
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenchmarking magnetic and spectroscopic properties on highly stable 3d metal complexes with tuneable bis(benzoxazol-2-yl)methanide ligands†
Christina M.
Legendre‡
,
Daniel
Lüert‡
,
Regine
Herbst-Irmer
 and
Dietmar
Stalke
*
and
Dietmar
Stalke
*
Institute for Inorganic Chemistry, University of Göttingen, Tammannstrasse 4, 37077 Göttingen, Germany. E-mail: dstalke@chemie.uni-goettingen.de
First published on 1st November 2021
Abstract
Two series a and b of 3d metal based complexes 1–4 [MII{(4-R-NCOC6H4)2CH}2], (with M = Mn (1), Fe (2), Co (3), Ni (4) and R = H (a) or Me (b)) were synthesised and structurally characterized. The complexes were found to crystallize differently depending on the dication ionic radius and the ligand substitution. All complexes showed remarkable X-ray diffraction resolution that will allow further advanced diffraction experiments. Subsequently, their spectroscopic and magnetic properties were analysed. Complexes 3a and 3b notably show slow magnetic relaxation of their magnetization and represent simple model systems relaxing through a phonon-bottleneck process (3a) or as a field-induced single-molecule magnet (3b, Ueff = 45.0 cm−1). Remarkably, the magnetic anisotropy in the manganese complex 1b results in induced slow magnetic relaxation. The influence of the dual 4-methylation of the ligands was investigated and found to generate important variations in the physical features of the corresponding complexes. Accessible via one-pot synthesis, these are highly robust against oxidation and moisture. Through smart ligand engineering, they represent stable and tuneable compounds for benchmarking purposes through standard and less-standard characterization methods.
Introduction
Benchmarking plays a central role in modern chemical research, as the interplay of experimental and theoretical advances allows to unveil the mechanisms of physical processes as well as to further develop both analytical and computational methods.1,2 To this aim, experimental chemists currently strive to provide suitable material to theoreticians and chemical engineers. In the field of molecular magnetism, there is currently a great demand for new single-molecule magnets (SMMs)3 – molecules that slowly relax their magnetization –, which are considered as a highly promising alternative to current data-storage devices.4 Since SMMs still show major drawbacks to their industrial applications,5 there is an increasing need to better understand how they function and how to better control their materials profile. Theoretical insights6,7 addressing the complicated relaxation pathways of the SMMs are numerous,8,9 on ideal models10 as well as on experimentally discovered molecules.11 Many existing compounds, however, are clusters with multiple metal centres,12 whose features are yet to be completely understood. Simpler compounds, such as bimetallic complexes or single-ion magnets,13 are more accessible for computational methods. In return, unfortunately, they are often air and moisture sensitive, preventing further investigation through less common experimental methods that currently focus on the analysis of stable compounds, such as advanced X-ray diffraction experiments,14 polarized neutron diffraction15 or synchrotron XAS/XMCD spectroscopy of coated surfaces.16 The complexity of the relaxation processes,9 responsible for the magnetic performances, highlights the tremendous need for magneto-structural correlations to better direct further experimental investigations. To this aim, we herein present a novel family of stable 3d metal complexes as an easily accessible and tuneable platform suitable for benchmarking magnetic properties. As these complexes are also spectroscopically interesting, we additionally investigated their optical properties, seeking for a structural link to their absorption characteristics. We designed and obtained by facile one-pot synthesis, based on the chelating bis(benzoxazol-2-yl) methanide (4-R-NCOC6H4)2CH− ligand17 (further abbreviated Box), the following complexes containing oxidation state +II first-row transition metals: [MII{(4-R-NCOC6H4)2CH}2], with M = Mn (1), Fe (2), Co (3) and Ni (4). Along the different substituents they are divided in two series a and b, where R = H (a) or Me (b). The ligand mimics the omnipresent β-diketiminate (nacnac) ligand system, where the N,N-chelation to the metal gives the six membered metallacycle.18 Furthermore, this system offers an additional oxygen donor site for various coordination motifs.19 The extension of the aromatic backbone leads to an electron rich system, where expanded conjugation provides a more rigid and almost planar coordination sphere. Additional substitution along the aromatic system allows steric and geometry control in the third dimension.19,20 Effective magnetic and steric shielding of the metal atom not only quenches the intermolecular metal–metal-coupling effects but also increases complex stability.21 Apart from implementation in main group chemistry,19,22,23 this system has so far neither been employed nor studied for d-metal chemistry. So herein we describe the syntheses and full characterization of the transition metal complexes 1–4. The influence of the 4-methylation of the ligand on the magnetic and optical properties of 1–4 is discussed.Results and discussion
Synthesis
We synthesized two quadruple series a and b of complexes 1–4 from the corresponding metal salt MCl2 (M = Mn, Fe, Co, Ni) and the potassium complex obtained via the deprotonation of the bis(4-R-benzoxazol-2-yl)methane ligand,22 as described in Scheme 1. The ligand is further denoted Box for R = H (series a) and Mebox for R = Me (series b). | ||
| Scheme 1 Synthesis route of complexes 1–4. | ||
The corresponding deprotonated ligand (KBox for the series a and KMebox for the series b, respectively) is dissolved in thf and added to a stirring thf solution of the metal salt at room temperature. A colour change is instantaneously observed in all cases. For 4a/b, subsequent heating for two hours gives a better yield. Upon short stirring, the colourful mixture is filtered to remove the precipitated potassium chloride and unreacted solids, and the solvent is removed to give the crude product in acceptable yields (from 28% 4b to 75% 3b). The syntheses must be carried out under inert atmosphere because of the sensitive character of the deprotonated ligand. After transmetalation however, the formed d-metal complexes are extremely stable, in solution for some weeks and in the solid-state for longer than 3 months. Except for manganese complexes 1a and 1b, all complexes are even air-stable for several weeks.
X-ray diffraction and solid-state structures of 1–4
Huge colourful, block-shaped crystals suitable for X-ray diffraction structure analyses are obtained overnight by slow evaporation of pentane into a concentrated thf solution at −35 °C. The crystals are yellow, orange, red-orange and blue, in the range of 1 to 4. The subsequent structure analyses revealed that all complexes contain a single divalent cation M2+ (M = Mn (1), Fe (2), Co (3) and Ni (4), respectively) N,N-chelated by two ligands (Fig. 1). Selected geometrical parameters are summarized in Table 1 and detailed structural data can be found in the Experimental section and the ESI.† | ||
| Fig. 1 Solid-state structures of (a) 1–2a, (b) 3–4a and (c) 1–2b, (d) 3–4b. | ||
| d N–M range | N1–M–N2/N3–M–N4 | N1–C1/N2–C3 | C1–C2/C2–C3 | Twist anglea | |
|---|---|---|---|---|---|
| a Angles between N(1)C(2)N(2) and N(3)C(5)N(4) planes, and between N(5)C(32)N(6) and N(7)C(35)N(8) planes for 3a and 4a. For detailed procedures see ESI.† | |||||
| 1a | 2.137–2.159 | 85.82(5) | 1.343(2) | 1.385(2) | 54.25(6) |
| 86.20(5) | 1.334(2) | 1.388(2) | |||
| 2a | 2.084–2.103 | 87.14(6) | 1.343 (16) | 1.385(2) | 56.07(6) |
| 87.22(5) | 1.333 (16) | 1.389(2) | |||
| 3a | 1.977–1.989 | 92.89(12) | 1.341(4) | 1.389(5) | 64.32(14) |
| 92.80(12) | 1.333(5) | 1.384(5) | 63.63(15) | ||
| 4a* | 1.949–1.975 | 91.7(2) | 1.353(8) | 1.356(10) | 62.3(3) |
| 91.3(2) | 1.339(9) | 1.394(10) | 61.3(4) | ||
| 1b | 2.101–2.110 | 92.01(5) | 1.3393(18) | 1.390(2) | 89.60(5) |
| 91.97(5) | 1.3393(18) | 1.387(2) | |||
| 2b | 2.037–2.043 | 93.73(4) | 1.3411(14) | 1.3857(16) | 89.51(5) |
| 93.71(4) | 1.3401(15) | 1.3890(16) | |||
| 2b′ | 2.034–2.036 | 95.57(4) | 1.3437(15) | 1.3869(16) | 88.82(5) |
| 94.04(4) | 1.3406(15) | 1.3926(17) | |||
| 3b | 1.985–1.990 | 95.50(6) | 1.342(2) | 1.378(2) | 87.66(6) |
| 96.72(6) | 1.339(2) | 1.382(2) | |||
| 4b | 1.975–1.977 | 93.43(6) | 1.336(2) | 1.386(2) | 87.69(6) |
| 94.53(5) | 1.3414(19) | 1.383(2) | |||
The two nitrogen atoms coordinate the metal ion almost equidistantly. The M–N distances (Table 1, 1.9 to 2.2 Å) are in the typical range for such compounds.24 The bite angles N1–M–N2 and N3–M–N4 are more acute than the ideal tetrahedral angle (Table 1, from 85.8° in 1a to 96.7° in 3b) and about 10–20° wider than the ideal N–M–N angle of 78° in distorted Co(N2R)2 complexes.25 The structural variations of the complexes from the ideal tetrahedron are further characterized by τ4 and τ4′, as reported in the ESI (see Table S11†).26,27 Series a clearly shows a distortion further away from a Td geometry than series b. Hence, better magnetic properties are expected from the latter.
We found that complexes 1a and 2a are isomorphous and crystallize in the monoclinic space group C2/c. In addition to the thf molecule coordinated to the metal centre, the structures contain two half lattice thf molecules located on symmetry elements. 1b and 2b share comparable similarities, both crystalizing in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one complex molecule and two lattice thf solvent molecules in the asymmetric unit. Noteworthy, we were able to obtain an additional molecular structure for 2b, without any solvent molecule, further denoted as 2b′. The complexes 2b′, 3b and 4b are also isostructural and all crystallize in the monoclinic space group P21/n, each asymmetric unit containing only one complex molecule without any solvent. Complex 3a and 4a contain two molecules in the asymmetric unit, but share the same space group as 2b′, 3b, and 4b. The bond lengths in the C3N2nacnac-like chelating unit strongly suggest that the π-system is fully conjugated and extends throughout the entire virtually planar ligands (Table 1). This feature was observed in previously reported alkali metal complexes of the bis-(benzoxazol-2-yl)methane derivatives.22 Their relative orientation differs. While almost orthogonal in 1–4b, the two ligand planes N(1)C(2)N(2) and N(3)C(5)N(4) are twisted of 60° in 1–4a, as shown in Table 1 and for more details see ESI.† The most remarkable feature is that all the X-ray datasets, except 4a reach at least 0.50 Å in resolution and that none of these molecular structures of the entire b series contains any disorder. Therefore, 1b–4b and 3a are in principle suitable for further experimental electron density analyses. In 1b and 2b the metal atoms are well separated (>8 Å) and solvent molecules seem to prevent any potential intermolecular π–π interactions between the ligands. Two nearest ligands are packed orthogonally to each other. 1a and 2a show solvent coordination at the metal atom due to the missing additional steric shielding of the methyl substituent.
with one complex molecule and two lattice thf solvent molecules in the asymmetric unit. Noteworthy, we were able to obtain an additional molecular structure for 2b, without any solvent molecule, further denoted as 2b′. The complexes 2b′, 3b and 4b are also isostructural and all crystallize in the monoclinic space group P21/n, each asymmetric unit containing only one complex molecule without any solvent. Complex 3a and 4a contain two molecules in the asymmetric unit, but share the same space group as 2b′, 3b, and 4b. The bond lengths in the C3N2nacnac-like chelating unit strongly suggest that the π-system is fully conjugated and extends throughout the entire virtually planar ligands (Table 1). This feature was observed in previously reported alkali metal complexes of the bis-(benzoxazol-2-yl)methane derivatives.22 Their relative orientation differs. While almost orthogonal in 1–4b, the two ligand planes N(1)C(2)N(2) and N(3)C(5)N(4) are twisted of 60° in 1–4a, as shown in Table 1 and for more details see ESI.† The most remarkable feature is that all the X-ray datasets, except 4a reach at least 0.50 Å in resolution and that none of these molecular structures of the entire b series contains any disorder. Therefore, 1b–4b and 3a are in principle suitable for further experimental electron density analyses. In 1b and 2b the metal atoms are well separated (>8 Å) and solvent molecules seem to prevent any potential intermolecular π–π interactions between the ligands. Two nearest ligands are packed orthogonally to each other. 1a and 2a show solvent coordination at the metal atom due to the missing additional steric shielding of the methyl substituent.
For 3a and 4a, no extra steric bulk is required to prevent solvent coordination at the metal centre for two reasons. First, these ions are smaller and there is simply no space for solvent coordination and, second, the tetrahedral coordination mode leaves no voids. In 2b′, 3b and 4b, the packing reveals the presence of long range π interactions between two parallel ligands (shortest distance of nearest neighbours: 3.34 Å). The metals, however, are still well separated (>8 Å), which is crucial for the magnetic properties.
UV-vis characterization
We further characterized the colourful complexes 1–4a and b by UV-vis spectroscopy both, in solution and the solid-state. Their absorption spectra in solution are shown in Fig. 2. | ||
| Fig. 2 UV-vis spectra of 1–4; thf solutions of 1a–4a (top) and 1b–4b (bottom). | ||
The high extinction coefficient values (Table 2) suggest that the intense absorption bands are due to a charge transfer from the metal to the ligand. Below 360 nm, the expected π → π* excitation of the aromatic backbone was observed. There are some substantial differences in absorption between the series a and b. Notably, the absorption patterns of the manganese and iron complexes (1 and 2) are drastically affected by the substituent variation. 1a does not absorb in the visible range, whereas 1b features several highly energetic absorption bands, which result in its bright yellow colour.
| # | λ exp (nm) | λ sim (nm) | λ max, ε (cm−1 mol−1 L) |
|---|---|---|---|
| a For 2b′, λsim = 442–4 nm (see details in ESI†) | |||
| 1a | 391 | 416 | 391 (ε = 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 762) 762) |
| 1b | 402, 429, 455 | 418 | 402 (ε = 19241) |
| 2a | 395, 492 | 410 | 395 (ε = 19136) |
| 2b | 399, 422, 516 | 443 | 422 (ε = 16251) |
| 3a | 404, 430, 513 | 403, 468, 721 | 404 (ε = 7579) |
| 3b | 408, 434, 490 | 414, 619, 749 | 408 (ε = 7680) |
| 4a | 406, 429, 560 | 429, 558 | 406 (ε = 2069) |
| 4b | 408, 431, 584 | 445, 564, 581 | 408 (ε = 1615) |
While 2a only weakly absorbs in the visible range, the absorption peak at λ = 516 nm for 2b is clearly responsible for its darker colour. Small changes are observable as well for cobalt and nickel, although to a lesser extent. For the cobalt complexes 3a and 3b, the absorption maxima are almost identical while the peaks are broader for 3b. Related tetrahedral NiII complexes with the β-diketimide motif exhibit comparable UV-vis data, while reported CoII complexes have a broader range.28 These changes from series a to b are probably due to the additional coordination with thf molecules on the metals, which might even be favoured in solution. Additionally, the bathochromic shift from the series b to a, which can be identified regardless of the metal (max Δλ = 30 nm for Ni), may be attributed to the potential hyperconjugation in series b.29 These experimental trends are reproduced in the theoretical calculations with the precise absorption wavelength values, however, frequently off the range. This is probably due to the different spatial arrangement of the molecules in solution and in the solid-state (since solid-state coordinates are used for the calculations). Solid-state UV-vis measurements are reported in the ESI† and show different results for 1a and 2a as the experiments in solution. The absorption maxima of 1a and 1b exhibit a strong bathochromic shift compared to solution data. Interestingly, both iron complexes (2a,2b) display a similar absorption pattern as observed for 2b in solution. For 3a and 3b, we also found similar results as in solution. These finding lead to the conclusion that the spectroscopic deviations between 1a,1b and 2a,2b are most likely attributed to the methyl substituent rather than to the additional thf coordination for 1a and 2a.
Magnetic properties
We measured the temperature dependency of the product χMT for complexes 1–4 (a and b) and they were found to be all paramagnetic, as reported in Fig. 3. The high temperature χMT values for the manganese and the nickel complexes are close to their expected spin-only values of 4.38 and 1.00 cm3 mol−1 K. Noteworthy, the χMT values of the manganese complexes 1a and 1b are further apart from each other, which might be attributed to the difficult weighing due to the presence of several solvent molecules (see S3† for more details). The iron and cobalt complexes, however, have χMT values of about 3.50 and 2.50 cm3 mol−1 K at high temperatures, which are larger than the corresponding spin-only systems (3.00 and 1.87 cm3 mol−1 K at 300 K, respectively). It indicates a significant orbital contribution. The product of the temperature and the static magnetic susceptibility is stable while decreasing the temperature and starts decreasing when lower temperatures are reached. While this behaviour is characteristic of magnetic saturation in the cases of the manganese and iron complexes, it seems that other effects may cause an earlier, smoother drop of χMT for the nickel complexes. It might be due to weak intermolecular interactions between the magnetic centres,30 which are closer to each other in 4a, b than in 1–2a, b. For 3a, b, it is likely due to the high anisotropies observable in these complexes. The χMT data was fitted simultaneously with the VTVH data (variable field variable temperature) using the Jul2s program,38 in order to extract accurate values for the static magnetic parameters, based on the following spin Hamiltonian: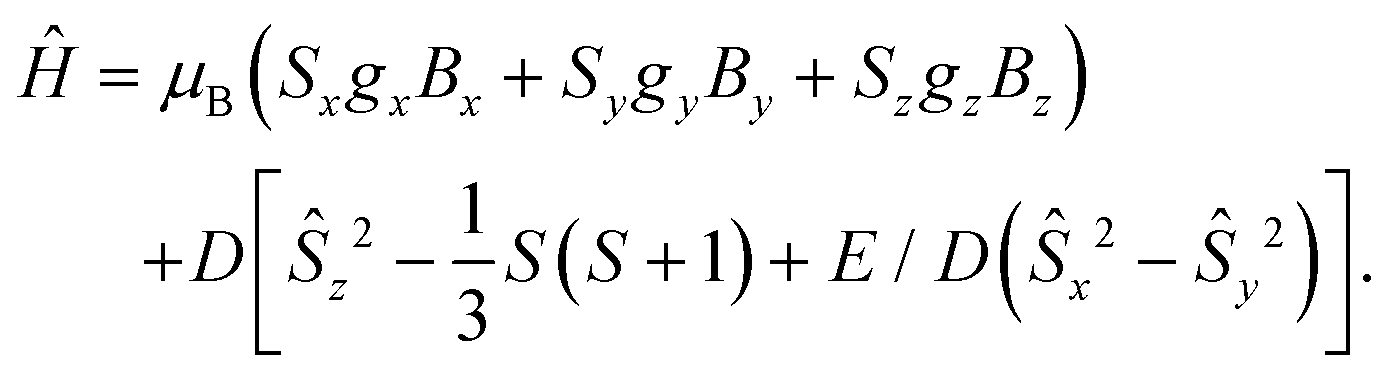 | (1) |
 | ||
| Fig. 3 Temperature dependency of χMT for 1–4 a and b complexes (from 210 K to 2 K). | ||
The manganese and iron complexes displayed very small and positive ZFS parameters and were successfully described as isotropic (gx = gy = gz). The nickel complexes possess larger ZFS values, however positive, which should prevent the presence of an effective energy barrier.31 The obtained dc data suggests that none of the six 1–2a, b and 4a, b complexes would feature SMM properties. With a negative D value (Table 3), only the cobalt complexes 3a and 3b were recognized as potential single-molecule magnets (SMM). The difference in the ZFS splitting in these two complexes might be attributed to their deviation from an ideal tetrahedral environment around the cobalt centre. The calculated τ4(3a) value is smaller than τ4(3b),26,27 (Table S11†) thus further apart from the ideal tetrahedron (τ4 = 1), which should result in larger magnetic anisotropy.1,25,32 Additionally, the best fit was obtained with the use of anisotropic g values (gx ≈ gy < gz), as expected for such distorted tetrahedral systems.
| # | D (cm−1) | E/D | g x, y, z | U eff (cm−1) | τ 0 (s) |
|---|---|---|---|---|---|
| 1a | −0.03 | 0 | 2.05 | — | — |
| 1b | 0.5 | 0 | 1.96 | 6.0 | 4.14 × 10−5 |
| 2a | 11.7 | 0.3 | 2.21 | — | — |
| 2b | 6.9 | 0 | 2.22 | — | — |
| 3a | −25.0 | 0 | 2.27, 2.27, 2.58 | 18 | 1.84 × 10−6 |
| 3b | −18.4 | 0.02 | 2.28, 2.21 2.51 | 45 | 2.47 × 10−11 |
| 4a | 67.5 | 0 | 2.00 | — | — |
| 4b | 55.0 | 0 | 2.00, 2.00, 2.71 | — | — |
Substantial transverse anisotropy was observable for 3a, while the E/D parameter was close to zero for 3b. For 1a and 1b, slow relaxation processes other than Orbach relaxation might still occur, as previously observed in Mn2+ systems with a non-zero, even positive, D value.33,34
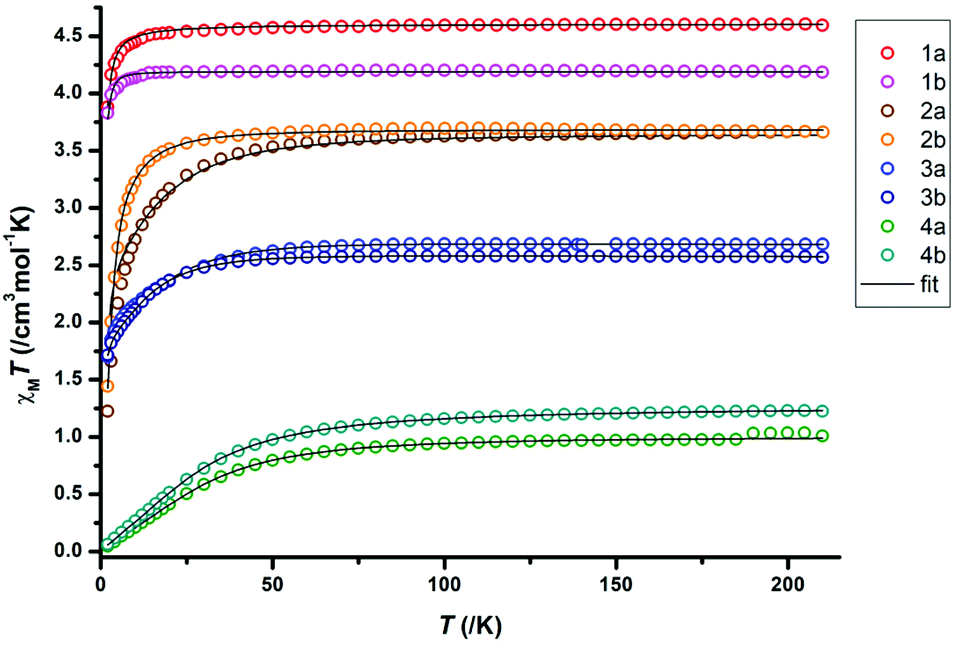
We further measured ac susceptibility data for all complexes, and the complexes 1b, 3a and 3b indeed showed field induced relaxation of their magnetization (Fig. 4–6). The ac data measurement process is thoroughly described in S3.†
 | ||
| Fig. 4 Frequency dependency of the real and imaginary parts of the ac dynamic susceptibility (lines are guide for the eyes), the corresponding Cole–Cole plot (black lines represent the fit to the data), and Arrhenius plot fitted with eqn (2) for 1b. | ||
 | ||
| Fig. 5 Frequency dependency of the real and imaginary parts of the ac dynamic susceptibility (lines: guide for the eyes), Cole–Cole plot (black lines: fit to the data), and Arrhenius plot for 3a. | ||
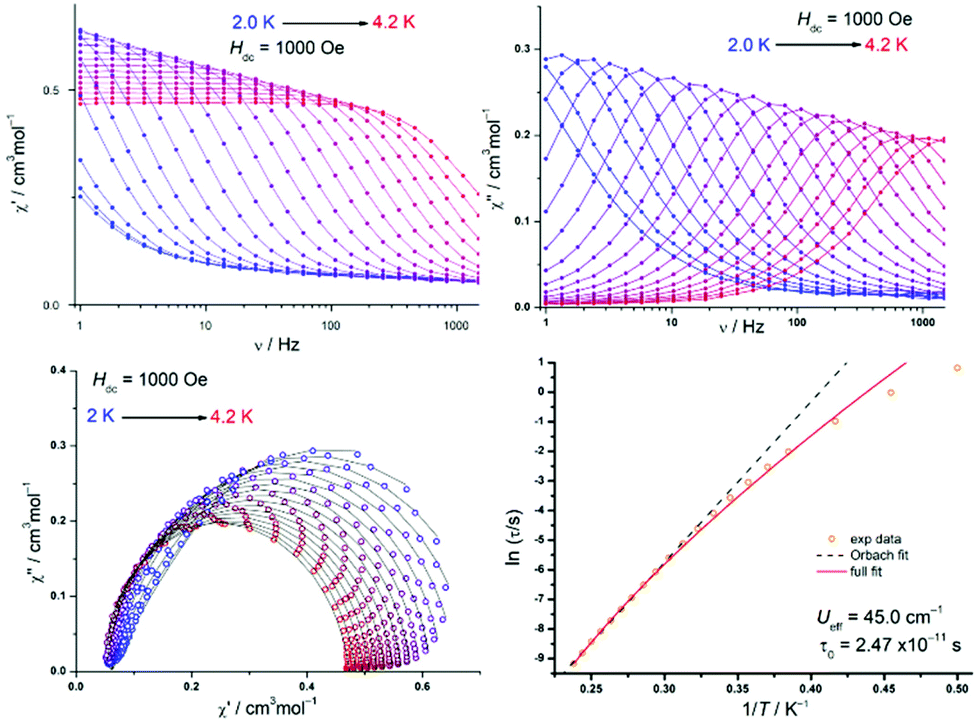 | ||
| Fig. 6 Frequency dependency of the real and imaginary parts of the ac dynamic susceptibility (lines are guide for the eyes), the corresponding Cole–Cole plot (black lines are fit to the data), and Arrhenius plot fitted with eqn (2) for 3b. | ||
For 1b, under an applied dc field of 1000 Oe, the typical maxima of the out-of-phase signal are detected between 2 K and 6 K. The maxima shift towards higher frequencies with the increase of temperature. This is characteristic of a temperature-dependent process. The Cole–Cole plot, however, features narrow-shaped curves instead of semi-circles, which suggest that the slow relaxation of the magnetization does not take place through a SMM-like Orbach process. The Arrhenius plot is constructed with the extracted relaxation times from the Cole–Cole plot and fitted according to the following equation, which takes Orbach and Raman processes into account:
| τ−1 = τ0−1e−Ueff/kBT + CTn. | (2) |
Fitting details and final parameters can be found in the ESI section S3.9, Table S13†. This fitting enables the identification of a phonon-bottleneck process (Raman parameter n = 2.28, close to an ideal value of 2).9 The presence of slow magnetic relaxation in 1b could be attributed to the relatively low coordination number of the Mn2+ ion, as previous reports suggest that the decrease of the coordination number results in the increase of D for complexes with only nitrogen ligands.35 The higher coordination number in 1a (5 instead of 4) would prevent the observation of comparable magnetic behaviour for 1a. The phonon bottleneck process is nevertheless compatible with both a positive or negative D value and explains the observation of slow magnetic relaxation in 1b despite its positive ZFS parameter value. The remarkably slow relaxation time of the magnetization is τ0 = 41 ms, only surpassed by two previously reported Mn2+ SIMs.33,36
A similar result is surprisingly found for the non-methyl-substituted cobalt compound 3a, as depicted in Fig. 5. Although we found here a negative D value of −25 cm−1, the corresponding Cole–Cole plot clearly indicates a relaxation process like this of 1b (Fig. 5). The corresponding Arrhenius plot revealed a small energy barrier of U = 18.8 cm−1 with an attempt relaxation time τ0 = 4.61 × 10−6 s provided by a full fit with all relaxation processes (eqn (2)). Further analysis of the Arrhenius plot revealed a n value of 1.98, confirming the occurrence of a phonon bottleneck process.
As for 3a, the cobalt analogue 3b, the out-of-phase signal of the dynamic susceptibility under 1000 Oe also shows the characteristic maxima moving towards higher frequencies with higher temperatures (Fig. 6). The Cole–Cole plot displays near-to-ideal semi-circles, suggesting this time an Orbach relaxation process. This is further supported by the Arrhenius plot, whose analysis gives an effective energy barrier U = 45.0 cm−1 with an attempt time τ0 = 2.47 × 10−11 s, while n is close to 9 (Kramers-Raman relaxation process).93b can therefore be classified as a true field-induced single-molecule magnet, while both 1b and 3a slowly relax their magnetization through a phonon-bottleneck process. The differences in dynamic magnetic behaviour in 3a and 3b are difficult to correlate with the structural analysis. However, for the static magnetic data, the more acute N–Co–N angle25 and smaller τ4 value1,32,37 in 3a is in good agreement with the higher ZFS value. Additional magnetic details for 3a, 3b and 1b, as well as for the other complexes, can be found in the ESI.†
Computational studies
To better understand the origin of the different relaxation processes in 3a and 3b, which both have a negative D value, we subsequently performed CASSCF-NEVPT2 calculations using the ORCA software,38 based on the geometries obtained from the single crystal X-ray diffraction. Ab initio ligand field theory (AILFT) provided the d orbital splitting and allowed us to quantify the energy gap between the d orbitals dxy and dx2−y2, which are responsible for the strength of the spin–orbit coupling (i.e. the magnetic anisotropy).25,39,40 The obtained parameters, shown in Table 4, are in relatively good agreement with the experiment, and confirm the lack of substantial axial magnetic anisotropy in complexes 3a and 3b.| D (cm−1) | E/D | g x, y, z | ΔE(dxy, dx2−y2) (cm−1) | |
|---|---|---|---|---|
| 3a | −43 | 0.12 | 2.07, 2.21, 2.64 | 1348 |
| 3b | −27 | 0.02 | 2.13, 2.14, 2.45 | 2358 |
While complex 3a shows a more favourable d orbital distribution and as found experimentally, a larger ZFS parameter D, the calculated rhombic parameter E/D is significantly larger than in 3b, which results in a decrease of the magnetic performances. This may explain the ultimately better magnetic performance of 3b over 3a and the variation in the preferred relaxation process.
Conclusions
We presented the successful synthesis and full characterization of eight novel robust compounds with interesting optical and magnetic properties. All paramagnetic and colourful, they crystallize without any disorder at the complex molecules. Additionally, complexes 1b, 3a and 3b were found to display interesting magnetic properties due to various spin–lattice relaxation processes. Notably, 1b is a rare example of a Mn2+ SIM, relaxing via phonon bottleneck with a remarkably slow relaxation time. The dual 4-methylation on the ligand backbone is shown to drastically affect the physical features of the complexes, both spectroscopically and magnetically. We are currently investigating the influence of further substitution variations on these properties.All complexes are also air-stable and easily accessible, offering a versatile and tuneable platform, which can be used to benchmark various analytical and computational methods. Advanced diffraction experiments on these complexes, including charge density investigations, are under way to evaluate further magneto-structural correlations.
Experimental section
General procedures
All reactions were carried out under an atmosphere of N2 and Ar by Schlenk techniques. All solvents were distilled from Na or K before using for synthesis. Starting materials were purchased commercially and used without further purification. The ligands were synthesized according to previously reported procedure. NMR spectroscopic data were recorded on a Bruker Advance 500 MHz and a Bruker Advance 300 MHz spectrometer. Deuterated solvents were dried over activated molecular sieves (3 Å) and stored in an argon dry box. Elemental analyses (C, H, N) were carried out on a Vario EL3 at the Mikroanalytisches Labor, Institut für Anorganische Chemie, University of Göttingen. LIFDI-MS spectra were measured on a Jeol AccuTOF spectrometer. Vis-spectra were recorded on an Agilent Cary 60 and an Agilent Cary 50 spectrometer using quartz cuvettes fitted with Young-type Teflon-valves from thf solutions. IR data were measured on neat samples with a Bruker ALPHA FT-IR spectrometer with Platinum ATR module and visualized with the Opus program.Diffraction data were collected with an Incoatec Mo–IμS microfocus source (structures 1b, 2a, 2b′, 3a, 3b, 4a) and a Bruker TXS-Mo rotating anode (structures 1a, 2b, 4b) with mirror optics and an APEX II detector with a D8 goniometer. All data were integrated with SAINT41,42 and the multiscan absorption and 3λ corrections42,43 were applied in SADABS.43 The structures were solved by direct methods in SHELXT44 and refined on F2 using the full-matrix least squares methods of SHELXL45 with the SHELXLE GUI.46 For the crystallographic Information Files (CIF), see the following referencing CCDC numbers: 2095985–2095993.†
Magnetic data were collected on a MPMS-XL-5 magnetometer and analyzed with the OriginPro 8.5, Jul2s47 and CC-Fit48 programs. The samples were prepared in an argon glovebox. The crystalline materials were crushed, covered with Fomblin oil in gelatine caps and inserted in plastic straws. The data were corrected for the diamagnetic contribution of the sample holder according to Mdia = χg × m × H, with experimentally obtained gram susceptibility of gelatin bucket (χg = −5.70 × 10−7 emu (g Oe)−1 and of the oil (χg = −3.51 × 10−7 emu (g Oe)−1). The molar susceptibility data were corrected for the diamagnetic contribution according to χM,dia(sample) = −0.5 M × 10−6 cm3 mol−1. Temperature-independent paramagnetism (TIP) was included according to χcalc = χ + TIP.49 Details regarding the determination of the optimal field and maximal temperature for ac data are given in the ESI section S3.†
CASSCF-NEVPT2/AILFT were performed in the ORCA suite38,50 on 3a and 3b using the Douglas–Kroll–Hess triple-ζ DKH-def2-TZVP basis-set with the d7 electronic configuration giving ten S = 3/2 electronic multiplets. Ab initio ligand field theory6,51 calculations were based on the wave functions obtained from CASSCF-NEVPT2 calculations using the actorbs dorbs command in the input file. To match the natural and molecular orbitals together, the molecule was rotated so that the origin of the coordinate system becomes the cobalt ion, while the z axis points at the centroid between the two nitrogen atoms of each ligand and the xy plane locates equidistantly from the four coordinating nitrogen atoms.40
General synthetic procedure
In an Argon-filled glovebox, a solution of the deprotonated ligand in THF was added dropwise to a stirring suspension of MCl2 in THF at room temperature. The obtained mixture was let to stir overnight, then filtered and the solvent was reduced to 1 mL. Crystals were obtained by slow evaporation of pentane in the THF solution at −35 °C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. M. L. and D. L. both thank the Fonds der Chemischen Industrie for financial support (PhD fellowships).References
- E. A. Suturina, J. Nehrkorn, J. M. Zadrozny, J. Liu, M. Atanasov, T. Weyhermüller, D. Maganas, S. Hill, A. Schnegg, E. Bill, J. R. Long and F. Neese, Inorg. Chem., 2017, 56, 3102–3118 CrossRef CAS PubMed.
- R. A. Mata and M. A. Suhm, Angew. Chem., Int. Ed., 2017, 56, 11011–11018 CrossRef CAS PubMed.
- (a) G. Christou, D. Gatteschi, D. N. Hendrickson and R. Sessoli, MRS Bull., 2000, 25, 66–71 CrossRef CAS; (b) P. Evans, D. Reta, C. A. P. Goodwin, F. Ortu, N. F. Chilton and D. P. Mills, Chem. Commun., 2020, 56, 5677–5680 RSC.
- (a) D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford Univ. Press, Oxford, 2006, vol. 5 CrossRef; (b) K. V. Raman, A. M. Kamerbeek, A. Mukherjee, N. Atodiresei, T. K. Sen, P. Lazić, V. Caciuc, R. Michel, D. Stalke, S. K. Mandal, S. Blügel, M. Münzenberg and J. S. Moodera, Nature, 2013, 493, 509–513 CrossRef CAS PubMed.
- L. Ungur and L. F. Chibotaru, Inorg. Chem., 2016, 55, 10043–10056 CrossRef CAS PubMed.
- M. Atanasov, J. M. Zadrozny, J. R. Long and F. Neese, Chem. Sci., 2013, 4, 139–156 RSC.
- M. K. Thomsen, A. Nyvang, J. P. S. Walsh, P. C. Bunting, J. R. Long, F. Neese, M. Atanasov, A. Genoni and J. Overgaard, Inorg. Chem., 2019, 58, 3211–3218 CrossRef CAS PubMed.
- (a) W. Wernsdorfer, N. Aliaga-Alcalde, D. N. Hendrickson and G. Christou, Nature, 2002, 416, 406–409 CrossRef PubMed; (b) Single-Molecule Magnets and Related Phenomena, ed. R. Winpenny, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006 Search PubMed; (c) R. Orbach, Proc.: Math., Phys. Eng. Sci., 1961, 264, 458–484 CAS.
- K. N. Shrivastava, Phys. Status Solidi B, 1983, 117, 437–458 CrossRef CAS.
- (a) J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078 RSC; (b) R. Orbach, Proc. Phys. Soc., 1961, 77, 821–826 CrossRef CAS.
- (a) D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 115, 278–309 CrossRef; (b) M. Craven, M. H. Nygaard, J. M. Zadrozny, J. R. Long and J. Overgaard, Inorg. Chem., 2018, 57, 6913–6920 CrossRef CAS PubMed; (c) K. S. Pedersen, J. Dreiser, H. Weihe, R. Sibille, H. V. Johannesen, M. A. Sørensen, B. E. Nielsen, M. Sigrist, H. Mutka, S. Rols, J. Bendix and S. Piligkos, Inorg. Chem., 2015, 54, 7600–7606 CrossRef CAS PubMed.
- (a) R. Sessoli, H. L. Tsai, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804–1816 CrossRef CAS; (b) A. Abhervé, T. Grancha, J. Ferrando-Soria, M. Clemente-León, E. Coronado, J. C. Waerenborgh, F. Lloret and E. Pardo, Chem. Commun., 2016, 52, 7360–7363 RSC.
- S. Gomez-Coca, E. Cremades, N. Aliaga-Alcalde and E. Ruiz, J. Am. Chem. Soc., 2013, 135, 7010–7018 CrossRef CAS PubMed.
- (a) M. S. Norre, C. Gao, S. Dey, S. K. Gupta, A. Borah, R. Murugavel, G. Rajaraman and J. Overgaard, Inorg. Chem., 2020, 59, 717–729 CrossRef CAS PubMed; (b) A. M. Thiel, E. Damgaard-Møller and J. Overgaard, Inorg. Chem., 2020, 59, 1682–1691 CrossRef CAS PubMed.
- (a) B. A. Dougan and Z. Xue, Sci. China, Ser. B: Chem., 2009, 52, 2083–2095 CrossRef CAS; (b) K. Ridier, A. Mondal, C. Boilleau, O. Cador, B. Gillon, G. Chaboussant, B. Le Guennic, K. Costuas and R. Lescouëzec, Angew. Chem., Int. Ed., 2016, 55, 3963–3967 CrossRef CAS PubMed.
- V. García-López, M. Palacios-Corella, V. Gironés-Pérez, C. Bartual-Murgui, J. A. Real, E. Pellegrin, J. Herrero-Martín, G. Aromí, M. Clemente-León and E. Coronado, Inorg. Chem., 2019, 58, 12199–12208 CrossRef PubMed.
- (a) D.-R. Dauer and D. Stalke, Dalton Trans., 2014, 43, 14432–14439 RSC; (b) D.-R. Dauer, M. Flügge, R. Herbst-Irmer and D. Stalke, Dalton Trans., 2016, 45, 6149–6158 RSC.
- I. Koehne, N. Graw, T. Teuteberg, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2017, 56, 14968–14978 CrossRef CAS PubMed.
- J. Kretsch, I. Koehne, M. Lõkov, I. Leito and D. Stalke, Eur. J. Inorg. Chem., 2019, 2019, 3258–3264 CrossRef CAS.
- (a) J. Kretsch, A. Kreyenschmidt, T. Schillmöller, C. Sindlinger, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2021, 60, 7389–7398 CrossRef CAS PubMed; (b) J. Kretsch, A.-K. Kreyenschmidt, T. Schillmöller, M. Lõkov, R. Herbst-Irmer, I. Leito and D. Stalke, Chem. – Eur. J., 2021, 27, 9858–9865 CrossRef CAS PubMed.
- I. Koehne, S. Bachmann, T. Niklas, R. Herbst-Irmer and D. Stalke, Chem. – Eur. J., 2017, 23, 13141–13149 CrossRef CAS PubMed.
- J. Kretsch, A. Kreyenschmidt, R. Herbst-Irmer and D. Stalke, Dalton Trans., 2018, 47, 12606–12612 RSC.
- (a) J. Kretsch, R. Herbst-Irmer and D. Stalke, Z. Anorg. Allg. Chem., 2018, 644, 657–660 CrossRef CAS; (b) J. Kretsch, A. Kreyenschmidt, T. Schillmöller, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2020, 59, 13690–13699 CrossRef CAS PubMed; (c) I. Koehne, R. Herbst-Irmer and D. Stalke, Eur. J. Inorg. Chem., 2017, 2017, 3322–3326 CrossRef CAS.
- S. Ziegenbalg, D. Hornig, H. Görls and W. Plass, Inorg. Chem., 2016, 55, 4047–4058 CrossRef CAS PubMed.
- C. M. Legendre, E. Damgaard-Møller, J. Overgaard and D. Stalke, Eur. J. Inorg. Chem., 2021, 3108–3114 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- (a) S. G. McGeachin, Can. J. Chem., 1968, 46, 1903–1912 CrossRef CAS; (b) Y. Nishida, N. Oishi and S. Kida, Inorg. Chim. Acta, 1979, 32, 7–10 CrossRef CAS; (c) L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3066 CrossRef CAS PubMed.
- V. A. Crawford, J. Chem. Soc., 1953, 2061 RSC.
- C. Rajnák, J. Titiš, O. Fuhr, M. Ruben and R. Boča, Inorg. Chem., 2014, 53, 8200–8202 CrossRef PubMed.
- A. Sarkar, S. Dey and G. Rajaraman, Chem. – Eur. J., 2020, 14036–14058 CrossRef CAS PubMed.
- T. Wu, Y.-Q. Zhai, Y.-F. Deng, W.-P. Chen, T. Zhang and Y.-Z. Zheng, Dalton Trans., 2019, 48, 15419–15426 RSC.
- C. Rajnák, J. Titiš, J. Moncol, R. Mičová and R. Boča, Inorg. Chem., 2019, 58, 991–994 CrossRef PubMed.
- C. Duboc, Chem. Soc. Rev., 2016, 45, 5834–5847 RSC.
- C. Duboc, M.-N. Collomb, J. Pécaut, A. Deronzier and F. Neese, Chemistry, 2008, 14, 6498–6509 CrossRef CAS PubMed.
- A. C. Benniston, S. Melnic, C. Turta, A. B. Arauzo, J. Bartolomé, E. Bartolomé, R. W. Harrington and M. R. Probert, Dalton Trans., 2014, 43, 13349–13357 RSC.
- S. Vaidya, S. Tewary, S. K. Singh, S. K. Langley, K. S. Murray, Y. Lan, W. Wernsdorfer, G. Rajaraman and M. Shanmugam, Inorg. Chem., 2016, 55, 9564–9578 CrossRef CAS PubMed.
- F. Neese, ORCA. An ab initio, DFT and semiempirical SCF-MO package Search PubMed.
- E. A. Suturina, D. Maganas, E. Bill, M. Atanasov and F. Neese, Inorg. Chem., 2015, 54, 9948–9961 CrossRef CAS PubMed.
- E. Damgaard-Møller, L. Krause, K. Tolborg, G. Macetti, A. Genoni and J. Overgaard, Angew. Chem., Int. Ed., 2020, 21203–21209 CrossRef PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- SAINT v8.30C. Bruker Apex CCD, Bruker AXS Inc., WI, USA, Madison, 2013 Search PubMed.
- L. Krause, R. Herbst-Irmer and D. Stalke, J. Appl. Crystallogr., 2015, 48, 1907–1913 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, SHELXL in SHELXTL v2014/7, WI, USA, Madison, 2014 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- E. Bill, JulX-2s, Max-Planck Institute for Chemical Energy Conversion, Mülheim/Ruhr, Germany, 2013 Search PubMed.
- D. Reta and N. F. Chilton, Phys. Chem. Chem. Phys., 2019, 21, 23567–23575 RSC.
- O. Kahn, Molecular Magnetism, VCH, 1993 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- M. Atanasov, D. Ganyushin, K. Sivalingam and F. Neese, in Molecular electronic structures of transition metal complexes, ed. D. M. P. Mingos, P. Day and J. P. Dahl, Springer, Heidelberg, Dordrecht, New York, 2012, vol. 143, pp. 149–220 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The synthesis, X-ray diffraction, magnetism, vis-spectroscopy and computations. CCDC 2095985–2095993. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03230e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |