left), the OH radical can abstract an H-atom from different sites of the molecule. The mechanism shown in Fig. 10 focuses on the H-abstraction from C-3, followed by the addition of dissolved O2 to form the corresponding RO2. The reaction with another RO2 produces an alkoxy radical that proceeds via β-scission to break the four-membered cyclic structure. The resulting peroxy radical can react with HO2 to form the corresponding carboxylic acid with m/z 187. The elemental composition and fragmentation pattern of the m/z 187 product was confirmed by the results presented in Sections 3.3 and 3.4. As shown in Fig. 9, the m/z 187 compound appears earlier than MBTCA, which is consistent with it being an intermediate for MBTCA formation. Additionally, the mechanism shown in Fig. 10 follows Enami and Sakamato's work on MBTCA formation from CPA oxidation,34 in which the authors also identified m/z 187 as a byproduct of MBTCA. As the last step, the aldehyde functional group of the m/z 187 can be converted into a carboxyl group following the same mechanism discussed in the previous paragraph. This reaction pathway agrees with the mechanism proposed by Witkowski et al.13 and Enami and Sakamato34 for MBTCA formation from the aqueous phase oxidation of CPA.
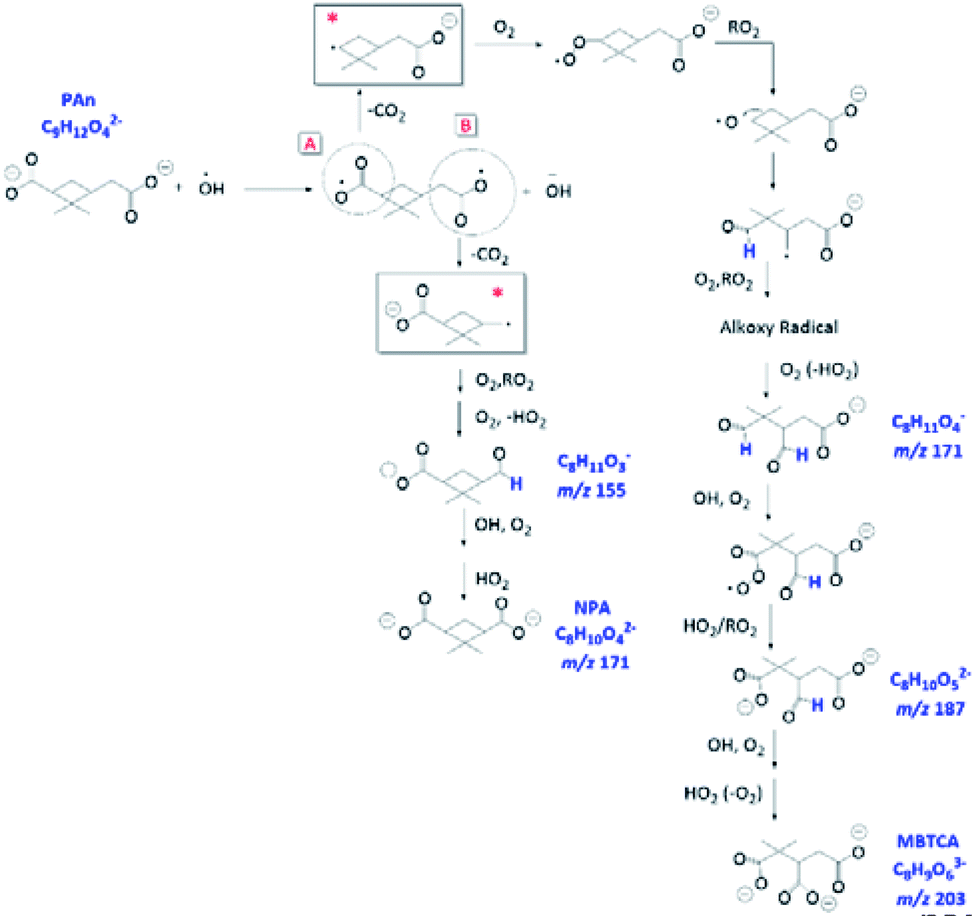 | ||
| Fig. 11 Proposed formation mechanism of NPA and MBTCA under the pH 8 condition via charge transfer reaction occurring to the carboxylate groups. The reaction is initiated by an electron-transfer reaction on the carboxylate A. | ||
Overall, we propose that the charge transfer reaction, followed by initial decarboxylation of PA and formation of the corresponding alkyl radicals (denoted with * in Fig. 11) are the key steps that serve as additional formation routes for NPA and MBTCA. The mechanisms proposed in Fig. 11 are unique to the aqueous-phase OH-oxidation of PA. In comparison, the molecular structure of CPA differs from PA at site A, in which CPA poses a ketone functional group, which does not allow the charge transfer reaction. The aqueous-phase formation of MBTCA from PA is, however, possible due to the additional carboxyl group under basic pH conditions. These results agree with the higher yields of MBTCA under pH 8 discussed in Section 3.5. Therefore, we have arrived at the following conclusion. Although reactions occurring at pH 8 lead to similar compounds, as in the case of pH 2, the charge transfer reaction serves as additional formation pathways and thus accelerates the formation of several key oxidation products, including NPA and MBTCA.
4. Conclusions
We have, for the first time, investigated the mechanism of aqueous-phase PA OH-oxidation in detail. Using both offline high mass resolution (−)ESI-LC-MS and a novel online monitoring system (PILS-ESI-MS), we detected a large number of oxidation products. Among them, NPA (m/z 171) and MBTCA (m/z 203) were well-established α-pinene oxidation products, and yet have not been identified as products of PA oxidation. Their identities were confirmed by observing their fragmentation patterns using MS/MS. Organic hydroperoxides with m/z 217 were also detected as major and first-generation products. An iodometry-assisted method was used to confirm their peroxide nature.Combining observations obtained using (−)ESI-LC-MS and PILS-ESI-MS, we proposed the possible reaction mechanisms for the targeted compounds. The compounds of m/z 155 and m/z 187 serve as intermediates for the products assigned to NPA (m/z 171), and MBTCA (m/z 203). The detection of MBTCA from PA is new, as the current understanding is that MBTCA arises only from CPA.42,43 We estimate that the molar yield of MBTCA from PA is at least an order of magnitude larger than that from CPA. We also observed a difference in the yield of NPA and MBTCA under pH 2 and pH 8, with the yield under basic conditions doubling that under acidic conditions. We propose that it is the unique charge transfer reaction occurring only in the aqueous phase that leads to the enhancement of MBTCA and NPA formation. Such an impact of pH on the photochemical products of OAs has never been reported in the literature.
More generally, our results present significant implications to atmospheric chemistry. Measured pH of cloud and fog water fluctuates significantly between 2 and 7, depending on the location and chemical composition of these hydrometeors.39 Over the past 40 years, the average pH of fog and cloudwater in Europe and the US has increased by two units.38 This trend arises from reduced emissions of acid precursors to the atmosphere. The pH dependence of cloudwater chemistry must be understood to predict the impact of a constantly changing acidity in atmospheric aqueous phases. From Amorim et al.24 and this work, a complex interplay between acid–base chemistry and photochemistry has become clear. The charge transfer reaction does not significantly change the overall OH reactivity of PA.24 However, the underlying reaction mechanisms can be altered, changing the yield of reaction products. In the case of PA oxidation, MBTCA is known to contribute significantly to SOA mass due to its low volatility.42,43 The high yield of MBTCA from PA, particularly under basic conditions, indicates that aqSOA can increase when the acidity in atmospheric aqueous phases is constantly reduced. Although our study has focused on a single compound, PA, we believe that it serves as a vivid demonstration of how pH can affect the reaction mechanisms of many other atmospherically relevant OAs under cloudwater-relevant conditions. Our results suggest the importance of further understanding the impact of pH on aqueous-phase photochemistry, particularly acids and bases that are sensitive to the highly variable pH of atmospheric aqueous phases.
Conflicts of interest
There is no conflict to declare.Acknowledgements
The authors thank the U of Alberta MS and NMR Facility for technical support and also thank NSERC, CFI, Undergraduate Research Initiative, and the U of Alberta for financial support.References
- B. Ervens, B. J. Turpin and R. J. Weber, Secondary Organic Aerosol Formation in Cloud Droplets and Aqueous Particles (AqSOA): A Review of Laboratory, Field and Model Studies, Atmos. Chem. Phys., 2011, 11(21), 11069–11102 CrossRef CAS.
- J. D. Blando and B. J. Turpin, Secondary Organic Aerosol Formation in Cloud and Fog Droplets: A Literature Evaluation of Plausibility, Atmos. Environ., 2000, 34(10), 1623–1632 CrossRef CAS.
- V. F. McNeill, Aqueous Organic Chemistry in the Atmosphere: Sources and Chemical Processing of Organic Aerosols, Environ. Sci. Technol., 2015, 49(3), 1237–1244 CrossRef CAS PubMed.
- R. Winterhalter, R. V. Dingenen, B. R. Larsen, N. R. Jensen and J. Hjorth, LC-MS Analysis of Aerosol Particles from the Oxidation of α-Pinene by Ozone and OH-Radicals, Atmos. Chem. Phys. Discuss., 2003, 3(1), 1–39 CrossRef.
- Y. Ma, T. Luciani, R. A. Porter, A. T. Russell, D. Johnson and G. Marston, Organic Acid Formation in the Gas-Phase Ozonolysis of α-Pinene, Phys. Chem. Chem. Phys., 2007, 9(37), 5084 RSC.
- D. Aljawhary, R. Zhao, A. K. Y. Lee, C. Wang and J. P. D. Abbatt, Kinetics, Mechanism, and Secondary Organic Aerosol Yield of Aqueous Phase Photo-Oxidation of α-Pinene Oxidation Products, J. Phys. Chem. A, 2016, 120(9), 1395–1407 CrossRef CAS PubMed.
- A. G. Carlton, B. J. Turpin, H.-J. Lim, K. E. Altieri and S. Seitzinger, Link between Isoprene and Secondary Organic Aerosol (SOA): Pyruvic Acid Oxidation Yields Low Volatility Organic Acids in Clouds, Geophys. Res. Lett., 2006, 33(6), L06822 CrossRef.
- T. Hoffmann, R. Bandur, U. Marggraf and M. Linscheid, Molecular Composition of Organic Aerosols Formed in the α-Pinene/O3 Reaction: Implications for New Particle Formation Processes, J. Geophys. Res.: Atmos., 1998, 103(D19), 25569–25578 CrossRef CAS.
- F. Holloway, H. J. Anderson and W. Rodin, Ozonolysis of Alpha-Pinene, Ind. Eng. Chem., 1955, 47(10), 2111–2113 CrossRef CAS.
- J. Chen and R. J. Griffin, Modeling Secondary Organic Aerosol Formation from Oxidation of α-Pinene, β-Pinene, and d-Limonene, Atmos. Environ., 2005, 39(40), 7731–7744 CrossRef CAS.
- D. D. Huang, Q. Zhang, H. H. Y. Cheung, L. Yu, S. Zhou, C. Anastasio, J. D. Smith and C. K. Chan, Formation and Evolution of AqSOA from Aqueous-Phase Reactions of Phenolic Carbonyls: Comparison between Ammonium Sulfate and Ammonium Nitrate Solutions, Environ. Sci. Technol., 2018, 52(16), 9215–9224 CrossRef CAS PubMed.
- D. A. Cortés and M. J. Elrod, Kinetics of the Aqueous Phase Reactions of Atmospherically Relevant Monoterpene Epoxides, J. Phys. Chem. A, 2017, 121(48), 9297–9305 CrossRef PubMed.
- B. Witkowski and T. Gierczak, Cis-Pinonic Acid Oxidation by Hydroxyl Radicals in the Aqueous Phase under Acidic and Basic Conditions: Kinetics and Mechanism, Environ. Sci. Technol., 2017, 51(17), 9765–9773 CrossRef CAS PubMed.
- H. Lignell, S. A. Epstein, M. R. Marvin, D. Shemesh, B. Gerber and S. Nizkorodov, Experimental and Theoretical Study of Aqueous Cis-Pinonic Acid Photolysis, J. Phys. Chem. A, 2013, 117(48), 12930–12945 CrossRef CAS.
- B. Witkowski, M. Al-sharafi and T. Gierczak, Kinetics of Limonene Secondary Organic Aerosol Oxidation in the Aqueous Phase, Environ. Sci. Technol., 2018, 52(20), 11583–11590 CAS.
- J. Yu, D. R. Cocker, R. J. Griffin, R. C. Flagan and J. H. Seinfeld, Gas-Phase Ozone Oxidation of Monoterpenes: Gaseous and Particulate Products, J. Atmos. Chem., 1999, 34(2), 207–258 CrossRef CAS.
- Bo. R. Larsen, D. Di Bella, M. Glasius, R. Winterhalter, N. R. Jensen and J. Hjorth, Gas-Phase OH Oxidation of Monoterpenes: Gaseous and Particulate Products, J. Atmos. Chem., 2001, 38(3), 231–276 CrossRef CAS.
- T. S. Christoffersen, J. Hjorth, O. Horie, N. R. Jensen, D. Kotzias, L. L. Molander, P. Neeb, L. Ruppert, R. Winterhalter, A. Virkkula, K. Wirtz and B. R. Larsen, Cis-Pinic Acid, a Possible Precursor for Organic Aerosol Formation from Ozonolysis of α-Pinene, Atmos. Environ., 1998, 32(10), 1657–1661 CrossRef CAS.
- M. Bilde and S. N. Pandis, Evaporation Rates and Vapor Pressures of Individual Aerosol Species Formed in the Atmospheric Oxidation of α- and β-Pinene, Environ. Sci. Technol., 2001, 35(16), 3344–3349 CrossRef CAS PubMed.
- A. Kołodziejczyk, P. Pyrcz, A. Pobudkowska, K. Błaziak and R. Szmigielski, Physicochemical Properties of Pinic, Pinonic, Norpinic, and Norpinonic Acids as Relevant α-Pinene Oxidation Products, J. Phys. Chem. B, 2019, 123(39), 8261–8267 CrossRef PubMed.
- F. Wania, Y. D. Lei, C. Wang, J. P. D. Abbatt and K.-U. Goss, Using the Chemical Equilibrium Partitioning Space to Explore Factors Influencing the Phase Distribution of Compounds Involved in Secondary Organic Aerosol Formation, Atmos. Chem. Phys., 2015, 15(6), 3395–3412 CrossRef CAS.
- M. E. Jenkin, D. E. Shallcross and J. N. Harvey, Development and Application of a Possible Mechanism for the Generation of Cis-Pinic Acid from the Ozonolysis of α- and β-Pinene, Atmos. Environ., 2000, 34(18), 2837–2850 CrossRef CAS.
- M. Glasius, A. Calogirou, N. R. Jensen, J. Hjorth and C. J. Nielsen, Kinetic Study of Gas-Phase Reactions of Pinonaldehyde and Structurally Related Compounds, Int. J. Chem. Kinet., 1997, 29(7), 527–533 CrossRef CAS.
- J. V. Amorim, S. Wu, K. Klimchuk, C. Lau, F. J. Williams, Y. Huang and R. Zhao, pH Dependence of the OH Reactivity of Organic Acids in the Aqueous Phase, Environ. Sci. Technol., 2020, 54(19), 12484–12492 CrossRef CAS PubMed.
- R. Zhao, A. K. Y. Lee, C. Wang, F. Wania, J. P. S. Wong, S. Zhou and J. P. D. Abbatt, The role of water in organic aerosol multiphase chemistry: focus on partitioning and reactivity, Advances in Atmospheric Chemistry, World Scientific, 2016; vol. 1, pp. 95–184 Search PubMed.
- H. Herrmann, T. Schaefer, A. Tilgner, S. A. Styler, C. Weller, M. Teich and T. Otto, Tropospheric Aqueous-Phase Chemistry: Kinetics, Mechanisms, and Its Coupling to a Changing Gas Phase, Chem. Rev., 2015, 115(10), 4259–4334 CrossRef CAS.
- D. van Pinxteren, A. Plewka, D. Hofmann, K. Müller, H. Kramberger, B. Svrcina, K. Bächmann, W. Jaeschke, S. Mertes, J. L. Collett and H. Herrmann, Schmücke Hill Cap Cloud and Valley Stations Aerosol Characterisation during FEBUKO (II): Organic Compounds, Atmos. Environ., 2005, 39(23–24), 4305–4320 CrossRef CAS.
- A. Bianco, M. Passananti, M. Brigante and G. Mailhot, Photochemistry of the Cloud Aqueous Phase: A Review, Molecules, 2020, 25(2), 423 CrossRef CAS PubMed.
- R. Zhao, C. M. Kenseth, Y. Huang, N. F. Dalleska and J. H. Seinfeld, Iodometry-Assisted Liquid Chromatography Electrospray Ionization Mass Spectrometry for Analysis of Organic Peroxides: An Application to Atmospheric Secondary Organic Aerosol, Environ. Sci. Technol., 2018, 52(4), 2108–2117 CrossRef CAS PubMed.
- A. Wonaschuetz, T. Haller, E. Sommer, L. Witek, H. Grothe and R. Hitzenberger, Collection of Soot Particles into Aqueous Suspension Using a Particle-into-Liquid Sampler, Aerosol Sci. Technol., 2019, 53(1), 21–28 CrossRef CAS PubMed.
- C. H. Clark, S. Nakao, A. Asa-Awuku, K. Sato, R. David and I. Cocker, Real-Time Study of Particle-Phase Products from α-Pinene Ozonolysis and Isoprene Photooxidation Using Particle into Liquid Sampling Directly Coupled to a Time-of-Flight Mass Spectrometer (PILS-ToF), Aerosol Sci. Technol., 2013, 47(12), 1374–1382 CrossRef CAS.
- A. Sorooshian, F. J. Brechtel, Y. Ma, R. J. Weber, A. Corless, R. C. Flagan and J. H. Seinfeld, Modeling and Characterization of a Particle-into-Liquid Sampler (PILS), Aerosol Sci. Technol., 2006, 40(6), 396–409 CrossRef CAS.
- R. J. Weber, D. Orsini, Y. Daun, Y.-N. Lee, P. J. Klotz and F. Brechtel, A Particle-into-Liquid Collector for Rapid Measurement of Aerosol Bulk Chemical Composition, Aerosol Sci. Technol., 2001, 35(3), 718–727 CrossRef CAS.
- S. Enami and Y. Sakamoto, OH-Radical Oxidation of Surface-Active Cis-Pinonic Acid at the Air–Water Interface, J. Phys. Chem. A, 2016, 120(20), 3578–3587 CrossRef CAS PubMed.
- J. Waser, Quantitative Chemistry, W.A. Benjamin Inc., New York, 1964, p. 183 Search PubMed.
- M. Blétry, Considerations on the Derivative of the Titration Curve of a Weak Diacid, preprint, 2019, DOI:10.26434/chemrxiv.8256047.
- H. Howell and G. S. Fisher, The Dissociation Constants of Some of the Terpene Acids, J. Am. Chem. Soc., 1958, 80(23), 6316–6319 CrossRef CAS.
- H. O. T. Pye, A. Nenes, B. Alexander, A. P. Ault, M. C. Barth, S. L. Clegg, J. L. Collett Jr, K. M. Fahey, C. J. Hennigan, H. Herrmann, M. Kanakidou, J. T. Kelly, I.-T. Ku, V. F. McNeill, N. Riemer, T. Schaefer, G. Shi, A. Tilgner, J. T. Walker, T. Wang, R. Weber, J. Xing, R. A. Zaveri and A. Zuend, The Acidity of Atmospheric Particles and Clouds, Atmos. Chem. Phys., 2020, 20(8), 4809–4888 CrossRef CAS PubMed.
- J. L. Collett, A. Bator, X. Rao and B. B. Demoz, Acidity Variations across the Cloud Drop Size Spectrum and Their Influence on Rates of Atmospheric Sulfate Production, Geophys. Res. Lett., 1994, 21(22), 2393–2396 CrossRef.
- F. Yasmeen, R. Szmigielski, R. Vermeylen, Y. Gómez-González, J. D. Surratt, A. W. H. Chan, J. H. Seinfeld, W. Maenhaut and M. Claeys, Mass Spectrometric Characterization of Isomeric Terpenoic Acids from the Oxidation of α-Pinene, β-Pinene, d-Limonene, and Δ3-Carene in Fine Forest Aerosol, J. Mass Spectrom., 2011, 46(4), 425–442 CrossRef CAS.
- F. Yasmeen, R. Vermeylen, N. Maurin, E. Perraudin, J.-F. Doussin and M. Claeys, Characterisation of Tracers for Aging of α-Pinene Secondary Organic Aerosol Using Liquid Chromatography/Negative Ion Electrospray Ionisation Mass Spectrometry, Environ. Chem., 2012, 9(3), 236 CrossRef CAS.
- R. Szmigielski, J. D. Surratt, Y. Gómez-González, P. V. der Veken, I. Kourtchev, R. Vermeylen, F. Blockhuys, M. Jaoui, T. E. Kleindienst, M. Lewandowski, J. H. Offenberg, E. O. Edney, J. H. Seinfeld, W. Maenhaut and M. Claeys, 3-Methyl-1,2,3-Butanetricarboxylic Acid: An Atmospheric Tracer for Terpene Secondary Organic Aerosol, Geophys. Res. Lett., 2007, 34(24), L24811 CrossRef.
- L. Müller, M.-C. Reinnig, K. H. Naumann, H. Saathoff, T. F. Mentel, N. M. Donahue and T. Hoffmann, Formation of 3-Methyl-1,2,3-Butanetricarboxylic Acid via Gas Phase Oxidation of Pinonic Acid – a Mass Spectrometric Study of SOA Aging, Atmos. Chem. Phys., 2012, 12(3), 1483–1496 CrossRef.
- M.-C. Reinnig, J. Warnke and T. Hoffmann, Identification of Organic Hydroperoxides and Hydroperoxy Acids in Secondary Organic Aerosol Formed during the Ozonolysis of Different Monoterpenes and Sesquiterpenes by On-Line Analysis Using Atmospheric Pressure Chemical Ionization Ion Trap Mass Spectrometry, Rapid Commun. Mass Spectrom., 2009, 23(11), 1735–1741 CrossRef CAS PubMed.
- A. Tilgner, T. Schaefer, B. Alexander, M. Barth, J. L. Collett Jr, K. M. Fahey, A. Nenes, H. O. T. Pye, H. Herrmann and V. F. McNeill, Acidity and the Multiphase Chemistry of Atmospheric Aqueous particles and Clouds, preprint, Aerosols/Laboratory Studies/Troposphere/Chemistry (chemical composition and reactions), 2021.
- R. Zhao, E. L. Mungall, A. K. Lee, D. Aljawhary and J. P. Abbatt, Aqueous-Phase Photooxidation of Levoglucosan–a Mechanistic Study Using Aerosol Time-of-Flight Chemical Ionization Mass Spectrometry (Aerosol ToF-CIMS), Atmos. Chem. Phys., 2014, 14(18), 9695–9706 CrossRef.
- R. Sander, Compilation of Henry's Law Constants (Version 4.0) for Water as Solvent, Atmos. Chem. Phys., 2015, 15(8), 4399–4981 CrossRef CAS.
- J. J. Orlando and G. S. Tyndall, Laboratory Studies of Organic Peroxy Radical Chemistry: An Overview with Emphasis on Recent Issues of Atmospheric Significance, Chem. Soc. Rev., 2012, 41(19), 6294–6317 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ea00031d |
| This journal is © The Royal Society of Chemistry 2021 |