
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIncorporation of iodine into uranium oxyhydroxide phases†
Gabriel L.
Murphy
 *,
Philip
Kegler
,
Martina
Klinkenberg
,
Andreas
Wilden
,
Maximilian
Henkes
,
Dimitri
Schneider
and
Evgeny V.
Alekseev
*
*,
Philip
Kegler
,
Martina
Klinkenberg
,
Andreas
Wilden
,
Maximilian
Henkes
,
Dimitri
Schneider
and
Evgeny V.
Alekseev
*
Institute of Energy and Climate Research, Forschungszentrum Jülich GmbH, 52428 Jülich, Germany. E-mail: g.murphy@fz-juelich.de; e.alekseev@fz-juelich.de
First published on 17th November 2021
Abstract
Herein, we have synthesised a novel uranium oxyhydroxide (UOH) phase, Rb2K2[(UO2)6O4(OH)6]·(IO3)2, under hydrothermal conditions which intercalates IO3−via a hybrid salt-inclusion and host–guest mechanism. The mechanism is based on favorable intermolecular bonding between disordered Rb+/K+ and IO3− ions and hydroxyl and layer void positions respectively. To examine whether the intercalation may occur ubiquitously for UOH phases, the known UOH mineral phases metaschoepite ([(UO2)8O2(OH)12]·12H2O), compreignacite (K2[(UO2)6O4(OH)6]·7H2O) and also related β-UO2(OH)2 were synthesised and exposed to aqueous I− and IO3− for 1 month statically at RT and 60 °C in air and the solid analysed using laser ablation inductively coupled plasma mass spectroscopy. Measurements indicate intercalation can occur homogeneously, but the affinity is dependent upon the structure of the UOH phases and temperature, where higher temperatures and when the interlayer space is free of initial moieties are favoured. It was also found that after repeated washing of the UOH samples with DI water the intercalated iodine was retained. UOH phases are known to form during the oxidative corrosion of spent nuclear fuel during an accident scenario in the near field, this work suggests they may help retard the transport of radiolytic iodine into the environment during a long-term release event.
Introduction
Uranium oxyhydroxide (UOHs) phases are important minerals which occur as weathering products of uraninite deposits and importantly, are encountered during the paragenetic sequence of secondary phases that form during an accident scenario involving spent nuclear fuel (SNF) oxidation in the near-field.1–9 Understanding the structural chemistry of these phases is of importance as via interaction mechanisms, they can potentially retard the transport of harmful fission daughters from SNF towards the biosphere.5,7 Of these, 129I (t1/2 = 1.7 × 107 years) is considered to be of significance, since it is one of the main contributors to the final dose in performance assessment calculations in SNF repository operations and can readily be incorporated into human metabolic processes.6,10–12 Chemically, it is expected upon long-term release from SNF under aqueous conditions, radiolytic iodine should occur in the near-field predominantly as iodide, I−,13 and also iodate, IO3−, in the case of more oxidative environments.14–16 Invariably within the near-field of SNF, the vast majority of backfill and mineral phases encountered possess anionic layered structures for instance bentonite,17 which are favourable for cationic, but inhibit anionic, fission daughter species retention to occur.5,18 Although some evidence is present for trace level intercalation of anionic iodine into layered mineral structures through ionic exchange mechanisms in model system studies19–21 the current consensus is the retention of radiolytic iodine in the near field is not proven and best considered in the far-field of SNF repository performance and accident scenario assessment.22 It has been previously suggested that UOH phases, considering their structural chemistry and proximity of formation to SNF, can intercalate fission species, particularly cationic, during unexpected release.7,8 However, there are at present no studies that have examined the potential retention of anionic fission species into UOH phases for instance radiolytic iodine, despite the renewed focus these materials have recently received23–26 coupled with the broader reinvigorated interest in actinide and actinide-iodine chemical science.27–37In the present investigation, we describe the synthesis and characterisation of a novel UOH structure which successfully intercalates IO3− into its structure, Rb2K2[(UO2)6O4(OH)6]·(IO3)2 under hydrothermal conditions. We further show that using laser ablation inductively coupled plasma mass spectroscopy (LA-ICPMS) that the known UOH mineral phases metaschoepite,38 [(UO2)8O2(OH)12]·12H2O, compreignacite,39 K2[(UO2)6O4(OH)6]·7H2O, and also related β-UO2(OH)240 are able to intercalate iodine at RT and 60 °C from aqueous solutions of KI and KIO3, although at a lesser degree, than that observed in Rb2K2[(UO2)6O4(OH)6]·(IO3)2. The results from this investigation are discussed in the context of the broader family of UOH phases and particularly their occurrence and interaction with radiolytic iodine species during long term release from SNF in an accident scenario. This investigation unveils the importance of the formation of UOH phases during the oxidative corrosion of SNF and their ability to interact and potentially impede the transport of radiolytic iodine into the biosphere.
Experimental
Synthesis
Single crystal X-ray diffraction
Suitable quality crystals of Rb2K2[(UO2)6O4(OH)6]·(IO3)2 and β-UO2(OH)2 were selected for analysis and structural refinement using single crystal X-ray diffraction (SC-XRD). Unfortunately, suitable crystals of metaschoepite and compreignacite could not be recovered for SC-XRD analysis due to insufficient size. Data for Rb2K2[(UO2)6O4(OH)6]·(IO3)2 and β-UO2(OH)2 were recorded with CrysAlisPro software on an Agilent Oxford Diffraction Super Nova diffractometer with a Mo Kα tube at 296 K. Absorption corrections for the raw data were performed using the multiscan method. The unit cell was determined, and background effects were processed by the CrysAlisPro software. The initial structures for Rb2K2[(UO2)6O4(OH)6]·(IO3)2 and β-UO2(OH)2 were refined using SHELXL-2018 within the WinGX (v1.80.05) software,42 and the ADDSYM algorithm of the PLATON program43 was used for the checking of possible higher symmetries. The structure of Rb2K2[(UO2)6O4(OH)6]·(IO3)2 has been submitted to the Cambridge Crystallographic Data Centre (CCDC) under the number 2094057.†Powder X-ray diffraction
Powder X-ray Diffraction (XRD) measurements were made at room temperature using a Bruker AXS D8 Endeavor diffractometer (40 kV/40 mA) in Bragg−Brentano geometry. The diffractometer has a copper X-ray tube and a primary nickel filter, producing Cu Kα1,2 radiation (λ = 1.541 87 Å). A linear silicon strip LynxEye detector (Bruker-AXS) was used. Data were collected in the range of 2θ = 10–120° with 10 s/step and a step width of 0.02°. The aperture of the fixed divergence slit was set to 0.2 mm, and the receiving slit was set to 8 mm. The discriminator of the detector was set to an interval of 0.16–0.25 V. Data were analysed by pattern matching using the program FullProf,44 where space group and lattice parameter information were obtained from SC-XRD measurements for Rb2K2[(UO2)6O4(OH)6]·(IO3)2 and β-UO2(OH)2 and from reference cif-files39 for metaschoepite and compreignacite as starting parameters. Analysis and refinement profiles are giving in the ESI S1.†Bond valence sums calculations
Bond valence sums (BVS) calculations were undertaken for the anions and cations in Rb2K2[(UO2)6O4(OH)6]·(IO3)2 using the parameters reported by Burns and co-workers45 for seven-coordinate U. The parameters provided by Brese and O'Keeffe46 were used for Rb, K, I and O.I− and IO3− intercalation experiments
Stock aqueous solutions of KI, KIO3 and K2SeO4 were generated with a target concentration of 0.015 M (actual 0.0181, 0.0135 and 0.0140 M respectively). Samples of metaschoepite, compreignacite and β-UO2(OH)2 were mixed with 2 ml aliquots of the KI, KIO3 and K2SeO4 aqueous stock solutions and were left to exchange for 1 month statically at RT and 60 °C in air. The exchange experiments were conducted with an U excess such that the U![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :I/Se ratio was 5:1 approximately. The choice of U excess was due to the initial intention to monitor the change of I/Se concentration in solution rather than solid. However corresponding measurement precision was not satisfactory and instead the solid was analysed via LA-ICPMS alternatively.
:I/Se ratio was 5:1 approximately. The choice of U excess was due to the initial intention to monitor the change of I/Se concentration in solution rather than solid. However corresponding measurement precision was not satisfactory and instead the solid was analysed via LA-ICPMS alternatively.
Laser ablation inductively coupled mass spectroscopy
Laser Ablation Inductively coupled Mass spectroscopy (LA-ICPMS) measurements were undertaken using PerkinElmer NexION 2000C ICPMS with New Wave Research, Inc. – UP266 UV YAG Laser Ablation System attachment. For the LA-ICPMS measurements, solid dried samples were mounted and dispersed on sample holders. The laser was slowly rastered across the holder surface and samples were ablated and ICPMS measurements collected successively for I-127 and U-235.Scanning electron microscopy
The morphology and elemental composition of the crystals were determined using a FEI Quanta 200F Environment Scanning Electron Microscope (SEM) fitted with an energy-dispersive X-ray spectrometer (EDS). This verified the sample compositions based on the SC-XRD structure solutions. Details of SEM/EDS measurements are given in the ESI.†Results and discussion
Structural chemistry – Rb2K2[(UO2)6O4(OH)6]·(IO3)2
The structure of Rb2K2[(UO2)6O4(OH)6]·(IO3)2, synthesised using a hydrothermal method, was found to be a trigonal structure via SC-XRD measurements in space group P31m with lattice parameters a = 7.1406(5), c = 7.4646(7) Å and V = 329.62(6) Å3. Full crystallographic information is given in Table 1 for Rb2K2[(UO2)6O4(OH)6]·(IO3)2. The structure consists of UO7 pentagonal bipyramids edge sharing to form infinite layers containing distinctive triangular voids (Fig. 1) consistent with an α-U3O8 topology8 and similar to the UOH phase becquerelite.47 The in-plane uranium-oxygen bond length was found to range from 2.2296(17) to 2.59(7) Å whereas the normal to the layer oxo-uranium-oxygen bond length was found to range from 1.75(4) to 1.77(6) Å consistent with the presence of the uranyl moiety in a hexavalent state.45 Bond valence sums (BVS) calculations supported the hexavalent uranium, pentavalent iodine and monovalent potassium/rubidium crystallographic sites with respective values of 5.81, 5.51 and 1.45.45 Between the [(UO2)6O4(OH)6]2− layers, intercalated disordered Rb+, K+, and IO3− are found (Fig. 1b). In the case of Rb+ and K+, SC-XRD indicated they were disordered about their positions whereas the IO3− ions were found to be disordered relative to the c axis. Pertinently when observed relative to the [(UO2)6O4(OH)6]2− layers, the Rb+, K+ and IO3− ions follow an ordered arrangement, such that IO3− ions are found in the void space whereas the K+/Rb+ coordinate to the hydroxyl groups (Fig. 1b). The observed disordering in the structure from the SC-XRD analysis was also reflected in the oxygen sub-lattice, for instance in O5 in the equatorial plane. Possible twining was ruled out based on using Platon software to check and analysing of reciprocal space of the full data sets collected from different crystals.43 SEM-EDS measurements support the assigned chemical formula of Rb2K2[(UO2)6O4(OH)6]·(IO3)2 (see ESI S2† for details). | ||
| Fig. 1 Structural representation of Rb2K2[(UO2)6O4(OH)6]·(IO3)2 (a) General view and (b) [001] direction illustrating the coordination configuration between the interlayer IO3- and Rb/K+ions with void and hydroxyl positions of the [(UO2)6O4(OH)6]2- layers respectively. The black lines and boxes are guides for the coordination motif. Yellow and purple polyhedra and light/dark green spheres respectively represent UO7, IO3 and disordered Rb/K units respectively. | ||
| Compound | Rb2K2[(UO2)6O4(OH)6]·(IO3)2 |
|---|---|
| Formula | Rb2K2[(UO2)6O4(OH)6]·(IO3)2 |
| Formula weight | 1189.53 |
| Crystal system | Trigonal |
| Space group | P31m |
| a (Å) | 7.1406(5) |
| b (Å) | 7.1406(5) |
| c (Å) | 7.4646(7) |
| α (°) | 90 |
| β (°) | 90 |
| γ (°) | 120 |
| Volume (Å3) | 329.62(6) |
| Z/μ (mm−1) | 1 |
| F(000) | 497.0 |
| d calcd (g cm−3) | 5.993 |
| GOF | 1.078 |
| Final R1 [I > 2σ(I)] | 0.0447 |
| Final wR2 [I > 2σ(I)] | 0.0968 |
A pertinent feature of the structure of Rb2K2[(UO2)6O4(OH)6]·(IO3)2 is the bonding motif of intercalated IO3− and Rb+/K+ units, which appears to occur via an intermolecular mechanism due to the disordering and lack of direct bonding to the layers of the UOH structure. Accordingly the intercalation mechanism is best described as salt-inclusion or as a host–guest interaction.48 Salt-inclusion structures typically contain 0D ionic moieties contained within a covalent network structure. Examples of these are often encountered in tellurite halides49 or rare earth selenites50 where the lone-pair possessing salt ion often coordinates to the covalent network structure. This is argued to occur in Rb2K2[(UO2)6O4(OH)6]·(IO3)2via the lone pair possessing IO3− units with the positive void space within the [(UO2)6O4(OH)6]2− layers. An important distinction for salt-inclusion structures is that the salt can be readily removed under ambient conditions with water.48,51 Crystals of Rb2K2[(UO2)6O4(OH)6]·(IO3)2 were washed repeatedly with DI water and analysed with SC-XRD and SEM/EDS where no change to the presence of IO3/Rb2K2 was detected. Subsequently the interaction and intercalation of (IO3)2/Rb2K2 ions between the [(UO2)6O4(OH)6]2− can be considered that of a hybrid between salt-inclusion and host–guest.52
Ion-exchange studies – uranium oxyhydroxides phases
That Rb2K2[(UO2)6O4(OH)6]·(IO3)2 is able to retain IO3− is pertinent in the context of SNF repository conditions, as a part of the larger family of UOH phases, as it suggests these phases may act to impede IO3− and other radiolytic iodine if they contact each other. To examine this hypothesis the known UOH phases metaschoepite,38 compreignacite39 and also related β-UO2(OH)238 were synthesised using hydrothermal methods. Consistent with previous investigations,38,53 metaschoepite, compreignacite and β-UO2(OH)2 were found to adopt orthorhombic structures in space groups Pbcn, Pnnm and Pbca respectively. The structures of metaschoepite, compreignacite and β-UO2(OH)2 are presented in Fig. 2 comparatively with Rb2K2[(UO2)6O4(OH)6]·(IO3)2 where the topology of the UOH layers of each is compared. Full details of structural characterization can be found in the ESI.† | ||
| Fig. 2 Comparison of UOH (i) layer topology and (ii) general structures for (a) Rb2K2[(UO2)6O4(OH)6]·(IO3)2, (b) metaschoepite, (c) compreignacite and (d) β-UO2(OH)2. Note in (b) and (c) blue and green spheres respectively represent H2O oxygen atoms and K cations. | ||
Contrasting the four UOH structures shows that they all possess distinctive void and hydroxyl positions, which were argued to facilitate the intercalation of IO3− into the structure of Rb2K2[(UO2)6O4(OH)6]·(IO3)2 previously. It is also noteworthy that the structure of compreignacite is more closely aligned to Rb2K2[(UO2)6O4(OH)6]·(IO3)2 since they both contain electronegative layers whereas that of metaschoepite and β-UO2(OH)2 the layers are electroneutral. To examine whether this may occur also for metaschoepite, compreignacite and β-UO2(OH)2, these three UOH phases were exposed to aqueous KI and KIO3 solutions for 1 month at RT and 60 °C and the solid analysed via LA-ICPMS. The results of the LA-ICPMS are presented in Fig. 3.
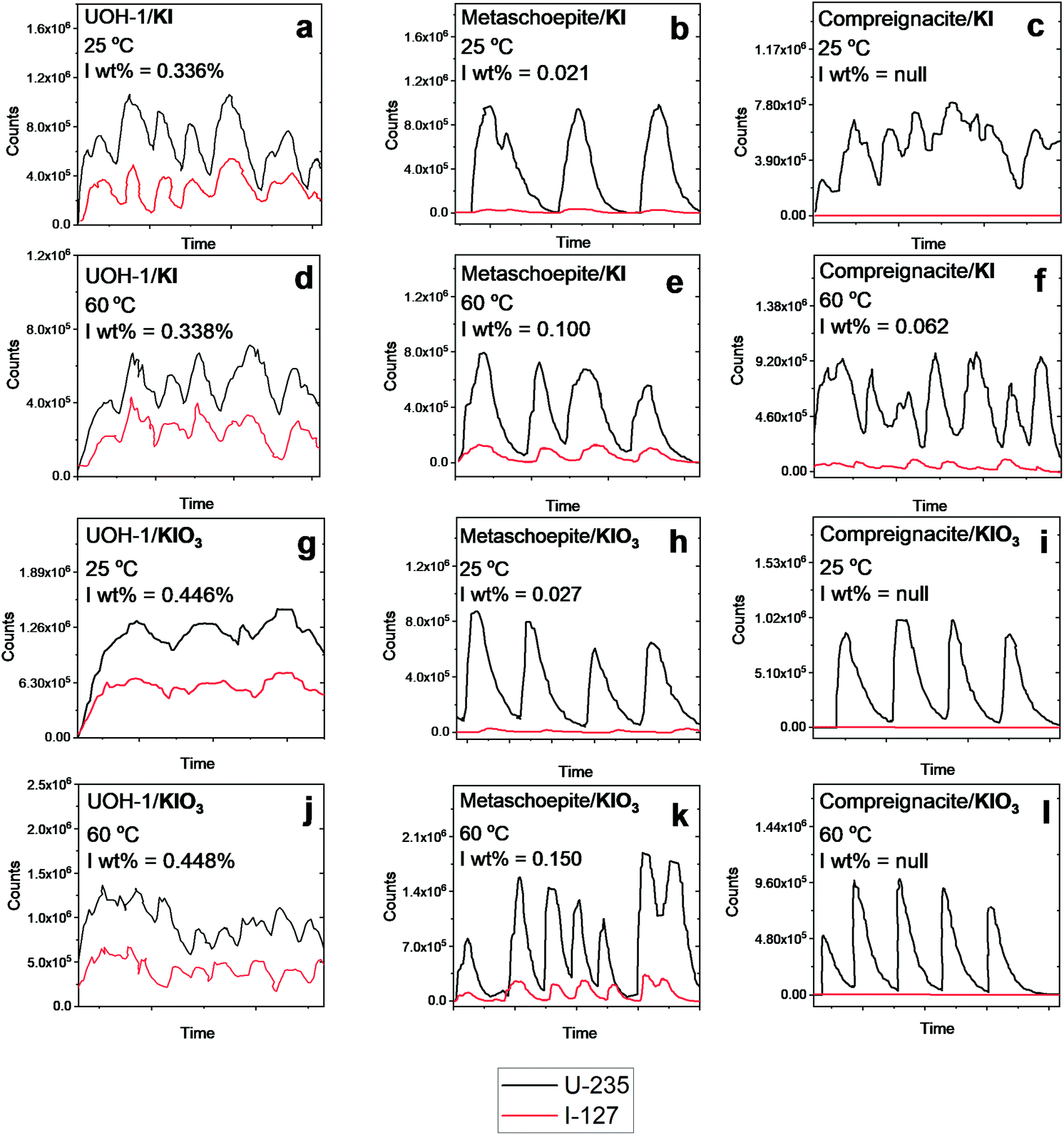 | ||
| Fig. 3 LA-ICPMS results for metaschoepite, compreignacite and β-UO2(OH)2 statically exposed to KI and KIO3 solutions at 25 °C and 60 °C in air for 1 month. Black and red lines correspond to ICPMS U-235 and I-127 signals respectively. | ||
Inspecting Fig. 3 intercalation of iodine is indeed observed from solutions containing I− and IO3− at RT and 60 °C for metaschoepite and β-UO2(OH)2. However, in the case of compreignacite intercalation of iodine is only observed from solution containing I− 60 °C. An important distinction between the compreignacite structure and that of metaschoepite and β-UO2(OH)2 is the presence of K+ cations between the former UOH phases layers of which instead in the latter two UOH phases is found H2O and free space respectively (see Fig. 2). Distinctive to the LA-ICPMS spectrum of the examined UOH phases where intercalation of iodine is observed is the near parallel line profile behaviour between that of the ICPMS U-235 and I-127 signals. That the profiles near mimic each other, particularly in terms of relatively intensities, suggests that the incorporation of iodine occurs homogenously across the UOH samples and it is not just a surface sorption effect. Although the relative degree of intercalation of iodine into the UOH phases is considerably lower than that observed for Rb2K2[(UO2)6O4(OH)6]·(IO3)2, it is pertinent that for the latter UOH phase intercalation occurred during hydrothermal synthesis at higher temperatures of 220 °C. Indeed, a general trend can be observed from Fig. 3 whereby increasing temperature results in higher intercalation affinity of which the rate of this is also dependent upon the presence of ions or moieties in the interlayer spacing the UOH phases. This is shown by the poor intercalation behavior of K+ interlaying possessing compreignacite compared to H2O and free interlayer metaschoepite and β-UO2(OH)2 respectively. It should be noted that chemical analysis for the iodine chemical state (I− and IO3−) was not performed on the examined UOH samples and as such in these intercalation experiments it is not clear if redox processes are present. Attempts were also made to examine the intercalation ability of these phases with SeO42− in the form of aqueous K2SeO4, however no intercalation was observed for any of the examined phases.
Discussion and implications
A plethora of UOH phases, both as minerals and synthetic compounds, have been previously examined particularly by Burns and co-workers, among others.7,23,25,39,53–55 A consistent trend among the UOH phases is their adoption of layered 2D structures which possess hydroxyl groups and distinctive voids in their layers (see Fig. 1b and 2). This consistent topology affords structural modifications of UOH phases containing alkali, alkaline earth, transition and also lanthanide cations in the interlayer space.7,8,25,53,54 Paragenetically it is understood that ternary UOH phases such as metaschoepite form prior to alkali or alkaline earth metal containing quaternary phases such as compreignacite or becquerelite.2,3,56 The inclusion of cations in the interlayer space of UOH phases generally involves them coordinating via the hydroxyl positions, where the void positions are vacant comparatively.24 This is consistent with Rb2K2[(UO2)6O4(OH)6]·(IO3)2 where the Rb/K cations from the SC-XRD model are observed to lie on the hydroxyl positions whereas the IO3− units are found near the voids. As discussed previously, the intercalation mechanism for IO3− in Rb2K2[(UO2)6O4(OH)6]·(IO3)2 is argued to be hybrid salt-inclusion and host–guest based and is mediated by intermolecular bonding. Such a bonding interaction suggests the intercalation is not structure specific and can occur ubiquitously across the UOH family of compounds due to their similar topologies of the layers. Indeed, evidence for this is demonstrated via LA-ICPMS measurements for metaschoepite, compreignacite and β-UO2(OH)2 with aqueous solutions of KI and KIO3 at RT and 60 °C. In the example of compreignacite the intercalation affinity is far less than that of metaschoepite and β-UO2(OH)2 and this is suspected to be related to the presence of K+ cations in the layers. Although the intercalation of IO3− in the Rb2K2[(UO2)6O4(OH)6]·(IO3)2 model requires counter charge balancing Rb+/K+ cations, in the case of compreignacite the K+ cations already present charge balance the UOH layers and as a result reduce the potential intercalation pathways for iodine anions to enter the structure, thus reducing intercalation affinity. In metaschoepite the initial intercalated neutral H2O molecules can be readily exchanged for iodine anions and K+ cations as their inclusion would be argued to be more preferred due to favourable interactions with respective positive void and negative hydroxyl positions.During the oxidative corrosion of SNF, the formation of UOH phases is understood to progressively occur through the interaction of ground water with irradiated fuel7via radiolysis and indeed this has been observed during accident scenarios,57 within interim storage58 and also under simulated laboratory conditions.59 Under a severe accident scenario involving SNF radiolytic iodine generally makes up a predominant component of the instant release fraction due to its volatile nature.60 However during prolonged storage conditions and long-term release, where SNF temperatures are moderate, the chemical state of radiolytic iodine is more inclined towards soluble and aqueous states particularly when contacted with ground water.61,62 Under this scenario the potential for radiolytic iodine to encounter UOH phases is plausible. It would be expected then that host–guest/salt-inclusion chemistry of UOH formed phases would be favourable for intercalation of some radiolytic I−/IO3− species. Such an effect would be more favourable for the earlier paragenetically formed UOH phases such as metaschoepite or those similar to β-UO2(OH)2 which don't possess initial interlayer salt cations for instance seen in compreignacite or becquerelite. It is not clear the effect of competing anions have upon the intercalation of iodine as this was out of the scope of the present study. Although it is shown counter cations are necessary for iodine intercalation to occur, so if other pertinent fission daughters such as caesium are present, they may be captured along with radiolytic iodine in UOH phases. Nevertheless, it would be expected over a long-term release scenario that UOH phases formed during the oxidative corrosion of SNF under aqueous conditions may assist in retarding the transport of radiolytic iodine species into the biosphere.
Conclusions
To summarise we have synthesised a novel uranium oxyhydroxide phase Rb2K2[(UO2)6O4(OH)6]·(IO3)2, which intercalates IO3−via a hybrid salt-inclusion and host–guest mechanism. The intercalation is observed to occur via favorable interactions between respectively the IO3− and Rb+/K+ anions and positive void and negative hydroxyl positions of the UOH layers based on intermolecular bonding interactions. The mechanism is described as a hybrid salt-inclusion and host–guest mechanism. Such a bonding motif suggests the intercalation of iodine species may occur ubiquitously across the family of UOH phases due to their similar layer topologies. To examine this the known UOH mineral phases metaschoepite and compreignacite and related β-UO2(OH)2 were synthesised and exposed to aqueous I− and IO3− for 1 month at RT and 60 °C in air. Intercalation of iodine in these phases was observed to occur homogeneously via LA-ICPMS measurements where the affinity is apparently temperature and structural dependant, such that it is favoured at higher temperatures and when the interlayer space of the UOH phase is free of initial chemical species. Consequently, it is argued that the topology and layer chemistry of UOH phases favours the intercalation of I−/IO3− anionic species into their structures and allows them to be retained even after washing with DI water. This work suggests that UOH phases that may occur during an accident scenario involving SNF under oxidising conditions can impact and potentially retard the transport of some radiolytic iodine from the near-field into the biosphere.Author contributions
GLM and EA conceived and designed the experiments, GLM and EA performed the SC-XRD, GLM and PK performed and analysed the XRD measurements, MK performed and analysed the SEM/EDS measurements, AW, MH, DS and GLM, performed and analysed the LA-ICPMS measurements. The manuscript was prepared by GLM and EA with input from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work has been supported by the Deutsche Forschungsgemeinschaf (DFG) grant AL1527/3-1.References
- E. C. Buck, D. J. Wronkiewicz, P. A. Finn and J. K. Bates, J. Nucl. Mater., 1997, 249, 70–76 CrossRef CAS.
- D. J. Wronkiewicz, J. K. Bates, S. F. Wolf and E. C. Buck, J. Nucl. Mater., 1996, 238, 78–95 CrossRef CAS.
- D. J. Wronkiewicz, J. K. Bates, T. J. Gerding, E. Veleckis and B. S. Tani, J. Nucl. Mater., 1992, 190, 107–127 CrossRef CAS.
- A. G. Sowder, S. B. Clark and R. A. Fjeld, Environ. Sci. Technol., 1999, 33, 3552–3557 CrossRef CAS.
- R. J. Baker, Coord. Chem. Rev., 2014, 266–267, 123–136 CrossRef CAS.
- B. J. Riley, J. D. Vienna, D. M. Strachan, J. S. McCloy and J. L. Jerden, J. Nucl. Mater., 2016, 470, 307–326 CrossRef CAS.
- P. C. Burns, R. C. Ewing and A. Navrotsky, Science, 2012, 335, 1184–1188 CrossRef CAS PubMed.
- P. C. Burns, R. C. Ewing and M. L. Miller, J. Nucl. Mater., 1997, 245, 1–9 CrossRef CAS.
- R. J. Finch and R. C. Ewing, J. Nucl. Mater., 1992, 190, 133–156 CrossRef CAS.
- I. Pointeau, N. Coreau and P. E. Reiller, Radiochim. Acta, 2008, 96, 367–374 CrossRef CAS.
- G. D. Thomas, S. M. Smith and J. A. Turcotte, Health Promot. Pract., 2009, 10, 92–101 CrossRef PubMed.
- J. R. Goldsmith, C. M. Grossman, W. E. Morton, R. H. Nussbaum, E. A. Kordysh, M. R. Quastel, R. B. Sobel and F. D. Nussbaum, Environ. Health Perspect., 1999, 107, 303–308 CrossRef CAS PubMed.
- S. A. Kulyukhin, Russ. Chem. Rev., 2012, 81, 960 CrossRef.
- H. Bruchertseifer, R. Cripps, S. Guentay and B. Jaeckel, Anal. Bioanal. Chem., 2003, 375, 1107–1110 CrossRef CAS PubMed.
- S. Zhang, K. A. Schwehr, Y. F. Ho, C. Xu, K. A. Roberts, D. I. Kaplan, R. Brinkmeyer, C. M. Yeager and P. H. Santschi, Environ. Sci. Technol., 2010, 44, 9042–9048 CrossRef CAS PubMed.
- S. Zhang, C. Xu, D. Creeley, Y.-F. Ho, H.-P. Li, R. Grandbois, K. A. Schwehr, D. I. Kaplan, C. M. Yeager, D. Wellman and P. H. Santschi, Environ. Sci. Technol., 2013, 47, 9635–9642 CrossRef CAS PubMed.
- E. Wieland, H. Wanner, Y. Albinsson, P. Wersin and O. Karnland, A surface chemical model of the bentonite-water interface and its implications for modelling the near field chemistry in a repository for spent fuel, Swedish Nuclear Fuel and Waste Management Co., 1994 Search PubMed.
- N. Daniels, C. Franzen, G. L. Murphy, K. Kvashnina, V. Petrov, N. Torapava, A. Bukaemskiy, P. Kowalski, H. Si, Y. Ji, A. Hölzer and C. Walther, Appl. Clay Sci., 2019, 176, 1–10 CrossRef CAS.
- S. Wu, S. Wang, A. Simonetti, F. Chen and T. E. Albrecht-Schmitt, Radiochim. Acta, 2011, 99, 573–579 CrossRef CAS.
- S. Wu, F. Chen, M. Kang, Y. Yang and S. Dou, Radiochim. Acta, 2009, 97, 459–465 CAS.
- S. J. Wu, F. R. Chen, A. Simonetti and T. E. Albrecht-Schmitt, Environ. Sci. Technol., 2010, 44, 3192–3196 CrossRef CAS PubMed.
- V. Metz, H. Geckeis, E. Gonzalez-Robles, A. Loida, C. Bube and B. Kienzler, Radiochim. Acta, 2012, 100, 699–713 CrossRef CAS.
- Y. Zhang, T. Wei, T. T. Tran, K. T. Lu, Z. Zhang, J. R. Price, I. Aharonovich and R. Zheng, Inorg. Chem., 2020, 59, 12166–12175 CrossRef CAS PubMed.
- K. T. Lu, Y. Zhang, T. Wei, J. Čejka and R. Zheng, Dalton Trans., 2020, 49, 5832–5841 RSC.
- Y. Zhang, R. D. Aughterson, Z. Zhang, T. Wei, K. Lu, J. Čejka and I. Karatchevtseva, Inorg. Chem., 2019, 58, 10812–10821 CrossRef CAS PubMed.
- J. Plášil, A. R. Kampf, T. A. Olds, J. Sejkora, R. Škoda, P. C. Burns and J. Čejka, Am. Mineral., 2020, 105, 561–568 CrossRef.
- G. L. Murphy, B. J. Kennedy, J. A. Kimpton, Q. Gu, B. Johannessen, G. Beridze, P. M. Kowalski, D. Bosbach, M. Avdeev and Z. Zhang, Inorg. Chem., 2016, 55, 9329–9334 CrossRef CAS PubMed.
- G. L. Murphy, P. Kegler, Y. Zhang, Z. Zhang, E. V. Alekseev, M. D. de Jonge and B. J. Kennedy, Inorg. Chem., 2018, 57, 13847–13858 CrossRef CAS PubMed.
- G. L. Murphy, C. H. Wang, Z. M. Zhang, P. M. Kowalski, G. Beridze, M. Avdeev, O. Muransky, H. E. A. Brand, Q. F. Gu and B. J. Kennedy, Inorg. Chem., 2019, 58, 6143–6154 CrossRef CAS PubMed.
- G. L. Murphy, E. M. Langer, O. Walter, Y. Wang, S. Wang and E. V. Alekseev, Inorg. Chem., 2020, 59, 7204–7215 CrossRef CAS PubMed.
- G. L. Murphy, P. Kegler, M. Klinkenberg, S. Wang and E. V. Alekseev, Dalton Trans., 2020, 49, 15843–15853 RSC.
- G. L. Murphy, Z. Zhang, R. Tesch, P. M. Kowalski, M. Avdeev, E. Y. Kuo, D. J. Gregg, P. Kegler, E. V. Alekseev and B. J. Kennedy, Inorg. Chem., 2021, 60, 2246–2260 CrossRef CAS PubMed.
- G. Murphy, B. J. Kennedy, B. Johannessen, J. A. Kimpton, M. Avdeev, C. S. Griffith, G. J. Thorogood and Z. M. Zhang, J. Solid State Chem., 2016, 237, 86–92 CrossRef CAS.
- P. M. Almond and T. E. Albrecht-Schmitt, Am. Mineral., 2004, 89, 976–980 CrossRef CAS.
- A. C. Bean, C. F. Campana, O. Kwon and T. E. Albrecht-Schmitt, J. Am. Chem. Soc., 2001, 123, 8806–8810 CrossRef CAS PubMed.
- A. C. Bean, S. M. Peper and T. E. Albrecht-Schmitt, Chem. Mater., 2001, 13, 1266–1272 CrossRef CAS.
- Y. Wang, Y. Wang, L. Zhang, L. Chen, Z. Liu, X. Yin, D. Sheng, J. Diwu, J. Wang, N. Liu, Z. Chai and S. Wang, Inorg. Chem., 2017, 56, 3702–3708 CrossRef CAS PubMed.
- R. J. Finch, F. C. Hawthorne and R. C. Ewing, Canad. Mineral., 1998, 36, 831–845 CAS.
- D. Gorman-Lewis, J. B. Fein, P. C. Burns, J. E. Szymanowski and J. Converse, J. Chem. Thermodyn., 2008, 40, 980–990 CrossRef CAS.
- J. Brugger, N. Meisser, B. Etschmann, S. Ansermet and A. Pring, Am. Mineral., 2011, 96, 229–240 CrossRef CAS.
- K.-A. Kubatko, K. Helean, A. Navrotsky and P. C. Burns, Am. Mineral., 2006, 91, 658–666 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- P. C. Burns, R. C. Ewing and F. C. Hawthorne, Can. Mineral., 1997, 35, 1551–1570 CAS.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef.
- M. K. Pagoaga, D. E. Appleman and J. M. Stewart, Am. Mineral., 1987, 72, 1230–1238 CAS.
- J. P. West and S.-J. Hwu, J. Solid State Chem., 2012, 195, 101–107 CrossRef CAS.
- I. D. Kharitonov, D. O. Charkin, P. S. Berdonosov, C. Black, L. J. Downie, P. Lightfoot and V. A. Dolgikh, Eur. J. Inorg. Chem., 2014, 2014, 3140–3146 CrossRef CAS.
- D. O. Charkin, C. Black, L. J. Downie, D. E. Sklovsky, P. S. Berdonosov, A. V. Olenev, W. Zhou, P. Lightfoot and V. A. Dolgikh, J. Solid State Chem., 2015, 232, 56–61 CrossRef CAS.
- G. Morrison and H.-C. zur Loye, Cryst. Growth Des., 2020, 20, 8071–8078 CrossRef CAS.
- S. N. Volkov, D. O. Charkin, M. Y. Arsent'ev, A. V. Povolotskiy, S. Y. Stefanovich, V. L. Ugolkov, M. G. Krzhizhanovskaya, V. V. Shilovskikh and R. S. Bubnova, Inorg. Chem., 2020, 59, 2655–2658 CrossRef CAS PubMed.
- P. C. Burns, Canad. Mineral., 1998, 36, 1061–1067 CAS.
- F. C. Hill and P. C. Burns, Canad. Mineral., 1999, 37, 1283–1288 CAS.
- M. Schindler, F. C. Hawthorne, P. C. Burns and P. A. Maurice, Canad. Mineral., 2006, 44, 1207–1225 CrossRef CAS.
- P. F. Weck and E. Kim, Dalton Trans., 2014, 43, 17191–17199 RSC.
- B. E. Burakov, E. E. Strykanova and E. B. Anderson, MRS Proc., 1996, 465, 1309 CrossRef.
- J. Abrefah, S. C. Marschman and E. D. Jenson, Examination of the surface coatings removed from K-East Basin fuel elements, United States, 1998 Search PubMed.
- B. D. Hanson, B. McNamara, E. C. Buck, J. I. Friese, E. Jenson, K. Krupka and B. W. Arey, Radiochim. Acta, 2005, 93, 159–168 CrossRef CAS.
- L. Johnson, C. Ferry, C. Poinssot and P. Lovera, J. Nucl. Mater., 2005, 346, 56–65 CrossRef CAS.
- B. Clément, L. Cantrel, G. Ducros, F. Funke, L. Herranz, A. Rydl, G. Weber and C. Wren, State of the art report on iodine chemistry, Organisation for Economic Co-Operation and Development, 2007 Search PubMed.
- R. C. Ewing, Nat. Mater., 2015, 14, 252–257 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2094057. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03237b |
| This journal is © The Royal Society of Chemistry 2021 |