
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neutral and cationic germanium(IV) fluoride complexes with phosphine coordination – synthesis, spectroscopy and structures†
Rhys P.
King
,
William
Levason
 and
Gillian
Reid
*
and
Gillian
Reid
*
School of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
First published on 16th November 2021
Abstract
The neutral complexes trans-[GeF4(PiPr3)2] and [GeF4(κ2-L)] (L = CH3C(CH2PPh2)3 or P(CH2CH2PPh2)3) are obtained from [GeF4(MeCN)2] and the ligand in CH2Cl2. Treatment of [GeF4(PMe3)2] with n equivalents of TMSOTf (Me3SiO3SCF3) leads to formation of the series [GeF4−n(PMe3)2(OTf)n] (n = 1, 2, 3), each of which contains six-coordinate Ge(IV) with trans PMe3 ligands and X-ray structural data confirm that the OTf groups interact with Ge(IV) to varying degrees. Unexpectedly, [GeF3(PMe3)2(OTf)] undergoes reductive defluorination in solution, forming the Ge(II) complex, [Ge(PMe3)3][OTf]2 (and [FPMe3]+). The bulkier PiPr3 leads to formation of the ionic [GeF3(iPr3P)2][OTf], containing a [GeF3(iPr3P)2]+ cation. [GeF4{o-C6H4(PMe2)2}], containing the cis-chelating diphosphine, also reacts with n equivalents of TMSOTf to generate [GeF4−n{o-C6H4(PMe2)2}(OTf)n] (n = 1, 2, 3). As for the PMe3 system, the trifluoride, [GeF3{o-C6H4(PMe2)2}(OTf)], is unstable to reductive defluorination in solution, producing the pyramidal Ge(II) complex [Ge{(o-C6H4(PMe2)2}(OTf)][OTf], whose crystal structure has been determined. The [GeF3{Ph2P(CH2)2PPh2}(OTf)] and [GeF2{Ph2P(CH2)2PPh2}(OTf)2], obtained similarly from the parent tetrafluoride complex, are poorly soluble, however their structures were confirmed crystallographically. The complexes in this work have been characterised via variable temperature 1H, 19F{1H} and 31P{1H} NMR studies in solution, IR spectroscopy and microanalysis and through single crystal X-ray analysis of representative examples across each series. Trends in the NMR and structural parameters are also discussed.
Introduction
P-block coordination and organometallic chemistry has seen a surge in research activity over recent years. There have been a variety of drivers for this, including, for example, our deeper understanding of the intrinsic chemical and structural diversity of the p-block acceptors compared to the d-block ions, the motivation to develop efficient metal-free catalysts, precursors for the growth of semiconductor materials for electronic and optical applications, as well as the range of radionuclides in the p-block that offer exciting prospects in medical imaging and therapy.1,2a Fluoride derivatives of several p-block elements, e.g. B, Al, Ga and P, have drawn significant interest as prospective carriers for the positron-emitting fluorine-18 isotope in new positron emitting tomography (PET) imaging agents.3 However, the wider coordination chemistry of the p-block fluorides is still relatively limited in scope, with the majority of examples featuring the Group 13 with hard N- and O-donor ligands.3–5 Recently we have reported several series of neutral and cationic Sn(IV) fluoride coordination complexes involving a range of hard and soft donor ligands.6Germanium tetrafluoride is a molecular monomer with boiling point = −36.5 °C, which has been shown to form a range of six-coordinate complexes either by the direct reaction of GeF4 with the neutral ligands, or by using an appropriate adduct, typically [GeF4(MeCN)2]. Other nitriles like NCCH2X (X = F and Cl) also form [GeF4(NCCH2X)2]; for X = F, the crystal structure shows that the nitrile ligands lie cis.7 Reacting germanium tetra-ethoxide, aqueous HPF6 and pyridine under solvothermal conditions forms trans-[GeF4(py)2].8 The reaction of 2,2′-bipyridine, 1,10-phenanthroline, or Me2NCH2CH2NMe2 with [GeF4(MeCN)2] in CH2Cl2 leads exclusively to the formation of the cis-isomer with the neutral ligand chelating, as expected.9,10 The tetra-aza macrocycle Me4[14]aneN4 (1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) forms [GeF4(κ2-Me4[14]aneN4)], where the ligand binds in an exocyclic bidentate fashion and the Ge(IV) retains the four bound fluorides.9
[GeF4(MeCN)2] also reacts with OPR3 (R = Me, Et, Ph) to form [GeF4(OPR3)2], which exist as a cis/trans mixture in solution,11 while bidentate phosphine oxides form only the cis isomer.12 While the heavier GeCl4 and GeBr4 analogues react with OPMe3 to form cationic or dicationic complexes of the form [GeCl3(OPMe3)3]2[GeCl6] and [GeX2(OPMe3)4][X]2 (X = Cl, Br), the latter through self-ionisation, this does not occur for GeF4, consistent with the Ge–X bonds becoming weaker as the halide becomes heavier.11
There are a few reports of germanium fluoride complexes with neutral, soft donor ligands.3 The reaction of [GeF4(MeCN)2] with two equivalents of PMe3 forms trans-[GeF4(PMe3)2], whereas using AsEt3 does not produce an isolable complex, and while AsEt3 reacts with GeCl4 to form trans-[GeCl4(AsEt3)2], this complex undergoes slow redox chemistry in solution, forming AsEt3Cl2 and GeCl2.13 [GeCl4(PMe3)2] can be isolated from the solvent-free reaction of GeCl4 and PMe3; although, if a solvent is employed, redox chemistry occurs and the product is [PMe3Cl][GeCl3] exclusively.13 A small number of diphosphine complexes, cis-[GeF4(diphosphine)] (diphosphine = o-C6H4(PR2)2 (R = Me, Ph), R2P(CH2)2PR2; R = Me, Et, Cy, Ph),13 and dithioether complexes, cis-[GeF4{RS(CH2)2SR}] (R = Me, Et, iPr) have also been described, with crystal structures reported for representative examples.14
Examples of cationic Ge(IV) fluoride complexes are few. The reaction of [GeF4(MeCN)2] with Me3[9]aneN3 (1,4,7-trimethyl-1,4,7-triazacyclononane) in CH2Cl2 leads to [GeF3(Me3[9]aneN3)]2[GeF6], with the strong preference for tridentate coordination of the macrocycle causing F− displacement; this cation has been confirmed crystallographically.9 Dicationic germanium fluoride complexes can be obtained by the fluorination of Ge(II) complexes. The reaction of [Ge(BIMEt3)][OTf]2 (BIMEt3 = tris(1-ethyl-benzoimidazol-2-ylmethyl)amine) with XeF2 leads to the clean formation of [GeF2(BIMEt3)][OTf]2, driven by the formation of the strong Ge–F bonds. The reaction of this difluoride complex with one equivalent of TMSOTf then leads to [GeF(OTf)(BIMEt3)][OTf]2. This reaction can be reversed by the addition of one equivalent of NaBF4. The structures of both complexes have been reported.15
We describe here the coordination chemistry of GeF4 with neutral mono-, bi-, tri- and tetra-phosphine ligands, together with the reactions of [GeF4(PR3)2] (R = Me or iPr), [GeF4{o-C6H4(PMe2)2}], and [GeF4{Ph2P(CH2)2PPh2}] with TMSOTf (Me3SiO3SCF3), revealing that fluoride ligands can be readily abstracted, in some cases sequentially, generating a range species with OTf− anions, which interact with the Ge(IV) centres to varying degrees. The new complexes have been characterised by variable temperature multinuclear NMR (1H, 19F{1H} and 31P{1H}) and IR spectroscopy, together with microanalyses, and their identities confirmed by X-ray crystal structures of representative examples.
Results and discussion
Germanium tetrafluoride phosphine complexes
Scheme 1 summarises the reactions of [GeF4(MeCN)2] with phosphines in this work.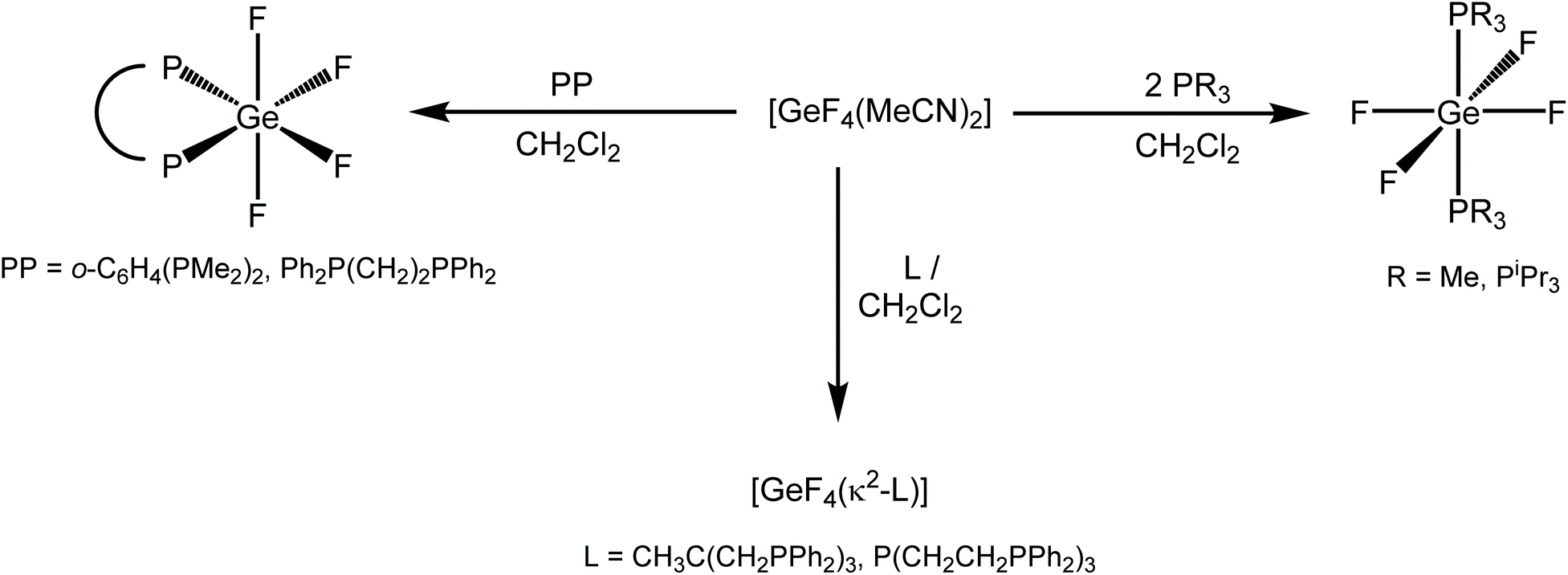 | ||
| Scheme 1 Summary of the neurtal phosphine complexes of GeF4 prepared in this work. | ||
Reaction of [GeF4(MeCN)2] with 2 equiv. of PR3 (R = Me, iPr) in CH2Cl2 affords the complexes [GeF4(PR3)2] in good yield as colourless solids. Spectroscopic data for [GeF4(PMe3)2] are in accord with those reported,13 and slow evaporation of a CH2Cl2 solution deposited crystals suitable for single crystal X-ray analysis. The structure is centrosymmetric (Fig. 1), with a distorted octahedral geometry and confirming the trans isomer. This is the first structurally characterised monodentate phosphine complex of germanium fluoride.
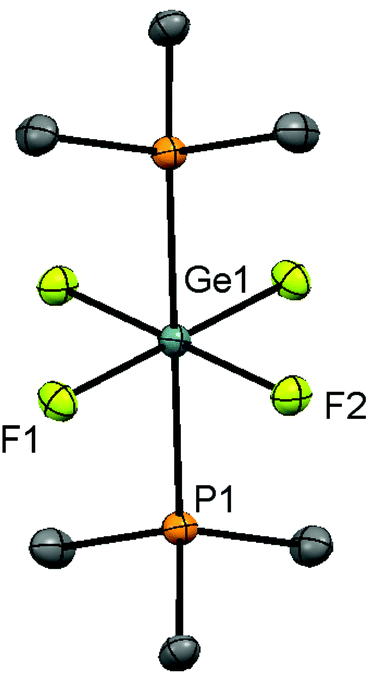 | ||
| Fig. 1 View of the crystal structure of [GeF4(PMe3)2] showing the atom labelling scheme. The ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Symmetry operation: −x, −y, −z. Selected bond lengths (Å) and angles (°) are: Ge1–P1 = 2.3717(6), Ge1–F1 = 1.8240(13), Ge1–F2 = 1.8158(13), F1–Ge1–F2 = 90.99(6). | ||
The analogous complex [GeF4(iPr3P)2] was prepared to allow the effect of the increased steric requirement of the phosphine on the fluoride abstraction chemistry (vide infra) to be explored. Its room temperature 31P{1H} and 19F{1H} NMR spectra contain broad resonances at +24.6 ppm and −65.0 ppm, respectively, indicating fast exchange on the NMR time scale. Low temperature NMR data support this (see Fig. S2.3 and S2.5†).
To expand the range of multidentate phosphine complexes of GeF4 and test the possibility of increasing the number of coordinated phosphine donors in this work, the tridentate ligand CH3C(CH2PPh2)3 was reacted with [GeF4(MeCN)2] in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio to form [GeF4(κ2-CH3C{CH2PPh2}3)], crystals of which were grown by layering a CH2Cl2 solution of the compound with hexane. The structure (Fig. 2) shows that the ligand coordinates in a cis-bidentate fashion, generating a six-membered chelate ring, with the third -PPh2 group remaining uncoordinated, hence the Ge(IV) retains all four fluorides.
:1 ratio to form [GeF4(κ2-CH3C{CH2PPh2}3)], crystals of which were grown by layering a CH2Cl2 solution of the compound with hexane. The structure (Fig. 2) shows that the ligand coordinates in a cis-bidentate fashion, generating a six-membered chelate ring, with the third -PPh2 group remaining uncoordinated, hence the Ge(IV) retains all four fluorides.
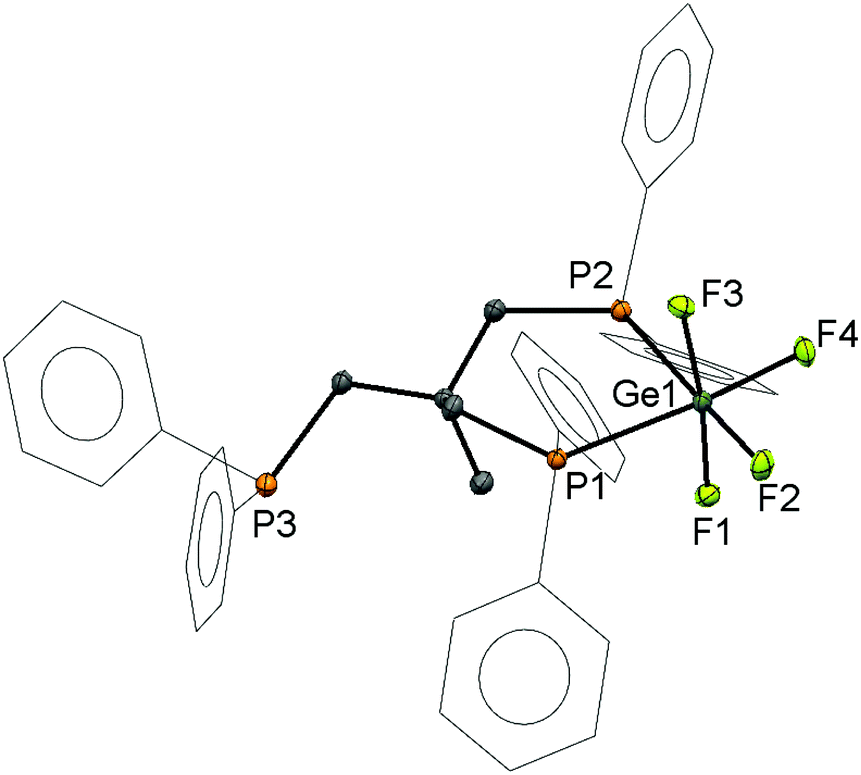 | ||
| Fig. 2 View of the crystal structure of [GeF4(κ2-CH3C{CH2PPh2}3)] showing the atom labelling scheme. The ellipsoids are drawn at the 50% probability level and H atoms and CH2Cl2 solvent molecule are omitted, and phenyl rings are displayed as wireframe for clarity. Selected bond lengths (Å) and angles (°) are: Ge1–P1 = 2.4731(4), Ge1–P2 = 2.5030(4), Ge1–F1 = 1.7872(9), Ge1–F2 = 1.7778(9), Ge1–F3 = 1.7976(9), Ge1–F4 = 1.7735(9), P1–Ge1–P2 = 92.886(13), F1–Ge1–F3 = 172.44(4), F2–Ge1–F4 = 92.47(4), P1–Ge1–F4 = 174.90(3), P2–Ge1–F2 = 178.75(3). | ||
In CD2Cl2 the room temperature 1H NMR spectrum from a freshly prepared and analytically pure sample of [GeF4(κ2-CH3C{CH2PPh2}3)] shows three sets of methylene resonances, corresponding to the two different environments within the chelated portion, as well as the uncoordinated arms. In the 19F{1H} spectrum (298 K) two broad resonances of equal intensity are seen at δ = −72.2 and −108.6, corresponding to the two environments in the cis isomer. Another sharper resonance at δ = −137.7 corresponds to GeF4. Consistent with this, the 31P{1H} NMR spectrum shows a broad resonance at δ = −4.0, as well as some uncoordinated triphosphine (−26.4 ppm). These data suggest that the complex is dynamic in solution and the phosphine is weakly bound, with both uncoordinated phosphine and GeF4 being seen in the NMR spectra. Upon cooling to 183 K the 31P{1H} NMR spectrum shows a tdd at −4.5 ppm and a singlet at −27.9 ppm in the 2:1 ratio expected for κ2-coordination.
This triphosphine complex has approximately C2v symmetry at Ge, so group theory predicts four IR active Ge–F stretching vibrations. However, it is common for these to overlap and in the present case only two broad peaks are seen at 517 and 603 cm−1.
The tripodal tetraphosphine, P(CH2CH2PPh2)3, was also reacted with [GeF4(MeCN)2] in a 1:1 molar ratio, leading to the formation of [GeF4{κ2-P(CH2CH2PPh2)3}]. At 298 K the 1H NMR spectrum shows two broad singlets in the methylene region at 2.03 and 2.28 ppm, and broad resonances in the aromatic region with a 1:1:5 integration, suggesting a dynamic process interchanging the terminal –PPh2 groups in solution. This is consistent with the room temperature 19F{1H} NMR data where a single broad resonance is seen at −94.8 ppm, while the 31P{1H} NMR spectrum shows broad, ill-defined multiplets to high frequency of the tetraphosphine itself. However, cooling to 183 K causes these broad resonances in the 31P{1H} NMR spectrum to sharpen into three resonances in a 1:2:1 ratio, shown in Fig. 3(a). The resonance corresponding to the pendent arms has an integral of two, consistent with bidentate coordination via the apical P donor and one –PPh2 pendant arm. The 31P{1H} NMR spectrum was also simulated using the SPINACH software package16 to confirm the coupling scheme (Fig. 3(b)).
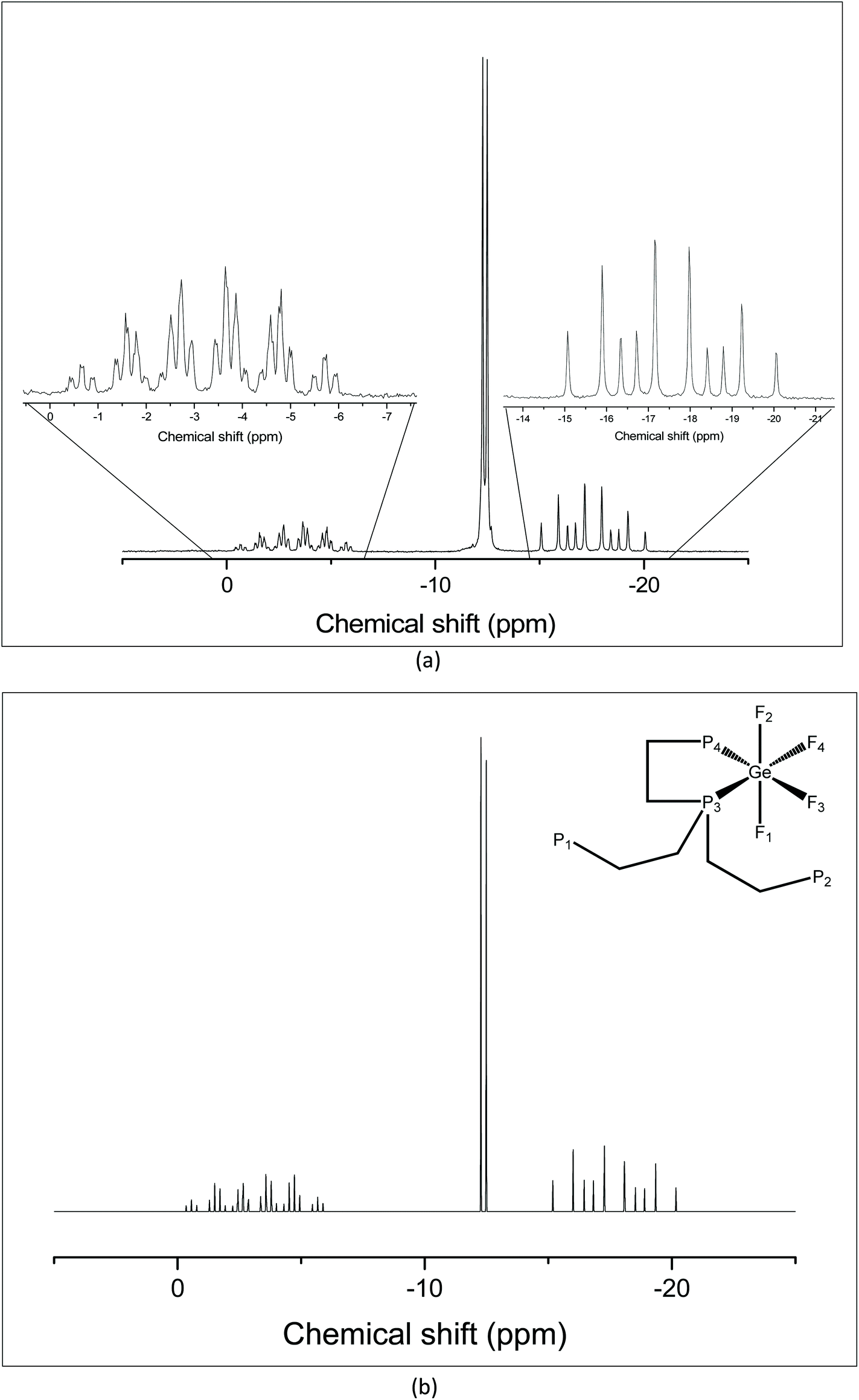 | ||
| Fig. 3 (a) 31P{1H} NMR spectrum of [GeF4{κ2-P(CH2CH2PPh2)3}] recorded at 183 K (CH2Cl2); (b) 31P{1H} NMR spectrum of [GeF4(κ2-P{CH2CH2PPh2})] simulated using the SPINACH16 package; the inset shows the NMR active nuclei giving rise to the coupling scheme. Spin system: JP1P3 = JP12P3 = 35 Hz; JP3P4 = 336 Hz; JP4F1 = JP4F2 = 133 Hz; JP3F1 = JP3F2 = 153 Hz; JF1F3 = JF1F4 = 54 Hz; JF2F3 = JF2F4 = 54 Hz; JF3F4 = 54 Hz; JP3F4 = 188 Hz; JP4F3 = 207 Hz. | ||
The 19F{1H} NMR spectrum at 213 K supports this (Fig. 4), showing three distinct resonances, a doublet of doublet of triplets at −80.4 ppm and two sets of what appear to be doublets of quartets, but which are in fact doublets of doublets of triplets (Fig. S11.2.5†). Overall, the solution data are therefore consistent with the isomer illustrated in Fig. 5, in which the apical P atom and one –PPh2 arm are coordinated.
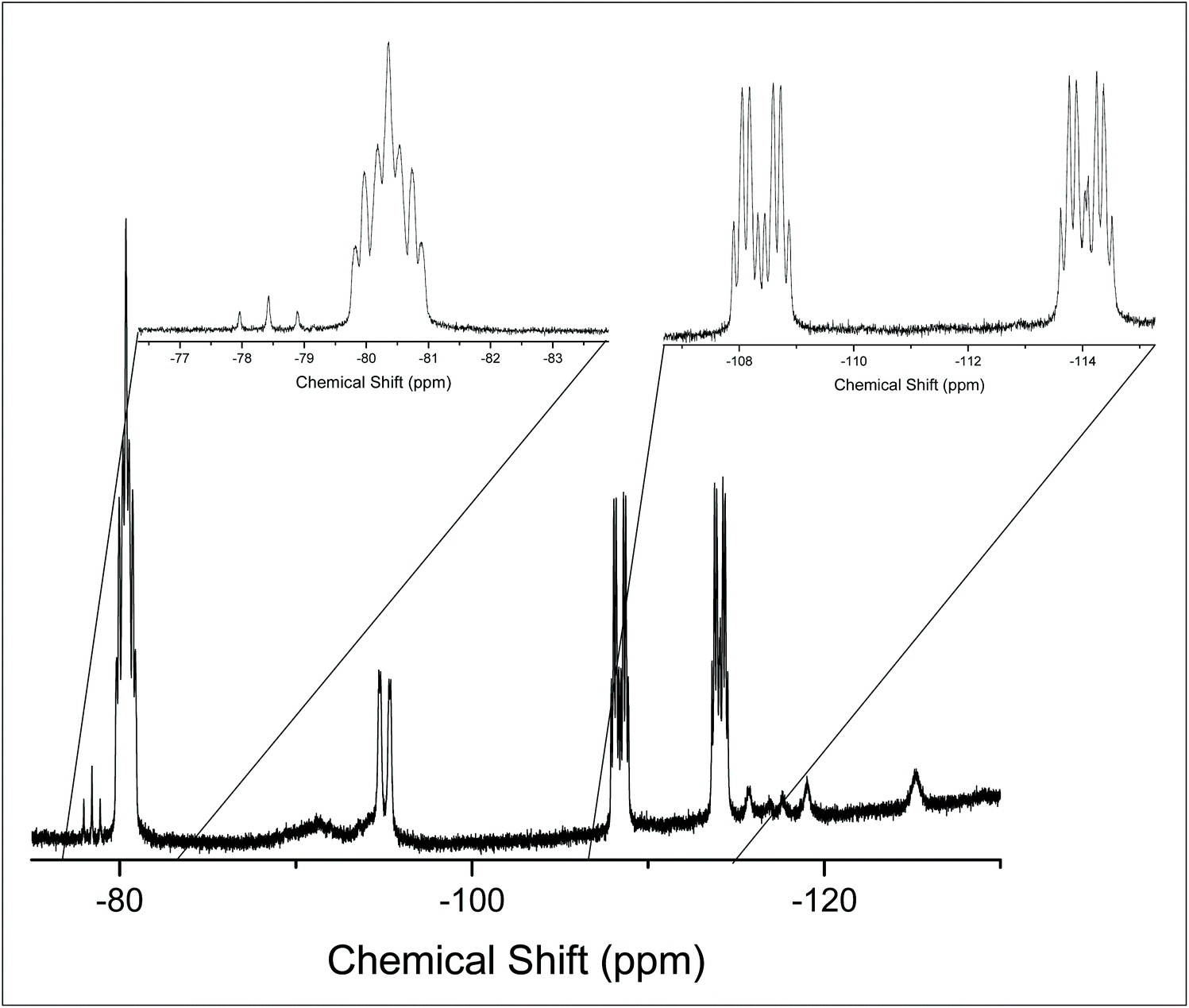 | ||
| Fig. 4 19F{1H} NMR spectrum of [GeF4{κ2-P(CH2CH2PPh2)3}] at 213 K (CH2Cl2). | ||
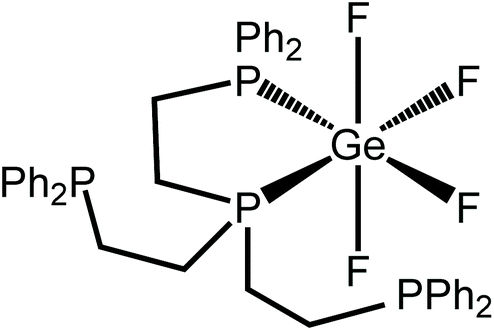 | ||
| Fig. 5 The isomer of [GeF4{κ2-P(CH2CH2PPh2)3}] present in CH2Cl2 at low temperature from the NMR data. | ||
Reactions of [GeF4(PMe3)2] with TMSOTf
Scheme 2 shows the products from reactions of [GeF4(PMe3)2] with different ratios of TMSOTf. | ||
| Scheme 2 Summary of the GeF4/PMe3/TMSOTf chemistry in this work. | ||
The reaction of [GeF4(PMe3)2] with one equivalent of TMSOTf (TMSOTf = Me3SiO3SCF3) in CH2Cl2 leads to the formation of [GeF3(PMe3)2(OTf)], which was isolated as a white powder. The 1H NMR spectrum shows that the doublet corresponding to the PMe3 groups at δ = 1.68 ppm, i.e. Δδ = +0.22 ppm vs. [GeF4(PMe3)2]. The room temperature 31P{1H} NMR spectrum shows a broad singlet resonance at 3.1 ppm, with Δδ = +15.5 ppm vs. [GeF4(PMe3)2]. At 183 K the singlet shifts to +5.2 ppm and splits into a triplet of doublets (2JPF = 192, 134 Hz) as shown in Fig. 6, consistent with the triflate being coordinated to germanium at this temperature; a quartet would be expected if it were not (assuming a trigonal bipyramidal geometry, as seen in [SnCl3(PMe3)2]+).17
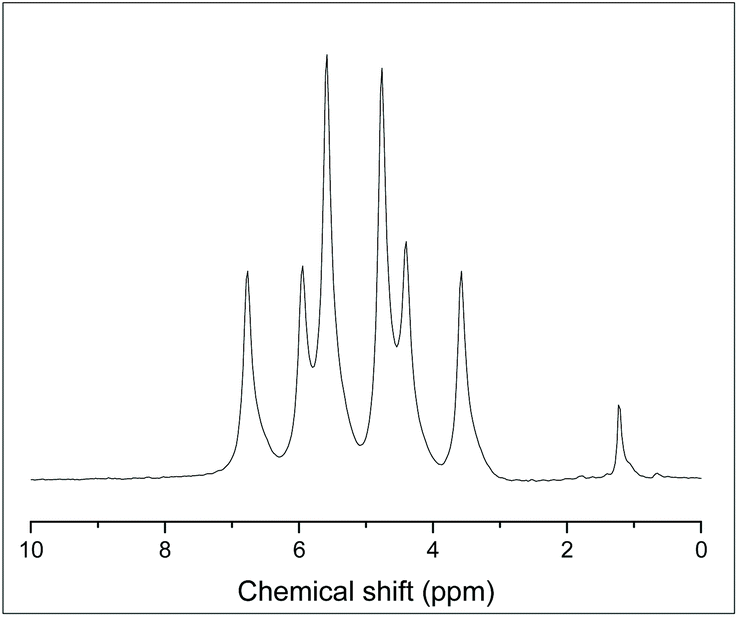 | ||
| Fig. 6 31P{1H} NMR spectrum of [GeF3(PMe3)2(OTf)] at 183 K (CD2Cl2). | ||
The 19F{1H} NMR data at room temperature show only the triflate resonance, whereas at 183 K three resonances are observed; a triplet of triplets at −123.4 ppm (2JPF = 134, 2JFF = 41 Hz) [F], due to the fluorine trans to the triflate, a triplet of doublets at −85.9 ppm [2F] (2JPF = 192, 2JFF = 41 Hz), from the fluorines cis to the triflate and a singlet at −78.9 ppm [3F] from the triflate. The couplings are only evident in the 19F{1H} spectrum at 183 K; around 210 K the couplings are lost and the peaks broaden significantly. Eventually, the peaks merge together, indicating fast exchange between the different fluorine environments. This is shown by the stacked variable temperature 19F{1H} NMR spectra in Fig. 7. The exchange of the fluorine environments is probably due to reversible triflate dissociation in solution.
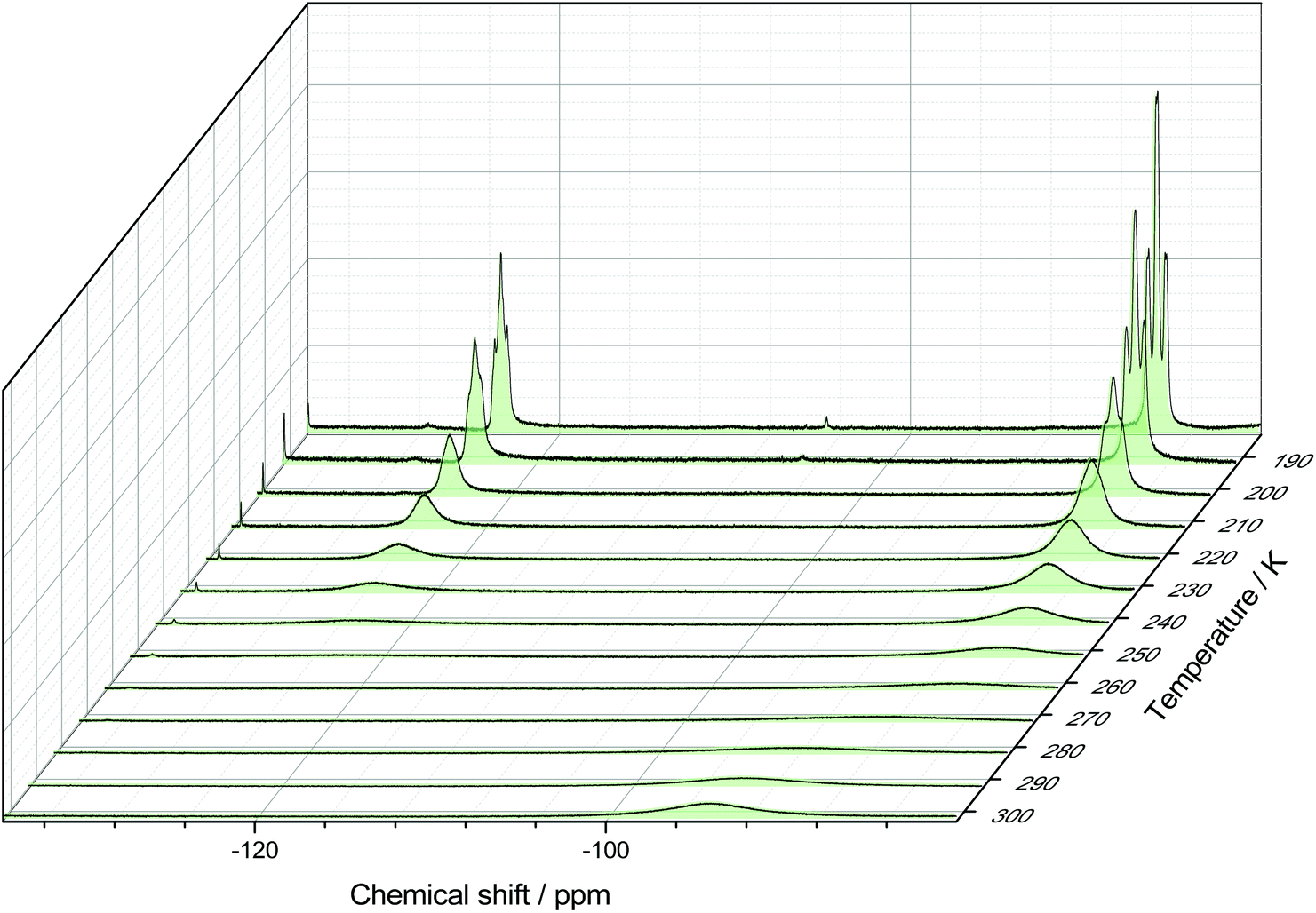 | ||
| Fig. 7 Stacked 19F{1H} NMR spectra of [GeF3(PMe3)2(OTf)] in CD2Cl2 showing the temperature dependant behaviour. | ||
Although the complex can be isolated as a white powder, it is relatively unstable even under inert atmosphere conditions. Over a period of 24 h the colour changes from white to dark brown, and this product is insoluble in CH2Cl2, hence all spectroscopic measurements on this complex were performed on a freshly synthesised sample. The complex is also unstable in solution and a CH2Cl2 solution of the compound left to evaporate overnight led to the deposition of crystals of the Ge(II) complex, [Ge(PMe3)3][OTf]2, containing a Ge(II) dication, the direct synthesis (from GeCl2(dioxane), PMe3 and TMSOTf) and crystal structure of which we have reported recently.18 This indicates that the [GeF3(PMe3)2(OTf)] is susceptible to redox chemistry and further analysis of its 19F{1H} NMR spectrum over time shows the appearance of a doublet at −135.7 ppm (JPF = 936 Hz) (Fig. S3.2.5†), consistent with the formation of [FPMe3]+,19 indicating that the Ge(II) dication is a result of reductive defluorination in solution. This can be compared to the reaction of GeCl4 with PMe3 in CH2Cl2, which also undergoes a redox process forming [PMe3Cl][GeCl3]. Notably, the germanium tetrafluoride phosphine complexes do not appear to exhibit any tendency to undergo redox behaviour in solution.
A CH2Cl2 solution of the Ge(IV) complex layered with hexane and stored at −78 °C (dry ice), however, produced crystals of [GeF3(PMe3)2(OTf)]. The structure shows (Fig. 8) that the triflate is coordinated in the solid state, with a distorted octahedral geometry at Ge, similar to the case for [SnX3(PMe3)2(OTf)] (X = F or Cl).6,17
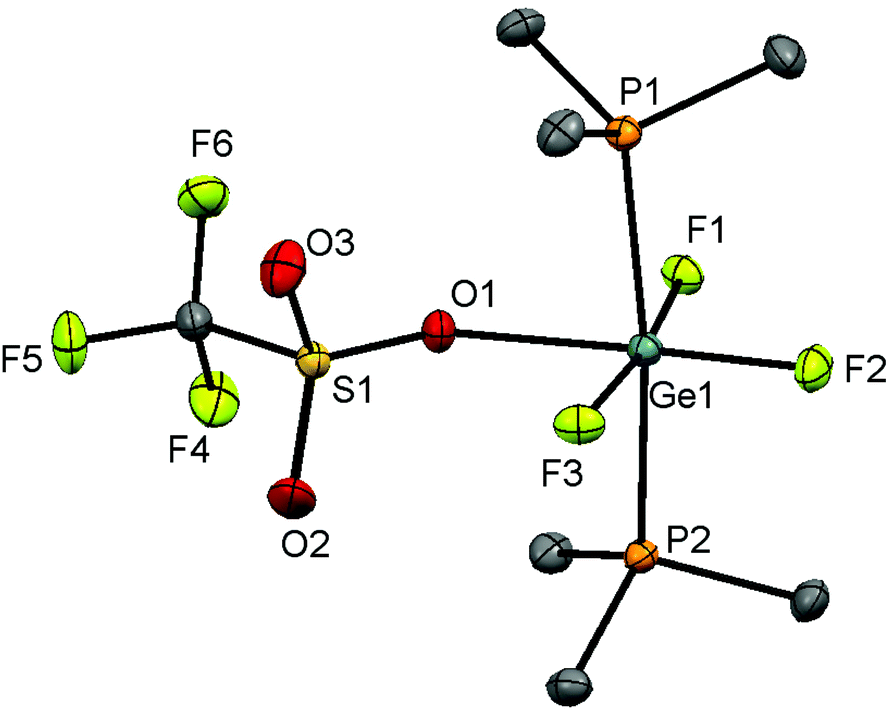 | ||
| Fig. 8 The structure of [GeF3(PMe3)2(OTf)] showing the atom labelling scheme. The ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are: Ge1–P1 = 2.3546(3), Ge1–P2 = 2.3655(3), Ge1–F1 = 1.8149(7), Ge1–F2 = 1.7753(7), Ge1–F3 = 1.8015(7), Ge1–O1 = 2.1878(9), P1–Ge1–P1 = 171.122(11), F1–Ge1–F3 = 170.48(3), O1–Ge1–F2 = 177.54(3). | ||
A similar reaction with TMSOTf was undertaken using [GeF4(iPr3P)2]. PiPr3 has a larger Tolman cone angle than PMe3 (160° vs. 118°),20 hence the increased steric requirement may reduce the likelihood of triflate coordination. The room temperature 31P{1H} NMR spectrum of the resulting [GeF3(iPr3P)2][OTf] shows a quartet 46.8 ppm (Fig. 9(a), contrasting with the behaviour of the PMe3 analogue, and the quartet remains at 183 K. The quartet coupling indicates that the three fluorides are in the same environment, therefore, the triflate is not bound in solution. In the 19F{1H} NMR spectrum a triplet is seen at −56.7 ppm, together with a sharp singlet at −79.0 ppm (OTf), with a 1:1 integration ratio. The large positive shift of the fluorine resonance is also consistent with a large increase in positive charge on the GeF3 unit, and hence supports the conclusion that the complex is ionic in solution, i.e. [GeF3(PiPr3)2][OTf] (Fig. 9(b)).
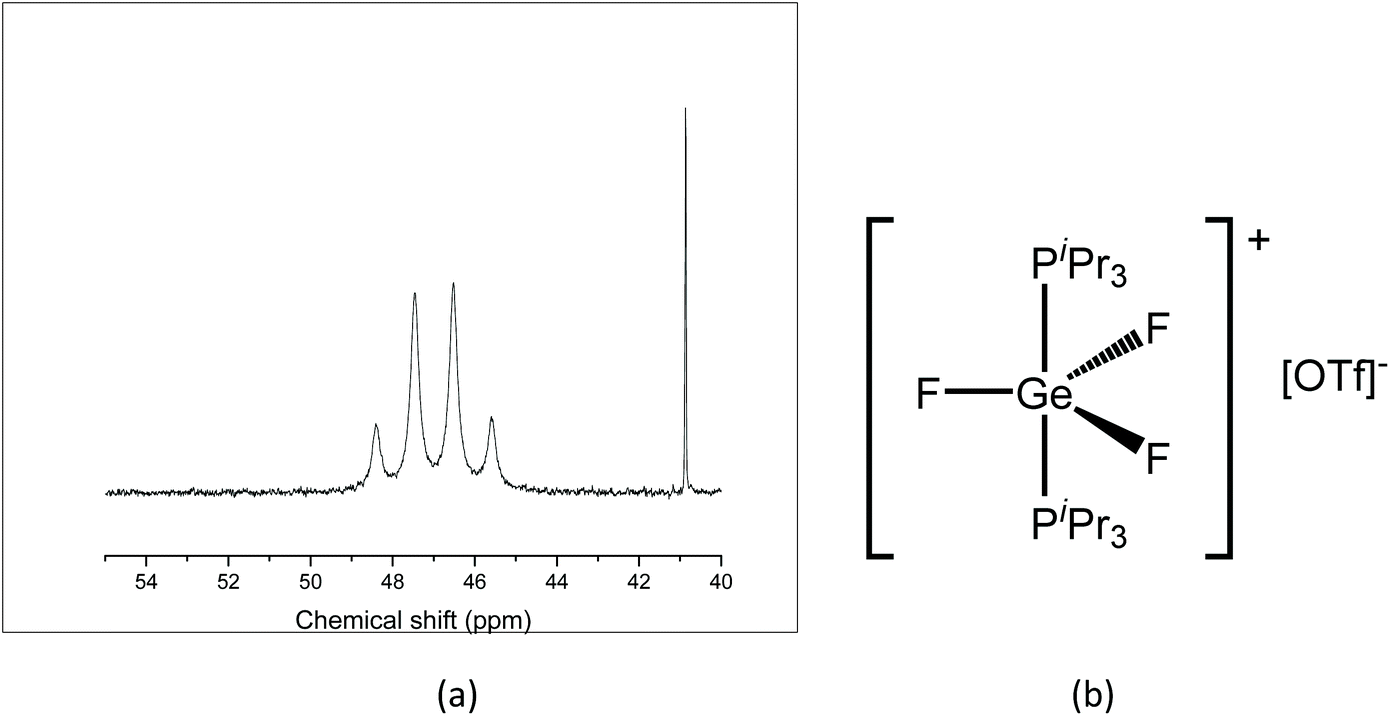 | ||
| Fig. 9 (a) 31P{1H} NMR spectrum of [GeF3(iPr3P)2][OTf] (298 K, CH2Cl2) showing the quartet resonance due to the [GeF3(iPr3P)2]+ cation; (b) proposed solution structure of [GeF3(iPr3P)2][OTf] (the singlet at ca. 41 is due to a small amount of [HPiPr3]+). | ||
The addition of two equivalents of TMSOTf to [GeF4(PMe3)2] in CH2Cl2 results in the formation of [GeF2(PMe3)2(OTf)2] as a white powder, which is stable for several weeks under inert atmosphere conditions. The room temperature 31P{1H} NMR spectrum shows a broad resonance at +25.8 ppm, which corresponds to Δδ = +22.6 ppm vs. the trifluoride, [GeF3(PMe3)2(OTf)]. At 233 K, this resonance shifts to +27.8 ppm and splits into a triplet (2JPF = 83 Hz). In the room temperature 19F{1H} NMR spectrum two resonances are observed, one at −78.3 ppm corresponding to triflate and a broad peak at −119.5 ppm due to the germanium bound fluorines. At 233 K this broad resonance sharpens into a triplet at −122.3 ppm (2JPF = 83 Hz). These data are consistent with the formation of the bis-triflate complex.
The evaporation of a concentrated CH2Cl2 solution of [GeF2(PMe3)2(OTf)2] deposited crystals, and the X-ray structure of this complex is shown in Fig. 10(a). It has pseudo-octahedral coordination with two relatively long contacts to the oxygen atoms of the κ1-triflates, Ge–OTf = 2.375(3) and 2.396(3) Å, which are ∼0.3 Å longer the Ge–OTf bond in [GeF3(PMe3)2(OTf)]. The P–Ge–P bond angle is 150.29(5)°, a significant deviation from an ideal octahedral complex. This, together with the large cis F–Ge–F bond angle of 97.4(14)°, suggests that core the ‘GeF2(PMe3)2’ unit could alternatively be described as a pseudo-tetrahedral dication, with the triflates interacting only weakly.
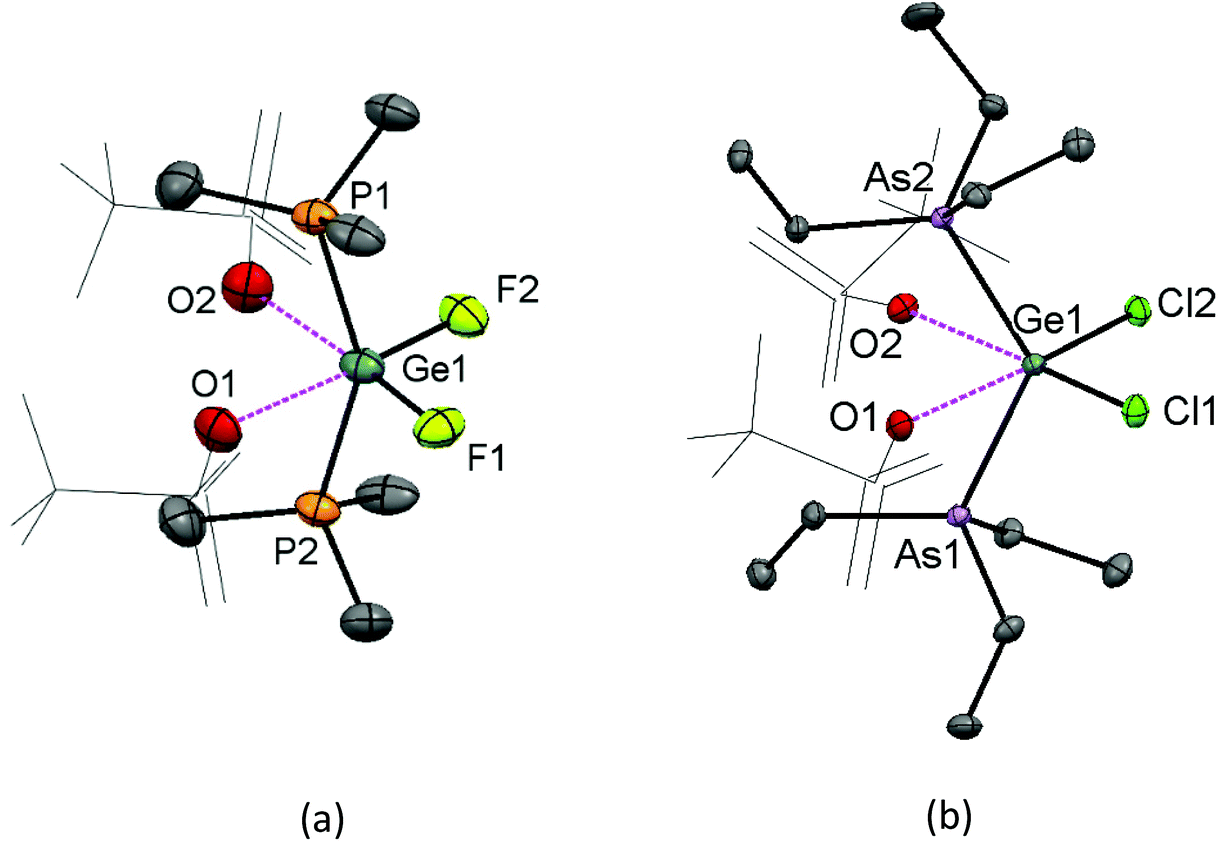 | ||
| Fig. 10 The structures of (a) [GeF2(PMe3)2(OTf)2] showing the atom labelling scheme (there are three independent molecules in the asymmetric unit). The ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity and only the closest oxygen of the triflate is drawn as an ellipsoid for clarity. Selected bond lengths (Å) and angles (°) for the Ge1-centred moiety are: Ge1–P1 = 2.3100(12), Ge1–P2 = 2.3074(12), Ge1–F1 = 1.747(3), Ge1–F2 = 1.748(3), Ge1⋯O1 = 2.374(3), Ge1⋯O2 = 2.396(3) P1–Ge1–P2 = 150.29(5), F1–Ge1–F2 = 97.40(14); (b) [GeCl2(AsEt3)2(OTf)2] showing the atom labelling scheme. The ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity and only the closest oxygen of the triflate is drawn as an ellipsoid for clarity. Selected bond lengths (Å) and angles (°) are: Ge1–As1 = 2.4048(4), Ge1–As2 = 2.4169(4), Ge1–Cl1 = 2.1453(7), Ge1–Cl2 = 2.1602(7), Ge1⋯O1 = 2.6848(19), Ge1⋯O2 = 2.7436(19), As1–Ge1–As2 = 125.186(16), Cl1–Ge1–Cl2 = 101.36(3). | ||
A concentrated solution containing GeCl4:2TMSOTf:2AsEt3 in CH2Cl2 left to evaporate slowly also afforded crystals, in this case of [GeCl2(AsEt3)2(OTf)2] (Fig. 10(b)). This complex is analogous to [GeF2(PMe3)2(OTf)2], although in the dichloro species Ge⋯OTf contacts are even longer, at 2.6848(19) and 2.7436(2) Å, indicating that the Ge–OTf interactions are even weaker. The As–Ge–As angle is 125.186(16)° and the Cl–Ge–Cl bond angle is 101.36(3)°, i.e. significantly closer to ideal angles for a tetrahedral species than an octahedron.
The addition of three equivalents of TMSOTf to [GeF4(PMe3)2] in CH2Cl2 forms [GeF(PMe3)2(OTf)3] as a stable white solid. In the 1H NMR spectrum a doublet of doublets can be seen at 2.02 ppm, which continues the trend of increasing chemical shift going from the tetrafluoride to the monofluoride species, and consistent with an increasing positive charge along the series.
The room temperature 31P{1H} spectrum shows a sharp doublet at 32.3 ppm (2JPF = 75 Hz) as expected for the monofluoride complex, while the room temperature 19F{1H} spectrum shows a sharp triplet at −107.2 ppm (2JPF = 75 Hz) as well as two broad triflate resonances at −77.6 and −78.1 ppm. The broad triflate peaks suggest that there is a triflate exchange process occurring in solution at this temperature (Table 1).
| Complex | δ 31P{1H}/ppm | δ 19F{1H}a/ppm | 2 J(31P–19F)/Hz | 2 J(19F–19F)/Hz |
|---|---|---|---|---|
| a Excluding the triflate resonances. | ||||
| trans-[GeF4(PMe3)2]13 (240 K) | −12.4 (quin) | −96.9 (t) | 196 | — |
| [GeF3(PMe3)2(OTf)] (183 K) | 5.2 (dt) | −85.9 (td), −123.4 (tt) | 192, 134 | 41 |
| [GeF2(PMe3)2(OTf)2] (233 K) | 27.4 (t) | −122.3 (t) | 83 | — |
| [GeF(PMe3)2(OTf)3] (298 K) | 32.3 (d) | −107 (t) | 75 | — |
For the phosphine complexes in Table 2, a decrease in both the Ge–P and Ge–F bond distances is observed as the fluorides are replaced by triflates, consistent with an increase in positive charge and Lewis acidity at the germanium centre. This picture is also supported by the increase in the average C–P–C bond angle of the phosphine ligand going down the series, with the bond angle in the bis-triflate complex being almost 10° larger than in the phosphine itself and almost 3° larger than in [GeF4(PMe3)2]. The same trends are seen with the germanium chloride arsine complexes. Scheme 1 summarises the chemistry of the monophosphine complexes with TMSOTf.
| Complex | d(Ge–E) (E = P or As)/Å | d(Ge–X) (X = F or Cl)/Å | E–Ge–E angle/° | Range of C–E–C anglesa/° |
|---|---|---|---|---|
| a C–P–C angle of free PMe3 = 99.46°, C–As–C angle of free AsEt3 = 98.50°. | ||||
| [GeF4(PMe3)2] | 2.3717(6) | 1.8158(13), 1.8240(13) | 180.0 | 105.83(13)–106.93(11) |
| [GeF3(PMe3)2(OTf)] | 2.3655(3), 2.3546(3) | 1.8015(7)(cis), 1.8149(7) (cis), 1.7753(7) (trans) | 171.122(11) | 106.27(6)–109.00(6) |
| [GeF2(PMe3)2(OTf)2] | 2.3074(12), 2.3100(12) | 1.747(3), 1.748(3) | 150.29(5) | 107.0(2)–110.9(3) |
| [GeCl4(AsEt3)2]13 | 2.4904(9) | 2.3233(19), 2.3296(19) | 180.0 | 105.1(4)–106.4(4) |
| [GeCl2(AsEt3)2][OTf]2 | 2.4048(4), 2.4169(4) | 2.1453(7), 2.1602(7) | 125.186(16) | 105.79(12)–113.81(12) |
Reactivity of GeF4 complexes bearing bi- and multi-dentate phosphines with TMSOTf
Scheme 3 summarises the reaction chemistry involving [GeF4(diphosphine)] with TMSOTf.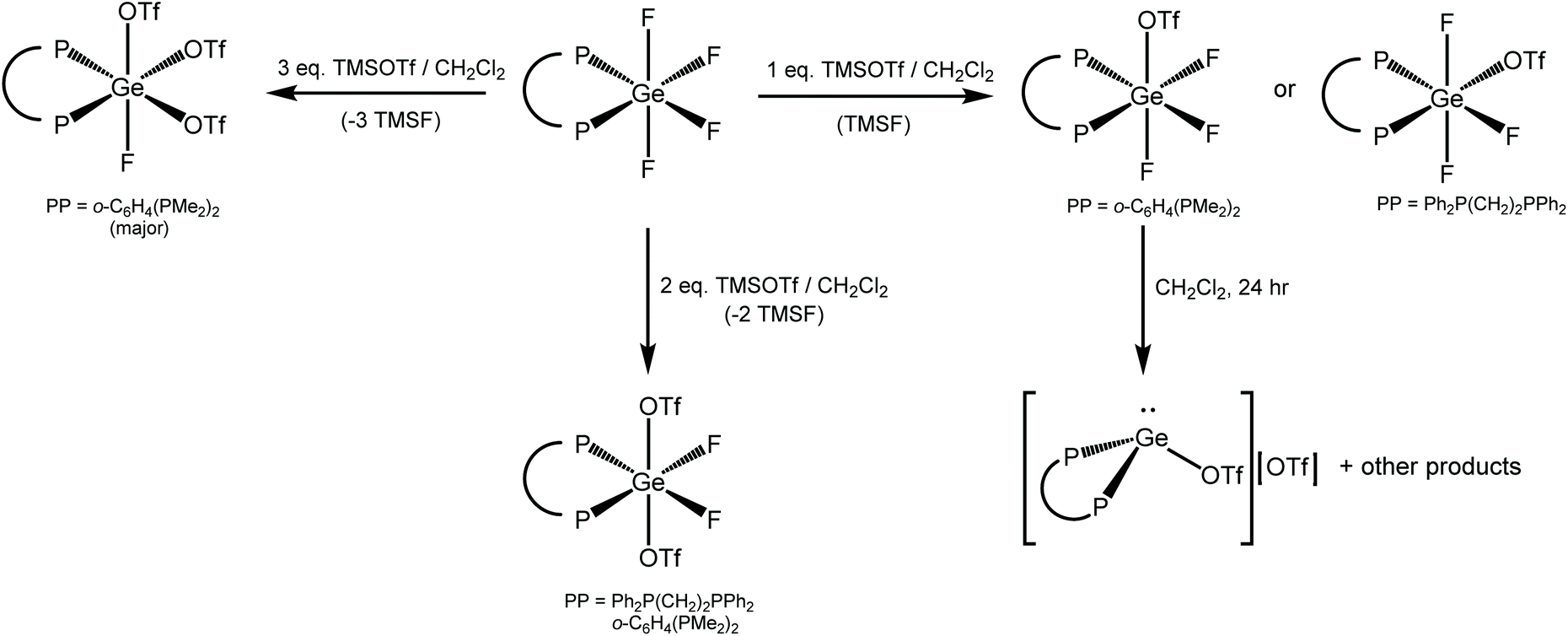 | ||
| Scheme 3 Reactions involving [GeF4(diphosphine)] with TMSOTf in this work. | ||
The reaction of [GeF4{o-C6H4(PMe2)2}] with one equivalent of TMSOTf in CH2Cl2 leads to the formation of [GeF3{o-C6H4(PMe2)2}(OTf)], with a small high frequency shift for the Me resonance in the 1H NMR spectrum. With one triflate bound, two isomers are possible, with mer or fac fluorines (Fig. 11).
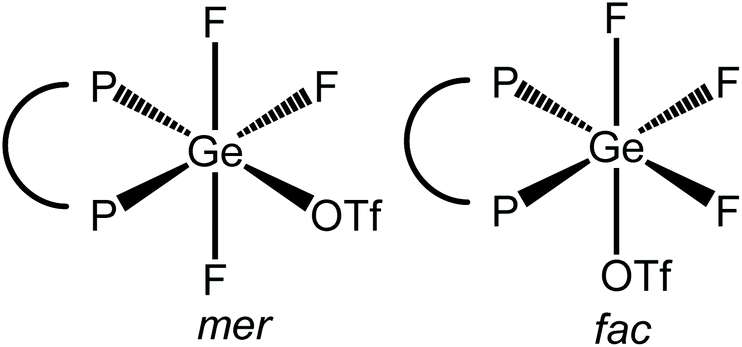 | ||
| Fig. 11 The two isomers possible for [GeF3{o-C6H4(PMe2)2}(OTf)] with the triflate coordinated. | ||
The 19F{1H} NMR spectrum of this complex at 183 K (Fig. 12) has a resonance at −109.5 ppm, which appears as a doublet of doublet of doublets with two different 2JPF couplings and one 2JFF coupling, as well as a triplet of triplets at −123.7 ppm with one 2JPF coupling and one 2JFF. In the 31P{1H} NMR spectrum (183 K) there is a resonance at −23.1 ppm, which is a doublet of doublet of doublets with three distinct 2JPF couplings, indicating that there is only one phosphorus chemical environment. These data are consistent with the fac isomer being present at this temperature.
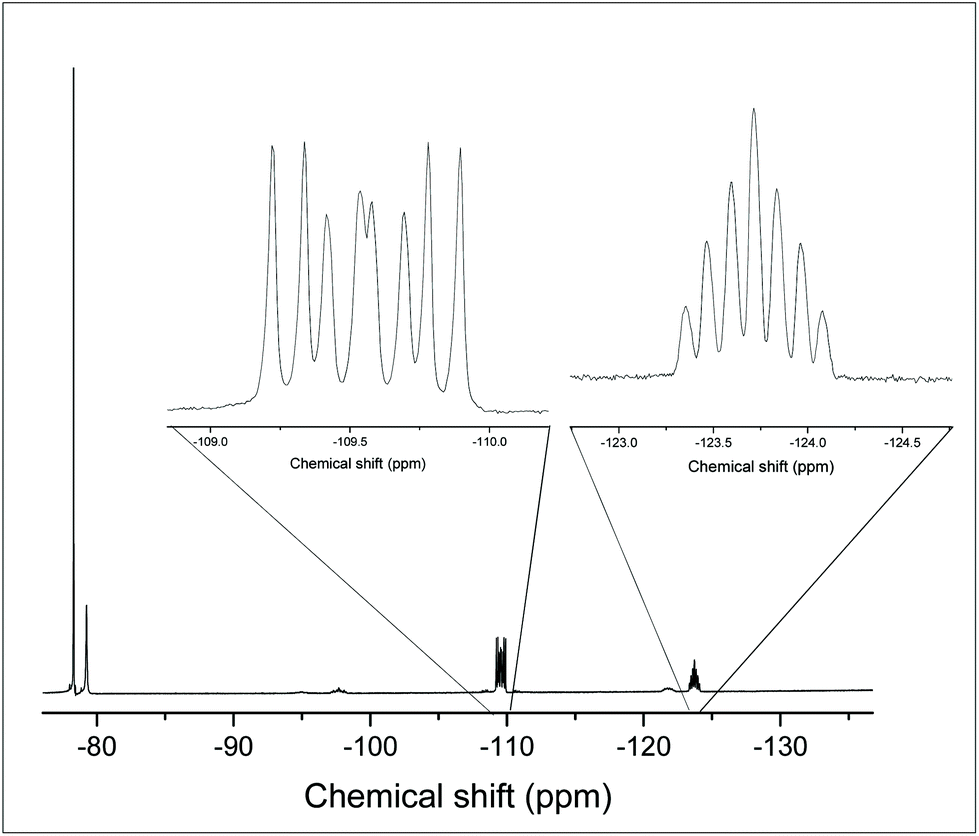 | ||
| Fig. 12 19F{1H} NMR spectrum of [GeF3{o-C6H4(PMe2)2}(OTf)] at 183 K (CH2Cl2). | ||
A CH2Cl2 solution of this complex layered with hexane deposited some crystals identified as [Ge{o-C6H4(PMe2)2}(OTf)][OTf], which is clearly not representative of the bulk product. The reduction from Ge(IV) to Ge(II) with concomitant complete defluorination was also seen in the PMe3 system (above). While in that case the doublet due to [FPMe3]+ was observed, the corresponding fluorophosphorane derived from o-C6H4(PMe3)2 is not known, and no obvious by-product of this type was evident. However, the reductive defluorination is also assumed to proceed via fluorination of the diphosphine, which may then undergo further reaction. The structure of [Ge{o-C6H4(PMe2)2}(OTf)][OTf] shows (Fig. S1, ESI†) one of the triflates coordinated to the Ge(II) centre, d(Ge–OTf) = 2.0968(15) Å. There are two longer contacts to the weakly interacting triflates at 2.6438(17) Å and 2.971 Å, completing an approximately five-coordinate geometry around the germanium, the complex exists as weakly associated dimers in the solid state. The complex has a similar geometry to the previously reported complex [GeCl(Me2PCH2CH2PMe2)][OTf], however the structural data in this case are not available so more detailed comparisons cannot be made.21
Using two equiv. of TMSOTf to [GeF4{o-C6H4(PMe2)2}] in CH2Cl2 led to the formation of [GeF2{o-C6H4(PMe2)2}(OTf)2]. Here the methyl 1H NMR resonance at 2.03 ppm, is to high frequency of both the tetrafluoride and trifluoride derivatives. In this difluoride complex three possible isomers exist if the triflates are bound (Fig. 13).
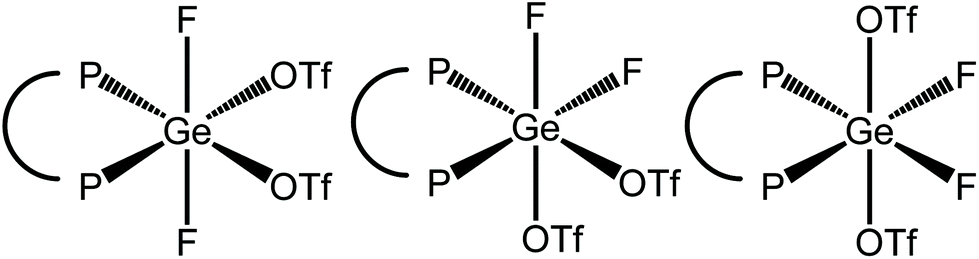 | ||
| Fig. 13 The three possible isomers of (six-coordinate) [GeF2{o-C6H4(PMe2)2}(OTf)2]. | ||
The room temperature 19F{1H} NMR spectrum shows a resonance at −100.5 ppm as doublet of doublets, correspondingly in the room temperature 31P{1H} NMR spectrum there is a doublet of doublets at −23.2 ppm, which both have the same coupling constants (2JPF = 121, 74 Hz). This is consistent with the 3rd isomer (rhs) being present in solution at room temperature.
Crystals from this product grown from a CH2Cl2 solution of the complex layered with hexane were indeed shown to be [GeF2{o-C6H4(PMe2)2}(OTf)2] and the structure is shown in Fig. 14(a). In the structure the triflates are trans to each other and the fluorines are cis, consistent with the NMR data.
 | ||
| Fig. 14 (a) The structure of [GeF2{o-C6H4(PMe2)2}(OTf)2] showing the atom labelling scheme. The ellipsoids are draw at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are: Ge1–P1 = 2.4033(16), Ge1–P2 = 2.3814(17) Ge1–F1 = 1.751(4), Ge1–F2 = 1.776(4), Ge1–O1 = 1.949(5), Ge1–O2 = 2.018(5), P1–Ge1–P2 = 86.33(6), O1–Ge1–O2 = 172.4(2), F1–Ge1–F2 = 92.7(2); (b) the structure of [GeF{o-C6H4(PMe2)2}(OTf)3] showing the atom labelling scheme. There are two [GeF{o-C6H4(PMe2)2}(OTf)3] in the asymmetric unit; only one is shown. The ellipsoids are drawn at the 50% probability level and H atoms and a CH2Cl2 solvent molecule are omitted for clarity. Selected bond lengths (Å) and angles (°) are: Ge1–P1 = 2.3900(9), Ge1–P2 = 2.3840(9) Ge1–F1 = 1.7681(18), Ge1–O1 = 1.948(2), Ge1–O2 = 1.939(2), Ge1–O3 = 1.917(2), P1–Ge1–P2 = 86.54(3), O2–Ge1–O3 = 83.67(10), O1–Ge1–F1 = 176.27(9). | ||
Addition of three equiv. of TMSOTf to [GeF4{o-C6H4(PMe2)2}] in CH2Cl2 leads to the formation of [GeF{o-C6H4(PMe2)2}(OTf)3]. There are two possible isomers in the case where the triflates are bound (Fig. 15).
 | ||
| Fig. 15 The two possible isomers of (six-coordinate) [GeF{o-C6H4(PMe2)2}(OTf)3]. | ||
The room temperature 19F{1H} NMR spectrum shows a triplet at −94.1 ppm, while in the 31P{1H} NMR spectrum a doublet at −13.5 ppm with a coupling constant of 2JPF = 73 Hz is evident, consistent with the fac isomer in solution. A CH2Cl2 solution of the complex layered with hexane deposited crystals and X-ray structure analysis (Fig. 14(b)) confirmed the fac geometry is maintained in the solid state.
The reaction of [GeF4{Ph2P(CH2)2PPh2}] with TMSOTf in CH2Cl2 leads to [GeF3{Ph2P(CH2)2PPh2}(OTf)]. Crystals of this compound were grown by layering a CH2Cl2 solution of the complex with hexane, and the structure is shown in Fig. 16(a).
 | ||
| Fig. 16 (a) The structure of [GeF3{Ph2P(CH2)2PPh2}(OTf)] showing the atom labelling scheme. The ellipsoids are draw at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are: Ge1–P1 = 2.4294(10), Ge1–P2 = 2.4321(11), Ge1–F1 = 1.791(2), Ge1–F2 = 1.762(2), Ge1–F3 = 1.775(2), Ge1–O1 = 1.965(3), P1–Ge1–P2 = 85.49(3), F1–Ge1–F3 = 173.69(10), P1–Ge1–O1 = 177.73(8), P2–Ge1–F2 = 173.04(8); (b) the structure of [GeF2{Ph2P(CH2)2PPh2}(OTf)2] showing the atom labelling scheme. The ellipsoids are draw at the 50% probability level and H atoms and a CH2Cl2 solvent molecule are omitted for clarity. Selected bond lengths (Å) and angles (°) are: Ge1–P1 = 2.4217(6), Ge1–F1 = 1.7641(11), Ge1–O1 = 1.934(7), P1–Ge1–P1 = 86.25(2), F1–Ge1–F1 = 91.43(8), O1–Ge1–O1 = 172.3(5). | ||
In the structure of [GeF3{Ph2P(CH2)2PPh2}(OTf)] the diphosphine ligand is chelating and the fluorines are mer, this is in contrast to [GeF3{o-C6H4(PMe2)2}(OTf)] where the NMR data suggests a fac arrangement. It is likely that the different electronic and steric properties of the ligands dictate the preferred isomer.
From the same batch of crystals, a crystal of [GeF2{Ph2P(CH2)2PPh2}(OTf)2] was also identified. The structure of this complex is shown in Fig. 16 (b). The geometry is analogous to the o-C6H4(PMe2)2 complex with both fluorides trans to the phosphine ligand and the triflates mutually trans.
As with monodentate phosphines, there is a decrease in d(Ge–P) and an increase in the P–Ge–P bond angles as fluoride is replaced by triflate, while the Ge–F distances remain largely unaffected (Table 3). The d(Ge–O) distances are much shorter for the diphosphine complexes compared to the monophosphine complexes.
| Complex | d(Ge–P)/Å | d(Ge–F)/Å | P–Ge–P angle/° | d(Ge–O)/Å |
|---|---|---|---|---|
| [GeF4{o-C6H4(PMe2)2}] 13 | 2.4273(12) | 1.815(2) | 85.61(4) | — |
| 2.4273(11) | 1.809(2) (cis) | |||
| 1.765(2) | ||||
| 1.772(2) (trans) | ||||
| [GeF2{o-C6H4(PMe2)2}(OTf)2] | 2.4033(16) | 1.751(4) | 86.33(6) | 1.949(5) |
| 2.3814(17) | 1.776(4) | 2.018(5) | ||
| [GeF{o-C6H4(PMe2)2}(OTf)3] | 2.3900(9) | 1.7681(18) | 86.54(3) | 1.948(2) |
| 2.3840(9) | 1.939(2) | |||
| 1.917(2) | ||||
| [GeF4{Ph2P(CH2)2PPh2}] 13 | 2.4636(7) | 1.7692(14) (trans) | 84.08(2) | — |
| 2.4822(7) | 1.7731(14) | |||
| 1.7829(13) (cis) | ||||
| 1.7987(14) | ||||
| [GeF3{Ph2P(CH2)2PPh2}(OTf)] | 2.4294(10) | 1.791(2) | 85.49(3) | 1.965(3) |
| 2.4321(11) | 1.775(2) (cis) | |||
| 1.762(2) (trans) | ||||
| [GeF2{Ph2P(CH2)2PPh2}(OTf)2] | 2.4217(6) | 1.7641(11) | 86.25(2) | 1.934(7) |
The complexes involving the Ph2P(CH2)2PPh2 ligand were very poorly soluble, hindering acquisition of solution NMR data. Finally, attempts to increase the number of phosphine donor groups at Ge(IV) through fluoride abstraction led to mixtures of species (from the NMR data) for both the triphosphine and tetraphosphine complexes, along with evidence of reduction to Ge(II). Hence this was not pursued further.
Experimental
The syntheses were carried out using standard Schlenk and vacuum line techniques, with samples handled and stored in a glove box under a dry dinitrogen atmosphere to exclude moisture. TMSOTf was obtained from Sigma-Aldrich and distilled before use. Germanium tetrafluoride was obtained from Fluorochem and GeCl4 from Acros Organics and were used as received. Phosphine ligands and AsEt3 were obtained from Sigma-Aldrich or Strem and used as received, except for o-C6H4(PMe2)2 which was made by the literature route.22 CH2Cl2 and MeCN were dried by distillation from CaH2 and n-hexane from sodium wire. [GeF4(PMe3)2], [GeF4{o-C6H4(PMe3)2}] and [GeF4{Ph2P(CH2)2PPh2}] were made as described.13Infrared spectra were recorded as Nujol mulls between CsI plates using a PerkinElmer Spectrum 100 spectrometer over the range 4000–200 cm−1. 1H, 19F{1H} and 31P{1H} NMR spectra were recorded from CH2Cl2/CD2Cl2 solutions unless otherwise stated, using a Bruker AV400 spectrometer and are referenced to TMS via the residual solvent resonance, CFCl3, and 85% H3PO4 respectively. Microanalyses were undertaken by London Metropolitan University or Medac.
[GeF4(PiPr3)2]
To a suspension of [GeF4(MeCN)2] in CH2Cl2 (5 mL) two equiv. of PiPr3 was added as a solution in CH2Cl2 (2 mL) and the reaction mixture stirred for 2 h to yield a clear colourless solution. The volatiles were removed in vacuo to leave a white solid which was washed with hexane (3 × 10 mL) and dried in vacuo. Yield: 0.941 g (76%). Satisfactory analytical data could not be obtained despite repeated attempts on different samples, with the elemental compositions varying from sample to sample. 1H NMR (CD2Cl2, 298 K): δ = 1.33 (dd, 3JHH = 15 Hz, 3JPH = 7 Hz, [6H]), 2.38 (m, [1H]), 19F{1H} NMR (CD2Cl2, 298 K): δ = –65.0 (s); (183 K): δ = −62.9 (2JPF = 162 Hz) 31P{1H} NMR (CD2Cl2, 298 K): δ = 24.6 (s); (183 K): δ = 24.9 (quint, 2JPF = 162 Hz). IR (Nujol/cm−1): ν = 590s (Ge–F).[GeF3(PMe3)2(OTf)]
To a solution of [GeF4(PMe3)2] (0.100 g, 0.33 mmol) in CH2Cl2 (2 mL), a solution of TMSOTf (0.074 g, 0.33 mmol) in CH2Cl2 (2 mL) was added dropwise to form a clear solution. The reaction was stirred for 2 h, when the volatiles were removed in vacuo leaving a solid that was washed with hexane (3 × 10 mL) and dried in vacuo to form a white powder. Yield: 0.080 g (56%). Required for C7H18F6GeO3P2S (430.86): C, 19.5; H, 4.2. Found: C, 19.6; H, 4.3%. 1H NMR (CD2Cl2, 298 K): δ = 1.68 (d, 2JHP = 13.2 Hz). 19F{1H} NMR (CD2Cl2, 298 K): δ = −78.76 (s, [3F], OTf), −94.74 (br, [3F]); (183 K): δ = –78.9 (s, [3F], OTf), −85.9 (td, [2F], 2JPF(cis-OTf) = 192 Hz, 2JFF = 41 Hz, GeF), −123.4 (tt, [F], 2JPF(trans-OTf) = 134 Hz, 2JFF = 41 Hz, GeF). 31P{1H} NMR (CD2Cl2, 298 K): δ = 5.2 (br s); (183 K): δ = 5.2 (td, 2JPF(cis-OTf) = 192 Hz, 2JPF(trans-OTf) = 134 Hz). IR (Nujol/cm−1): 572 (br, m), 515 (m) Ge–F.[GeF2(PMe3)2(OTf)2]
Method as above using [GeF4(PMe3)2] (0.100 g, 0.33 mmol) and TMSOTf (0.148 g, 0.66 mmol). White solid. Yield: 0.140 g (75%). Required for C8H18F8GeO6P2S2 (560.63): C, 17.1; H, 3.2. Found: C, 17.0; H, 3.3%. 1H NMR (CD2Cl2, 298 K): 1.96 (m); (233 K): δ = 1.98 (d, 2JHP = 12 Hz). 19F{1H} NMR (CD2Cl2, 298 K): δ = −78.3 (s, [6F], OTf), −119.5 (br s, [2F], GeF); (233 K): δ = –78.3 (br, [6F], OTf), −122.3 (t, [2F], 2JPF = 83 Hz, GeF). 31P{1H} NMR (CD2Cl2, 298 K): δ = 25.8 (br); (233 K): δ = 27.8 (t, 2JPF = 83 Hz). IR (Nujol/cm−1): ν = 573 (br m) (Ge–F).[GeF(PMe3)2(OTf)3]
Method as above using [GeF4(PMe3)2] (0.100 g, 0.33 mmol) and TMSOTf (0.222 g, 1.00 mmol). White solid. Yield: 0.175 g (76%). Required for C9H18F10GeO9P2S3 (691.00): C, 15.6; H, 2.6. Found: C, 15.8; H, 2.6%. 1H NMR (CD2Cl2, 298 K): δ = 2.03 (dd, 2JHP = 13.8 Hz, 4JFH = 1 Hz). 19F{1H} NMR (CD2Cl2, 298 K): δ = –77.6 (br s, OTf), −78.1 (br s, OTf, total OTf integral [9F]), −107.2 (t, 2JPF = 75 Hz, [F], Ge–F). 31P{1H} NMR (CD2Cl2, 298 K): δ = 32.3 (d, 2JPF = 75 Hz). IR (Nujol/cm−1): 583 (br, m) Ge–F.[GeF3(PiPr3)2][OTf]
Method as above using [GeF4(PiPr3)2] (0.100 g, 0.33 mmol) and TMSOTf (0.074 g, 0.33 mmol). White powder. Yield: 0.188 g (48%). Required for C18H42F6GeO3P2S·1/2CH2Cl2 (629.63): C, 35.3; H, 6.9. Found: C, 35.3; H, 6.9%. 1H NMR (CD2Cl2, 298 K): δ = 1.43 (dd, 3JHH = 16 Hz, 3JPH = 7 Hz, [6H]), 2.69 (m, [1H]). 19F{1H} NMR (CD2Cl2, 298 K): δ = –79.0 (s, [3F], OTf), −56.7 (t, 2JPF = 153 Hz, [3F], Ge–F). 31P{1H} NMR (CD2Cl2, 298 K): δ = 46.8 (q, 2JPF = 153 Hz). IR (Nujol/cm−1): 572 m (br m) Ge–F.[GeF3{o-C6H4(PMe3)2}(OTf)]
Method as above using [GeF4{o-C6H4(PMe3)2}] (0.100 g, 0.288 mmol) and TMSOTf (0.064 g, 0.288 mmol) and stirring for 30 min. White solid. Yield: 0.084 g (61%). Required for C11H16F6GeO3P2S (476.86): C, 27.7; H, 3.4. Found: C, 27.5; H, 3.6%. 1H NMR (CD2Cl2, 298 K): δ = 1.90 (m, [12H], Me), 7.84 (m, [4H], Ar–H). 19F{1H} NMR (CD2Cl2, 298 K): −78.3 (s, OTf), −113.3 (br s, GeF); (183 K): δ = −78.3 (s, [3F], [OTf]), −109.5 (ddd, [2F], 2JPF = 135, 76 Hz; 2JFF = 43 Hz), −123.7 (tt, [1F], 2JPF = 94 Hz; 2JFF = 43 Hz). 31P{1H} NMR (CD2Cl2, 298 K): δ = –24.1 (br s); (183 K): δ = –23.1 (ddd, 2JPF = 135, 94, 76 Hz). IR (Nujol/cm−1): 518 (m), 579 (m), 602 (m) Ge–F.[GeF2{o-C6H4(PMe3)2}(OTf)2]
Method as above using [GeF4{o-C6H4(PMe3)2}] (0.100 g, 0.288 mmol) and TMSOTf (0.128 g, 0.576 mmol) and stirring for 30 min. White solid. Yield: 0.102 g (58%). Required for C12H16F8GeO6P2S2·CH2Cl2 (691.84): C, 22.6; H, 2.6. Found: C, 22.7; H, 3.1%. 1H NMR (CD2Cl2, 298 K): δ = 2.03 (m, [12H], Me), 7.91 (m, [4H], Ar–H). 19F{1H} NMR (CD2Cl2, 298 K): δ = −77.5 (s, OTf), −78.2 (s, OTf), −100.5 (dd, 2JPF = 121 Hz, 74 Hz); (183 K): δ = −77.7 (s, OTf), −78.0 (s, OTf), −78.3 (s, OTf), −79.1 (s, OTf), −101.1 (dd, 2JPF = 126, 72 Hz), 31P{1H} NMR (CD2Cl2, 298 K): δ = –23.2 (dd, 2JPF = 121 Hz, 74 Hz); (183 K): δ = −19.9 (dd, 2JPF = 126, 72 Hz) IR (Nujol/cm−1): 637 (m) Ge–F.[GeF{o-C6H4(PMe3)2}(OTf)3]
Method as above using [GeF4{o-C6H4(PMe3)2}] (0.100 g, 0.288 mmol) and TMSOTf (0.192 g, 0.864 mmol) and stirring for 30 min., then the solution was layered with hexane (10 mL). After 3 days the product crystallised. Crystals were collected by filtration, washed with hexane (3 × 10 mL), and dried in vacuo. Yield: 0.086 g (40%). Required for C13H16F10GeO9P2S3 (736.98): C, 21.2; H, 2.2. Found: C, 22.1; H, 3.8%. 1H NMR (CD2Cl2, 298 K): δ = 2.03 (m, Me), 2.16 (m, Me), 2.25 (m, Me) (sum of methyl resonances [12H]), 7.97 (m, [4H], Ar–H). 19F{1H} NMR (CD2Cl2, 298 K): δ = −77.1 (s, OTf), −77.3 (s, OTf), −77.6 (s, OTf), −78.2 (br, OTf), −81.8 (t, 2JPF = 82 Hz)+, −91.9 (t, 2JPF = 69 Hz)+, −94.0 (t, 2JPF = 73 Hz, GeF)*. 31P{1H} NMR (CD2Cl2, 298 K): δ = –13.5 (d, 2JPF = 73 Hz)*, −8.53 (d, 2JPF = 69 Hz); (* = major species; + = minor species). IR (Nujol/cm−1): 633 (m) Ge–F.[GeF3{Ph2P(CH2)2PPh2}(OTf)]
Method as above using [GeF4{Ph2P(CH2)2PPh2}] (0.100 g, 0.183 mmol) and TMSOTf (0.041 g, 0.184 mmol). White solid. Yield: 0.045 g (36%). Required for C27H24F6GeO3P2S (677.12): C, 47.9; H, 3.6 Found: C, 47.7; H, 3.7%. The product was not sufficiently soluble in CH2Cl2 or CD3NO2 to obtain reliable solution NMR data. IR (Nujol/cm−1): 519 (m), 526 (m), 573 (m) Ge–F.[GeF2{Ph2P(CH2)2PPh2}(OTf)2]
Method as above using [GeF4{Ph2P(CH2)2PPh2}] (0.100 g, 0.183 mmol) and TMSOTf (0.081 g, 0.364 mmol). White solid. Yield: 0.119 g (81%). Required for C28H24F8GeO6P2S2·1/2CH2Cl2 (849.60): C, 40.3; H, 3.0. Found: C, 39.9; H, 3.0%. The product was not sufficiently soluble in CH2Cl2 or CD3NO2 to obtain reliable solution NMR data. IR (Nujol/cm−1): 502 (m), 527 (m) Ge–F.[GeF4{κ2-CH3C(CH2PPh2)3}]
To a suspension of [GeF4(MeCN)2] (0.200 g, 0.867 mmol) in CH2Cl2 (2 mL), CH3C(CH2PPh2)3 (0.541 g, 0.867 mmol) was added as a solid and the resulting solution is stirred for 2 h forming a clear colourless solution. Volatiles were removed in vacuo to yield a white solid, which was washed with hexane (3 × 10 mL) and dried in vacuo. Yield: 0.413 g (62%) Required for C41H39F4GeP3·1/2 CH2Cl2 (815.70): C, 61.1; H, 4.9. Found: C, 61.2; H, 5.1%. 1H NMR (CD2Cl2, 298 K): δ = 0.87 (s, [3H] CH3), 2.69 (br s, [6H], CH2), 7.28–7.58 (m, ArH); (183 K): δ = 0.73 (s, [3H]), 2.01 (br m, [2H]), 2.79 (br s, [2H]), 2.90 (br s, [2H]), 7.28–7.58 (m, ArH). 19F{1H} NMR (CD2Cl2, 298 K): δ = –108.6 (br s, GeF), −72.2 (br s, GeF); (243 K): δ = –74.4 (overlapping multiplets, [2F]), −109.3 (ddt, 2JPF = 125, 67 Hz, 2JFF = 62 Hz); (183 K): δ = –77.7 (br m, [2F]), −110.3 (br m, [2F]). 31P{1H} NMR (CD2Cl2, 298 K): δ = –4.0 (br s), −23.5(s); (183 K): δ = –4.5 (tdd, [2P],2J = 142, 125, 67 Hz), −27.9 (s, [1P]). IR (Nujol/cm−1): 517 (br), 603 (br) Ge–F.[GeF4{κ2-P(CH2CH2PPh2)3}]
To a suspension of [GeF4(MeCN)2] (0.200 g, 0.867 mmol) in CH2Cl2 (2 mL), P(CH2CH2PPh2)3 (0.581 g, 0.867 mmol) was added as a solid and the resulting solution was stirred for 2 h to give a clear colourless solution, from which volatiles were removed in vacuo to yield a white solid. The resulting solid was washed with hexane (3 × 10 mL) and dried in vacuo. Yield 0.408 (57%). Required for C42H42F4GeP4 (819.25): C, 61.6; H, 5.2. Found: C, 62.1; H, 5.5%. 1H NMR (CD2Cl2, 298 K): δ = 2.03 (br s, [6H], CH2), 2.28 (br s, [6H], CH), 7.38–7.48 (br m, [30H], ArH). 19F{1H} NMR (CD2Cl2, 298 K): δ = –94.8 (br s), −114.2 (br s); (213 K): δ = –80.4 (ddt, 2JPF = 153, 133 Hz, 2JFF = 54 Hz, [2F]), −108.4 (ddt, 2JPF = 205, 2JFF = 54, 54 Hz, [F]), −114.1 (ddt, 2JPF = 188 Hz, 2JFF = 54, 54 Hz, [F]), 31P{1H} NMR (CD2Cl2, 183 K): δ = –3.2 (ddtt, [P], 2JPP = 336, 2JPF = 188, 153,3JPP = 35 Hz), −12.4 (d, [2P], 3JPP = 35 Hz), −17.6 (ddt, [P], 2JPF = 336, 207, 133 Hz). IR (Nujol/cm−1): 508 (m), 517 (m), 589 (m), 608 (m) Ge–F.Reaction of GeCl4 two eq. of AsEt3 and two eq. of TMSOTf
To a solution of GeCl4 (0.134 g, 0.625 mmol) in CH2Cl2 (2 mL), AsEt3 (0.200 g, 1.23 mmol) was added as a solution in CH2Cl2 (2 mL). To this mixture TMSOTf (0.274 g, 1.23 mmol) in CH2Cl2 (2 mL) was added and the reaction mixture was stirred for 1 h, during which the solution remained colourless. The solution was concentrated to 1 mL, layered with hexane (3 mL) and stored at −18 °C. After a few days a colourless crystalline material formed, which was collected by filtration and dried in vacuo to yield a white powder. Yield: 0.261 g. 1H NMR (CD2Cl2, 298 K): indicates a complex mixture of species, which could not be identified with certainty.X-Ray experimental
Crystals were grown as described above. Data collections used a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with VHF Varimax optics (70 μm focus) with the crystal held at 100 K. Structure solution and refinement were performed using SHELX(S/L)97, SHELX-2013, or SHELX-2014/7.41 and OLEX.23 H atoms bonded to C were placed in calculated positions using the default C–H distance and refined using a riding model. [GeF2{Ph2P(CH2)2PPh2}(OTf)2] contains some substitutional disorder between OTf and F on the axial positions (ratio 85:15), which was modelled satisfactorily. The structures of [GeF2(PMe3)2(OTf)2] and [Ge{o-C6H4(PMe2)2}(OTf)][OTf] contained disordered triflate which was modelled using split occupancies. Details of the crystallographic parameters are given in Table S1 (ESI†). CCDC reference numbers for the crystallographic information files in cif format are 2113230 [GeF4(PMe3)2], 2113231 [GeF3(PMe3)2(OTf)], 2113232 [GeF2(PMe3)2(OTf)2], 2113233 [GeF2{o-C6H4(PMe2)2}(OTf)2], 2113234 [GeF{o-C6H4(PMe2)2}(OTf)3], 2113235 [GeCl2(AsEt3)2)(OTf)2], 2113236 [Ge{o-C6H4(PMe2)2}(OTf)][OTf], 2113237 [GeF3{Ph2P(CH2)2PPh2}(OTf)], 2113415 [GeF4(κ2-CH3C(CH2PPh2)3}], 2113416 [GeF2{Ph2P(CH2)2PPh2}(OTf)2].†
Conclusions
This work shows that neutral complexes of GeF4 can be extended to the tri- and tetraphosphine ligands CH3C(CH2PPh2)2 and P(CH2CH2PPh2)3, both of which bond in a bidentate κ2-mode only. The treatment of [GeF4(PMe3)2] with n equivalents of TMSOTf leads to formation of the series of complexes, [GeF4−n(PMe3)2][OTf]n (n = 1, 2, 3), each based on six-coordinate Ge(IV). [GeF3(PMe3)2(OTf)] is unstable in solution, with the Ge(II) complex, [Ge(PMe3)3][OTf]2, crystallising from the solution. The observation of [FPMe3]+ in the NMR spectrum strongly suggests the occurrence of reductive defluorination in solution. Using the bulkier PiPr3 allows formation of [GeF3(iPr3P)2][OTf], whose variable temperature NMR spectra strongly indicate is a triflate salt of the [GeF3(iPr3P)2]+ monocation.[GeF4{o-C6H4(PMe2)2}] reacts with n equivalents of TMSOTf to generate the [GeF4−n{o-C6H4(PMe2)2}][OTf]n (n = 1, 2, 3) series, and again the trifluoride, [GeF3{o-C6H4(PMe2)2}(OTf)], was shown to be unstable to reductive defluorination in solution, producing [Ge{(o-C6H4(PMe2)2}(OTf)][OTf], which features a Ge(II) monocation.
The 19F and 31P NMR chemical shifts and couplings and the X-ray structural trends observed with sequential fluoride removal across the series are consistent with an increase in positive charge at germanium as fluoride is replaced with triflate.
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We thank EPSRC for funding via the ADEPT Programme Grant (EP/N035437/1) and a studentship to R. P. K. (EP/N509747/1). We also thank Dr M. E. Light for helpful discussions regarding aspects of the crystallographic data and the Crystallographic Reviewer for very helpful suggestions regarding treatment of some of the disorder observed.References
- For examples see: (a) Coordination Chemistry of the s, p, and f metals, Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, 2003, vol. 3 Search PubMed; (b) Modern Main Group Chemistry (special issue), Chem. Soc. Rev, ed. S. Aldridge and C. Jones, 2016, vol. 45, pp. 763–764 and references therein Search PubMed; (c) G. E. Smith, H. L. Sladen, S. C. G. Biagini and P. J. Blower, Dalton Trans., 2011, 40, 6196–6205 RSC; (d) S. L. Benjamin and G. Reid, Coord. Chem. Rev., 2015, 297–298, 168–180 CrossRef CAS.
- (a) J. Parr, Germanium, tin and lead, Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, 2003, vol. 3, 545–608 Search PubMed; (b) T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, J. Am. Chem. Soc., 2014, 136, 3028–3031 CrossRef CAS PubMed; (c) R. D. Rittinghaus, J. Tremmel, A. Růžička, C. Conrads, P. Albrecht, A. Hoffman, A. N. Ksiazkiewicz, A. Pich, R. Jambor and S. Herres-Pawlis, Chem. – Eur. J., 2020, 26, 212–221 CrossRef CAS PubMed; (d) M. M. D. Roy, A. A. Omaña, A. S. S. Wilson, M. S. Hill, S. Aldridge and E. Rivard, Chem. Rev., 2021, 121, 12784–12965 CrossRef PubMed.
- (a) S. L. Benjamin, W. Levason and G. Reid, Chem. Soc. Rev., 2013, 42, 1460–1499 RSC; (b) W. Levason, F. M. Monzittu and G. Reid, Coord. Chem. Rev., 2019, 391, 90–130 CrossRef CAS; (c) K. Chansaenpak, B. Vabre and F. P. Gabbaï, Chem. Soc. Rev., 2016, 45, 954–971 RSC.
- J. Burt, W. Levason and G. Reid, Coord. Chem. Rev., 2014, 260, 65–115 CrossRef CAS.
- W. Levason, G. Reid and W. Zhang, Coord. Chem. Rev., 2011, 255, 1319–1341 CrossRef CAS.
- R. P. King, M. S. Woodward, G. McRobbie, J. Grigg, W. Levason and G. Reid, Dalton Trans., 2021, 50, 14400–14410 RSC.
- A. W. Waller, N. M. Weiss, D. A. Decato and J. A. Phillips, J. Mol. Struct., 2017, 1130, 984–993 CrossRef CAS PubMed.
- D. T. Tran, P. Y. Zavalij and S. R. J. Oliver, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, m742–m743 CrossRef CAS.
- F. Cheng, M. F. Davis, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Eur. J. Inorg. Chem., 2007, 4897–4905 CrossRef CAS.
- A. D. Adley, P. H. Bird, A. R. Fraser and M. Onyszchuk, Inorg. Chem., 1972, 11, 1402–1409 CrossRef CAS.
- F. Cheng, M. F. Davis, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Eur. J. Inorg. Chem., 2007, 2488–2495 CrossRef CAS.
- K. George, A. L. Hector, W. Levason, G. Reid, G. Sanderson, M. Webster and W. Zhang, Dalton Trans., 2011, 40, 1584–1593 RSC.
- M. F. Davis, W. Levason, G. Reid and M. Webster, Dalton Trans., 2008, 2261–2269 RSC.
- M. F. Davis, W. Levason, G. Reid, M. Webster and W. Zhang, Dalton Trans., 2008, 533–538 RSC.
- R. Suter, A. Swidan, C. L. B. Macdonald and N. Burford, Chem. Commun., 2018, 54, 4140–4143 RSC.
- H. J. Hogben, M. Krzystyniak, G. T. P. Charnock, P. J. Hore and I. Kuprov, J. Magn. Reson., 2011, 208, 179–194 CrossRef CAS PubMed.
- V. K. Greenacre, R. P. King, W. Levason and G. Reid, Dalton Trans., 2019, 48, 17097–17105 RSC.
- R. P. King, V. K. Greenacre, W. Levason, J. M. Dyke and G. Reid, Inorg. Chem., 2021, 60, 12100–12108 CrossRef CAS PubMed.
- A. M. Forster and A. J. Downs, Polyhedron, 1985, 4, 1625–1635 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- E. MacDonald, PhD Thesis, Dalhousie University, Canada, 2013.
- E. P. Kyba, S. T. Liu and R. L. Harris, Organometallics, 1983, 2, 1877–1879 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (c) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. L. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic parameters for the structures reported (Table S1), the crystal structure of [Ge{o-C6H4(PMe2)2}(OTf)][OTf] (Fig. S1), together with multinuclear NMR and IR spectra associated with each of the new compounds described (Fig. S2–S14). CCDC 2113230, 2113231, 2113232, 2113233, 2113234, 2113235, 2113236, 2113237, 2113415 and 2113416. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03339e |
| This journal is © The Royal Society of Chemistry 2021 |