
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDihypoiodites stabilised by 4-ethylpyridine through O–I–N halogen bonds†
Eric
Kramer
,
Shilin
Yu
 ,
Jas S.
Ward
* and
Kari
Rissanen
*
,
Jas S.
Ward
* and
Kari
Rissanen
*
University of Jyvaskyla, Department of Chemistry, Jyväskylä 40014, Finland. E-mail: james.s.ward@jyu.fi; kari.t.rissanen@jyu.fi
First published on 14th October 2021
Abstract
Four bis(O–I–N) compounds have been synthesised from various dihypoiodites and 4-ethylpyridine. The compounds were characterised in both the solution and solid states by NMR spectroscopy (1H, 15N), X-ray diffraction, and computational calculations.
Halogen bonding, recently defined as the attraction between an electrophilic halogen atom and a neutral or anionic nucleophile,1 has continued to rapidly expand as a field of research as demonstrated by the enormity of published works on the topic in recent years.2–7 As a non-covalent interaction, halogen bonding has been implemented for a broad array of uses such as the generation of phosphorescent, magnetic, and porous functional materials.2,5 Whilst not as ubiquitous as the closely related hydrogen bonding, halogen bonding enjoys advantages such as being far more directional than hydrogen bonding due to its electronic origins as a σ-hole interaction.4 This reliability of a high degree of linear directionality has been implemented in self-assembling processes toward generating supramolecular architectures,8–10 and very recently has been incorporated into the realm of coordination polymers as halogen-bonded organic frameworks (XOFs).11
Neutral N–I⋯N and N–I⋯O–N halogen-bonded systems have been extensively studied, thanks to their ease of preparation from appropriate pairings of halogen bond (XB) donors (e.g., haloimides, halosulfonimides) and acceptors (e.g., pyridine derivatives, pyridine-N-oxides), by Fourmigué et al. and Rissanen et al.12–17
As first reported in the 1960s,18,19 the sub-group of halogen(I) (or halonium) ions, [L–X–L]+ (X = Cl, Br, I; L = predominantly N-heterocyclic Lewis bases, but also includes oxygen- and sulfur-based donor compounds), represent the case of when a halogen atom is fully ionised to a formally cationic state, X+, and stabilised by a pair of Lewis bases. The study of halogen(I) ions was reinvigorated in the 1990s by Barluenga, who demonstrated a myriad of effective organic transformations using his eponymous reagent, [I(pyridine)2]BF4.20–22 Comprehensive solution-state and computational studies by Erdélyi and co-workers have confirmed the symmetric nature of the [N–I–N]+ group,23,24 with the iodine atom lying in the centre between the nitrogen atoms of the Lewis bases. The analogous anionic dioxoiodane complexes, [O–I–O]−, are also known and have found applications as organic reagents.25
The field of halogen(I) chemistry has seen numerous advancements in recent years, with the observation of the first asymmetric,10 unrestrained heteroleptic,26 and nucleophilic interactions of iodine(I) complexes being reported,27–29 creating renewed interest in other overlooked halogen bonded motifs. Compounds demonstrating the first stabilised benzoyl hypoiodite, PhC(O)O–I-pyridine (CSD code BZPRIA), and the analogous compound based on the dicarboxylic acid, (phthaloyl-OI)(pyridine)2 (CSD code BZPRIB), were first reported 40 years ago.30 However, their full potential was not realised until recent studies revealed that the un-stabilised trifluoroacetyl hypoiodite has the highest σ-hole value, VS,Max, of +234.55 kJ mol–1 when compared to N-iodosaccharin (VS,Max = +226.36 kJ mol−1), making it, to date, the strongest known neutral halogen bond donor molecule,31 which was determined computationally based on the solid-state data of the 4-dimethylaminopyridine-stabilised trifluoroacetyl hypoiodite. Encouraged by the promising results already observed for the stabilised carbonyl hypoiodites, a continued study into the bis-stabilised carbonyl hypoiodites is reported herein.
Four bis(O–I–N) complexes were straightforwardly synthesised from the silver(I) dicarboxylate salts of phthalic acid, tetrafluorophthalic acid, isophthalic acid, or terephthalic acid, with 4-ethylpyridine (4-Etpy) and elemental iodine (Schemes 1 and 2), to give (phthaloyl-OI)(4-Etpy)2 (1), (tetrafluorophthaloyl-OI)(4-Etpy)2 (1F), (isophthaloyl-OI)(4-Etpy)2 (2), and (terephthaloyl-OI)(4-Etpy)2 (3).
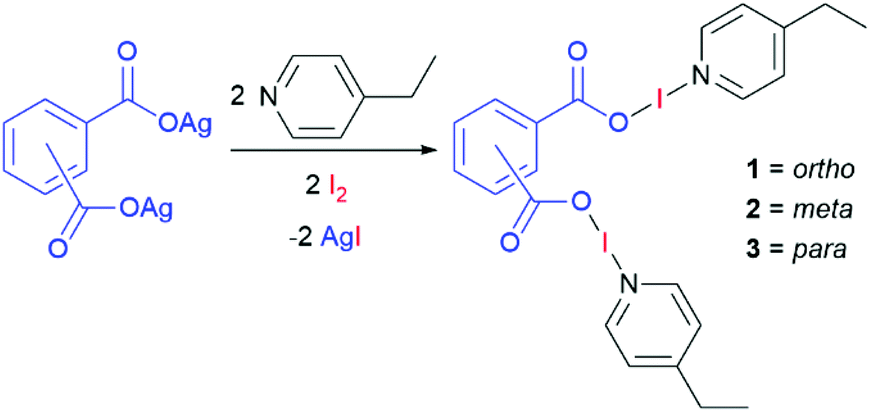 | ||
| Scheme 1 The general synthetic procedure for the synthesis of compounds 1, 2, and 3. | ||
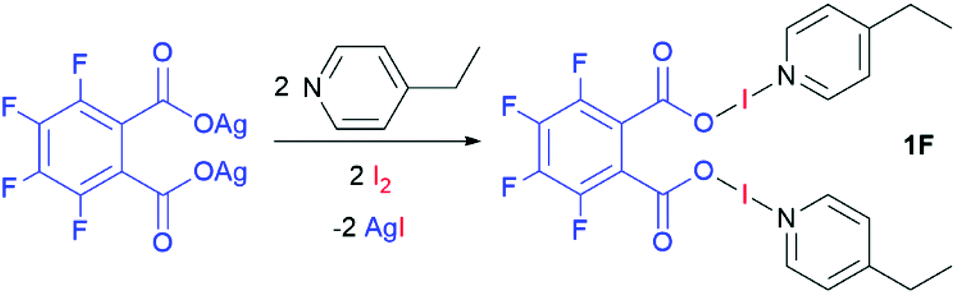 | ||
| Scheme 2 The synthetic procedure to prepare compound 1F. | ||
The compounds were characterised in solution by NMR spectroscopy, however, the 1H NMR spectra were straightforward and as expected in that the installation of the iodine caused the chemical shifts to move to higher frequencies. The same pattern has been observed in the formation of [N–I–N]+ complexes,31 and therefore warrants no further comment. The more informative 15N NMR chemical shifts of the stabilising 4-Etpy Lewis bases were also recorded to study the effect of varying the geometric and electronic properties of the oxygen components of the O–I–N groups.
Upon inclusion into the neutral O–I–N compounds, the 4-Etpy experiences a drastic change in its 15N NMR chemical shift from −75.6 ppm to approximately −160 ppm.26 Between the three isomers, the 15N NMR chemical shifts showed only minor variations within a 3.4 ppm range for 1 (−160.7 ppm), 2 (−159.5 ppm), and 3 (−162.9 ppm). The previously unreported mono(O–I–N) compound (benzoyl-OI)(4-Etpy) (4) was also synthesised to obtain the 15N NMR chemical shift, for the purpose of comparison to the bis(O–I–N) analogues, which also fell into the range of values observed for 1–3 with a chemical shift of −161.6 ppm. Compound 1F (−171.2 ppm), which incorporated a far more electron withdrawing dicarboxylate backbone, understandably demonstrated a more noticeable upfield shift compared to 1, with a difference of 10.5 ppm. In comparison to the formally positive halogen(I) analogue, [I(4-Etpy)2]PF6 (δN −182.9),26 compounds 1–3 are less shielded by 20–23 ppm in the 15N NMR spectra, and by only 11 ppm for 1F, demonstrating the effect of the different Lewis basicities of the neutral 4-Etpy and an anionic carboxylate.
Single crystals were obtained for 1, 1F, and 2 (Fig. 1), permitting solid-state comparisons to be made. The O–I and I–N bond lengths possess an inversely reciprocal relationship in all cases, owing to the consistent O⋯N distances always lying in the narrow range of 4.41(1)–4.45(1) Å. The shortest O–I (2.153(5) Å) and longest I–N (2.305(5) Å) bond lengths were found in 1, whilst the longest O–I (2.223(6) Å) and shortest I–N (2.208(6) Å) bond lengths were observed in 1F. Between 1 and 2, no crystallographically significant differences were apparent. Similarly, no crystallographically significant differences were observed between 1 and (phthaloyl-OI)(pyridine)2 (cf. O–I = 2.136, 2.183 Å; I–N = 2.282, 2.324 Å), though it should be noted that the latter was collected 40 years ago at room temperature,30 which should be taken into account when directly comparing these values. The O–I–N angles in all cases were within the effectively linear range of 173.2(2)–176.2(6)°, as imposed by the iodine atom which has a strong preference toward linear coordination. When the I–N bond lengths are compared to the known crystallographic parameters for the [I(4-Etpy)2]+ iodine(I) ion (2.25(1) and 2.26(1) Å; reported as a solvate of a larger silver(I) polymeric complex),28 it can be seen that 1 and 2 have slightly elongated bond lengths, and 1F has slightly shorter values, with the most extreme in each case being crystallographically significant in terms of their respective esds. On the other hand, when the O–I bond lengths are compared to the formally negative [O–I–O]− complex [NnBu4] [I(OC(O)C6H4F-3)2] (cf. 2.193(5) Å; crystallographically equivalent),25 compounds 1 and 2 are only slightly shorter, with the bonds lengths mostly being within the error of measurements (to a 3σ2 tolerance) except for the 2.153(5) Å O–I bond length of compound 1; when the same [O–I–O]− complex is compared to compound 1F, the O–I bond lengths are slightly longer in 1F, though again only the 2.223(6) Å O–I bond length can be considered outside the error of the measurement. As expected for the highly directional halogen bonding of the O–I–N group,31 and previously observed for other iodine(I) motifs (e.g., [N–I–N]+),26,31,32 the solid-state packing of compounds 1, 1F, and 2 were unremarkable with respect to the O–I–N groups, with no apparent involvement of the iodine atoms in the observed intermolecular packing interactions. A short intramolecular I⋯I distance of 4.0170(7) Å and intermolecular distance of 4.2768(6) Å were observed in 1F (Fig. 2), both of which are approaching the sum of the van der Waals radii for two iodine atoms (3.96 Å), though shorter I⋯I distances have been previously reported between two molecules of [I(pyridine)(4-dimethylaminopyridine)]PF6 (3.78 Å; no esd due to the iodine atom lying on a crystallographic special position).26
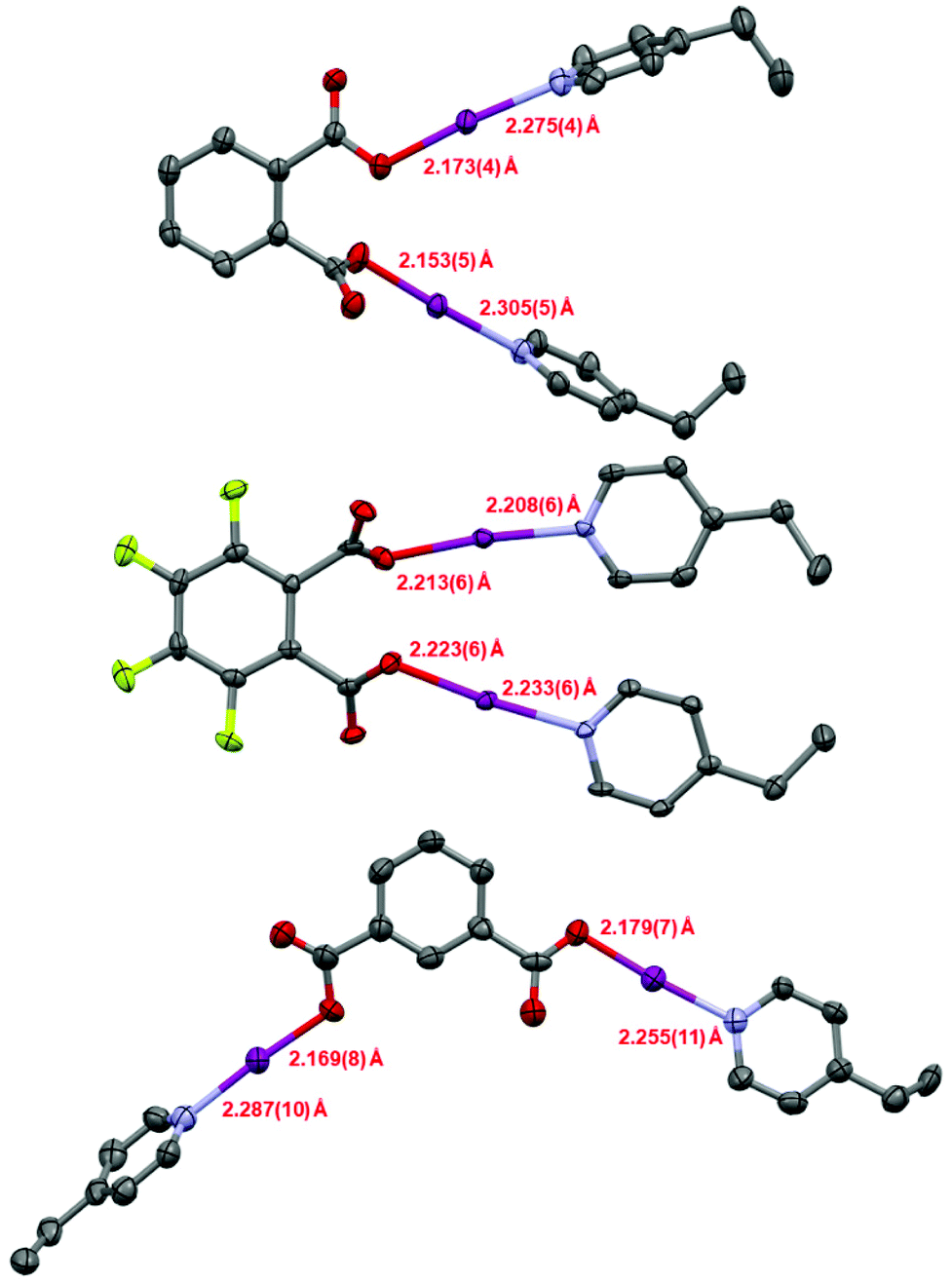 | ||
| Fig. 1 The molecular structures of 1 (top), 1F (middle), and 2 (bottom), as determined by X-ray diffraction, with the O–I and I–N bond lengths annotated (hydrogen atoms omitted for clarity; thermal ellipsoids at 50% probability). | ||
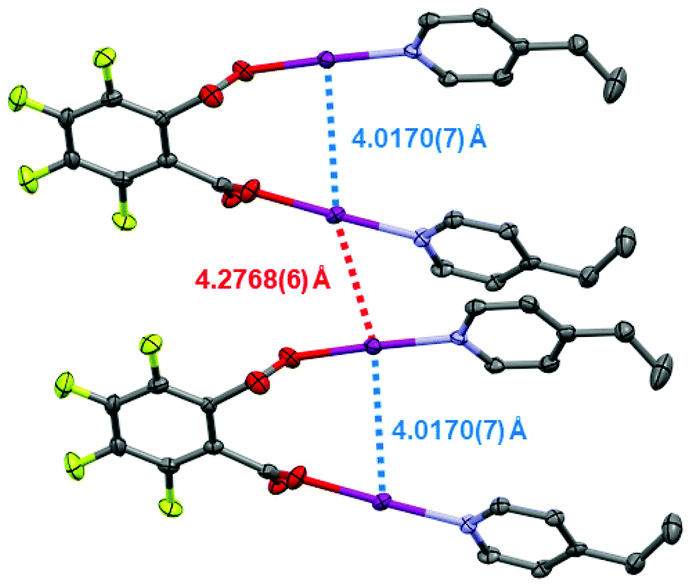 | ||
| Fig. 2 The I⋯I intramolecular (annotated in blue) and shortest I⋯I intermolecular (annotated in red) distances between two molecules of 1F (hydrogen atoms omitted for clarity; thermal ellipsoids at 50% probability). | ||
Overall, the solid-state results demonstrate that a change in geometry of the stabilising ligands has only a negligible effect on the nature of the O–I–N groups, whilst the effects of electronic changes are much more pronounced. These observations are consistent with studies for [N–I–N]+ halogen(I) complexes,33 which have been reported to be similarly resilient toward such aspects.
The DFT geometries of 1, 1F, 2, and the literature compound (phthaloyl-OI)(pyridine)2 (Fig. S14–S16 and S18†),30 were calculated using the experimentally determined coordinates for the purpose of comparison, as had been reported previously for similar Lewis base-stabilised carbonyl hypoiodite compounds.31 These calculations were performed to the M06-2X (def2-TZVP) level of theory, including a non-polar (DCM) solvent model, using the Spartan 18 software package.34 The calculated models were all in good agreement with the experimentally determined structures obtained by X-ray crystallography, with differences in the range of 0.003–0.052 Å for the O–I bond lengths, and 0.000–0.052 Å for the I–N bond lengths. The computational models did show much better consistency between the two O–I–N groups within each compound than was observed for the experimental data, however, this can be attributed to packing effects in the experimental X-ray diffraction structures, an additional influence that was not accounted for in the computational calculations. The experimental-computational comparison with the largest errors was those of 1F, as has been demonstrated previously with other strongly electron withdrawing carboxylates, e.g., trifluroracetate.31 Despite repeated attempts, satisfactory single crystal X-ray diffraction data could not be obtained for compound 3, however, a calculated structural model for 3 (Fig. 3) was found to be effectively unchanged with respect to those calculated for 1 and 2 with respect to the O–I and I–N bond lengths, which themselves had good agreement to the experimentally determined structures of 1 and 2, and is also supported by the 15N NMR data (vide supra).
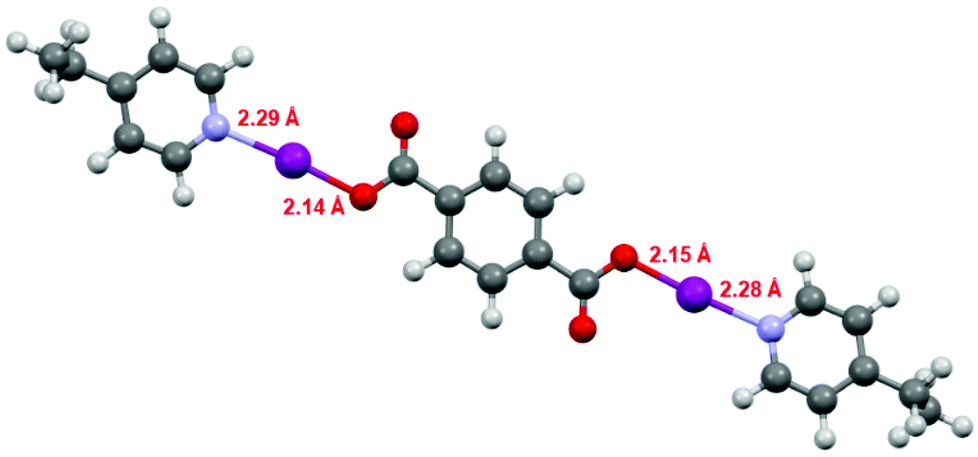 | ||
| Fig. 3 The computationally generated structure for compound 3, with the O–I and I–N bond lengths annotated. | ||
In conclusion, four compounds incorporating two O–I–N groups have been synthesised, and characterised in both the solution (1H and 15N NMR) and solid (X-ray) states, as well as by computational methods. These compounds demonstrate that the O–I–N groups are effectively unchanged between different isomers (compounds 1, 2 and 3), though they are affected by electronic changes of the incorporated carboxylate (compounds 1 and 1F), as determined by 15N NMR chemical shifts and in the solid-state by X-ray diffraction. Computational calculations were in good agreement with experimentally determined structures, demonstrating their use of accurately predicting structural models for compounds for which X-ray diffraction data could not be obtained, such as with compound 3. In the Cambridge Structural Database, iodine dominates out of the halogen elements (group 7A) in such O-7A-N systems, probably due to the more stabilised hypoiodite moiety in comparison to the analogous hypobromites and hypochlorites. No corresponding X-ray structurally characterised examples of hypobromites and hypochlorites currently exist in the literature, presenting a tempting challenge for future research endeavours.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Finnish Cultural Foundation Central Fund (J.S.W. grant number 00201148), the Magnus Ehrnrooth Foundation (J.S.W.), the Academy of Finland (K.R. grant no. 317259), and the University of Jyvaskyla, Finland for financial support.Notes and references
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- M. H. Kolář and P. Hobza, Chem. Rev., 2016, 116, 5155–5187 CrossRef PubMed.
- J. Pancholi and P. D. Beer, Coord. Chem. Rev., 2020, 416, 213281 CrossRef CAS.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- H. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef CAS PubMed.
- M. Saccone and L. Catalano, J. Phys. Chem. B, 2019, 123, 9281–9290 CrossRef CAS PubMed.
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033–14036 CrossRef CAS PubMed.
- L. Turunen, U. Warzok, C. A. Schalley and K. Rissanen, Chem, 2017, 3, 861–869 CAS.
- A. Vanderkooy, A. K. Gupta, T. Földes, S. Lindblad, A. Orthaber, I. Pápai and M. Erdélyi, Angew. Chem., Int. Ed., 2019, 58, 9012–9016 CrossRef CAS PubMed.
- G. Gong, S. Lv, J. Han, F. Xie, Q. Li, N. Xia, W. Zeng, Y. Chen, L. Wang, J. Wang and S. Chen, Angew. Chem., Int. Ed., 2021, 60, 14831–14835 CrossRef CAS PubMed.
- O. Makhotkina, J. Lieffrig, O. Jeannin, M. Fourmigué, E. Aubert and E. Espinosa, Cryst. Growth Des., 2015, 15, 3464–3473 CrossRef CAS.
- E. Aubert, E. Espinosa, I. Nicolas, O. Jeannin and M. Fourmigué, Faraday Discuss., 2017, 203, 389–406 RSC.
- K. Raatikainen and K. Rissanen, CrystEngComm, 2011, 13, 6972–6977 RSC.
- K. Raatikainen and K. Rissanen, Chem. Sci., 2012, 3, 1235–1239 RSC.
- R. Puttreddy, O. Jurček, S. Bhowmik, T. Mäkelä and K. Rissanen, Chem. Commun., 2016, 52, 2338–2341 RSC.
- R. Puttreddy, J. M. Rautiainen, T. Mäkelä and K. Rissanen, Angew. Chem., Int. Ed., 2019, 58, 18610–18618 CrossRef CAS PubMed.
- J. A. Creighton, I. Haque and J. L. Wood, Chem. Commun., 1966, 229 RSC.
- I. Haque and J. L. Wood, J. Mol. Struct., 1968, 2, 217–238 CrossRef CAS.
- J. Barluenga, J. M. González, M. A. Garcia-Martin, P. J. Campos and G. Asensio, J. Chem. Soc., Chem. Commun., 1992, 1016–1017 RSC.
- J. Ezquerra, C. Pedregal, C. Lamas, J. Barluenga, M. Pérez, M. A. García-Martín and J. M. González, J. Org. Chem., 1996, 61, 5804–5812 CrossRef CAS.
- G. Espuña, G. Arsequell, G. Valencia, J. Barluenga, M. Pérez and J. M. González, Chem. Commun., 2000, 1307–1308 RSC.
- A.-C. C. Carlsson, J. Gräfenstein, J. L. Laurila, J. Bergquist and M. Erdélyi, Chem. Commun., 2012, 48, 1458–1460 RSC.
- A.-C. C. Carlsson, J. Gräfenstein, A. Budnjo, J. L. Laurila, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdélyi, J. Am. Chem. Soc., 2012, 134, 5706–5715 CrossRef CAS PubMed.
- K. Muñiz, B. García, C. Martínez and A. Piccinelli, Chem. – Eur. J., 2017, 23, 1539–1545 CrossRef PubMed.
- J. S. Ward, G. Fiorini, A. Frontera and K. Rissanen, Chem. Commun., 2020, 56, 8428–8431 RSC.
- S. Yu, P. Kumar, J. S. Ward, A. Frontera and K. Rissanen, Chem, 2021, 7, 948–958 CAS.
- J. S. Ward, A. Frontera and K. Rissanen, Inorg. Chem., 2021, 60, 5383–5390 CrossRef CAS PubMed.
- J. S. Ward, A. Frontera and K. Rissanen, Chem. Commun., 2021, 57, 5094–5097 RSC.
- H. Hartl and M. Hedrich, Z. Naturforsch. B, 1981, 36b, 922–928 CrossRef CAS.
- S. Yu, J. S. Ward, K.-N. Truong and K. Rissanen, Angew. Chem., Int. Ed., 2021, 60, 20739–20743 CrossRef CAS PubMed.
- K. Rissanen and M. Haukka, Top. Curr. Chem., 2015, 359, 77–90 CrossRef CAS PubMed.
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Gräfenstein, K. Rissanen and M. Erdélyi, J. Am. Chem. Soc., 2016, 138, 9853–9863 CrossRef CAS PubMed.
- Spartan'18, Wavefunction Inc., Irvine CA, USA, 2018 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, NMR and computational details. CCDC 2105108–2105110. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03324g |
| This journal is © The Royal Society of Chemistry 2021 |