
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the complexity of atmospheric brown carbon produced by smoldering boreal peat using size-exclusion chromatography with selective mobile phases†
Ming
Lyu
 a,
Dan K.
Thompson‡
b,
Nianci
Zhang
a,
Chad W.
Cuss§
c,
Cora J.
Young
d and
Sarah A.
Styler
*ae
a,
Dan K.
Thompson‡
b,
Nianci
Zhang
a,
Chad W.
Cuss§
c,
Cora J.
Young
d and
Sarah A.
Styler
*ae
aDepartment of Chemistry, University of Alberta, 11227 Saskatchewan Drive, Edmonton, Alberta T6G 2G2, Canada
bNatural Resources Canada – Northern Forestry Centre, Edmonton, Alberta, Canada
cSWAMP Lab, Department of Renewable Resources, University of Alberta, Edmonton, Alberta, Canada
dDepartment of Chemistry, York University, Toronto, Ontario, Canada
eDepartment of Chemistry & Chemical Biology, McMaster University, Hamilton, Ontario, Canada. E-mail: stylers@mcmaster.ca
First published on 18th June 2021
Abstract
Boreal wildfires are a significant source of atmospheric brown carbon (BrC), a complex mixture of thousands of light-absorbing organic compounds that contributes to the warming effects of combustion particulate matter. Here, we use size-exclusion chromatography (SEC) coupled with photodiode array detection to characterize BrC collected from the controlled combustion of boreal peat. Importantly, rather than attempting to estimate the molecular weight of BrC chromophores through the minimization and correction of secondary interactions, we instead exploit these interactions to systematically explore BrC hydrophobicity, lability, and size-dependent light absorption properties. Using this new approach, which we corroborate using independent asymmetric flow field-flow fractionation (AF4) analysis, we show that the components of fresh wildfire BrC span a wide range of sizes, polarities, and light absorption characteristics. Unlike atmospherically aged wildfire BrC, which has previously been shown to resemble terrestrial humic substances in both its absorption profile and its retention behaviour, the fresh BrC sample studied here contains both higher-MW chromophores with “humic-like” featureless absorption and smaller-MW chromophores with structured absorption, and is more susceptible to hydrophobic interactions with the column matrix. Interestingly, we find that the contribution of the low-MW fraction to overall BrC absorption increases with increasing mobile phase acetonitrile content, which suggests that the high-MW fraction consists of metastable aggregates held together by easily disrupted intermolecular forces. Together, these results highlight the compositional diversity of atmospheric BrC and the challenge and potential of SEC for the characterization of complex, and poorly defined, environmental matrices.
Environmental significanceAtmospheric brown carbon (BrC) is the dominant light-absorbing component of wildfire particulate matter. Our ability to quantify the climate impact of BrC emissions is currently limited by gaps in our understanding of its composition, properties, and atmospheric fate. Here, we use size-exclusion chromatography to examine the size, polarity, and lability of fresh BrC produced by the combustion of boreal peat. Through the careful examination of solvent-dependent elution behaviour, we show that this BrC sample is more diverse in both size and polarity than atmospherically aged BrC examined in previous studies, and that its large-MW components consist largely of smaller units aggregated together by weak intermolecular interactions. These results demonstrate the importance of studying the composition and light-absorbing properties of BrC over its entire atmospheric lifetime, as well as the need for careful consideration of extraction and analysis conditions when studying this complex class of atmospheric particulate matter. |
1 Introduction
Combustion processes release large amounts of carbonaceous particulate matter (PM) into the atmosphere,1,2 which both absorbs and scatters solar radiation and influences global climate in complex ways.3,4 Light-absorbing carbonaceous PM can be divided into two categories: soot, also known as black carbon (BC), which absorbs light efficiently from the UV region to the infrared;5 and a complex mixture of organic compounds known as brown carbon (BrC), which exhibits a sharp decrease in absorption with increasing wavelength from the UV region to the visible.6,7 Although measurements and models have suggested that the warming effect of BrC is substantial,8,9 inclusion of BrC light absorption in climate simulations is challenging,10 primarily because of the complexity of BrC formation11,12 and light absorption mechanisms,13,14 chemical composition,15 and atmospheric fate.16,17One major BrC source is wildfires, which directly release BrC as a result of incomplete combustion and indirectly lead to BrC formation via atmospheric photolysis, oxidation, and gas-to-particle partitioning of co-emitted volatile organic compounds (VOCs).18 An understanding of the molecular characteristics of BrC chromophores in wildfire PM, as well as their susceptibility to chemical transformations in the atmosphere,19,20 is necessary for the accurate prediction of the overall climate impact of wildfires. Measured increases in the frequency and intensity of boreal wildfire activity,21 coupled with projected increases in extreme fire weather22 and overall wildfire risk,23 lend particular urgency to these investigations.
Molecular-level BrC characterization is usually accomplished via offline analysis of PM extracts using high-performance liquid chromatography (HPLC) coupled with photodiode array (PDA) and high-resolution mass spectrometric (HRMS) detection; however, summing the contribution of chromophores identified by MS (e.g., N-containing aromatic compounds, aromatic carbonyls, and aromatic carboxylic acids) typically cannot account for all BrC absorption.24–29 This discrepancy likely reflects qualitative and quantitative challenges associated with the application of HPLC-PDA-HRMS to the analysis of this highly complex yet poorly characterized substrate, including the need to account for (largely unknown) variations in component ionization efficiency,25,30 distinguish which of a set of co-eluting compounds is responsible for HPLC-PDA absorption signals in complex chromatograms,24 and apply multiple ionization methods to achieve more comprehensive analyte coverage.25 In addition, the analysis of the high-molecular-weight (MW) component of BrC (often referred to as “humic-like substances”, or HULIS), which consists of macromolecular aggregates composed of highly conjugated aromatic structures with polar functional groups containing oxygen and nitrogen (e.g., carboxyl and nitro groups),31 may be complicated by fragmentation in the MS32 and by multiple charging effects.33
A promising technique for the analysis of the high-MW component of BrC is size-exclusion chromatography (SEC),34 which has been widely employed to estimate the MW and MW distributions of dissolved organic matter (DOM),35 humic substances,36 and the HULIS fraction of ambient atmospheric PM.37,38 In recent years, this technique has also been coupled with PDA detection to estimate the MW and characterize the light absorbing properties of BrC chromophores in ambient39–41 and laboratory-generated42,43 PM samples. One major challenge associated with SEC separation is the hydrophobic and/or electrostatic interaction of analytes with the column matrix, which can lead to changes in elution profiles and thus introduce large uncertainties to MW estimates.34 Additional uncertainties arise from intra- and intermolecular interactions, which can influence analyte hydrodynamic volume (e.g., via coiling, expansion, and aggregation/agglomeration), thereby leading to either early or delayed elution and corresponding overestimates or underestimates in MW.36,44 Although the challenges associated with these secondary effects have been acknowledged in the broader atmospheric literature,45,46 studies of BrC using this technique have largely assumed that size exclusion is the dominant separation mechanism. However, as BrC consists of a complex mixture of compounds with a wide range of polarities and sizes,7 and with the potential to undergo aggregation and/or dissociation with changes in solvent environment,47,48 this assumption warrants critical assessment.
In this study, we used HPLC-SEC-PDA to characterize BrC produced by combustion of boreal peat, an important yet poorly understood source of biomass burning PM.49,50 Importantly, rather than attempting to obtain accurate absolute MW estimates for BrC samples through the use of mobile phase solvents chosen to minimize secondary effects, we instead exploited these effects to systematically investigate the hydrophobicity and lability of the samples. In particular, we examined their elution behaviour as a function of mobile phase composition, including organic solvent content and ionic strength, and compared this behaviour to that of Suwannee River Humic Acid (SRHA), which has been previously used as a proxy for biomass burning BrC.39 In addition, we compared these results to those obtained using a complementary technique, asymmetric flow field-flow fractionation (AF4).51–53 Our results highlight the complex nature of secondary interactions in the analysis of fresh wildfire BrC using SEC and, at the same time, show that a sound understanding of these interactions can be used to provide new insights into the properties of this important PM class, including distributions of molecular size and polarity. Using this new approach, this study proceeds to reveal the aggregation and dissociation of water-soluble BrC components for the first time.
2 Methods
2.1 Boreal peat sampling
Boreal peat was sampled in the traditional territory of the Bigstone Cree Nation (Calling Lake, northern Alberta, Canada; 55.092, −113.272) in June 2018. To capture the typical depth of burn (∼10 cm) during wildfires,54 the bulk peat was sampled from the ground surface to ∼40 cm. A BrC sample generated from the combustion of the peat surface layer (0–5 cm) was used in this study; the remaining samples were used for a comprehensive study of the influence of sample depth and moisture content on the light absorption properties of BrC generated from peat combustion.552.2 Peat combustion and BrC collection
Biomass burning PM was generated from the peat sample described above at the laboratory combustion facility in the Northern Forestry Centre (Edmonton, Alberta). The peat sample was ignited by placing a quartz tube infrared electric heater (210 V, 1000 W; Re-Verber-Ray BAH-25) operating at ∼800 °C over it for 1 min. The combustion phase (i.e., flaming versus smoldering) was estimated using the modified combustion efficiency (MCE = ΔCO2/(ΔCO2 + ΔCO)),56 which was calculated using concentrations of CO (ULTRAMAT 23, Siemens) and CO2 (ULTRAMAT 6, Siemens) measured using an inlet at the intake of the overhead exhaust, ∼3 m above the sample; here, this value was 0.84, which is consistent with smoldering combustion.57Water-soluble BrC was collected ∼1 m above the combustion site using a particle-into-liquid sampler (PILS; Model 4001, Brechtel) system equipped with an inlet for the selective collection of particles with diameters smaller than 2.5 μm (PM2.5), an activated carbon VOC denuder, and an auto-collection system (3.5 min per sample).58 The aerosol sampling volumetric flow rate was 13.5–13.7 L min−1; the quartz impactor plate wash flow rate was 0.42–0.46 mL min−1. PILS sample vials were stored at 277 K until analysis, at which time samples were diluted (×3) with deionized water (18 MΩ) and filtered (PTFE, 0.2 μm, 13 mm, Fisherbrand Basix).
2.3 HPLC-SEC-PDA analysis
The system adopted in this paper is based on the previous work of Di Lorenzo et al.39–41 A HPLC (Agilent 1100) system equipped with an aqueous gel filtration column (300 × 7.8 mm, 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–250 Da, Polysep GFC P-3000, Phenomenex) and coupled to a PDA (Agilent G1315B) detector was employed for the separation and detection of samples. The mobile phase consisted of phosphate buffer (NaH2PO4 + Na2HPO4, pH = 6.8, 20–100 mM) mixed with varying concentrations of acetonitrile (ACN) and/or methanol (MeOH) as organic modifier; all mixing ratios of buffer and organic phase are reported here as volume percentages, and all separations were performed under isocratic conditions. The flow rates and column temperatures employed were either 1 mL min−1 and 40 °C (50% and 25% ACN) or 0.8 mL min−1 and 55 °C (50% MeOH, 40% MeOH/10% ACN), with conditions selected to avoid overpressure of the column. Sample-to-sample reproducibility was assessed using the retention times of acetone and SRHA (Table S2†); on a given day, the variation in retention time for each marker was <0.015 min. The sample injection volume was 15 μL.
000–250 Da, Polysep GFC P-3000, Phenomenex) and coupled to a PDA (Agilent G1315B) detector was employed for the separation and detection of samples. The mobile phase consisted of phosphate buffer (NaH2PO4 + Na2HPO4, pH = 6.8, 20–100 mM) mixed with varying concentrations of acetonitrile (ACN) and/or methanol (MeOH) as organic modifier; all mixing ratios of buffer and organic phase are reported here as volume percentages, and all separations were performed under isocratic conditions. The flow rates and column temperatures employed were either 1 mL min−1 and 40 °C (50% and 25% ACN) or 0.8 mL min−1 and 55 °C (50% MeOH, 40% MeOH/10% ACN), with conditions selected to avoid overpressure of the column. Sample-to-sample reproducibility was assessed using the retention times of acetone and SRHA (Table S2†); on a given day, the variation in retention time for each marker was <0.015 min. The sample injection volume was 15 μL.
2.4 Asymmetric flow field-flow fractionation (AF4) analysis
To measure the size-resolved optical properties of BrC in the absence of a stationary phase and thereby verify that our results were not specific to SEC analysis, BrC and Suwannee River natural organic matter (SRNOM; International Humic Substances Society) were also analyzed using AF4 (AF2000 Multiflow, Postnova Analytics) with UV-Vis absorbance and fluorescence detection (G4212 DAD and G1321B FLD, Agilent). Fluorescence emission (280–450 nm, excitation at 230 nm) was collected with <1 s time resolution over the course of the separation. The relationship between retention time and molecular mass was calibrated before and after the sample separations using a mixture of poly(styrene sulfonate) sodium salts (Polymer Standards) and a molecular standard (bromophenol blue; Sigma Aldrich) that together spanned the range of 0.67–20.7 kDa. This relationship was used to determine the molecular mass at peak maximum (Mp) of the BrC and SRNOM, measured at an absorbance wavelength of 254 nm. The AF4 flow program was optimized to maximize resolution in the lower size range, and a polyethersulfone (PES) membrane with the smallest available pore size (300 Da) was used to maximize retention of the smallest molecules. The carrier fluid consisted of an ammonium carbonate buffer (2 mM, pH 8). Additional details regarding the analysis method are published elsewhere.532.5 Materials and chemicals
Suwannee River humic acid (SRHA; analyzed as a saturated aqueous solution) was obtained from the International Humic Substances Society. MeOH (Optima grade, 99.9%), ACN (Optima grade, 99.9%), acetone (ACS grade), and monosodium phosphate (ACS grade, 100.2%) were purchased from Fisher Chemical. Dibasic sodium phosphate (ACS grade, 99.0%) was purchased from Sigma Aldrich. Sodium poly(styrene sulfonate) standards for SEC analysis (PSS; Mw: 1690 Da, 5580 Da, 7540 Da, 16 kDa, 33.4 kDa, 68.3 kDa, 78.4 kDa) were purchased from Scientific Polymer Products, Inc.; aqueous standard solutions (∼50 mg L−1) were stored at 277 K until use. Deionized water (18.2 MΩ) was obtained from a Millipore Synergy UV ultrapure water system.2.6 Safety statement
The primary hazard associated with this work was the peat combustion process itself; however, this was carried out in a dedicated combustion facility, which minimized risk.3 Results and discussion
3.1 Challenges associated with application of SEC to absolute MW determinations
Given the chemical complexity of BrC, we first characterized our size-exclusion column using SRHA as a BrC proxy;39 PSS (Mw: 1690 Da to 78.4 kDa), a linear anionic polyelectrolyte that has a similar charge density to NOM,59 as calibrant; and acetone as a marker for the total permeation volume (Vt). Retention times for SRHA, PSS standards, and acetone are presented in Table S1.†As discussed in the Introduction, SEC MW estimates are subject to biases from hydrophobic and/or electrostatic interactions of analytes with the column matrix; these biases can be mitigated using organic and/or ionic mobile phase modifiers,34 which suppress analyte–column interactions. In order to assess the importance of secondary interactions in this study, therefore, we prepared PSS calibration curves at a range of phosphate buffer (20–100 mM, pH 6.8) and organic modifier (0–50% ACN, 0–10% MeOH) concentrations. As shown in Fig. S1 and Table S1,† the elution behaviors of the PSS standards and SRHA responded differently to modifications in mobile phase composition; as a result, the estimated MW values of SRHA over our range of mobile phase compositions ranged from ∼1000 to >10000 Da, despite being obtained using mobile phase-specific PSS calibration curves.
In summary, because SEC separation is based on analyte hydrodynamic volume rather than MW and ideal analyte behaviour is often compromised by non-size effects, accurate estimation of MW using SEC requires the selection of calibrants that are chemically and structurally similar to the analytes of interest.34,38 Owing to their different molecular properties, PSS and SRHA are subject to different non-size effects and undergo different conformational changes in response to mobile phase compositional changes; we discuss this in detail in the ESI.† As a result, although analysis conditions can be optimized for calibrant and analyte separately, conditions that are near-ideal for both are challenging to find. This challenge is magnified for BrC, which is a complex mixture of hundreds to thousands of organic compounds7 that differ in their three-dimensional structure, charge density, and hydrophobicity. Given these limitations, we do not use our SEC results to provide BrC MW estimates; instead, as described in the following sections, we use the identity and magnitudes of non-size effects observed under different mobile phase conditions to gain insight into BrC composition and properties.
3.2 Insights into BrC composition and properties from changes in mobile phase composition
As shown in Fig. 1b, the BrC chromatogram exhibited a “smeared” light absorption at 25% ACN, which persisted >15 min after the total permeation volume marker. Similar column retention was observed by Wong et al.,42 who subjected fresh BrC from laboratory pyrolysis of wood to SEC analysis using a 25 mM ammonium acetate buffer with 10% MeOH as mobile phase organic modifier. In our study, this smearing was significantly reduced at 50% ACN (Fig. 1d), which suggests that it arose from hydrophobic adsorption of BrC components at the surface of the column matrix. As illustrated in Fig. 1a and c, SRHA did not exhibit this smeared absorption, and was not influenced by these changes in mobile phase composition, which clearly highlights its limitations as a proxy for fresh biomass burning BrC.
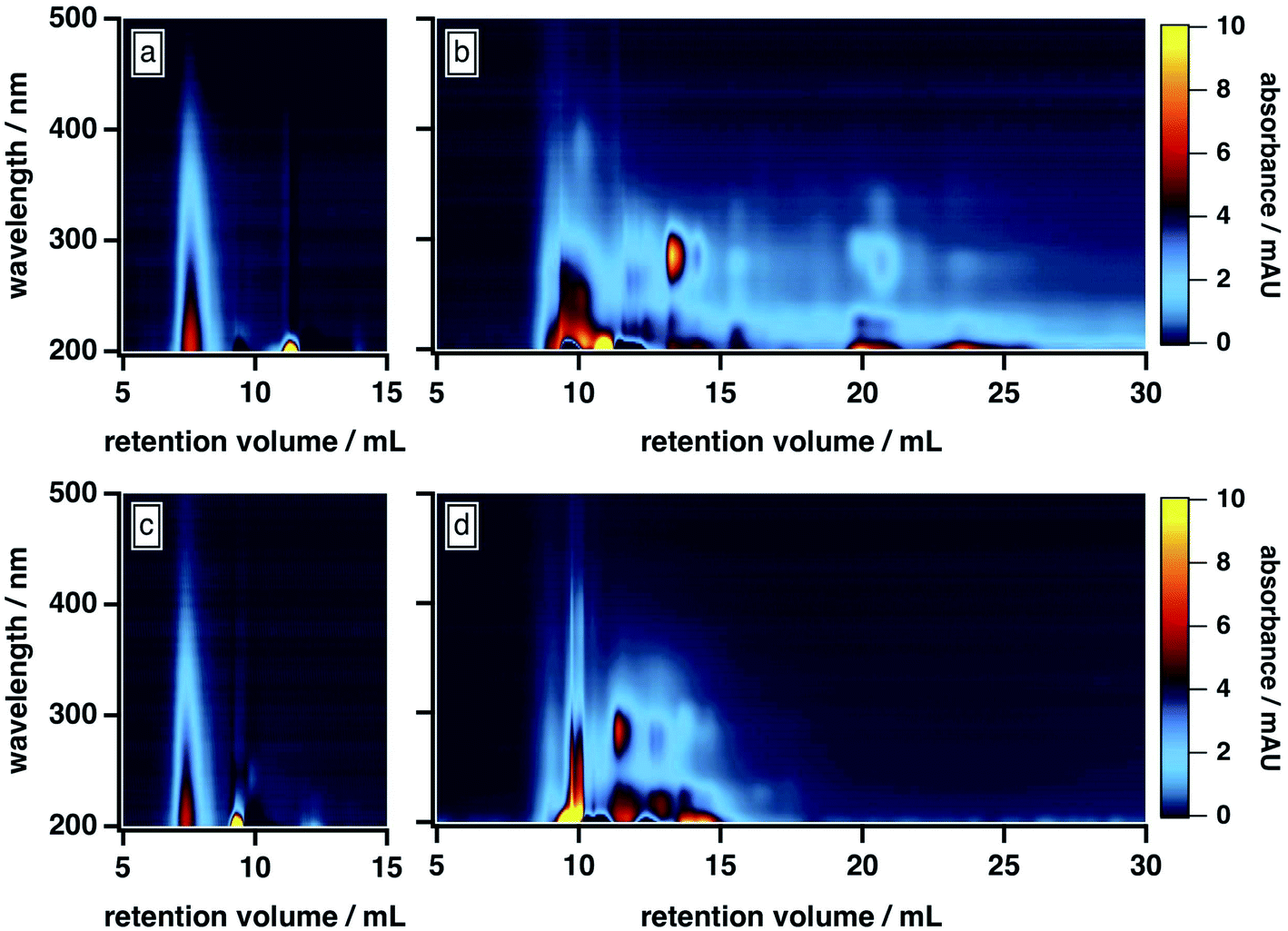 | ||
| Fig. 1 Absorption density plots of SRHA and fresh biomass burning BrC (sampled via PILS, as described in the main text) as a function of mobile phase ACN content: (a) SRHA, 25% ACN; (b) BrC, 25% ACN; (c) SRHA, 50% ACN; (d) BrC, 50% ACN. In all cases, the remainder of the mobile phase was 20 mM phosphate buffer (pH 6.8). | ||
Further comparison of the elution behaviour of our BrC sample to that of SRHA reveals that these samples also differ in their molecular size, compositional complexity, and light absorption profiles. At 50% ACN, for example, SRHA eluted as a single component, with featureless light absorption that decreased sharply with increasing wavelength (Fig. 1c). By contrast, under these conditions, our BrC sample eluted as two distinct fractions, both of which eluted later than SRHA: a high-MW fraction, the absorption profile of which resembled SRHA, and a low-MW fraction, which exhibited a structured absorption profile in the UV region (Fig. 1d). Since, as discussed above, this mobile phase composition effectively mitigated hydrophobic interactions between BrC and the column matrix, these observations suggest that the MW of our BrC sample is smaller than that of SRHA, and again underscores that SRHA is an unsuitable calibrant for fresh BrC.
To measure the size-resolved optical properties of BrC in the absence of a stationary phase and thereby verify that our conclusions were not specific to SEC analysis, we also analyzed BrC and Suwannee River natural organic matter (SRNOM) standard using a complementary technique, AF4. As shown in Fig. 2, although performed using a different mobile phase and with SRNOM rather than SRHA as comparative standard, AF4 shows similar results to those obtained using SEC. In particular, the measured molecular size of BrC (0.81 ± 0.02 kDa) determined via AF4 analysis was significantly smaller than that of SRNOM (1.20 ± 0.03 kDa). In addition, whereas the SRNOM eluted as a single peak with a relatively broad and featureless fluorescence emission maximum near 440 nm, the BrC sample again eluted as two distinct fractions. The first fraction, which eluted over a narrow size range, had a comparably lower MW and a narrow fluorescence maximum near 305 nm. In terrestrial/marine environments, this emission profile is typically attributed to tyrosine-like62 and/or polyphenolic63 fluorophores, but this may not be the case for atmospheric samples.64 The second fraction, which eluted over a broad size range, had a comparably larger MW and a broad fluorescence emission profile centred at 390 nm, the latter of which is consistent with emission by HULIS in ambient PM samples collected in Malaysia during periods of intense haze resulting from peatland fire emissions.65 Although further investigation of BrC using AF4 is certainly warranted, these results broadly verify the intrinsic complexity of BrC, as revealed by our SEC results.
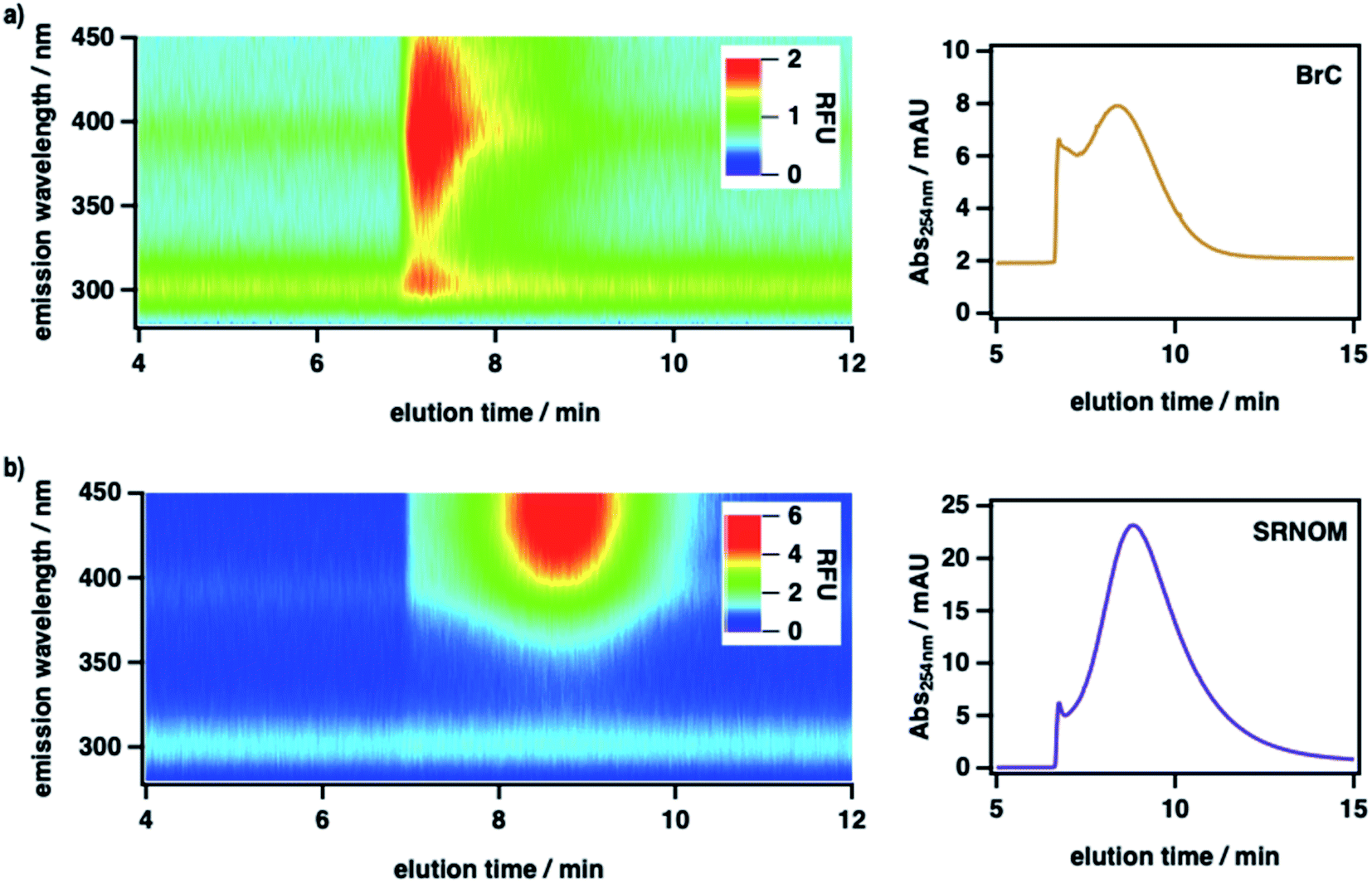 | ||
| Fig. 2 Asymmetric flow field-flow fractionation (AF4; UV-Vis and fluorescence detection) characterization of (a) fresh peat BrC and (b) Suwannee River natural organic matter (SRNOM) standard. For each sample, the leftmost plot shows the fluorescence emission spectrum (excitation at 230 nm) as a function of AF4 elution time, and the rightmost plot shows the corresponding single-wavelength absorption chromatogram (254 nm). | ||
All of these results differ from those of Di Lorenzo and co-workers,39,40 who used HPLC-SEC-PDA to study light absorption by ambient BrC sampled from atmospherically aged biomass burning plumes (∼10 h to >3 days). Specifically, their BrC samples displayed only minor evidence of hydrophobic interactions with the column matrix, even in the absence of organic mobile phase modifiers. In addition, the light absorption of their samples was similar to SRHA, in that humic-like absorption was observed over the entire chromatogram, without the structured absorption we observed for our fresh BrC; finally, their samples eluted at a similar time as SRHA.
These differences could reflect fundamental differences in sample types: whereas our BrC sample is solely from peat combustion, the BrC samples studied by Di Lorenzo et al. were collected from a variety of locations, and would therefore be expected to include contributions from multiple fuel types—and, in some cases, urban and industrial emissions.40 These differences could also reflect changes in BrC properties during atmospheric transport: for example, previous aircraft66 and laboratory67 studies of water uptake by biomass burning PM have shown an increase in particle hygroscopicity with increasing particle oxidation. Additional evidence for the role of atmospheric aging is provided in the Di Lorenzo et al. study itself, which showed that the contribution of high-MW chromophores to overall BrC absorption increased with plume age; therein, the authors attributed this change to the in-plume (photo)oxidation and/or oligomerization of low-MW species.40 Since our BrC sample was collected immediately after its release from the combustion source, we would expect it to be less oxidized, more hydrophobic, and less depleted in low-MW chromophores than even the “freshest” BrC sample in the aforementioned work, all of which is consistent with our observations. To fully understand these differences, however, investigation of the aging behaviour of our specific BrC sample would be required.
To further explore the compositional diversity of our BrC sample, we examined its elution behaviour as a function of mobile phase solvent strength. In these experiments, we employed a 50:50 mixture of 20 mM phosphate buffer and an organic modifier blend consisting of varying ratios of MeOH and ACN; the resulting BrC elution profiles are shown in Fig. 3a. To quantify the relative contribution of the high-MW and low-MW fractions described above to the total BrC absorption for each mobile phase composition, we integrated the absorbance values obtained for each fraction (see the ESI† for details); these results are shown in Fig. 3b.
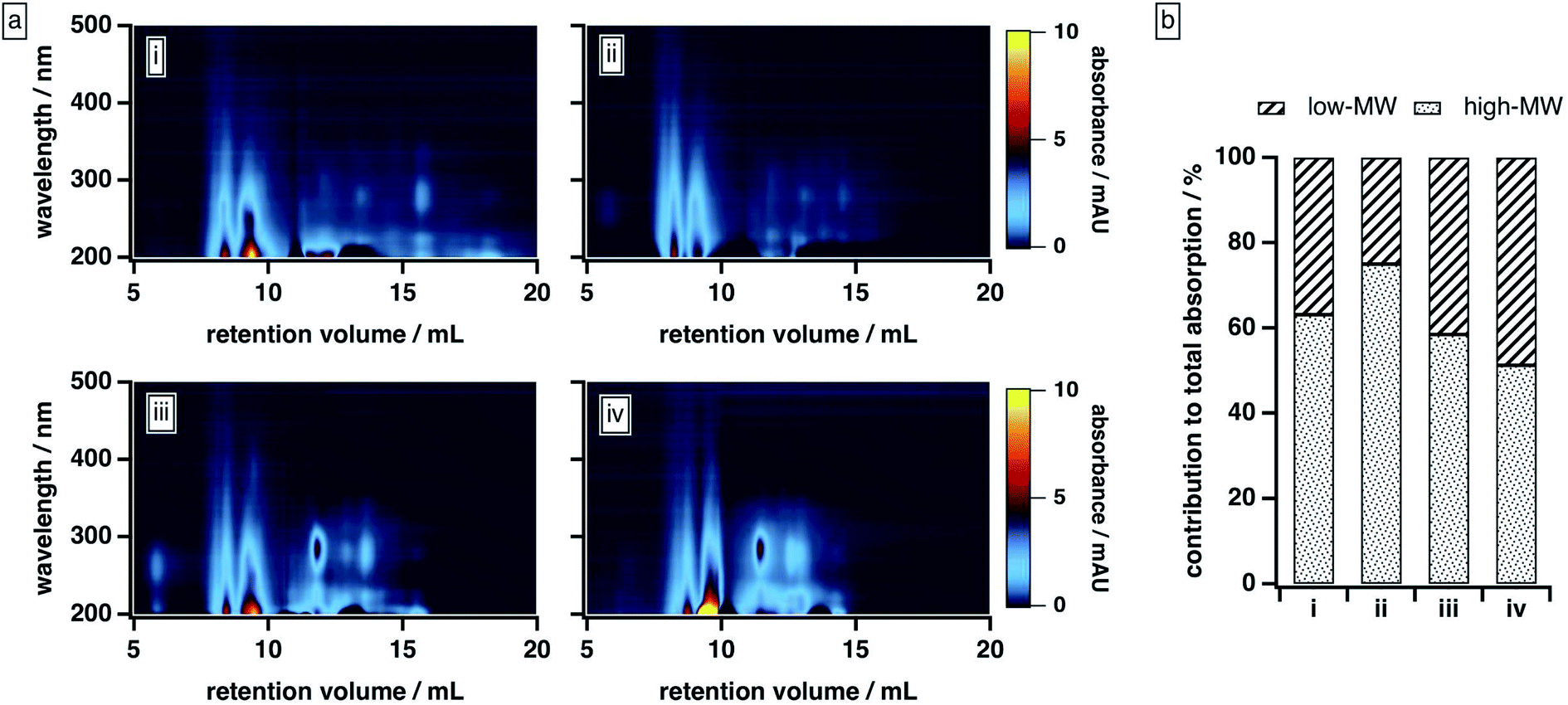 | ||
| Fig. 3 BrC elution behaviour as a function of mobile phase solvent strength: (a) absorption density plots and (b) absorption contributed by high-MW and low-MW BrC fractions, with the latter calculated as described in the ESI.† Experiments were conducted under the following mobile phase organic modifier conditions: (i) 50% MeOH/0% ACN, (ii) 40% MeOH/10% ACN, (iii) 25% MeOH/25% ACN, (iv) 0% MeOH/50% ACN. In all cases, the remainder of the mobile phase was 20 mM phosphate buffer (pH 6.8). | ||
As the composition of the organic blend shifted from MeOH to ACN, the elution profile of BrC changed substantially. At 40% MeOH/10% ACN, absorption by the low-MW fraction was barely visible, and ∼75% of the total absorption was contributed by the high-MW fraction. As the ACN content of the organic blend increased, however, the absorption of the low-MW fraction became more apparent, especially around 250–300 nm, and its contribution to the integrated absorbance increased significantly: at 0% MeOH/50% ACN, for example, the low-MW fraction contributed nearly half of the total absorption. We note that the results obtained at 50% MeOH/0% ACN diverged from this trend: under these conditions, the BrC sample exhibited the same delayed elution and “smeared” absorption profile observed at 25% ACN (see Section 3.2.1); as a result, the absorption contributed by the low-MW fraction was biased high. These results, which are unsurprising given the relative elution strengths of these solvents in reversed-phase partition chromatography (i.e., ACN > MeOH), suggest that MeOH is less effective than ACN at disrupting hydrophobic interactions between the sample and the column matrix.
In principle, the increased contribution of the low-MW fraction to the total BrC absorption at higher ACN contents could reflect a change in column–analyte interactions. However, if these interactions were dominant here, we would have expected to see an increase in the high-MW fraction at elevated ACN/MeOH ratios as a result of suppression of hydrophobic adsorption, which is the opposite of what we observed. Instead, we attribute our observations to the disruption of weak intermolecular forces present in the large-MW BrC fraction, presumably resulting from changes in the relative strengths of solute–solute and solute–solvent interactions in mobile phases with different amounts of ACN and MeOH. Specifically, in the case of MeOH, which is both a hydrogen bond donor and acceptor, we suggest that the attractive forces between the BrC and the solvent are not strong enough to overcome the solvent–solvent hydrogen bonding network and the intermolecular forces between the BrC components. By contrast, because ACN is only a hydrogen bond acceptor, it weakens the solvent–solvent hydrogen bonding network; in addition, it can interact with π-electrons, thereby disrupting π–π interactions between BrC components.68
Our observations are consistent with two possible roles for ACN: first, promoting the liberation of adsorbed smaller molecules from large molecules and/or stable aggregates; second, mediating the partial dissociation of labile aggregates. To investigate the importance of the second pathway, we examined our sample's elution behavior at even higher ACN contents (>50%), which we anticipated would further disrupt the intermolecular forces between BrC components. As shown in Fig. 4, at higher ACN contents, the BrC elution profile became narrower: specifically, as the ACN content increased and the ionic strength of the mobile phase correspondingly decreased, the high-MW fraction tended to elute later and the low-MW fraction tended to elute earlier. We attribute the early elution of the low-MW fraction at elevated ACN contents to further suppression of hydrophobic interactions between the sample and the column matrix. If the elution behaviour of the high-MW fraction were dominated by hydrophobic interactions (or, alternatively, by either of ionic exclusion or intramolecular electrostatic repulsions), the high-MW fraction should also elute earlier at higher ACN contents; however, we observed the opposite trend. At 70% ACN, all BrC components eluted together with acetone at the total permeation volume (i.e., the lower-MW limit of the column separation), with the structured absorption still remaining as an overlay on the broad, featureless absorption. These observations suggest that the high-MW fraction of the fresh BrC sample consists of aggregates susceptible to disruption by ACN; in addition, the preservation of this fraction's broad, featureless absorption extending into the visible region indicates that this disaggregation process does not affect the sample's chromophoric properties.
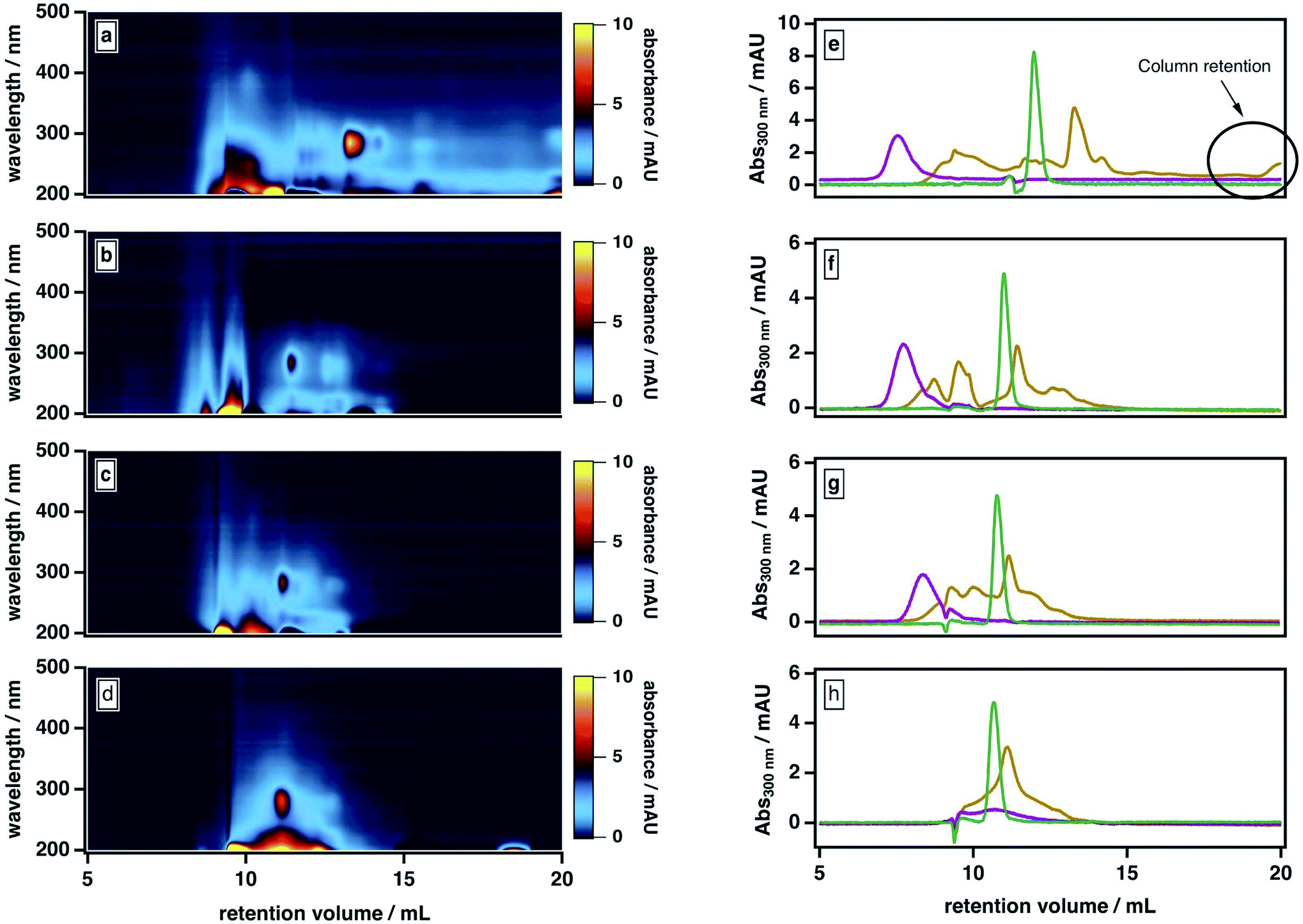 | ||
Fig. 4 BrC and SRHA elution behaviour as a function of mobile phase solvent strength: (a–d) BrC absorption density plots and (e–h) single-wavelength (300 nm) chromatograms ( SRHA, SRHA,  acetone, acetone,  BrC). Experiments were conducted under the following mobile phase organic modifier conditions: 25% ACN (a and e), 50% ACN (b and f), 60% ACN (c and g), and 70% ACN (d and h). In all cases, the remainder of the mobile phase was 20 mM phosphate buffer (pH 6.8). BrC). Experiments were conducted under the following mobile phase organic modifier conditions: 25% ACN (a and e), 50% ACN (b and f), 60% ACN (c and g), and 70% ACN (d and h). In all cases, the remainder of the mobile phase was 20 mM phosphate buffer (pH 6.8). | ||
To further verify this conclusion, we subjected SRHA to the same mobile phase protocol (Fig. S2†). Previous studies36 have shown that humic materials consist of supramolecular associations of relatively small fractions connected by nonbonding intermolecular forces. Similar to the high-MW BrC fraction, SRHA eluted later and with a broader size distribution at higher ACN contents (Fig. 4e–g), and co-eluted with acetone at 70% ACN (Fig. 4h), indicating dissociation of SRHA aggregates into smaller subunits. These observations support our characterization of the high-MW component of BrC as small BrC components held together via hydrophobic interactions.
:50 mixture of buffer and an organic modifier (25% ACN/25% MeOH), the latter of which we chose to minimize the influence of hydrophobic interactions between our samples and the column matrix, and varied the concentration of the buffer from 10 to 40 mM.
As shown in Fig. 5, the high-MW BrC fraction eluted later at 20 mM versus 10 mM; the same delay was also observed for SRHA (Fig. S3†). In principle, ionic strength may have influenced the elution in two ways here: first, via suppressing electrostatic interactions between the column matrix and polar and charged functional groups (e.g., hydroxyl, carbonyl, amide)7 present in the BrC sample (i.e., ionic exclusion); second, via reducing intramolecular repulsions between these same charged functional groups, resulting in a more “compacted” molecule. Because both of these effects would delay elution, we cannot definitively distinguish between them. However, evidence supporting the existence of the second effect is provided by previous studies of humic substance conformation, which reported appreciable decreases in the size of humic aggregates at elevated ionic strengths.44,47 We also observed an enhanced absorption for the low-MW fraction at higher ionic strengths: in particular, as shown in Fig. 5, the absorption at ∼300 nm was higher in the presence of 20 mM buffer than in the presence of 10 mM buffer. As well, at 40 mM, the high-MW and low-MW fractions were no longer resolved. These results could be explained by a decrease in the hydrophobic sorption capacities of BrC aggregates at higher ionic strengths,70 leading to liberation of some absorbed low-MW BrC subunits.
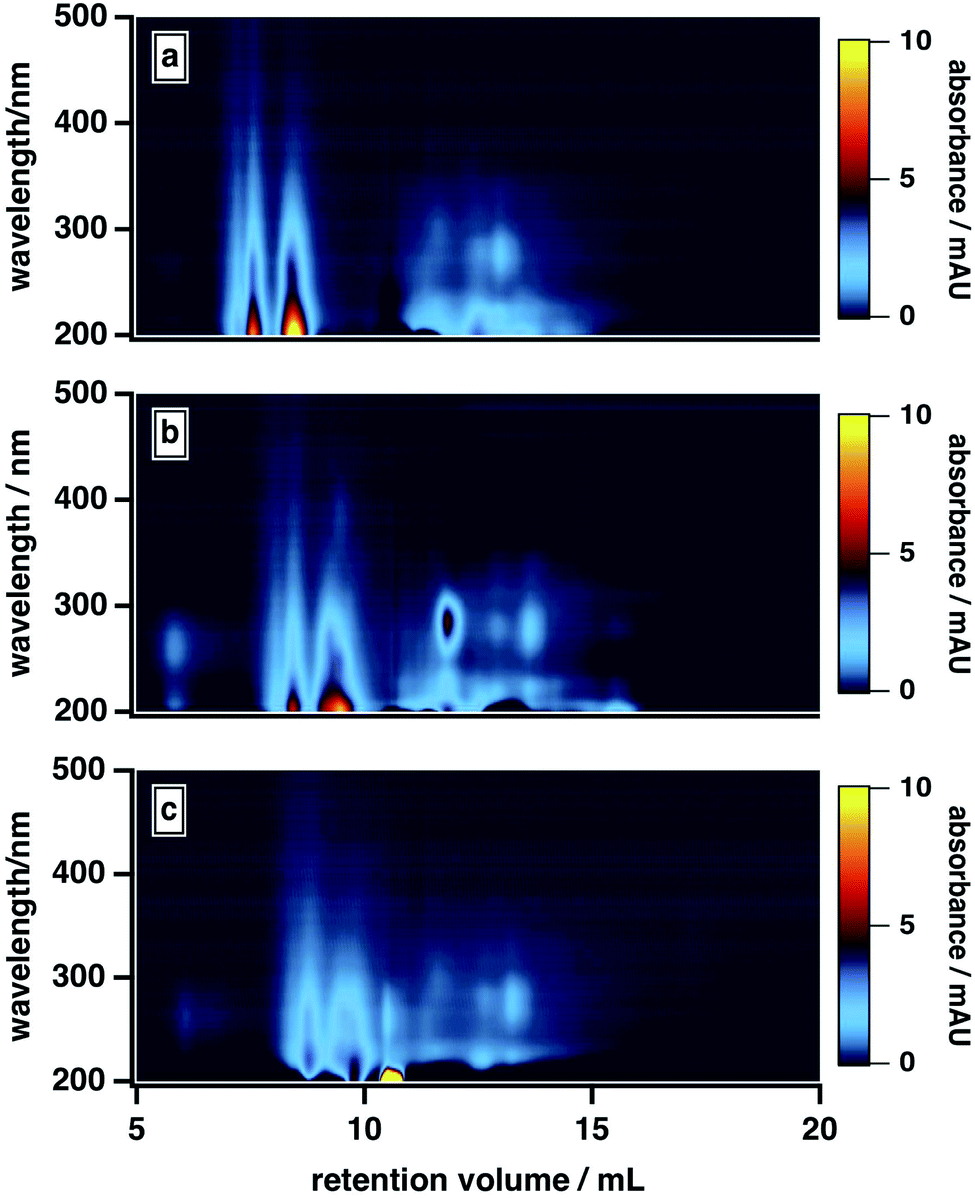 | ||
| Fig. 5 Absorption density plots for fresh peat BrC as a function of mobile phase phosphate buffer concentration (i.e., ionic strength). Mobile phases were prepared using three concentrations of phosphate buffer (pH 6.8): (a) 10 mM, (b) 20 mM, (c) 40 mM. In all cases, the mobile phase composition was 50% phosphate buffer, 25% MeOH, and 25% ACN. | ||
4 Conclusions and implications for analysis of complex atmospheric samples
This study represents the first systematic investigation of the challenges and benefits associated with the application of HPLC-SEC-PDA to the characterization of atmospheric brown carbon (BrC), an important class of light-absorbing PM and the dominant contributor to the absorption profile of wildfire PM. In the following paragraphs, we highlight the implications of our results for SEC analysis of atmospheric samples and provide recommendations for future work in this area.A major conclusion of this study is that careful consideration of solvent–analyte interactions is crucial when identifying appropriate extraction and chromatographic/spectroscopic analysis conditions for natural samples. Previous work has shown that the composition of PM extracts can be influenced by reactions of specific organic functional groups with sample extraction solvents (e.g., MeOH).71 Here, we show that organic solvents can influence BrC properties even without reacting directly with BrC components: in particular, we show that ACN not only mitigates hydrophobic interactions between BrC and the SEC column matrix but also disrupts metastable BrC aggregates, thereby leading to changes in measured BrC size distributions. This aggregation and dissociation behaviour could potentially contribute to discrepancies between MW values inferred from SEC and those determined using mass spectrometry for the same sample.38,39,46,72
Importantly, and more broadly, our findings imply a need for caution when using chromatographic elution behaviour (e.g., in reversed-phase HPLC) to make inferences regarding the hydrophobicity and other properties of BrC, as the relationship between retention time and mobile phase identity for a given BrC component may reflect both changes in its distribution constant and changes in its intrinsic properties. For example, increases in ACN mobile phase content (e.g., in gradient methods) could potentially lead not only to changes in partitioning behaviour but also to dissociation of BrC aggregates, with concomitant indirect changes in retention behaviour.
Successful application of SEC to MW estimations of complex mixtures is a challenging task, as it requires both selection of calibration standards with comparable chemical and structural properties to the analytes of interest and careful consideration of biases resulting from differences in the magnitudes of secondary effects experienced by mixture components compared to the chosen calibrant suite. For example, previous studies applying SEC to analysis of atmospheric PM have employed both commercial macromolecules and atmospherically relevant small molecules (e.g., phenols and aromatic acids) for MW calibration.38,43,73,74 However, given that we would expect these calibrants to be differently susceptible to secondary effects than BrC, this strategy has the potential to lead to dramatic changes in estimated MWs with changing solvent composition. In the case of humic substances, these issues have occupied analytical chemists for decades.75 In the case of fresh BrC, which we show here is made up of components with a wide range of sizes, polarities, and susceptibility to conformational changes—and therefore subject to different secondary effects—achieving size-based separation of all BrC components at any single mobile phase composition, no matter how optimized, is likely an impossible task. This challenge has the potential to be especially important in cases where SEC is coupled with complementary techniques to investigate the chemical properties of specific analyte size fractions,37,73,74 as a given elution volume could potentially contain contributions not only from “ideal” analytes (i.e., those for which secondary interactions have been minimized) but also from many other analyte classes (e.g., higher-MW but more hydrophobic, smaller-MW but susceptible to intramolecular electrostatic repulsions).
In this context, we suggest a more expansive approach to the SEC characterization of complex atmospheric mixtures, in which its apparent limitations are instead reconceptualized as advantages. For example, in this study, by subjecting our BrC sample to different mobile phase conditions, we were able to explore not only its molecular size but also its hydrophobicity, conformation, and intermolecular associations.36 This approach, which is analogous in some respects to the use of chromatographic retention behaviour to estimate the properties of single analytes (e.g., vapour pressure76 and KOW77), could also be applied in other ways. For example, if coupled with refractive index detection, this strategy would enable the characterization of non-chromophoric PM components. In addition, the fluorescence detection approach described here could be expanded through the use of excitation–emission matrix analysis,64 which has the potential to aid in the chemical characterization of the organic fluorophores present in BrC fractions. More broadly, the investigation of mobile phase-dependent sample behaviour has the potential to facilitate the prediction of analyte properties under the elevated organic content and ionic strength conditions characteristic of atmospheric PM, which are challenging and/or impossible to explore directly in the laboratory.
Author contributions
M. L., D. K. T., and S. A. S. designed the combustion experiments with assistance from C. J. Y., M. L., N. Z., S. A. S., and D. K. T. collected the boreal peat samples and performed the combustion experiments. M. L. performed all HPLC-SEC-PDA analyses; C. W. C. conducted the AF4 analysis. M. L. and S. A. S. interpreted experimental data, with assistance from C. J. Y., and wrote the manuscript, with critical comments from D. K. T., C. W. C., and C. J. Y.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the Department of Chemistry and the Faculty of Science at the University of Alberta (U of A) for start-up funding, the Natural Sciences and Engineering Research Council of Canada (NSERC) for funding through the Discovery Grant and Research Tools and Instruments programs, the Canada Foundation for Innovation (CFI) for funding through the John R. Evans Leaders Fund, the Alberta Ministry of Jobs, Economy and Innovation for funding through the Small Equipment Grants stream of the Research Capacity Program, and Ed Fu in the Analytical and Instrumentation Laboratory at U of A for the loan of the HPLC system employed in these experiments. This research was undertaken, in part, thanks to funding from the Canada Research Chairs Program. Funding for M. L. was provided by the CRC Tier 2 research stipend, as well as a CRC research supplement from the Faculty of Science at U of A; funding for N. Z. was provided by the MITACS Globalink program. C. W. C.'s participation in this study was supported by the SWAMP Lab at U of A; at the time of this study, he was partially financially supported by a NSERC Collaborative Research and Development (CRD) grant with Alberta Innovates and the Canadian Oil Sands Innovation Alliance. The authors acknowledge Ginny Marshall for assistance with combustion experiments; Dr Fred Brechtel for PILS troubleshooting assistance; Dirk Kelm and Vincent Bizon for PILS inlet line modifications; Wayne Moffat, Mario Schmidt, and Drs Matthew Ross and Randy Whittal for assistance with HPLC-SEC-PDA system set-up; and Scott Krepich (Phenomenex) and Dr Robert Di Lorenzo (SCIEX) for helpful SEC discussions.References
- T. C. Bond, D. G. Streets, K. F. Yarber, S. M. Nelson, J.-H. Woo and Z. Klimont, A technology-based global inventory of black and organic carbon emissions from combustion, J. Geophys. Res.: Atmos., 2004, 109, D14203 CrossRef
.
- G. R. van der Werf, J. T. Randerson, L. Giglio, G. J. Collatz, M. Mu, P. S. Kasibhatla, D. C. Morton, R. S. DeFries, Y. Jin and T. T. van Leeuwen, Global fire emissions and the contribution of deforestation, savanna, forest, agricultural, and peat fires (1997–2009), Atmos. Chem. Phys., 2010, 10, 11707–11735 CrossRef CAS
-
IPCC, Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. M. Midgley, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2013, pp. 1535 Search PubMed
- D. Liu, C. He, J. P. Schwarz and X. Wang, Lifecycle of light-absorbing carbonaceous aerosols in the atmosphere, npj Clim. Atmos. Sci., 2020, 3, 1–18 CrossRef
- T. C. Bond, S. J. Doherty, D. W. Fahey, P. M. Forster, T. Berntsen, B. J. DeAngelo, M. G. Flanner, S. Ghan, B. Kärcher, D. Koch, S. Kinne, Y. Kondo, P. K. Quinn, M. C. Sarofim, M. G. Schultz, M. Schulz, C. Venkataraman, H. Zhang, S. Zhang, N. Bellouin, S. K. Guttikunda, P. K. Hopke, M. Z. Jacobson, J. W. Kaiser, Z. Klimont, U. Lohmann, J. P. Schwarz, D. Shindell, T. Storelvmo, S. G. Warren and C. S. Zender, Bounding the role of black carbon in the climate system: A scientific assessment, J. Geophys. Res.: Atmos., 2013, 118, 5380–5552 CAS
- M. O. Andreae and A. Gelencsér, Black carbon or brown carbon? The nature of light-absorbing carbonaceous aerosols, Atmos. Chem. Phys., 2006, 6, 3131–3148 CrossRef CAS
- A. Laskin, J. Laskin and S. A. Nizkorodov, Chemistry of atmospheric brown carbon, Chem. Rev., 2015, 115, 4335–4382 CrossRef CAS
- G. Lin, J. E. Penner, M. G. Flanner, S. Sillman, L. Xu and C. Zhou, Radiative forcing of organic aerosol in the atmosphere and on snow: Effects of SOA and brown carbon, J. Geophys. Res.: Atmos., 2014, 119, 7453–7476 Search PubMed
- R. Bahadur, P. S. Praveen, Y. Xu and V. Ramanathan, Solar absorption by elemental and brown carbon determined from spectral observations, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17366–17371 CrossRef CAS
- R. Saleh, From measurements to models: toward accurate representation of brown carbon in climate calculations, Curr. Pollut. Rep., 2020, 6, 90–104 CrossRef CAS
- M. H. Powelson, B. M. Espelien, L. N. Hawkins, M. M. Galloway and D. O. De Haan, Brown carbon formation by aqueous-phase carbonyl compound reactions with amines and ammonium sulfate, Environ. Sci. Technol., 2014, 48, 985–993 CrossRef CAS
- H. A. Al-Abadleh, M. S. Rana, W. Mohammed and M. I. Guzman, Dark iron-catalyzed reactions in acidic and viscous aerosol systems efficiently form secondary brown carbon, Environ. Sci. Technol., 2021, 55, 209–219 CrossRef CAS
- S. M. Phillips and G. D. Smith, Light absorption by charge transfer complexes in brown carbon aerosols, Environ. Sci. Technol. Lett., 2014, 1, 382–386 CrossRef CAS
- A. Trofimova, R. F. Hems, T. Liu, J. P. D. Abbatt and E. G. Schnitzler, Contribution of charge-transfer complexes to absorptivity of primary brown carbon aerosol, ACS Earth Space Chem., 2019, 3, 1393–1401 CrossRef CAS
-
A. Laskin, P. Lin, J. Laskin, L. T. Fleming and S. Nizkorodov, in Multiphase Environmental Chemistry in the Atmosphere, ACS Symposium Series, ed. S. R. Hunt, A. Laskin and S. A. Nizkorodov, American Chemical Society, Washington, D.C, 2018, vol. 1299, pp. 261–274 Search PubMed
- R. F. Hems and J. P. D. Abbatt, Aqueous phase photo-oxidation of brown carbon nitrophenols: reaction kinetics, mechanism, and evolution of light absorption, ACS Earth Space Chem., 2018, 2, 225–234 CrossRef CAS
- S. Dasari, A. Andersson, S. Bikkina, H. Holmstrand, K. Budhavant, S. Satheesh, E. Asmi, J. Kesti, J. Backman, A. Salam, D. S. Bisht, S. Tiwari, Z. Hameed and Ö. Gustafsson, Photochemical degradation affects the light absorption of water-soluble brown carbon in the South Asian outflow, Sci. Adv., 2019, 5, eaau8066 CrossRef PubMed
- B. B. Palm, Q. Peng, C. D. Fredrickson, B. H. Lee, L. A. Garofalo, M. A. Pothier, S. M. Kreidenweis, D. K. Farmer, R. P. Pokhrel, Y. Shen, S. M. Murphy, W. Permar, L. Hu, T. L. Campos, S. R. Hall, K. Ullmann, X. Zhang, F. Flocke, E. V. Fischer and J. A. Thornton, Quantification of organic aerosol and brown carbon evolution in fresh wildfire plumes, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 29469–29477 CrossRef CAS
- L. T. Fleming, P. Lin, J. M. Roberts, V. Selimovic, R. Yokelson, J. Laskin, A. Laskin and S. A. Nizkorodov, Molecular composition and photochemical lifetimes of brown carbon chromophores in biomass burning organic aerosol, Atmos. Chem. Phys., 2020, 20, 1105–1129 CrossRef CAS
- H. Forrister, J. Liu, E. Scheuer, J. Dibb, L. Ziemba, K. L. Thornhill, B. Anderson, G. Diskin, A. E. Perring, J. P. Schwarz, P. Campuzano-Jost, D. A. Day, B. B. Palm, J. L. Jimenez, A. Nenes and R. J. Weber, Evolution of brown carbon in wildfire plumes, Geophys. Res. Lett., 2015, 42, 4623–4630 CrossRef CAS
- C. C. Hanes, X. Wang, P. Jain, M.-A. Parisien, J. M. Little and M. D. Flannigan, Fire-regime changes in Canada over the last half century, Can. J. For. Res., 2018, 49(3), 256–269 CrossRef
- X. Wang, D. K. Thompson, G. A. Marshall, C. Tymstra, R. Carr and M. D. Flannigan, Increasing frequency of extreme fire weather in Canada with climate change, Clim. Change, 2015, 130, 573–586 CrossRef
-
IPCC, Climate Change and Land: an IPCC special report on climate change, desertification, land degradation, sustainable land management, food security, and greenhouse gas fluxes in terrestrial ecosystems, ed. P. R. Shukla, J. Skea, E. Calvo Buendia, V. Masson-Delmotte, H.-O. Pörtner, D. C. Roberts, P. Zhai, R. Slade, S. Connors, R. van Diemen, M. Ferrat, E. Haughey, S. Luz, S. Neogi, M. Pathak, J. Petzold, J. Portugal Pereira, P. Vyas, E. Huntley, K. Kissick, M. Belkacemi and J. Malley, 2019, in press Search PubMed
- P. Lin, P. K. Aiona, Y. Li, M. Shiraiwa, J. Laskin, S. A. Nizkorodov and A. Laskin, Molecular characterization of brown carbon in biomass burning aerosol particles, Environ. Sci. Technol., 2016, 50, 11815–11824 CrossRef CAS PubMed
- P. Lin, L. T. Fleming, S. A. Nizkorodov, J. Laskin and A. Laskin, Comprehensive molecular
characterization of atmospheric brown carbon by high resolution mass spectrometry with electrospray and atmospheric pressure photoionization, Anal. Chem., 2018, 90, 12493–12502 CrossRef CAS PubMed
- M. Claeys, R. Vermeylen, F. Yasmeen, Y. Gómez-González, X. Chi, W. Maenhaut, T. Mészáros and I. Salma, Chemical characterization of humic-like substances from urban, rural and tropical biomass burning environments using liquid chromatography with UV/vis photodiode array detection and electrospray ionization mass spectrometry, Environ. Chem., 2012, 9, 273–284 CrossRef CAS
- Y. Desyaterik, Y. Sun, X. Shen, T. Lee, X. Wang, T. Wang and J. L. Collett, Speciation of “brown” carbon in cloud water impacted by agricultural biomass burning in eastern China, J. Geophys. Res.: Atmos., 2013, 118, 7389–7399 CAS
- P. Lin, N. Bluvshtein, Y. Rudich, S. A. Nizkorodov, J. Laskin and A. Laskin, Molecular chemistry of atmospheric brown carbon inferred from a nationwide biomass burning event, Environ. Sci. Technol., 2017, 51, 11561–11570 CrossRef CAS
- M. Xie, X. Chen, M. D. Hays and A. L. Holder, Composition and light absorption of N-containing aromatic compounds in organic aerosols from laboratory biomass burning, Atmos. Chem. Phys., 2019, 19, 2899–2915 CrossRef CAS PubMed
- K. Tang, J. S. Page and R. D. Smith, Charge competition and the linear dynamic range of detection in electrospray ionization mass spectrometry, J. Am. Soc. Mass Spectrom., 2004, 15, 1416–1423 CrossRef CAS PubMed
- C. Mohr, F. D. Lopez-Hilfiker, P. Zotter, A. S. H. Prévôt, L. Xu, N. L. Ng, S. C. Herndon, L. R. Williams, J. P. Franklin, M. S. Zahniser, D. R. Worsnop, W. B. Knighton, A. C. Aiken, K. J. Gorkowski, M. K. Dubey, J. D. Allan and J. A. Thornton, Contribution of nitrated phenols to wood burning brown carbon light absorption in Detling, United Kingdom during winter time, Environ. Sci. Technol., 2013, 47, 6316–6324 CrossRef CAS PubMed
- T. Reemtsma and A. These, On-line coupling of size exclusion chromatography with electrospray ionization-tandem mass spectrometry for the analysis of aquatic fulvic and humic Acids, Anal. Chem., 2003, 75, 1500–1507 CrossRef CAS PubMed
- A. Gaspar, E. V. Kunenkov, R. Lock, M. Desor, I. Perminova and P. Schmitt-Kopplin, Combined utilization of ion mobility and ultra-high-resolution mass spectrometry to identify multiply charged constituents in natural organic matter, Rapid Commun. Mass Spectrom., 2009, 23, 683–688 CrossRef CAS PubMed
-
S. Mori and H. G. Barth, Size Exclusion Chromatography, Springer Laborator, Springer, Berlin, Heidelberg ,p. 1999 Search PubMed
- B. C. McAdams, G. R. Aiken, D. M. McKnight, W. A. Arnold and Y.-P. Chin, High pressure size exclusion chromatography (HPSEC) determination of dissolved organic matter molecular weight revisited: accounting for changes in stationary phases, analytical standards, and isolation methods, Environ. Sci. Technol., 2018, 52, 722–730 CrossRef CAS PubMed
- A. Piccolo, The supramolecular structure of humic substance, Soil Sci., 2001, 166, 810–832 CrossRef CAS
- T. Spranger, D. van Pinxteren and H. Herrmann, Atmospheric “HULIS” in different environments: polarities, molecular sizes, and sources suggest more than 50% are not “humic-like”, ACS Earth Space Chem., 2020, 4, 272–282 CrossRef CAS
- V. Samburova, R. Zenobi and M. Kalberer, Characterization of high molecular weight compounds in urban atmospheric particles, Atmos. Chem. Phys., 2005, 5, 2163–2170 CrossRef CAS
- R. A. Di Lorenzo and C. J. Young, Size separation method for absorption characterization in brown carbon: application to an aged biomass burning sample, Geophys. Res. Lett., 2016, 43(1), 458–465 CrossRef CAS
- R. A. Di Lorenzo, R. A. Washenfelder, A. R. Attwood, H. Guo, L. Xu, N. L. Ng, R. J. Weber, K. Baumann, E. Edgerton and C. J. Young, Molecular-size-separated brown carbon absorption
for biomass-burning aerosol at multiple field sites, Environ. Sci. Technol., 2017, 51, 3128–3137 CrossRef CAS PubMed
- R. A. Di Lorenzo, B. K. Place, T. C. VandenBoer and C. J. Young, Composition of size-resolved aged boreal fire aerosols: brown carbon, biomass burning tracers, and reduced nitrogen, ACS Earth Space Chem., 2018, 2, 278–285 CrossRef CAS
- J. P. S. Wong, A. Nenes and R. J. Weber, Changes in light absorptivity of molecular weight separated brown carbon due to photolytic aging, Environ. Sci. Technol., 2017, 51, 8414–8421 CrossRef CAS PubMed
- J. P. S. Wong, M. Tsagkaraki, I. Tsiodra, N. Mihalopoulos, K. Violaki, M. Kanakidou, J. Sciare, A. Nenes and R. J. Weber, Atmospheric evolution of molecular-weight-separated brown carbon from biomass burning, Atmos. Chem. Phys., 2019, 19, 7319–7334 CrossRef CAS
- H. D. Haan, R. I. Jones and K. Salonen, Does ionic strength affect the configuration of aquatic humic substances, as indicated by gel filtration?, Freshwater Biol., 1987, 17, 453–459 CrossRef
- Y. Wang, C.-A. Chiu, P. Westerhoff, K. T. Valsaraj and P. Herckes, Characterization of atmospheric organic matter using size-exclusion chromatography with inline organic carbon detection, Atmos. Environ., 2013, 68, 326–332 CrossRef CAS
- Z. Krivácsy, G. Kiss, B. Varga, I. Galambos, Z. Sárvári, A. Gelencsér, Á. Molnár, S. Fuzzi, M. C. Facchini, S. Zappoli, A. Andracchio, T. Alsberg, H. C. Hansson and L. Persson, Study of humic-like substances in fog and interstitial aerosol by size-exclusion chromatography and capillary electrophoresis, Atmos. Environ., 2000, 34, 4273–4281 CrossRef
- P. Conte and A. Piccolo, Conformational arrangement of dissolved humic substances. Influence of solution composition on association of humic molecules, Environ. Sci. Technol., 1999, 33, 1682–1690 CrossRef CAS
- R. Baigorri, M. Fuentes, G. González-Gaitano and J. M. García-Mina, Analysis of molecular aggregation in humic substances in solution, Colloids Surf., A, 2007, 302, 301–306 CrossRef CAS
- O. B. Popovicheva, G. Engling, I.-T. Ku, M. A. Timofeev and N. K. Shonija, Aerosol emissions from long-lasting smoldering of boreal peatlands: chemical composition, markers, and microstructure, Aerosol Air Qual. Res., 2019, 19, 484–503 CrossRef CAS
- R. K. Chakrabarty, M. Gyawali, R. L. N. Yatavelli, A. Pandey, A. C. Watts, J. Knue, L.-W. A. Chen, R. R. Pattison, A. Tsibart, V. Samburova and H. Moosmüller, Brown carbon aerosols from burning of boreal peatlands: microphysical properties, emission factors, and implications for direct radiative forcing, Atmos. Chem. Phys., 2016, 16, 3033–3040 CrossRef CAS
-
Field-Flow Fractionation Handbook, ed. M. E. Schimpf, K. Caldwell and J. C. Giddings, John Wiley & Sons, New York, 2000 Search PubMed
- C. Guéguen and C. W. Cuss, Characterization of aquatic dissolved organic matter by asymmetrical flow field-flow fractionation coupled to UV-visible diode array and excitation emission matrix fluorescence, J. Chromatogr. A, 2011, 1218, 4188–4198 CrossRef PubMed
- C. W. Cuss, I. Grant-Weaver and W. Shotyk, AF4-ICPMS with the 300 Da membrane to resolve metal-bearing “colloids” < 1 kDa: optimization, fractogram deconvolution, and advanced quality control, Anal. Chem., 2017, 89, 8027–8035 CrossRef CAS PubMed
- S. L. Wilkinson, P. A. Moore, M. D. Flannigan, B. M. Wotton and J. M. Waddington, Did enhanced afforestation cause high severity peat burn in the Fort McMurray Horse River wildfire?, Environ. Res. Lett., 2018, 13, 014018 CrossRef
- M. Lyu, C. J. Young, D. K. Thompson and S. A. Styler, Laboratory combustion experiments performed in Summer, 2018, unpublished work..
- R. J. Yokelson, D. W. T. Griffith and D. E. Ward, Open-path Fourier transform infrared studies of large-scale laboratory biomass fires, J. Geophys. Res.: Atmos., 1996, 101, 21067–21080 CrossRef CAS
- Y. Hu, N. Fernandez-Anez, T. E. L. Smith and G. Rein, Review of emissions from smouldering peat fires and their contribution to regional haze episodes, Int. J. Wildland Fire, 2018, 27, 293–312 CrossRef CAS
- A. Sorooshian, F. J. Brechtel, Y. Ma, R. J. Weber, A. Corless, R. C. Flagan and J. H. Seinfeld, Modeling and characterization of a particle-into-liquid sampler (PILS), Aerosol Sci. Technol., 2006, 40, 396–409 CrossRef CAS
- Q. Zhou, S. E. Cabaniss and P. A. Maurice, Considerations in the use of high-pressure size exclusion chromatography (HPSEC) for determining molecular weights of aquatic humic substances, Water Res., 2000, 34, 3505–3514 CrossRef CAS
- C. M. Carrico, M. D. Petters, S. M. Kreidenweis, A. P. Sullivan, G. R. McMeeking, E. J. T. Levin, G. Engling, W. C. Malm and J. L. Collett, Water uptake and chemical composition of fresh aerosols generated in open burning of biomass, Atmos. Chem. Phys., 2010, 10, 5165–5178 CrossRef CAS
- J. Chen, S. H. Budisulistiorini, M. Itoh, W.-C. Lee, T. Miyakawa, Y. Komazaki, L. D. Q. Yang and M. Kuwata, Water uptake by fresh Indonesian peat burning particles is limited by water-soluble organic matter, Atmos. Chem. Phys., 2017, 17, 11591–11604 CrossRef CAS
- P. G. Coble, Characterization of marine and terrestrial DOM in seawater using excitation–emission matrix spectroscopy, Mar. Chem., 1996, 51, 325–346 CrossRef CAS
- N. Maie, N. M. Scully, O. Pisani and R. Jaffé, Composition of a protein-like fluorophore of dissolved organic matter in coastal wetland and estuarine ecosystems, Water Res., 2007, 41, 563–570 CrossRef CAS PubMed
- Q. Chen, Y. Miyazaki, K. Kawamura, K. Matsumoto, S. Coburn, R. Volkamer, Y. Iwamoto, S. Kagami, Y. Deng, S. Ogawa, S. Ramasamy, S. Kato, A. Ida, Y. Kajii and M. Mochida, Characterization of chromophoric water-soluble organic matter in urban, forest, and marine aerosols by HR-ToF-AMS analysis and excitation–emission matrix spectroscopy, Environ. Sci. Technol., 2016, 50, 10351–10360 CrossRef CAS PubMed
- Y. Fujii, S. Tohno, K. Ikeda, M. Mahmud and N. Takenaka, A preliminary study on humic-like substances in particulate matter in Malaysia influenced by Indonesian peatland fires, Sci. Total Environ., 2021, 753, 142009 CrossRef CAS PubMed
- A. E. Perring, J. P. Schwarz, M. Z. Markovic, D. W. Fahey, J. L. Jimenez, P. Campuzano-Jost, B. D. Palm, A. Wisthaler, T. Mikoviny, G. Diskin, G. Sachse, L. Ziemba, B. Anderson, T. Shingler, E. Crosbie, A. Sorooshian, R. Yokelson and R.-S. Gao,
In situ measurements of water uptake by black carbon-containing aerosol in wildfire plumes, J. Geophys. Res.: Atmos., 2017, 122, 1086–1097 CAS
- M. Martin, T. Tritscher, Z. Jurányi, M. F. Heringa, B. Sierau, E. Weingartner, R. Chirico, M. Gysel, A. S. H. Prévôt, U. Baltensperger and U. Lohmann, Hygroscopic properties of fresh and aged wood burning particles, J. Aerosol Sci., 2013, 56, 15–29 CrossRef CAS
- M. Yang, S. Fazio, D. Munch and P. Drumm, Impact of methanol and acetonitrile on separations based on π–π interactions with a reversed-phase phenyl column, J. Chromatogr. A, 2005, 1097, 124–129 CrossRef CAS PubMed
- H. Herrmann, T. Schaefer, A. Tilgner, S. A. Styler, C. Weller, M. Teich and T. Otto, Tropospheric aqueous-phase chemistry: kinetics, mechanisms, and Its coupling
to a changing gas phase, Chem. Rev., 2015, 115, 4259–4334 CrossRef CAS PubMed
- C.-L. Lee, L.-J. Kuo, H.-L. Wang and P.-C. Hsieh, Effects of ionic strength on the binding of phenanthrene and pyrene to humic substances: three-stage variation model, Water Res., 2003, 37, 4250–4258 CrossRef CAS PubMed
- A. P. Bateman, M. L. Walser, Y. Desyaterik, J. Laskin, A. Laskin and S. A. Nizkorodov, The effect of solvent on the analysis of secondary organic aerosol using electrospray ionization mass spectrometry, Environ. Sci. Technol., 2008, 42, 7341–7346 CrossRef CAS PubMed
- V. Samburova, S. Szidat, C. Hueglin, R. Fisseha, U. Baltensperger, R. Zenobi and M. Kalberer, Seasonal variation of high-molecular-weight compounds in the water-soluble fraction of organic urban aerosols, J. Geophys. Res.: Atmos., 2005, 110, D23210 CrossRef
- A. P. Sullivan and R. J. Weber, Chemical characterization of the ambient organic aerosol soluble in water: 2. Isolation of acid, neutral, and basic fractions by modified size-exclusion chromatography, J. Geophys. Res.: Atmos., 2006, 111, D05315 CrossRef
- T. Spranger, D. van Pinxteren and H. Herrmann, Two-dimensional offline chromatographic fractionation for the characterization of humic-like substances in atmospheric aerosol particles, Environ. Sci. Technol., 2017, 51, 5061–5070 CrossRef CAS PubMed
- M. De Nobili and Y. Chen, Size exclusion chromatography of humic substances: limits, perspectives and prospectives, Soil Sci., 1999, 164, 825–833 CrossRef CAS
- B. Koutek, T. Mahnel, P. Šimáček, M. Fulem and K. Růžička, Extracting vapor pressure data from GLC retention times. Part 1: Analysis of single reference approach, J. Chem. Eng. Data, 2017, 62, 3542–3550 CrossRef CAS
- R. Hackenberg, A. Schütz and K. Ballschmiter, High-resolution gas chromatography retention data as basis for the estimation of Kow values using PCB congeners as secondary standards, Environ. Sci. Technol., 2003, 37, 2274–2279 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: A comprehensive discussion of our SEC column characterization results; selection/design of quality control experiments; details regarding the collection and extraction of a complementary BrC sample collected on quartz fiber filters (QFF); a description of our integration procedure for SEC absorption density plots; and a summary of asymmetric flow field-flow fractionation (AF4) analysis conditions. Eight supporting figures, which present SEC calibration curves; supplementary characterization information for SRHA; SEC absorption density plots for quality control samples, including the QFF sample described above; and complementary AF4 characterization of our BrC sample. Two supporting tables, which present calibration curve equations (and associated SRHA MW estimates) and retention time reproducibility data for acetone and SRHA. See DOI: 10.1039/d1ea00011j |
| ‡ Now at Natural Resources Canada – Great Lakes Forestry Centre, Sault Ste. Marie, Canada. |
| § Now at School of Science and the Environment, Memorial University of Newfoundland (Grenfell Campus), Corner Brook, Newfoundland and Labrador, Canada. |
| This journal is © The Royal Society of Chemistry 2021 |