
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into HONO sources from observations during a solar eclipse†
Ajit
Singh
 a,
Leigh R.
Crilley
b,
Francis D.
Pope
a and
William J.
Bloss
*a
a,
Leigh R.
Crilley
b,
Francis D.
Pope
a and
William J.
Bloss
*a
aSchool of Geography, Earth and Environmental Sciences, University of Birmingham, Birmingham, B15 2TT, UK. E-mail: W.J.Bloss@bham.ac.uk; a.singh.2@bham.ac.uk; F.Pope@bham.ac.uk
bDepartment of Chemistry, York University, Toronto, Canada. E-mail: lcrilley@yorku.ca
First published on 20th July 2021
Abstract
Nitrous acid (HONO) is a major, and often the dominant, precursor to primary OH radical production in the daytime boundary layer, driving the removal of many primary pollutants and formation of secondary species such as ozone and many aerosol components. A number of photochemical HONO production mechanisms have been proposed, alongside homogeneous gas-phase reactions, to account for field observations of daytime HONO. The range of production mechanisms show varying dependencies upon precursor species such as NO2, available surfaces for heterogeneous reactions, and dark/photoenhanced aspects. Here, we exploit measurements of HONO and related species during a near-total solar eclipse as a natural perturbation to the atmospheric photochemistry to assess the characteristics of the production mechanisms occurring at an urban background location. Little variation in HONO abundance was observed in response to changing light levels during the eclipse, pointing to relatively balanced photochemical source and (well-understood) sink terms. We employ a series of simple kinetic simulations to explore the consistency of different potential source mechanisms with the observations, finding evidence for a dominant role for photochemical processing of traffic-derived NO2 upon surfaces producing HONO, alongside indications of a smaller contribution from direct vehicular emissions. Other mechanisms involving dark heterogeneous reactions were not, in isolation, consistent with the observations. The critical role of NO2, ultimately derived overwhelmingly from local road traffic emissions at this location, points to significant future reductions in daytime HONO production with vehicle fleet evolution and reduction of tailpipe emissions.
Environmental significanceNitrous acid (HONO) is a major, often the dominant, boundary layer precursor to the key daytime atmospheric oxidant OH; however sources of HONO are poorly understood, with a number of candidate formation mechanisms advanced. Here, we use a natural perturbation – a solar eclipse – to identify the photochemical factors governing HONO formation, from their variation with this large-scale change in solar intensity. We demonstrate that HONO is not controlled by a single source, but that photoenhanced production (i.e. reactions accelerated by sunlight) are a major component of HONO production, suggesting a dominant role for photochemical processing of traffic-derived NO2 upon surfaces producing HONO, alongside a smaller contribution from direct vehicular emissions. In both cases, association with (anthropogenically derived) NO2 indicates that HONO formation may fall with future vehicle fleet evolution and tailpipe emissions reduction. |
1 Introduction
Atmospheric chemical processing in the sunlit troposphere is driven primarily by the OH radical, which initiates the removal of most organic compounds, and drives the formation of ozone, and secondary organic and inorganic aerosols.1,2 While the dominant primary source of OH in the free troposphere is the photolysis of ozone and subsequent reaction of electronically excited oxygen atoms with water vapour, a series of recent field campaigns have shown that the photolysis of nitrous acid (HONO) is a major/the dominant primary OH precursor in the continental boundary layer.3,4| HONO + hν (λ < 440 nm) → NO + OH | (R1) |
HONO is formed from the slow homogeneous reaction between OH and NO, and in this sense acts as a photolabile reservoir for OH (and NO) with (R1) and (R2) forming a null cycle; however observed levels of HONO during daytime are typically one order of magnitude higher than (R1) and (R2) in isolation predict, indicating the presence of additional HONO formation mechanisms or emissions (and hence net OH production).5–7 The dark formation of HONO through the heterogeneous hydrolysis of nitrogen dioxide ((R3) below) has been known for a number of years;8 however recent laboratory and field students have identified a number of additional candidate HONO formation mechanisms and/or emission sources.
| OH + NO + M → HONO + M | (R2) |
| 2NO2 + H2O → HONO + HNO3 | (R3) |
Primary sources of HONO include vehicle emissions,9,10 biomass burning,11–13 microbial activities in soils14,15 and associated with biocrusts.16 Laboratory studies have shown that photoenhanced HONO production can occur following NO2 uptake to surfaces including soot,17 aromatic species,18 humic acids19,20 and TiO2.21 Photoenhanced NO2 heterogeneous reactions on ‘urban grime’ on building surfaces22 have also been shown to generate appreciable levels of HONO. Further potential sources are photolysis of nitric acid,23 nitrophenols24 and of particulate nitrate.25–27
Many field and laboratory studies have been carried out to investigate the sources of HONO in different environments, e.g. urban, rural, coastal, forest and vehicle tunnels.5,9,28–34 In a recent study, Tong, et al.35 performed measurements at urban and suburban environments in Beijing during winter, and observed that direct emission and homogenous gas phase sources made a larger contribution in urban areas, while heterogeneous sources were suggested to be more significant away from urban centres. A recurrent challenge remains quantitatively reconciling daytime HONO concentrations in many urban environments with the current known sources.5–7
Here, we take advantage of a near-total solar eclipse (∼90% attenuation) as a natural short-term perturbation to atmospheric photochemistry to explore the chemical processes affecting HONO abundance. The reduction in photolysis frequencies during an eclipse is effectively uniform over a large area (relative to the chemical lifetime and hence spatial footprint of HONO, NOx and related species), of relevance to the challenges of heterogeneity.36,37 Comparison of observed temporal behaviour during the eclipse therefore provides a unique test of our understanding of HONO photochemistry. We report the temporal variation of HONO, NOx, O3 and aerosol characteristics during the eclipse, and use these measurements to explore the nature of sources of HONO which are consistent with their behaviour.
2 Observations during a solar eclipse
HONO, NOx, O3 and particle number concentration were measured at an urban background monitoring station situated (52.45° N, 1.93° W) within the Edgbaston campus of the University of Birmingham, UK. Possible anthropogenic emission sources located nearby include a suburban rail line (north-west, ca. 90 m away), and a suburban road (east, 125 m away). HONO was measured using a long-path absorption photometer (LOPAP).38 Briefly, the LOPAP is a wet chemical technique, where gas phase HONO is sampled, using a stripping coil, into an acidic solution and is derivatived into an azo dye. The light absorption at 550 nm of the azo dye is then measured with a spectrometer using an optical path length of 2.4 m. The LOPAP was operated and calibrated according to the standard procedures,39 with a sampling height of 3 m above ground level, and data acquired at 5 min time resolution. Baseline (zero) measurements, obtained by sampling zero air, were taken at frequent intervals (8 hours). The detection limit (2σ) under the instrument operating conditions of these measurements was determined to be 6 pptV. In addition to the LOPAP, co-located measurements of NO, NO2 and NOx (Thermo Scientific 42c-Mo convertor for NO2 measurement, hence potentially responding to NOy interferences), and ozone (Thermo Scientific 49i) were performed. An optical particle spectrophotometer (TSI 3330) measured the particle number size distribution, and converted from particle number concentration (PNC) to total surface area (TSA) via the TSI AIM software (version 9), which assumes particle sphericity. NOx, O3 and particle data were obtained at a 1 min time resolution. Meteorological data (relative humidity, solar intensity, air temperature, and wind speed and direction) was obtained at 1 min time resolution from a nearby weather station operated by the University of Birmingham/UK Met Office, located within 100 m of the EROS site. At the University of Birmingham site, the eclipse (20th March 2015) occurred from 08:25 to 10:40 GMT (first/last contact) with the maximum coverage (89% of the sun's face) occurring at 9:31.403 Approach to exploring the anticipated response of HONO during the solar eclipse
HONO abundance is expected to respond to rapid changes in light conditions (i.e. with and without the shading condition of the solar eclipse), to an extent dependent upon the dominant HONO source and sink mechanisms, and their respective variation with solar radiation levels. To explore the variation in HONO (and other species) abundance anticipated across the eclipse timeframe (ca. 2 hours) a series of simple simulations were performed representing different scenarios (mechanisms) for potential atmospheric processes/emissions that may form and remove HONO. We do not attempt to reproduce the detailed gas- or heterogeneous chemistry using a formal model or chemical mechanism – as many of the species that would be required were not measured (notably, no VOC data were available, precluding modelling HOx sources or sinks). However, with some assumptions (below), over the limited duration of the eclipse event, the time evolution of HONO can be calculated from its initial abundance, the time variation of various possible source term(s), and its chemical removal which is dominated (≥95%, see below) by photolysis. This allows us to explore the response of potential HONO source(s), parameterised in terms of the measured variation of sunlight, NOx abundance and aerosol particle parameters, by comparison with the observed HONO variation over the course of the eclipse.The attenuation of photolysis frequencies was approximated using values obtained from the TUV model41 for clear-sky conditions, with the reduction in j values modelled as a 1-π sine wave. We note that this is an approximation, both to the actual photolysis frequencies and the geometric coverage of the solar disk, but one which is acceptable in the context of the analysis which follows. The calculated jNO2 values prior to and subsequent to the eclipse were consistent with those that would be estimated using photo-stationary steady state (NOx/O3 abundance, neglecting HO2/RO2 reactions) to within 5.6 ± 1.5% (pre-eclipse), 21 ± 1.9% (during the eclipse) and 6.4 ± 2.0% (post-eclipse). The different scenarios examined are described in Table 1. In each case, the calculation was initiated at 07:30 GMT, prior to the start of the eclipse, and the subsequent evolution of HONO calculated according to the various alternative mechanisms presented below. In general, each case may be represented viaeqn (1), where Sx is the source term for HONO formation in each scenario:
| d[HONO]/dt = Sx − jHONO [HONO] | (1) |
| Cases | Potential sources | HONO source strength (S)/ppb s−1 | Scale factor (C) | Initial HONO ([HONO]t=0)/ppb | Fit RMS residual/ppb |
|---|---|---|---|---|---|
| Case 1 | No source term | S 1 = 0 | N/A | 1.2 | 0.325 |
| Case 2 | Photolysis-related | S 2 = C × jNO2 | 7.2 × 10−2 ppb | 1.04 | 0.118 |
| Case 3 | NO2-related | S 3 = C × [NO2] | 1.57 × 10−5 s−1 | 0.71 | 0.067 |
| Case 4 | Source(s) related to photolysis and NO2 | S 4 = C × jNO2 × [NO2] | 3.99 × 10−3 | 1.0 | 0.096 |
| Case 5 | Source(s) related to aerosol surface area (TSA) | S 5 = C × TSA | 1.65 × 10−6 ppb s−1 (μm2 cm−3)−1 | 0.68 | 0.078 |
| Case 6 | Source(s) related to photolysis and TSA | S 6 = C × jNO2 × TSA | 4.42 × 10−4 ppb (μm2 cm−3)−1 | 0.97 | 0.090 |
| Case 7 | Source(s) related to NO2 and TSA | S 7 = C × [NO2] × TSA | 7.61 × 10−8 s−1 (μm2 cm−3)−1 | 0.67 | 0.105 |
| Case 8 | Source(s) related to photolysis, NO2 and TSA | S 8 = C × jNO2 × [NO2] × TSA | 2.31 × 10−5 (μm2 cm−3)−1 | 0.94 | 0.075 |
The different HONO sources considered, Sx, are listed in Table 1. In each case HONO concentrations were calculated by numerically evaluating the differential equations for HONO formation/removal, using time-dependent observed values of the input parameters (jNO2, [NO2] etc), on a 60 second timestep.
A number of assumptions are inherent in this approach: eqn (1) neglects HONO sinks other than photolysis (i.e., reaction with OH). Using OH measurements performed near to the measurement site during a previous campaign,42,43 the rate of loss of HONO due to reaction with OH is calculated to be 1.1–5.0% (using [OH] of 2 × 106 to 9 × 106 mol cm−3) of that due to photolysis, at the lowest photolysis frequency corresponding to the eclipse maximum, hence this approximation is reasonable. Eqn (1) also neglects homogeneous sources of HONO – from the (slow) termolecular OH + NO chemistry, which we estimate would contribute <7% of the HONO production necessary to maintain the observed levels (Section 4.2 below) and would, to an extent, offset the HONO + OH reaction. We also discount the proposed, but uncertain, 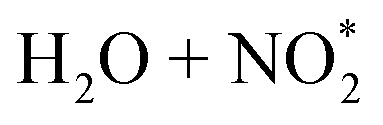 mechanism.44–46 A number of assumptions regarding advection/mixing are made: we assume air mass homogeneity over the timescale of the eclipse period, and neglect vertical mixing. The former assumption is likely justified considering the background nature of the measurement site and limited timespan of the analysis; recent measurements of vertical profiles of HONO in urban areas47 point to a broadly uniform profile near to the ground (<200 m), with some increase near ground level attributed to traffic source mixing and ground surface production – although the measurement period spans the early morning time when boundary layer breakup/enhanced vertical mixing may occur. Finally, each potential HONO source mechanism is considered in isolation, while in reality multiple mechanisms likely occur in parallel. These and other assumptions are considered further in the discussion, below.
mechanism.44–46 A number of assumptions regarding advection/mixing are made: we assume air mass homogeneity over the timescale of the eclipse period, and neglect vertical mixing. The former assumption is likely justified considering the background nature of the measurement site and limited timespan of the analysis; recent measurements of vertical profiles of HONO in urban areas47 point to a broadly uniform profile near to the ground (<200 m), with some increase near ground level attributed to traffic source mixing and ground surface production – although the measurement period spans the early morning time when boundary layer breakup/enhanced vertical mixing may occur. Finally, each potential HONO source mechanism is considered in isolation, while in reality multiple mechanisms likely occur in parallel. These and other assumptions are considered further in the discussion, below.
In each case, the initial HONO concentration [HONO]t=0, and the source mechanism scaling constant C (Table 1) was determined from the measured values of HONO, NOx, jNO2 and total aerosol surface area (TSA) as appropriate. Both [HONO]t=0 and C were optimised independently, for each source scenario, by minimising the sum of squares of residuals between calculated and observed HONO concentrations for the two 1 hour periods perior to and immediately following the eclipse period (from 07:30–08:30 and 10:40–11:40 respectively). We adopt the pre/post eclipse period to allow prediction of hypothetical, no-eclipse HONO concentrations; we test the sensitivity of this time period selection by repeating the fitting including both the pre/post and eclipse periods (ESI, Fig. S1†); while the optimised parameters vary slightly the conclusions regarding which mechanism(s) are consistent/inconsistent with the data are unchanged). All other species were set to their actual observed levels for the relevant point in time. It is important to note that we do not assume HONO is in steady state/equilibrium with its production and removal terms. For each source scenario, we repeat the analysis under two conditions – firstly for the actual (eclipse influenced) reduction in solar intensity (photolysis frequencies), and secondly, for comparison, a hypothetical scenario where no eclipse occurred and clear-sky photolysis (as derived from the changing SZA) applied throughout the 2 hour time period. The resulting predicted and observed HONO concentrations are shown in Fig. 2, and discussed below.
4 Results & discussion
4.1 Observations
During the eclipse, weather conditions were calm (wind speed of 0.5 ± 0.4 m s−1) with a mean relative humidity of 76 ± 6% and temperature of 6.7 ± 1.7 °C, which are typical for the time of year in the UK. In particular, the usual rise in temperature with daytime was observed, which may have begun the process of boundary layer break-up, although there was little evidence for change in NOx or PM levels associated with this. Relatively little change in HONO concentration was observed during the eclipse period (Fig. 1a), suggesting that closely balanced photochemical source and photochemical sink mechanisms may be dominating HONO abundance. Ozone and NOx were observed to be tightly coupled during eclipse period (08:25 am to 10:40 am GMT, Fig. 1b), and to follow their expected photochemistry,48 with NO and O3 levels falling and NO2 rising. During the eclipse, little variation was observed in the particle TSA (Fig. 1c), which was found to be reasonably well correlated with the HONO concentration (R2 = 0.63), possibly suggesting a common source or dilution process. Vehicle emissions have been identified as a significant source of HONO in urban environments, where a low HONO/NOx ratio is commonly used as a proxy parameter to evaluate HONO emissions from traffic.3,49,50 In the present study, high HONO/NO2 (1.9–4.1%) and HONO/NOx (1.0–1.8%) ratios were observed before, during and after the eclipse. Previous empirically determined emission ratios of HONO/NOx based on tunnel and car exhaust studies are between 0.3% and 1.0%,9,51 with recent evaluations at the high end of this range, e.g. 1.24 ± 0.35%50 and 0.72–1.01%33 (determined in Birmingham). Thus the measured HONO/NOx ratio is at the top end of/in excess of the ratio of expected for vehicle emissions – suggesting that these are not the sole source of HONO at this site.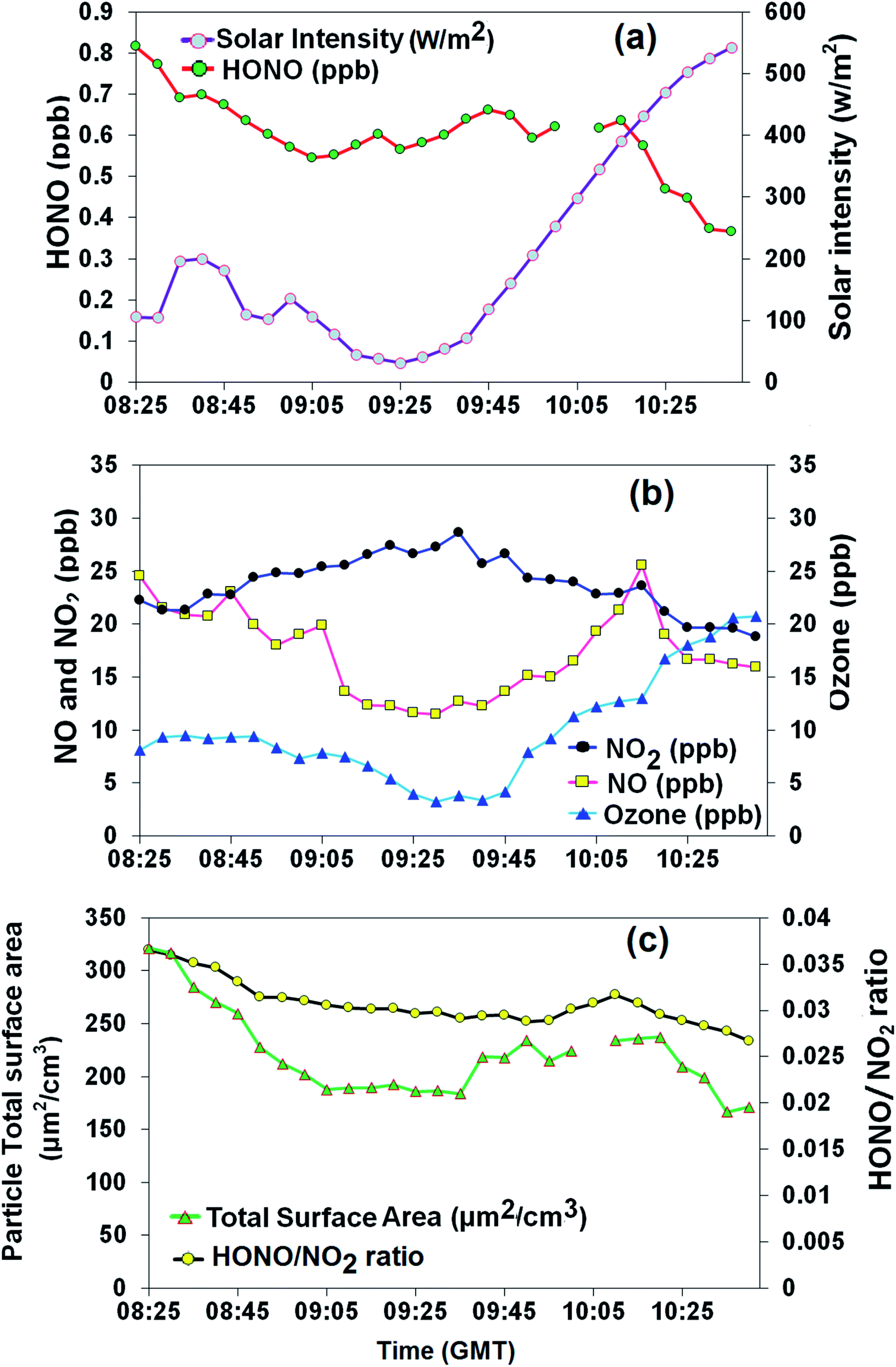 | ||
| Fig. 1 Time series of (a) measured HONO and solar intensity, (b) NOx and ozone, and (c) particle total surface area (TSA) and HONO/NO2 ratio. | ||
4.2 Evaluation of HONO source scenarios
Fig. 2 compares the observed (red line/points) and simulated (actual, eclipse condition – solid blue line; hypothetical no eclipse condition – dashed blue line) time evolution of the HONO abundance for each scenario considered. Case 1 (Fig. 2A) considers a scenario where no HONO source exists; this clearly diverges from the observed behaviour, indicating that additional HONO sources are in fact present. Note that the fit (minimised RMS residual) is affected by inclusion of the second fitting period, after the eclipse – but in which there is little sensitivity to the initial HONO level, due to the rapid photolytic decay. The reaction of OH radicals with NO forms HONO (via(R2)); no OH data were available during these experiments, but OH levels may be estimated from prior observations at this location (Heard, et al.42 – taking the mean OH level for 09:30 from these measurements (∼5 × 105 mol cm−3), a steady state HONO level of 0.04 ppb is obtained, approximately a factor of 15 lower than that observed mid-eclipse, indicating that OH + NO is not responsible for HONO formation in isolation, and rather forms a small contributor to the total HONO source. The role of OH + NO as a secondary HONO production term, alongside other, larger mechanisms, is considered further below.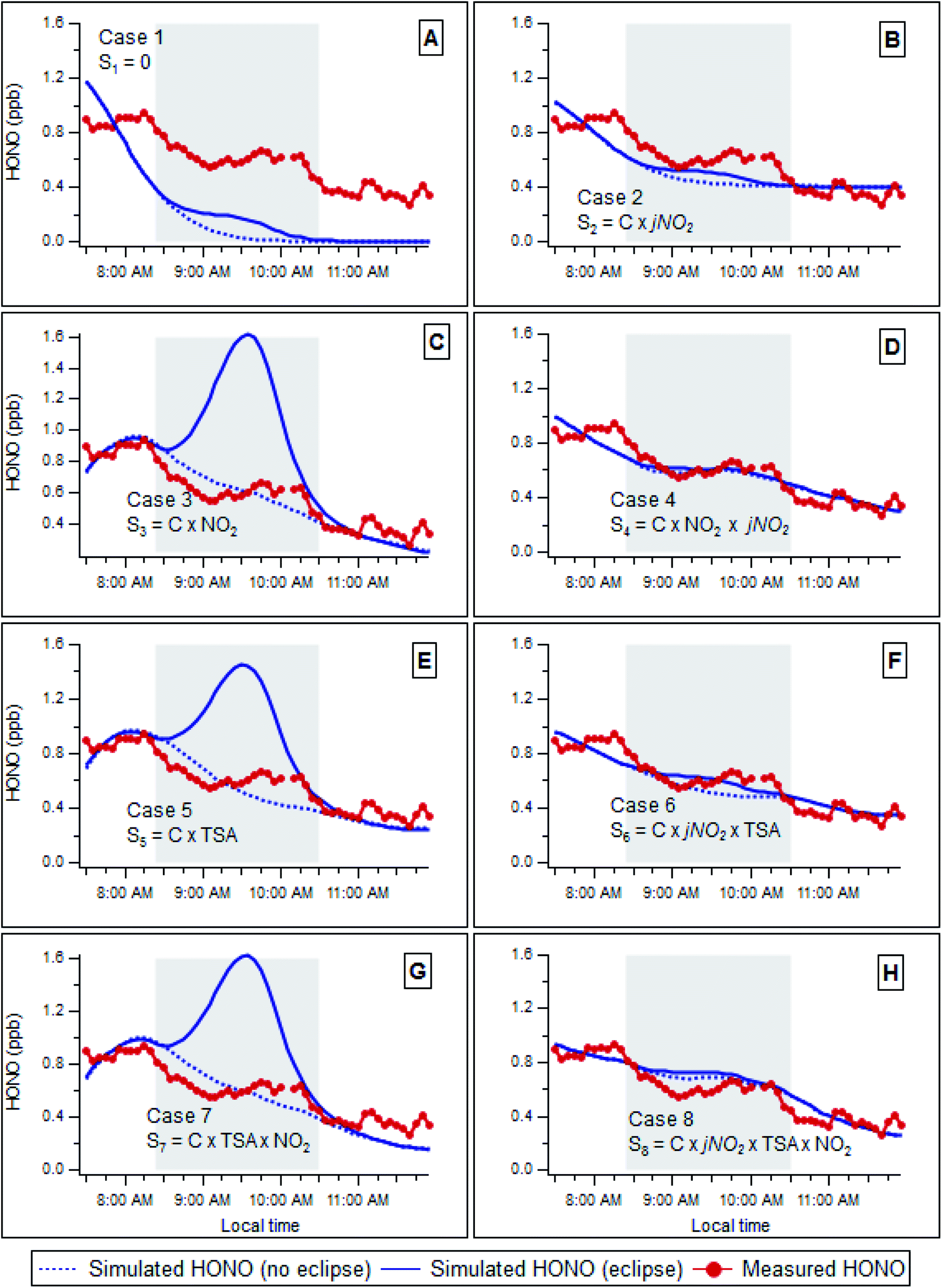 | ||
| Fig. 2 Comparison between measured (red points) and calculated HONO mixing ratio during the solar eclipse, using actual eclipse photolysis frequencies (solid blue line) and (hypothetical) non-eclipse photolysis frequencies (dotted blue line). Shading indicates eclipse duration from first to last contact. Source scenarios (panels A–H) as defined in Table 1. | ||
Previous work in urban areas has suggested a daytime source of HONO that scaled with NO2 levels and sunlight, specifically jNO2.7,52 Case 2 considers a HONO source term which is directly proportional to jNO2. The predicted HONO behaviour broadly matches that observed, indicating that in reality, the predominant source is likely to scale with jNO2 or some similar measure of solar insolation. Dark conversion of NO2 to HONO upon moist surfaces is a long-established HONO formation mechanism (e.g. Finlayson-Pitts, et al.53), and case 3 considers a HONO source term which is directly proportional to NO2 – however this scenario shows a poor fit of predicted to observed HONO levels, indicating that the predominant source does not scale with [NO2] in isolation. Photosensitized conversion of NO2 on surfaces containing organics has been identified as a significant daytime HONO source in a number of studies.18,19 To explore the effect of such reactions, a HONO source which scaled with the product of jNO2 and the NO2 mixing ratio was explored in case 4 (Fig. 2D), where a significantly better fit was observed, indicating that a source term which scaled with jNO2 × [NO2] could account for the majority of HONO production in this environment.
Direct emission of HONO from vehicles is another potential source in urban areas.9,33,50,51 In an urban area, particle total surface area (TSA) can be used as a reasonably conserved tracer for vehicle emissions.54 Case 5 (Fig. 2E) considers a scenario where the HONO source scales with the TSA; however this also does not match the measured HONO levels during the eclipse, implying that a source scaled to particle TSA alone – or direct vehicular emission of HONO in isolation – cannot explain the observed HONO concentrations in this environment. Similar results were observed when particle number concentration was used as a metric of traffic particulate matter emissions, as opposed to TSA (not shown). However, HONO production proportional to the product of jNO2 and TSA was a better match to observed (Case 6, Fig. 2F).
Cases 7 and 8 (Fig. 2G and H respectively) represent HONO formation via NO2 conversion on the surface of aerosol particles, without (case 7/Fig. 2G) and with (case 8/Fig. 2H) photo-enhancement respectively. These represent secondary chemical (photochemical) sources involving aerosol particles. In these cases, the HONO production rate is assumed to scale with TSA × [NO2] and jNO2 × TSA × [NO2], respectively. Case 7 did not reproduce the observed behaviour, indicating that such heterogeneous aerosol conversion of NO2 to HONO, without photoenhancement, could not, in isolation, account for HONO production in this environment. Case 8 showed significantly closer reproduction of the measured HONO evolution under the eclipse conditions, giving the best fit (lowest RMS residual) of all the scenarios considered, and indicating that a mechanism dependent upon solar insolation (jNO2), NO2 abundance and aerosol surface area is able to reproduce the observed HONO behaviour across the eclipse. The similarity of eclipse- and non-eclipse simulations reflects the balanced photolytic dependence – as observed for the actual HONO. While we obtain the optimal fit for case 8, in comparison to (e.g.) case 2, the geometric surface area of ground surfaces significantly exceeds that of aerosol particles (by at least a factor of 16-fold, assuming an aerosol surface area of 300 μm2 cm−3 and boundary layer height of 200 m, which may be representative of the measurement location early in the morning – low winds, under an eclipse condition), and hence it may be likely that ground surface-mediated photoenhanced conversion of NO2 dominated HONO formation at this location, potentially augmented by contributions from aerosol surfaces.
In reality, it is likely that a combination of mechanisms occur in parallel, and addition of the OH + NO reaction, with OH levels assumed equal to those measured42 and scaled to the relative solar intensity expected during the eclipse (which may underestimate OH, given the importance of non-photolytic sources such as alkene ozonolysis at this location in wintertime reported previously by Heard, et al.42 resulted in a slight improvement to the statistical fit – although increasing degrees of freedom would be expected to improve agreement. As a further evaluation of the performance of scenario 8, we applied this analysis approach to data from the days preceding and following the eclipse event (i.e. 19th and 21st March respectively); the simulation was able to satisfactorily reproduce the observed HONO levels (ESI, Fig. S2†). We note that in cases 3, 4, 7 and 8 the NOx would vary due to changing PSS in the (hypothetical) non-eclipse case. This approximation has the effect of biasing the non-eclipse (hypothetical) simulations high (as the real NO2 would be lower, in the absence of the eclipse-derived photolysis attenuation).
4.3 Insight from temporal variation of HONO production
A further analysis was undertaken to derive the cumulative HONO production with time, i.e. to remove the effect of the loss of HONO through photolysis under both the real (eclipse) and hypothetical (non-eclipse) conditions. This quantity – hypothetically conserved HONO, [HONO]conserved, was calculated using eqn (2):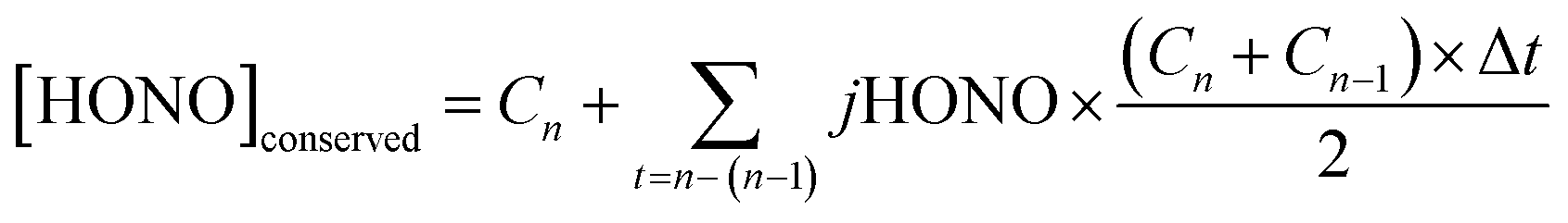 | (2) |
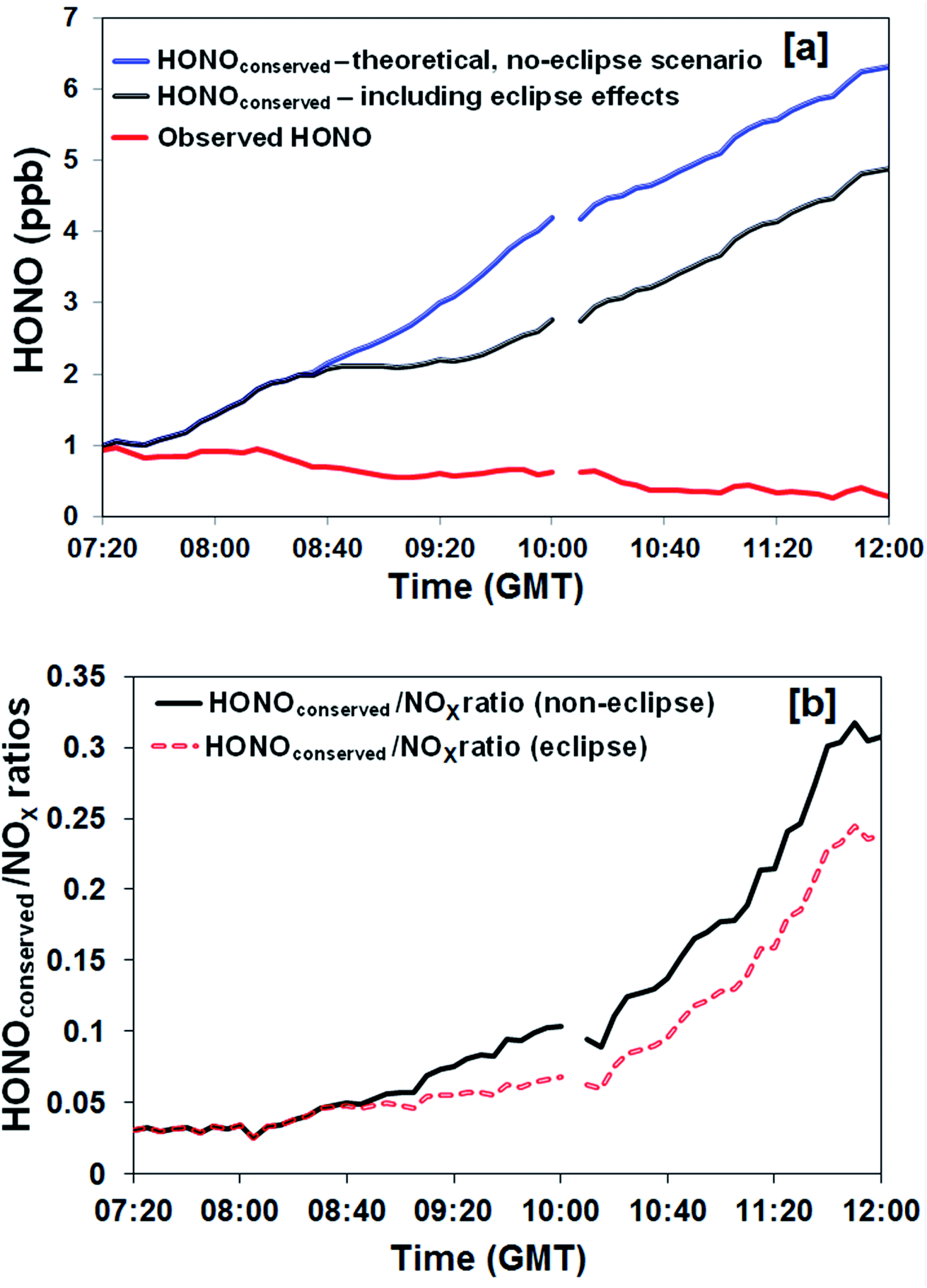 | ||
| Fig. 3 (a) Comparison between observed HONO mixing ratios, and HONOconserved (the HONO abundance calculated after accounting for photolytic loss) under eclipse and (hypothetical) non-eclipse scenarios, (b) variation in HONOconserved/NOx ratio under actual (eclipse) and hypothetical (non-eclipse) conditions. | ||
5 Concluding remarks
HONO, NOx, O3 and particle number size distributions were measured at an urban background location over the course of a near-total solar eclipse in Birmingham, UK. The observed NO, NO2 and O3 responded as anticipated from the well-understood atmospheric photochemistry. HONO levels showed limited variation across the eclipse period; given HONO removal is overwhelmingly dominated by photolysis, this observation indicates that photolytic sources predominate in producing HONO at this location. A simplified approach was adopted to explore which factors were required within the HONO source mechanism, assuming direct and multiplicative proportionality, by comparing the observed and predicted HONO levels across the eclipse perturbation. Source terms lacking a photolytic component – proportional to NO2 concentration, aerosol particle number or aerosol particle surface area – were not consistent with the observed behaviour. Source terms with an insolation dependence, here represented as jNO2, better replicated the measurements, with strong evidence for the involvement of NO2, and the best agreement (in isolation) obtained for a combination of jNO2, NO2 concentration and aerosol surface area. Both the observed source behaviour, and the temporal variation in the HONOconserved levels and the HONOconserved/NOx ratio, were inconsistent with primary traffic emissions dominating HONO production in this urban background location – although of course road traffic is ultimately the dominant emission source for NOx, and hence the key HONO precursor NO2 identified here. Accordingly, future vehicle fleet evolution, which is expected to lead to significantly lower urban NOx levels through improved emissions technologies and penetration of electric vehicles, will also lead to lower HONO concentrations, and hence lower local OH radical production and local atmospheric oxidation in urban boundary layer locations such as that considered here.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Birmingham for supporting Ajit Singh through the Elite Scholarship Scheme. The authors would also like to acknowledge UK Natural Environmental Research Council project SNAABL (Sources of Nitrous Acid in the Atmospheric Boundary Layer; NE/M013545/1) for funding this research.References
- B. W. LaFranchi, G. M. Wolfe, J. A. Thornton, S. A. Harrold, E. C. Browne, K. E. Min, P. J. Wooldridge, J. B. Gilman, W. C. Kuster, P. D. Goldan, J. A. de Gouw, M. McKay, A. H. Goldstein, X. Ren, J. Mao and R. C. Cohen, Closing the peroxy acetyl nitrate budget: observations of acyl peroxy nitrates (PAN, PPN, and MPAN) during BEARPEX 2007, Atmos. Chem. Phys., 2009, 9, 7623–7641, DOI:10.5194/acp-9-7623-2009.
- X. Ren, H. Gao, X. Zhou, J. D. Crounse, P. O. Wennberg, E. C. Browne, B. W. LaFranchi, R. C. Cohen, M. McKay, A. H. Goldstein and J. Mao, Measurement of atmospheric nitrous acid at Bodgett Forest during BEARPEX2007, Atmos. Chem. Phys., 2010, 10, 6283–6294, DOI:10.5194/acp-10-6283-2010.
- J. Kleffmann, Daytime sources of nitrous acid (HONO) in the atmospheric boundary layer, ChemPhysChem, 2007, 8, 1137–1144, DOI:10.1002/cphc.200700016.
- F. Spataro and A. Ianniello, Sources of atmospheric nitrous acid: state of the science, current research needs, and future prospects, J. Air Waste Manage. Assoc., 2014, 64, 1232–1250, DOI:10.1080/10962247.2014.952846.
- J. Kleffmann, T. Gavriloaiei, A. Hofzumahaus, F. Holland, R. Koppmann, L. Rupp, E. Schlosser, M. Siese and A. Wahner, Daytime formation of nitrous acid: a major source of OH radicals in a forest, Geophys. Res. Lett., 2005, 32, L05818, DOI:10.1029/2005gl022524.
- V. Michoud, A. Colomb, A. Borbon, K. Miet, M. Beekmann, M. Camredon, B. Aumont, S. Perrier, P. Zapf, G. Siour, W. Ait-Helal, C. Afif, A. Kukui, M. Furger, J. C. Dupont, M. Haeffelin and J. F. Doussin, Study of the unknown HONO daytime source at a European suburban site during the MEGAPOLI summer and winter field campaigns, Atmos. Chem. Phys., 2014, 14, 2805–2822, DOI:10.5194/acp-14-2805-2014.
- J. D. Lee, L. K. Whalley, D. E. Heard, D. Stone, R. E. Dunmore, J. F. Hamilton, D. E. Young, J. D. Allan, S. Laufs and J. Kleffmann, Detailed budget analysis of HONO in central London reveals a missing daytime source, Atmos. Chem. Phys., 2016, 16, 2747–2764, DOI:10.5194/acp-16-2747-2016.
- B. J. Finlayson-Pitts, L. M. Wingen, A. L. Sumner, D. Syomin and K. A. Ramazan, The heterogeneous hydrolysis of NO2 in laboratory systems and in outdoor and indoor atmospheres: an integrated mechanism, Phys. Chem. Chem. Phys., 2003, 5, 223–242, 10.1039/b208564J.
- R. Kurtenbach, K. H. Becker, J. A. G. Gomes, J. Kleffmann, J. C. Lörzer, M. Spittler, P. Wiesen, R. Ackermann, A. Geyer and U. Platt, Investigations of emissions and heterogeneous formation of HONO in a road traffic tunnel, Atmos. Environ., 2001, 35, 3385–3394, DOI:10.1016/s1352-2310(01)00138-8.
- B. Rappenglück, G. Lubertino, S. Alvarez, J. Golovko, B. Czader and L. Ackermann, Radical precursors and related species from traffic as observed and modeled at an urban highway junction, J. Air Waste Manage. Assoc., 2013, 63, 1270–1286, DOI:10.1080/10962247.2013.822438.
- R. J. Yokelson, J. D. Crounse, P. F. DeCarlo, T. Karl, S. Urbanski, E. Atlas, T. Campos, Y. Shinozuka, V. Kapustin, A. D. Clarke, A. Weinheimer, D. J. Knapp, D. D. Montzka, J. Holloway, P. Weibring, F. Flocke, W. Zheng, D. Toohey, P. O. Wennberg, C. Wiedinmyer, L. Mauldin, A. Fried, D. Richter, J. Walega, J. L. Jimenez, K. Adachi, P. R. Buseck, S. R. Hall and R. Shetter, Emissions from biomass burning in the Yucatan, Atmos. Chem. Phys., 2009, 9, 5785–5812, DOI:10.5194/acp-9-5785-2009.
- J. M. Roberts, P. Veres, C. Warneke, J. A. Neuman, R. A. Washenfelder, S. S. Brown, M. Baasandorj, J. B. Burkholder, I. R. Burling, T. J. Johnson, R. J. Yokelson and J. de Gouw, Measurement of HONO, HNCO, and other inorganic acids by negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS): application to biomass burning emissions, Atmos. Meas. Tech., 2010, 3, 981–990, DOI:10.5194/amt-3-981-2010.
- N. Theys, R. Volkamer, J. F. Müller, K. J. Zarzana, N. Kille, L. Clarisse, I. De Smedt, C. Lerot, H. Finkenzeller, F. Hendrick, T. K. Koenig, C. F. Lee, C. Knote, H. Yu and M. Van Roozendael, Global nitrous acid emissions and levels of regional oxidants enhanced by wildfires, Nat. Geosci., 2020, 13, 681–686, DOI:10.1038/s41561-020-0637-7.
- H. Su, Y. Cheng, R. Oswald, T. Behrendt, I. Trebs, F. X. Meixner, M. O. Andreae, P. Cheng, Y. Zhang and U. Pöschl, Soil nitrite as a source of atmospheric HONO and OH radicals, Science, 2011, 333, 1616–1618, DOI:10.1126/science.1207687.
- R. Oswald, T. Behrendt, M. Ermel, D. Wu, H. Su, Y. Cheng, C. Breuninger, A. Moravek, E. Mougin, C. Delon, B. Loubet, A. Pommerening-Röser, M. Sörgel, U. Pöschl, T. Hoffmann, M. O. Andreae, F. X. Meixner and I. Trebs, HONO emissions from soil bacteria as a major source of atmospheric reactive nitrogen, Science, 2013, 341, 1233–1235, DOI:10.1126/science.1242266.
- B. Weber, D. Wu, A. Tamm, N. Ruckteschler, E. Rodríguez-Caballero, J. Steinkamp, H. Meusel, W. Elbert, T. Behrendt, M. Sörgel, Y. Cheng, P. J. Crutzen, H. Su and U. Pöschl, Biological soil crusts accelerate the nitrogen cycle through large NO and HONO emissions in drylands, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15384–15389, DOI:10.1073/pnas.1515818112.
- M. E. Monge, B. D'Anna, L. Mazri, A. Giroir-Fendler, M. Ammann, D. Donaldson and C. George, Light changes the atmospheric reactivity of soot, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6605–6609, DOI:10.1073/pnas.0908341107.
- C. George, R. S. Strekowski, J. Kleffmann, K. Stemmler and M. Ammann, Photoenhanced uptake of gaseous NO2 on solid organic compounds: a photochemical source of HONO?, Faraday Discuss., 2005, 130, 195–210, 10.1039/b417888m.
- K. Stemmler, M. Ammann, C. Donders, J. Kleffmann and C. George, Photosensitized reduction of nitrogen dioxide on humic acid as a source of nitrous acid, Nature, 2006, 440, 195–198, DOI:10.1038/nature04603.
- S. Laufs, M. Cazaunau, P. Stella, R. Kurtenbach, P. Cellier, A. Mellouki, B. Loubet and J. Kleffmann, Diurnal fluxes of HONO above a crop rotation, Atmos. Chem. Phys., 2017, 17, 6907–6923, DOI:10.5194/acp-17-6907-2017.
- J. M. Langridge, R. J. Gustafsson, P. T. Griffiths, R. A. Cox, R. M. Lambert and R. L. Jones, Solar driven nitrous acid formation on building material surfaces containing titanium dioxide: a concern for air quality in urban areas?, Atmos. Environ., 2009, 43, 5128–5131, DOI:10.1016/j.atmosenv.2009.06.046.
- A. M. Baergen and D. J. Donaldson, Photochemical renoxification of nitric acid on real urban grime, Environ. Sci. Technol., 2013, 47, 815–820, DOI:10.1021/es3037862.
- X. Zhou, H. Gao, Y. He, G. Huang, S. B. Bertman, K. Civerolo and J. Schwab, Nitric acid photolysis on surfaces in low-NOx environments: significant atmospheric implications, Geophys. Res. Lett., 2003, 30, 2217, DOI:10.1029/2003gl018620.
- I. Bejan, Y. A. El Aal, I. Barnes, T. Benter, B. Bohn, P. Wiesen and J. Kleffmann, The photolysis of ortho-nitrophenols: a new gas phase source of HONO, Phys. Chem. Chem. Phys., 2006, 8, 2028–2035, 10.1039/b516590c.
- L. Wang, L. Wen, C. Xu, J. Chen, X. Wang, L. Yang, W. Wang, X. Yang, X. Sui, L. Yao and Q. Zhang, HONO and its potential source particulate nitrite at an urban site in North China during the cold season, Sci. Total Environ., 2015, 538, 93–101, DOI:10.1016/j.scitotenv.2015.08.032.
- C. Ye, X. Zhou, D. Pu, J. Stutz, J. Festa, M. Spolaor, C. Tsai, C. Cantrell, R. L. Mauldin, T. Campos, A. Weinheimer, R. S. Hornbrook, E. C. Apel, A. Guenther, L. Kaser, B. Yuan, T. Karl, J. Haggerty, S. Hall, K. Ullmann, J. N. Smith, J. Ortega and C. Knote, Rapid cycling of reactive nitrogen in the marine boundary layer, Nature, 2016, 532, 489–491, DOI:10.1038/nature17195.
- C. Reed, M. J. Evans, L. R. Crilley, W. J. Bloss, T. Sherwen, K. A. Read, J. D. Lee and L. J. Carpenter, Evidence for renoxification in the tropical marine boundary layer, Atmos. Chem. Phys., 2017, 17, 4081–4092, DOI:10.5194/acp-17-4081-2017.
- J. Stutz, B. Alicke, R. Ackermann, A. Geyer, S. Wang, A. B. White, E. J. Williams, C. W. Spicer and J. D. Fast, Relative humidity dependence of HONO chemistry in urban areas, J. Geophys. Res.: Atmos., 2004, 109, D03307, DOI:10.1029/2003jd004135.
- X. Li, T. Brauers, R. Häseler, B. Bohn, H. Fuchs, A. Hofzumahaus, F. Holland, S. Lou, K. D. Lu, F. Rohrer, M. Hu, L. M. Zeng, Y. H. Zhang, R. M. Garland, H. Su, A. Nowak, A. Wiedensohler, N. Takegawa, M. Shao and A. Wahner, Exploring the atmospheric chemistry of nitrous acid (HONO) at a rural site in Southern China, Atmos. Chem. Phys., 2012, 12, 1497–1513, DOI:10.5194/acp-12-1497-2012.
- J. Kleffmann, R. Kurtenbach, J. Lörzer, P. Wiesen, N. Kalthoff, B. Vogel and H. Vogel, Measured and simulated vertical profiles of nitrous acid—Part I: Field measurements, Atmos. Environ., 2003, 37, 2949–2955, DOI:10.1016/S1352-2310(03)00242-5.
- K. Acker, A. Febo, S. Trick, C. Perrino, P. Bruno, P. Wiesen, D. Möller, W. Wieprecht, R. Auel, M. Giusto, A. Geyer, U. Platt and I. Allegrini, Nitrous acid in the urban area of Rome, Atmos. Environ., 2006, 40, 3123–3133, DOI:10.1016/j.atmosenv.2006.01.028.
- H. Meusel, U. Kuhn, A. Reiffs, C. Mallik, H. Harder, M. Martinez, J. Schuladen, B. Bohn, U. Parchatka, J. N. Crowley, H. Fischer, L. Tomsche, A. Novelli, T. Hoffmann, R. H. H. Janssen, O. Hartogensis, M. Pikridas, M. Vrekoussis, E. Bourtsoukidis, B. Weber, J. Lelieveld, J. Williams, U. Pöschl, Y. Cheng and H. Su, Daytime formation of nitrous acid at a coastal remote site in Cyprus indicating a common ground source of atmospheric HONO and NO, Atmos. Chem. Phys., 2016, 16, 14475–14493, DOI:10.5194/acp-16-14475-2016.
- L. J. Kramer, L. R. Crilley, T. J. Adams, S. M. Ball, F. D. Pope and W. J. Bloss, Nitrous acid (HONO) emissions under real-world driving conditions from vehicles in a UK road tunnel, Atmos. Chem. Phys., 2020, 20, 5231–5248, DOI:10.5194/acp-20-5231-2020.
- W. J. Bloss, L. Kramer, L. R. Crilley, T. Vu, R. M. Harrison, Z. Shi, J. D. Lee, F. A. Squires, L. K. Whalley, E. Slater, R. Woodward-Massey, C. Ye, D. E. Heard, S. Tong, S. Hou, Y. Sun, J. Xu, L. Wei and P. Fu, Insights into air pollution chemistry and sulphate formation from nitrous acid (HONO) measurements during haze events in Beijing, Faraday Discuss., 2021, 226, 223–238, 10.1039/d0fd00100g.
- S. Tong, S. Hou, Y. Zhang, B. Chu, Y. Liu, H. He, P. Zhao and M. Ge, Exploring the nitrous acid (HONO) formation mechanism in winter Beijing: direct emissions and heterogeneous production in urban and suburban areas, Faraday Discuss., 2016, 189, 213–230, 10.1039/c5fd00163c.
- L. R. Crilley, L. Kramer, F. D. Pope, L. K. Whalley, D. R. Cryer, D. E. Heard, J. D. Lee, C. Reed and W. Bloss, On the interpretation of in situ HONO observations via photochemical steady state, Faraday Discuss., 2016, 189, 191–212, 10.1039/c5fd00224a.
- B. H. Lee, E. C. Wood, S. C. Herndon, B. L. Lefer, W. T. Luke, W. H. Brune, D. D. Nelson, M. S. Zahniser and J. W. Munger, Urban measurements of atmospheric nitrous acid: a caveat on the interpretation of the HONO photostationary state, J. Geophys. Res.: Atmos., 2013, 118, 12,274–12,281, DOI:10.1002/2013jd020341.
- J. Heland, J. Kleffmann, R. Kurtenbach and P. Wiesen, A New Instrument To Measure Gaseous Nitrous Acid (HONO) in the Atmosphere, Environ. Sci. Technol., 2001, 35, 3207–3212, DOI:10.1021/es000303t.
- J. Kleffmann and P. Wiesen, Technical Note: Quantification of interferences of wet chemical HONO LOPAP measurements under simulated polar conditions, Atmos. Chem. Phys., 2008, 8, 6813–6822, DOI:10.5194/acp-8-6813-2008.
- E. Hanna, J. Penman, T. Jónsson, G. R. Bigg, H. Björnsson, S. Sjúrðarson, M. A. Hansen, J. Cappelen and R. G. Bryant, Meteorological effects of the solar eclipse of 20 March 2015: analysis of UK Met Office automatic weather station data and comparison with automatic weather station data from the Faroes and Iceland, Philos. Trans. R. Soc., A, 2016, 374, 20150212, DOI:10.1098/rsta.2015.0212.
- S. Madronich, S. Flocke, J. Zeng, I. Petropavlovskikh and J. Lee-Taylor, Tropospheric Ultraviolet and Visible (TUV) Radiation Model, National Center for Atmospheric Research, Boulder, CO, USA, 2003 Search PubMed.
- D. E. Heard, L. J. Carpenter, D. J. Creasey, J. R. Hopkins, J. D. Lee, A. C. Lewis, M. J. Pilling, P. W. Seakins, N. Carslaw and K. M. Emmerson, High levels of the hydroxyl radical in the winter urban troposphere, Geophys. Res. Lett., 2004, 31 DOI:10.1029/2004gl020544.
- K. M. Emmerson, N. Carslaw and M. J. Pilling, Urban Atmospheric Chemistry During the PUMA Campaign 2: Radical Budgets for OH, HO2 and RO2, J. Atmos. Chem., 2005, 52, 165–183, DOI:10.1007/s10874-005-1323-2.
- S. Li, J. Matthews and A. Sinha, Atmospheric hydroxyl radical production from electronically excited NO2 and H2O, Science, 2008, 319, 1657–1660, DOI:10.1126/science.1151443.
- S. Carr, D. E. Heard and M. A. Blitz, Comment on “Atmospheric Hydroxyl Radical Production from Electronically Excited NO2 and H2O”, Science, 2009, 324, 336, DOI:10.1126/science.1166669.
- T. J. Dillon and J. N. Crowley, Reactive quenching of electronically excited NO2* and NO3* by H2O as potential sources of atmospheric HOx radicals, Atmos. Chem. Phys., 2018, 18, 14005–14015, DOI:10.5194/acp-18-14005-2018.
- F. Meng, M. Qin, K. Tang, J. Duan, W. Fang, S. Liang, K. Ye, P. Xie, Y. Sun, C. Xie, C. Ye, P. Fu, J. Liu and W. Liu, High-resolution vertical distribution and sources of HONO and NO2 in the nocturnal boundary layer in urban Beijing, China, Atmos. Chem. Phys., 2020, 20, 5071–5092, DOI:10.5194/acp-20-5071-2020.
- P. A. Leighton, Photochemistry of air pollution, Academic Press, New York, 1960 Search PubMed.
- Y. Elshorbany, B. Steil, C. Brühl and J. Lelieveld, Impact of HONO on global atmospheric chemistry calculated with an empirical parameterization in the EMAC model, Atmos. Chem. Phys., 2012, 12, 9977–10000, DOI:10.5194/acp-12-9977-2012.
- Y. Liang, Q. Zha, W. Wang, L. Cui, K. H. Lui, K. F. Ho, Z. Wang, S.-c. Lee and T. Wang, Revisiting Nitrous Acid (HONO) Emission from On-road Vehicles: A Tunnel Study with a Mixed Fleet, J. Air Waste Manage. Assoc., 2017, 67, 797–805, DOI:10.1080/10962247.2017.1293573.
- T. W. Kirchstetter, R. A. Harley and D. Littlejohn, Measurement of nitrous acid in motor vehicle exhaust, Environ. Sci. Technol., 1996, 30, 2843–2849, DOI:10.1021/es960135y.
- K. Wong, C. Tsai, B. Lefer, C. Haman, N. Grossberg, W. Brune, X. Ren, W. Luke and J. Stutz, Daytime HONO vertical gradients during SHARP 2009 in Houston, TX, Atmos. Chem. Phys., 2012, 12, 635–652, DOI:10.5194/acp-12-635-2012.
- B. Finlayson-Pitts, L. Wingen, A. Sumner, D. Syomin and K. Ramazan, The heterogeneous hydrolysis of NO2 in laboratory systems and in outdoor and indoor atmospheres: an integrated mechanism, Phys. Chem. Chem. Phys., 2003, 5, 223–242, 10.1039/b208564j.
- G. Lv, C.-L. Song, S.-Z. Pan, J.-H. Gao and X.-F. Cao, Comparison of number, surface area and volume distributions of particles emitted from a multipoint port fuel injection car and a gasoline direct injection car, Atmos. Pollut. Res., 2014, 5, 753–758, DOI:10.5094/apr.2014.084.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ea00010a |
| This journal is © The Royal Society of Chemistry 2021 |