
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-step, kit-based radiopharmaceuticals for molecular SPECT imaging: a versatile diphosphine chelator for 99mTc radiolabelling of peptides†
Ingebjørg N.
Hungnes
a,
Fahad
Al-Salemee
 a,
Peter J.
Gawne
a,
Thomas
Eykyn
a,
R. Andrew
Atkinson
bc,
Samantha Y. A.
Terry
a,
Fiona
Clarke
d,
Philip J.
Blower
a,
Paul G.
Pringle
e and
Michelle T.
Ma
*a
a,
Peter J.
Gawne
a,
Thomas
Eykyn
a,
R. Andrew
Atkinson
bc,
Samantha Y. A.
Terry
a,
Fiona
Clarke
d,
Philip J.
Blower
a,
Paul G.
Pringle
e and
Michelle T.
Ma
*a
aKing's College London, School of Biomedical Engineering and Imaging Sciences, 4th Floor Lambeth Wing, St Thomas’ Hospital, London, UK. E-mail: michelle.ma@kcl.ac.uk
bKing's College London, Randall Centre for Cell and Molecular Biophysics, and Centre for Biomolecular Spectroscopy, London, UK
cInstitut de Pharmacologie et de Biologie Structurale, IPBS, Université de Toulouse, CNRS, Université Paul Sabatier, 31077 Toulouse, France
dKing's College London, Centre for Inflammation Biology and Cancer Immunology, Faculty of Life Sciences and Medicine, London, UK
eUniversity of Bristol, School of Chemistry, Cantock's Close, Bristol, UK
First published on 15th October 2021
Abstract
Radiotracers labelled with technetium-99m (99mTc) enable accessible diagnostic imaging of disease, provided that radiotracer preparation is simple. Whilst 99mTc radiopharmaceuticals for imaging perfusion are routinely prepared from kits, and regularly used in healthcare, there are no 99mTc-labelled receptor-targeted radiopharmaceuticals in widespread clinical use. This is in part due to the multistep radiosyntheses required for the latter. We demonstrate that the diphosphine, 2,3-bis(diphenylphosphino)maleic anhydride (BMA), is an excellent platform for preparation of kit-based, receptor-targeted 99mTc-labelled radiotracers: its conjugates are simple to prepare and can be easily labelled with 99mTc using one-step, kit-based protocols. Here, reaction of BMA with the αvβ3-integrin receptor targeted cyclic peptide, Arg-Gly-Asp-DPhe-Lys (RGD), provided the first diphosphine-peptide conjugate, DP-RGD. DP-RGD was incorporated into a “kit”, and addition of a saline solution containing 99mTcO4− to this kit, followed by heating, furnished the radiotracer [99mTcO2(DP-RGD)2]+ in consistently high radiochemical yields (>90%). The analogous [ReO2(DP-RGD)2]+ compound was prepared and characterised, revealing that both [99mTcO2(DP-RGD)2]+ and [ReO2(DP-RGD)2]+ consist of a mixture of cis and trans geometric isomers. Finally, [99mTcO2(DP-RGD)2]+ exhibited high metabolic stability, and selectively targeted αvβ3-integrin receptors, enabling in vivo SPECT imaging of αvβ3-integrin receptor expression in mice.
Introduction
The γ-emitting radionuclide, technetium-99 m (99mTc, t1/2 = 6 h, 90% γ, 140 keV), is used in over 30 million routine nuclear medicine SPECT/γ-scintigraphy procedures every year, for diagnostic imaging of perfusion and anatomical processes.1,299mTc is produced by bench-top generators, enabling this widespread access. Despite the availability of 99mTc and the high prevalence of SPECT and γ-scintigraphy infrastructure, few receptor-targeted 99mTc molecular imaging agents have entered late stage clinic trials, and none are used routinely. In contrast, modern PET imaging with peptide-based, receptor-targeted radiotracers has had significant clinical impact. 68Ga-labelled peptides that target receptors over-expressed in prostate and neuroendocrine cancers have resulted in better disease management for patients and are now used in routine clinical practice.3,499mTc radiopharmaceuticals for imaging heart, kidney and brain perfusion are based on one-step, kit-based radiosyntheses, in which generator-produced 99mTcO4− is simply added to commercially available “kit” vials that contain a reducing agent, chelator and other reagents.2,5 These simple radiosynthetic procedures allow staff in hospital radiopharmacies to routinely prepare patient doses of 99mTc radiopharmaceuticals on a daily basis.
Several 99mTc-labelled chelator–peptide conjugates have recently demonstrated clinical utility in SPECT imaging of receptor expression. These include 99mTc-MIP-1404 and derivatives, and 99mTc-PSMA-I&S, which target PSMA (prostate specific membrane antigen) receptors that are overexpressed in prostate cancer.6,7 In 99mTc-MIP-1404, the tridentate N3 chelator (Chart 1) coordinates to a fac-[99mTc(CO)3]+ moiety.6 In 99mTc-PSMA-I&S (Chart 1), a modified tripeptide, mercaptoacetyl-D-Ser-D-Ser-D-Ser, coordinates to the [99mTcO]3+ motif, via a thiol and three deprotonated amide groups.7 These PSMA-targeted radiopharmaceuticals are prepared from kits. However, whilst 99mTc-PSMA-I&S is prepared in a single step at high radiochemical yields, preparation of 99mTc-MIP-1404 involves two “kits” – the first to generate the labile fac-[99mTc(CO)3(H2O)3]+ precursor and the second to form the 99mTc complex of the targeting chelator–peptide bioconjugate MIP-1404. Other molecular 99mTc radiopharmaceuticals are based on 6-hydrazinopyridine-3-carboxylic acid (HYNIC, Chart 1), which coordinates to 99mTc and acts as an attachment point for targeting peptides.8 Co-ligands such as ethylenediamine or tricine occupy remaining coordination sites on the Tc. Some of these HYNIC-based radiopharmaceuticals can be prepared from a single kit,9,10 but their structures remain ill-defined: it is unknown whether HYNIC coordinates to Tc via the hydrazino group only, or as a bidentate ligand, via the hydrazino and pyridyl groups.11,12
 | ||
| Chart 1 Existing chelators and complexes used for 99mTc radiopharmaceuticals. | ||
The radiopharmaceutical “Myoview” is used to image cardiac perfusion. In Myoview, two bidentate diphosphines coordinate to a trans-[TcO2]+ motif (Chart 1).13 Myoview is also prepared using a single step: 99mTcO4− is added to a kit containing sodium gluconate, tin chloride, sodium bicarbonate and diphosphine chelator, followed by incubation at room temperature for 15 min to produce Myoview in >90% radiochemical yield and purity. It is then administered to patients without further processing.14 Other chelators containing phosphines, notably a P2S2 chelator (Chart 1), have also exhibited efficient radiolabelling properties when reacted with [TcO2]+ derivatives.15–17
We aim to identify new diphosphine chemical platforms that enable simple, one-step, kit-based 99mTc-radiolabelling of receptor-targeted peptides, to provide structurally well-defined 99mTc radiotracers. Prior work has shown that primary amines react with 2,3-bis(diphenylphosphino)maleic anhydride (BMA, Scheme 1) to form a ring-opened amide species.18,19 We have therefore selected BMA as a potentially versatile chemical platform for preparing a diphosphine-peptide conjugate. We have also selected the cyclic peptide, Arg-Gly-Asp-DPhe-Lys (RGD), which targets the αvβ3-integrin receptor over-expressed in neovasculature, inflammation processes and cancer cells. RGD has been used extensively in receptor-targeted imaging,9,20 and it contains a single primary amine suitable for bioconjugation. We have chosen the [99mTcVO2]+ motif for radiolabelling of diphosphine-peptide conjugates because well-defined phosphine complexes based on [99mTcVO2]+ (e.g. Myoview) can be prepared from aqueous solutions of 99mTcO4− in a single step. Additionally, in comparison to the commonly used [99mTc(CO)3]+ motif, the [99mTcVO2]+ group is relatively hydrophilic, and this property favours rapid radiotracer clearance from circulation via a renal pathway, potentially enabling high contrast SPECT imaging of target disease.
 | ||
| Scheme 1 Preparation of [MO2(DP-RGD)2]+ (M = Re, 99mTc). | ||
Results and discussion
Synthesis and radiolabelling
Reaction of RGD with BMA21,22 in a basic solution of DMF followed by semi-preparative reverse-phase C18 HPLC provided the diphosphine conjugate DP-RGD (Scheme 1) in ≥95% purity and 86% yield. DP-RGD was characterised by 1H, 13C and 31P NMR, analytical HPLC and HR-ESI-MS (Fig. S1–S6 and Tables S1, S2†). Whilst DP-RGD slowly oxidised in solution to phosphine oxide derivatives under normal atmospheric conditions, in the solid state, DP-RGD was stable to oxidation: it can be handled either as a dry powder, in basic organic solutions, or in aqueous solutions at near-neutral pH. However, in acidic solutions, the reverse reaction was observed, and the DP-RGD conjugate decomposed to re-form RGD and BMA.The chemistry of Re and Tc are closely similar. As Tc has no stable isotopes, it was convenient to prepare [ReO2(DP-RGD)2]+ in order to obtain full characterisation. Reaction of [ReO2I(PPh3)2] with an excess of DP-RGD furnished geometric isomers of [ReO2(DP-RGD)2]+ (Scheme 1), which are labelled cis and trans to denote the relative positions of the RGD moieties, and which were formed in the ratio of 54% and 46% respectively. The isomers were separated by reverse phase C18 HPLC. For both species, the most intense signals in the ESI-MS at m/z = 785.92 and 1178.38, corresponded to the ions [M + 2H]3+ and [M + H]2+ where M = [ReO2(DP-RGD)2]+ (see Fig. S1†).
In the 31P{H} NMR spectrum of DP-RGD (Fig. 1A), the two inequivalent P atoms produce an AB pattern with δ(PA) = −12.04 and δ(PB) = −12.89 and 2JPP = 168.4 Hz. The 31P{H} NMR spectra of the isomers of [ReO2(DP-RGD)2]+ might be expected to show complex patterns associated with the AA′XX′ spin systems. However the spectra for the two isomers are shown in Fig. 1A and the upper spectrum is tentatively assigned to the cis isomer on the basis of the pseudo-AB pattern with a large 2J(PAPB) (356.1 Hz) expected for trans-inequivalent P atoms.23,24 In the 1H and 13C NMR spectra, the PPh2 signals shift upon ReV binding and become more complex, but RGD peptide resonances are not changed significantly (Fig. S3–S5 and Tables S1, S2†).
 | ||
| Fig. 1 (A) 31P{H} NMR of DP-RGD, cis-[ReO2(DP-RGD)2]+ and trans-[ReO2(DP-RGD)2]+. The full 31P NMR spectra are included in Fig. S6.† (B) Radio-HPLC trace of trans-/cis-[99mTcO2(DP-RGD)2]+ (red) prepared from an aqueous solution of 99mTcO4− and an optimised kit formulation (Kit 3, Table 1), and HPLC traces (λ220) of trans- and cis-[ReO2(DP-RGD)2]+ (blue). The full chromatograms are included in Fig. S7.† | ||
To assess the feasibility of 99mTc radiolabelling of DP-RGD with a “kit” formulation, lyophilised mixtures of DP-RGD, stannous chloride, sodium bicarbonate, and sodium gluconate were initially prepared (Table 1, Kit 1). The amounts of stannous chloride, sodium bicarbonate and sodium gluconate reagents used in Kit 1 replicate those in the Myoview kit. Stannous chloride reduces 99mTcO4− to 99mTc(V), sodium bicarbonate buffers the solution at pH 8 and sodium gluconate is a weak chelator that stabilises reduced 99mTc intermediates, and also coordinates Sn2+ in solution, to prevent formation and precipitation of stannous hydroxide species during radiolabelling reactions. All radiolabelling reactions were undertaken in a mixture of saline and ethanol to dissolve DP-RGD; lower amounts of ethanol were required for kits containing lower amounts of DP-RGD.
| Kit | Kit components | Radiochemical yielda |
|---|---|---|
| a Reactions were undertaken in duplicate to ensure reproducibility of radiochemical yields, except for radiolabelling reactions with Kit 3, where the reaction was replicated four times to give an average radiochemical yield of 93.0 ± 1.0%. | ||
| 1 | DP-RGD: 1.0 mg (0.93 μmol) | ≤34% |
| Sodium gluconate (NaC6H11O7): 1.0 mg (4.58 μmol) | ||
| SnCl2·2H2O: 50 μg (0.22 μmol) | ||
| NaHCO3: 1.8 mg (21.43 μmol) | ||
| 99mTcO4− in 150 μL saline/150 μL EtOH | ||
| 2 | DP-RGD: 500 μg (0.47 μmol) | 85% |
| Sodium tartrate (Na2C4H4O6): 1.05 mg (4.58 μmol) | ||
| SnCl2·2H2O: 50 μg (0.22 μmol) | ||
| NaHCO3: 1.8 mg (21.4 μmol) | ||
| 99mTcO4− in 150 μL saline/150 μL EtOH | ||
| 3 | DP-RGD: 125 μg (0.12 μmol) | ≥90% |
| Sodium tartrate: 0.26 mg (1.15 μmol) | ||
| SnCl2·2H2O: 25 μg (0.11 μmol) | ||
| NaHCO3: 0.9 mg (10.71 μmol) | ||
| 99mTcO4− in 250 μL saline/50 μL EtOH | ||
| 4 | DP-RGD: 64 μg (0.06 μmol) | 65% |
| Sodium tartrate: 0.26 mg (1.15 μmol) | ||
| SnCl2·2H2O: 25 μg (0.11 μmol) | ||
| NaHCO3: 0.9 mg (10.71 μmol) | ||
| 99mTcO4− in 260 μL saline/40 μL EtOH | ||
| Myoview (single dose kits) | Diphosphine: 250 μg (0.65 μmol) | Routinely > 90% |
| Sodium gluconate: 1.0 mg (4.6 μmol) | ||
| SnCl2·2H2O: 50 μg (0.22 μmol) | ||
| NaHCO3: 1.8 mg (21.4 μmol) | ||
| 99mTcO4− in saline | ||
Addition of generator-produced 99mTcO4− in saline solution (20–55 MBq) to the contents of Kit 1, followed by heating at 60 °C for 30 min, resulted in formation of [99mTcO2(DP-RGD)2]+ in radiochemical yields of up to 34%, as determined by radio-HPLC (vide infra) and TLC. Replacing sodium gluconate with sodium tartrate in the “kit” mixture whilst lowering the amount of DP-RGD conjugate from 1 mg to 0.5 mg, increased radiochemical yields to 85% (Kit 2). In Kit 3, radiochemical yields of 93.0 ± 1.0% (n = 4) were achieved, with 45–65 MBq of 99mTcO4− and only 125 μg of DP-RGD, providing specific activities of 375–540 MBq μmol−1. In Kit 3, sodium tartrate and stannous chloride amounts were also reduced. However, further decreasing DP-RGD, to 64 μg in Kit 4, reduced radiochemical yields to 65%.
These kit-based reactions were analysed using reverse phase C18 radio-HPLC. A very gradual mobile phase gradient allowed separation and observation of two radioactive products (Fig. 1B, see Experimental section, Method 4 for method details). The first is attributed to trans-[99mTcO2(DP-RGD)2]+ (52%, retention time (RT) = 41.0 min) and the second is attributed to cis-[99mTcO2(DP-RGD)2]+ (46%, RT = 44.1). These species exhibit similar chromatographic properties to trans-[ReO2(DP-RGD)2]+ (RT = 38.3 min) and cis-[ReO2(DP-RGD)2]+ (RT = 42.6 min). The difference in retention times between analogous 99mTc and Re complexes is at least in part due to the configuration of the UV and radioactivity (scintillation) detectors in series. The only other discernable radioactive species (<1%) eluted with the solvent front, and corresponds to either unreacted 99mTcO4− or 99mTc intermediates bound to other kit-based components (e.g. tartrate ligand).
Lastly, to unambiguously assign the stoichiometry of the [99mTcO2(DP-RGD)2]+ compounds, experiments with long-lived technetium-99g (99gTc, t1/2 = 211![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years) were undertaken. A sample of [N(C4H9)4][99gTcOCl4] was reacted with 3 equivalents of DP-RGD in methanol, and analysed by reverse phase C18 HPLC methods, which revealed the formation of two major Tc product complexes, with closely similar retention times. LC-ESI-LRMS analysis (Fig. 2) of [99gTcO2(DP-RGD)2]+ showed that these two major species possessed LRMS signals consistent with the stoichiometry of [99TcO2(DP-RGD)2]+ (m/z = 757.6 and 1136.0, corresponding to the ions [M + 2H]3+ and [M + H]2+ where M = [99TcO2(DP-RGD)2]+). Additionally, these two major products, tentatively assigned as trans-[99gTcO2(DP-RGD)2]+ and cis-[99gTcO2(DP-RGD)2]+ co-eluted with radioactive signals of [99mTcO2(DP-RGD)2]+ (Fig. S8†).
000 years) were undertaken. A sample of [N(C4H9)4][99gTcOCl4] was reacted with 3 equivalents of DP-RGD in methanol, and analysed by reverse phase C18 HPLC methods, which revealed the formation of two major Tc product complexes, with closely similar retention times. LC-ESI-LRMS analysis (Fig. 2) of [99gTcO2(DP-RGD)2]+ showed that these two major species possessed LRMS signals consistent with the stoichiometry of [99TcO2(DP-RGD)2]+ (m/z = 757.6 and 1136.0, corresponding to the ions [M + 2H]3+ and [M + H]2+ where M = [99TcO2(DP-RGD)2]+). Additionally, these two major products, tentatively assigned as trans-[99gTcO2(DP-RGD)2]+ and cis-[99gTcO2(DP-RGD)2]+ co-eluted with radioactive signals of [99mTcO2(DP-RGD)2]+ (Fig. S8†).
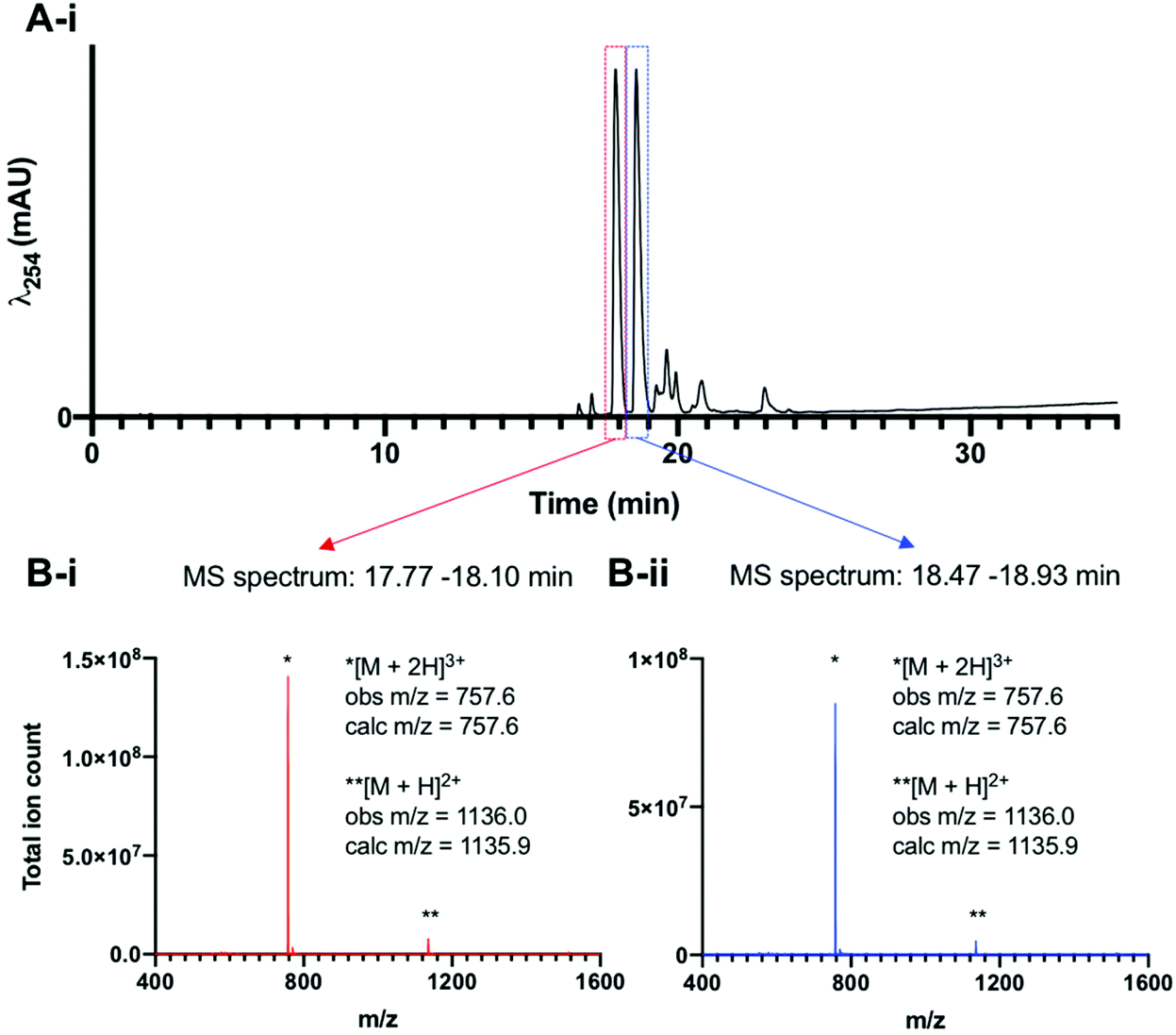 | ||
| Fig. 2 (A) UV (λ524) HPLC trace of [99gTcO2(DP-RGD)2]+. (B-i and B-ii) The ESI-LRMS corresponding to the two major HPLC signals indicates that the stoichiometry of both of these species corresponds to [99TcO2(DP-RGD)2]+ (M = C110H122N18O22P499Tc+). | ||
Biological characterisation of [99mTcO2(DP-RGD)2]+
For all subsequent experiments, including in vivo experiments, [99mTcO2(DP-RGD)2]+ was prepared from generator-produced 99mTcO4− and “Kit 3”, using our newly established one-step radiolabelling protocol. Solutions containing both the desired cis-[99mTcO2(DP-RGD)2]+ and trans-[99mTcO2(DP-RGD)2]+ products, as well as unreacted DP-RGD ligand, were used without further purification.The stability of [99mTcO2(DP-RGD)2]+ was assessed, by incubating [99mTcO2(DP-RGD)2]+ in human serum. C18 radio-HPLC analysis revealed that only 3% 99mTc dissociated from [99mTcO2(DP-RGD)2]+ over 4 h (Fig. 3), presumably forming 99mTcO4− (vide infra). The logDOCT/PBS of [99mTcO2(DP-RGD)2]+ measured −1.64 ± 0.04. In a cell-free solid phase αvβ3-integrin receptor binding assay,20 [99mTcO2(DP-RGD)2]+ bound to αvβ3-integrin receptor, with the binding inhibited by RGD peptide in a concentration-dependent manner (Fig. 4A).
 | ||
| Fig. 3 (A–C) [99mTcO2(DP-RGD)2]+ was incubated in human serum for 4 h. Analytical radio-HPLC analysis showed that 0.5% 99mTc dissociated from [99mTcO2(DP-RGD)2]+ over 1 h, and 3% 99mTc dissociated from [99mTcO2(DP-RGD)2]+ over 4 h. (D) Radio-HPLC analysis of urine, collected from a mouse administered [99mTcO2(DP-RGD)2]+, showed that [99mTcO2(DP-RGD)2]+ was excreted intact. N.B. In these experiments, a short HPLC method, with a relatively steep mobile phase gradient was used: under these conditions, cis-[99mTcO2(DP-RGD)2]+ and trans-[99mTcO2(DP-RGD)2]+ were not separated from one another, and eluted as a single peak. | ||
 | ||
| Fig. 4 (A) [99mTcO2(DP-RGD)2]+ exhibits binding to αvβ3 integrin receptor, which can be inhibited by increasing concentrations of RGD peptide. (B) Biodistribution of [99mTcO2(DP-RGD)2]+ in healthy mice 1 h PI: co-injection of 400 μg RGD inhibits [99mTcO2(DP-RGD)2]+ uptake in αvβ3 integrin-expressing tissue. Error bars correspond to 95% confidence interval. (C) Quantification of radioactivity distribution from SPECT/CT imaging (Fig. S9†) of a single healthy Balb/c mouse administered [99mTcO2(DP-RGD)2]+ intravenously. | ||
To assess biodistribution, a group of healthy Balb/c mice was intravenously administered with [99mTcO2(DP-RGD)2]+ (4.3–5.3 MBq). The αvβ3-integrin receptor is known to be expressed at low levels in normal vasculature. A second group of mice was co-administered with [99mTcO2(DP-RGD)2]+ (2.7–4.8 MBq) and a large “excess” of RGD peptide (400 μg), to saturate αvβ3-integrin receptors and “block” receptor-mediated radiotracer accumulation. All mice were culled and their organs harvested for ex vivo tissue counting, 1 h post injection (PI). Between the two groups of mice there were statistically significant differences in biodistribution: co-administration of RGD peptide significantly decreased 99mTc radioactivity concentration in the heart, liver, spleen, pancreas, muscles, stomach and intestines (Fig. 4B and Table S3†). This is consistent with the known expression pattern of αvβ3-integrin, and evidences the affinity and specificity of [99mTcO2(DP-RGD)2]+ for αvβ3-integrin receptors.20 [99mTcO2(DP-RGD)2]+ was cleared from circulation via a renal pathway, as evidenced by high concentrations of 99mTc in kidneys. Quantitative SPECT/CT image analysis of [99mTcO2(DP-RGD)2]+ in a healthy mouse (Fig. 4C and S9†) confirmed that [99mTcO2(DP-RGD)2]+ was indeed excreted renally: at 30 min PI, 35% of the injected radioactivity was in the bladder; at 2 h PI, 56% was in the bladder. This was consistent with ex vivo tissue counting data. Notably, radio-HPLC analysis of urine showed that [99mTcO2(DP-RGD)2]+ was excreted intact (Fig. 3D), consistent with the observed high serum stability.
Finally, to demonstrate that this new radiotracer can image αvβ3-integrin receptor expression in disease, [99mTcO2(DP-RGD)2]+ (4.3–5.2 MBq) was administered to mice induced with rheumatoid arthritis (RA), in which the expression of αvβ3-integrin receptor is associated with inflammation.20,25 This is a “heterogeneous” RA model: the degree of arthritis and symptomatic swelling differs between mice, and even between joints of the same animal.20,26 Quantitative analysis (Fig. 5A and Fig. S10†) of SPECT images (e.g.Fig. 5B) obtained 1 h PI revealed that 99mTc radioactivity accumulation and concentration in wrists and ankles correlated with the degree of symptomatic arthritic swelling, measured with calipers. 99mTc activity was also observed in thyroid tissue: 99mTcO4− acts as an iodide “mimic” in vivo, and is well-documented to be a substrate for the sodium iodide symporter expressed in the thyroid.27 It is likely that the observation of 99mTc activity in the thyroid is a result of small amounts of 99mTc dissociating from [99mTcO2(DP-RGD)2]+ and forming 99mTcO4−, consistent with the serum stability data.
 | ||
| Fig. 5 (A) In mice with rheumatoid arthritis, radioactivity accumulation in ankles and wrists correlates with joint swelling. (B) Maximum intensity projection of a SPECT/CT image of a mouse with rheumatoid arthritis, showing accumulation of [99mTcO2(DP-RGD)2]+ in an arthritic ankle (RA). B = bladder, K = kidneys, T = thyroid. | ||
Concluding remarks
New chemical platforms that enable one-step, kit-based 99mTc-radiolabelling of peptides could allow increased and widespread clinical use of receptor-targeted 99mTc radiopharmaceuticals. We have shown that BMA is an excellent candidate for this purpose: its conjugates are simple to prepare and can be easily labelled with 99mTc using one-step, kit-based protocols. The biological properties of its peptide conjugates are favourable, as exemplified by [99mTcO2(DP-RGD)2]+.Phosphines have previously been successfully incorporated into chelator–peptide bioconjugates for 99mTc binding.28–32 This includes derivatives of a tetradentate P2S2 ligand (Chart 1), which contains two tertiary alkyl-substituted phosphine groups, for coordinating [TcO2]+.15–17 Small amounts (<1 μmol) of P2S2-peptide compounds can be radiolabelled with 99mTcO4− in the presence of stannous chloride to yield 99mTc-labelled peptides of formula [99mTcO2(P2S2-peptide)]+, in high radiochemical yields (80–98%),15 similar to the radiochemical yields we achieve for [99mTcO2(DP-RGD)2]+. However, compared to the P2S2-peptide compounds, bioconjugates of BMA are more synthetically accessible. BMA itself is prepared in two steps21,22 from readily available starting materials, and the diphosphine-peptide derivatives of BMA are prepared simply by addition of a peptide containing a single primary amine group to BMA in the presence of base, followed by reverse-phase chromatographic purification.
99mTc-labelled diphosphine-peptide radiotracers based on BMA have advantages over existing 99mTc receptor-targeted radiopharmaceuticals. The radiochemical synthesis of [99mTcO2(DP-RGD)2]+ is achieved in a single step, from a single vial, in radiochemical yields >90%. This contrasts with existing molecular 99mTc radiotracers that have recently entered late-stage clinical trials, such as 99mTc-MIP-1404, which requires (i) radiochemical synthesis of fac-[99mTc(CO)3(H2O)3]+, prior to (ii) reaction with the tridentate MIP-1404 chelator–peptide, and (iii) further purification and formulation before administration.6 Additionally, the coordination sphere of [99mTcO2(DP-RGD)2]+ is structurally well-defined; the structures of other recently described 99mTc radiopharmaceuticals based on the HYNIC chelator (such as 99mTc-3PRGD29 and 99mTc-HYNIC-PSMA,10 which have both demonstrated clinical utility), are more ambiguous.11,12 We are currently developing new diphosphine-peptide bioconjugates based on BMA, and further optimising kit-based 99mTc-radiolabelling of such derivatives.
The presence of two isomeric radiolabelled products for DP-peptide conjugates, cis-[99mTcO2(DP-peptide)2]+ and trans-[99mTcO2(DP-peptide)2]+, is potentially disadvantageous. It is possible that prior to any clinical application, cis and trans isomers would require separate biological evaluation, to assess whether their target affinities, pharmacokinetics and stabilities are equivalent to each other. Notably, the 68Ga-labelled prostate cancer radiotracer, 68Ga-HBED-PSMA, consists of at least two distinguishable (and as yet, undefined) chemical species.33,34 However, the biological profiles of each separate 68Ga-HBED-PSMA species have not been elucidated, and this has not prevented widespread and routine use of 68Ga-HBED-PSMA, and its recent FDA approval, for clinical prostate cancer imaging.4,35
Lastly, 99mTc-radiolabelled peptide derivatives of BMA possess a significant advantage over existing receptor-targeted 99mTc radiotracers: upon radiolabelling with the [TcO2]+ motif, there are two copies of the peptide per molecule. Radiotracers containing two or more peptide copies typically demonstrate higher tumour uptake compared to their monomeric homologues, due to their higher affinity for target receptors.9,20,36–40 These examples include 99mTc-labelled compounds that incorporate two copies of a targeting peptide into a single coordinating ligand.9,38 However there are only a handful of examples in which 99mTc coordination by two or more copies of a ligand results in formation of radiotracers containing multiple copies of a targeting motif.41–43 We have shown that it is feasible to apply diphosphine-peptide bioconjugates for this purpose.
Experimental methods
General
All chemicals were supplied by Sigma-Aldrich or Fisher Scientific if not otherwise specified. Sodium [99m/99Tc]pertechnetate in saline was supplied by Guy's and St Thomas’ Hospital Nuclear Medicine Services. Cyclic RGD peptide (Arg-Gly-Asp-D-Phe-Lys, cyclised via the peptide backbone) was purchased from Peptide Synthetics (Hampshire, UK).NMR data (1H, 13C{H} and 31P{H} 1D spectra and COSY, TOCSY and HSQC spectra) were acquired on a Bruker Avance III 400 spectrometer equipped with a QNP probe or a Bruker Avance III 700 spectrometer equipped with an AVIII console and a quadruple-resonance QCI cryoprobe. High resolution mass spectrometry (MS) was performed by the King's College London Mass Spectrometry Facilities, using a high resolution Thermo Exactive mass spectrometer in positive electrospray mode. Samples were infused to the ion source at a rate of 10 μl min−1 using a syringe pump. High performance liquid chromatography (HPLC) was carried out on an Agilent 1200 LC system with the Laura software, a Rheodyne sample loop (200 μL) and UV spectroscopic detection at 220 nm or 254 nm. The HPLC was attached to a LabLogic Flow-Count detector with a sodium iodide probe (B-FC-3200) for radiation detection. LC-ESI-LRMS was carried out on an Agilent 1260 Infinity II HPLC system coupled to an Advion Expression Compact Mass Spectrometer using an ESI source. Semi-preparative (9.4 × 250 mm, 5 μm) and analytical (4.6 × 150 mm, 5 μm) Agilent Zorbax Eclipse XDB-C18 columns were used with purified water (A) and acetonitrile (B) containing 0.005% and 0.1% TFA as mobile phases for semi-preparative and analytical runs, respectively. Method 1 (semi-preparative): 100 minutes, 1% min−1 linear increase from 100% A to 100% B, flow rate = 3 mL min−1. Method 2 (analytical): 20 minutes, 5% min−1 linear increase from 100% A to 100% B, flow rate = 1 mL min−1. Method 3 (semi-preparative): 200 minutes, 0.5% min−1 linear increase from 95% A to 100% B, flow rate = 3 mL min−1. Method 4 (analytical): 55 minutes, 2.5% min−1 linear increase from 100% A to 75%A/25% B over 10 min, followed by 0.33% min−1 linear increase from 75% A/25% B to 60%A/40% B over 45 min, flow rate of 1 mL min−1. Method 5 (analytical, 0.1% formic acid in either water (A) or acetonitrile (B)): 0–5 min: 95% A/5% B; 5–35 min: linear increase from 95% A/5% B to 5% A/95% B; flow rate of 1 mL min−1.
Synthesis
Preparation and characterisation of [99m/99gTcO2(DP-RGD)2]+
Two separate iTLC analyses were undertaken, to enable quantification of 99mTc-colloids, unreacted 99mTcO4− and [99mTcO2(DP-RGD)2]+. To quantify amounts of unreacted 99mTcO4−, acetone was used as a mobile phase: Rf values: 99mTcO4− >0.9, 99mTc colloids <0.1, [99mTcO2(DP-RGD)2]+ <0.1. To quantify 99mTc-colloid formation, a 1:1 mixture of methanol and 2 M aqueous ammonium acetate solution was used as a mobile phase: 99mTcO4− >0.9, 99mTc colloids <0.1, [99mTcO2(DP-RGD)2]+ >0.9.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) D (pH 7.4).
The following procedure was carried out in triplicate. A solution containing [99mTcO2(DP-RGD)2]+ (1 MBq in 7.5 μL) was combined with phosphate buffered saline (pH 7.4, 500 μL) and octanol (500 μL), and the mixture was agitated for 30 min. The mixture was then centrifuged (10000 rpm, 10 minutes), and aliquots of octanol and aqueous PBS were analysed for radioactive using a gamma counter. LogD = −1.64 ± 0.04.
000 nM, 50 μL in binding buffer) were added simultaneously to wells, and left to incubate for 1 h at room temperature, before being washed twice as before. Finally, the amount of activity bound to the wells was counted. Binding of [99mTcO2(DP-RGD)2]+ to αvβ3 integrin was displaced by RGD peptide in a concentration-dependent manner. The pseudo-IC50 value of 8.54 ± 3.45 nM (95% CI: 1.67–15.41 nM) was calculated using a non-linear regression model (Binding/Saturation, one site – total) in GraphPad Prism (n = 6 from one experiment).
D (pH 7.4).
The following procedure was carried out in triplicate. A solution containing [99mTcO2(DP-RGD)2]+ (1 MBq in 7.5 μL) was combined with phosphate buffered saline (pH 7.4, 500 μL) and octanol (500 μL), and the mixture was agitated for 30 min. The mixture was then centrifuged (10000 rpm, 10 minutes), and aliquots of octanol and aqueous PBS were analysed for radioactive using a gamma counter. LogD = −1.64 ± 0.04.
000 nM, 50 μL in binding buffer) were added simultaneously to wells, and left to incubate for 1 h at room temperature, before being washed twice as before. Finally, the amount of activity bound to the wells was counted. Binding of [99mTcO2(DP-RGD)2]+ to αvβ3 integrin was displaced by RGD peptide in a concentration-dependent manner. The pseudo-IC50 value of 8.54 ± 3.45 nM (95% CI: 1.67–15.41 nM) was calculated using a non-linear regression model (Binding/Saturation, one site – total) in GraphPad Prism (n = 6 from one experiment).
Pre-clinical imaging and in vivo biodistribution studies of [99mTcO2(DP-RGD)2]+
Animal imaging studies were ethically reviewed and carried out in accordance with the Animals (Scientific Procedures) Act 1986 (ASPA) UK Home Office regulations governing animal experimentation. SPECT/CT imaging was accomplished using a pre-clinical nanoScan SPECT/CT Silver Upgrade instrument (Mediso) calibrated for technetium-99m. All scans were acquired by helical SPECT (4-head scanner with 4 × 9 [1.4 mm] pinhole collimators), and helical CT with 1.4 mm aperture collimators. All acquired images were reconstructed using a full 3D Monte Carlo-based iterative algorithm (Tera-Tomo; Mediso) and further processed and analysed using VivoQuant software (inviCRO, USA).Female balb/c mice (2 months old) were anaesthetised (2–3% v/v isofluorane in oxygen) and injected intravenously (tail vein) with [99mTcO2(DP-RGD)2]+ (2.7–5.3 MBq containing 5 μg of DP-RGD). For blocking studies, animals were co-injected with RGD peptide (400 μg). Mice remained under anaesthetic for 1 h, after which they were culled (pentabarbitone by i.v. injection). Tissues and organs were harvested and weighed, and radioactivity counted using a Gamma Counter (Wallac 1282 CompuGamma Universal Gamma Counter).
Mice were anesthetised (2.5–3% v/v isofluorane) and their paws were measured using microcallipers. Mice were then injected intravenously with [99mTcO2(DP-RGD)2]+ (approx. 5 MBq containing 5 μg of DP-RGD) and allowed to recover from anaesthetic administration. At 1 h post-injection of radiotracer, mice were culled (sodium pentabarbitone), and underwent SPECT/CT scanning post-mortem for 60–180 min. Finally, tissues and organs were harvested and weighed, and radioactivity counted using a Gamma Counter (Wallac 1282 CompuGamma Universal Gamma Counter). The acquired images were processed to units of %ID and the regions of interest (ROIs) delineated by CT using VivoQuant software (inviCRO, USA). Radioactivity in ankle and wrist ROIs were obtained in units of %ID and %ID/cm−3. Each “ankle” ROI was defined as the area between the tibiofibula joint and the base of phalanx V. Each “wrist” ROI was defined as the area between the narrowest point of the wrist (ulna and radius) and the end of the forepaw.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by a Cancer Research UK Career Establishment Award (C63178/A24959), King's College London & Imperial College London EPSRC Centre for Doctoral Training in Medical Imaging (EP/L015226/1), the EPSRC programme for Next Generation Molecular Imaging and Therapy with Radionuclides (EP/S032789/1, “MITHRAS”), Rosetrees Trust (M685, M606), the Wellcome Multiuser Equipment Radioanalytical Facility funded by Wellcome Trust (212885/Z/18/Z), and the Centre for Medical Engineering funded by the Wellcome Trust and the Engineering and Physical Sciences Research Council (WT088641/Z/09/Z), and the King's College London Centre for Biomolecular Spectroscopy funded by Wellcome Trust (202762/Z/16/Z) and British Heart Foundation (IG/16/2/32273).References
- J. A. Jackson, I. N. Hungnes, M. T. Ma and C. Rivas, Bioconjugate Chem., 2020, 31, 483–491 CrossRef CAS.
- C. Rivas, J. A. Jackson, I. N. Hungnes and M. T. Ma, Radioactive Metals in Imaging and Therapy, in Comprehensive Coordination Chemistry III, ed. E. C. Constable, G. Parkin, L. Que Jr. and N. J. Long, 2021, ch. 17, vol. 9, pp. 706–740, DOI:10.1016/B978-0-08-102688-5.00010-6.
- J. Strosberg, G. El-Haddad, E. Wolin, A. Hendifar, J. Yao, B. Chasen, E. Mittra, P. L. Kunz, M. H. Kulke, H. Jacene, D. Bushnell, T. M. O'Dorisio, R. P. Baum, H. R. Kulkarni, M. Caplin, R. Lebtahi, T. Hobday, E. Delpassand, E. Van Cutsem, A. Benson, R. Srirajaskanthan, M. Pavel, J. Mora, J. Berlin, E. Grande, N. Reed, E. Seregni, K. Öberg, M. L. Sierra, P. Santoro, T. Thevenet, J. L. Erion, P. Ruszniewski, D. Kwekkeboom and E. Krenning, N. Engl. J. Med., 2017, 376, 125–135 CrossRef CAS PubMed.
- M. S. Hofman, N. Lawrentschuk, R. J. Francis, C. Tang, I. Vela, P. Thomas, N. Rutherford, J. M. Martin, M. Frydenberg, R. Shakher, L. M. Wong, K. Taubman, S. Ting Lee, E. Hsiao, P. Roach, M. Nottage, I. Kirkwood, D. Hayne, E. Link, P. Marusic, A. Matera, A. Herschtal, A. Iravani, R. J. Hicks, S. Williams and D. G. Murphy, Lancet, 2020, 395, 1208–1216 CrossRef CAS.
- S. Liu and S. Chakraborty, Dalton Trans., 2011, 40, 6077–6086 RSC.
- S. M. Hillier, K. P. Maresca, G. Lu, R. D. Merkin, J. C. Marquis, C. N. Zimmerman, W. C. Eckelman, J. L. Joyal and J. W. Babich, J. Nucl. Med., 2013, 54, 1369–1376 CrossRef CAS.
- S. Robu, M. Schottelius, M. Eiber, T. Maurer, J. Gschwend, M. Schwaiger and H.-J. Wester, J. Nucl. Med., 2017, 58, 235–242 CrossRef CAS PubMed.
- M. J. Abrams, M. Juweid, C. I. TenKate, D. A. Schwartz, M. M. Hauser, F. E. Gaul, A. J. Fuccello, R. H. Rubin, H. W. Strauss and A. J. Fischman, J. Nucl. Med., 1990, 31, 2022–2028 CAS.
- Z. Zhu, W. Miao, Q. Li, H. Dai, Q. Ma, F. Wang, A. Yang, B. Jia, X. Jing, S. Liu, J. Shi, Z. Liu, Z. Zhao, F. Wang and F. Li, J. Nucl. Med., 2012, 53, 716–722 CrossRef PubMed.
- J. Zhang, J. Zhang, X. Xu, L. Lu, S. Hu, C. Liu, J. Cheng, S. Song, Y. Zhang and L. Q. Shi, Sci. Rep., 2020, 10, 1–9 CrossRef PubMed.
- L. K. Meszaros, A. Dose, S. C. G. Biagini and P. J. Blower, Inorg. Chim. Acta, 2010, 363, 1059–1069 CrossRef CAS.
- R. C. King, M. B.-U. Surfraz, S. C. G. Biagini, P. J. Blower and S. J. Mather, Dalton Trans., 2007, 4998–5007 RSC.
- J. D. Kelly, A. M. Forster, B. Higley, C. M. Archer, F. S. Booker, L. R. Canning, K. W. Chiu, B. Edwards, H. K. Gill, M. McPartlin, K. R. Nagle, I. A. Latham, R. D. Pickett, A. E. Storey and P. M. Webbon, J. Nucl. Med., 1993, 34, 222–227 CAS.
- J. Veggeland, G. K. Madsen and S. Hemsted, Radiopharmaceutical Composition, US9549999B, 2017 Search PubMed.
- H. Gali, T. J. Hoffman, G. L. Sieckman, N. K. Owen, K. V. Katti and W. A. Volkert, Bioconjugate Chem., 2001, 12, 354–363 CrossRef CAS PubMed.
- S. R. Karra, R. Schibli, H. Gali, K. V. Katti, T. J. Hoffman, C. Higginbotham, G. L. Sieckman and W. A. Volkert, Bioconjugate Chem., 1999, 10, 254–260 CrossRef CAS.
- H. Gali, S. R. Karra, V. S. Reddy and K. V. Katti, Angew. Chem., Int. Ed., 1999, 38, 2020–2023 CrossRef CAS.
- J. S. Lewis, S. L. Heath, A. K. Powell, J. Zweit and P. J. Blower, J. Chem. Soc., Dalton Trans., 1997, 855–861 RSC.
- J. S. Lewis, J. Zweit, J. L. J. Dearling, B. C. Rooney and P. J. Blower, Chem. Commun., 1996, 1093–1094 RSC.
- C. Imberti, S. Y. A. Terry, C. Cullinane, F. Clarke, G. H. Cornish, N. K. Ramakrishnan, P. Roselt, A. P. Cope, R. J. Hicks, P. J. Blower and M. T. Ma, Bioconjugate Chem., 2017, 28, 481–495 CrossRef CAS.
- D. Fenske and H. J. Becher, Chem. Ber., 1974, 107, 117–122 CrossRef CAS.
- M. Fei, S. K. Sur and D. R. Tyler, Organometallics, 1991, 10, 419–423 CrossRef CAS.
- R. G. Cavell, R. W. Hilts, H. Luo and R. McDonald, Inorg. Chem., 1999, 38, 897–905 CrossRef CAS.
- H. Luo, I. Setyawati, S. J. Rettig and C. Orvig, Inorg. Chem., 1995, 34, 2287–2299 CrossRef CAS.
- J. D. Young, V. Abbate, C. Imberti, L. K. Meszaros, M. T. Ma, S. Y. A. Terry, R. C. Hider, G. E. Mullen and P. J. Blower, J. Nucl. Med., 2017, 58, 1270–1277 CrossRef CAS PubMed.
- P. A. Monach, D. Mathis and C. Benoist, Curr. Protoc. Immunol., 2008, 81, 15.22.1–15.22.12 Search PubMed.
- A. Khoshnevisan, K. Chuamsaamarkkee, M. Boudjemeline, A. Jackson, G. E. Smith, A. D. Gee, G. O. Fruhwirth and P. J. Blower, J. Nucl. Med., 2017, 58, 156–161 CrossRef CAS PubMed.
- K. K. Kothari, H. Gali, K. R. Prabhu, N. Pillarsetty, N. K. Owen, K. V. Katti, T. J. Hoffman and W. A. Volkert, Nucl. Med. Biol., 2002, 29, 83–89 CrossRef CAS.
- C. Bolzati, A. Caporale, S. Agostini, D. Carta, M. Cavazza-Ceccato, F. Refosco, F. Tisato, E. Schievano and G. Bandoli, Nucl. Med. Biol., 2007, 34, 511–522 CrossRef CAS PubMed.
- C. Bolzati, E. Malagò, A. Boschi, A. Cagnolini, M. Porchia and G. Bandoli, New J. Chem., 1999, 23, 807–809 RSC.
- R. Kannan, N. Pillarsetty, H. Gali, T. J. Hoffman, C. L. Barnes, S. S. Jurisson, C. J. Smith and W. A. Volkert, Inorg. Chem., 2011, 50, 6210–6219 CrossRef CAS PubMed.
- K. K. Kothari, K. Raghuraman, N. K. Pillarsetty, T. J. Hoffman, N. K. Owen, K. V. Katti and W. A. Volkert, Appl. Radiat. Isot., 2003, 58, 543–549 CrossRef CAS PubMed.
- M. I. Tsionou, C. E. Knapp, C. A. Foley, C. R. Munteanu, A. Cakebread, C. Imberti, T. R. Eykyn, J. D. Young, B. M. Paterson, P. J. Blower and M. T. Ma, RSC Adv., 2017, 7, 49586–49599 RSC.
- M. Eder, O. Neels, M. Müller, U. Bauder-Wüst, Y. Remde, M. Schäfer, U. Hennrich, M. Eisenhut, A. Afshar-Oromieh, U. Haberkorn and K. Kopka, Pharmaceuticals, 2014, 7, 779–796 CrossRef CAS.
- U. Hennrich and M. Eder, Pharmaceuticals, 2021, 14, 713 CrossRef CAS.
- N. A. Zia, C. Cullinane, J. K. Van Zuylekom, K. Waldeck, L. E. McInnes, G. Buncic, M. B. Haskali, P. D. Roselt, R. J. Hicks and P. S. Donnelly, Angew. Chem., Int. Ed., 2019, 58, 14991–14994 CrossRef CAS PubMed.
- J. Notni, K. Pohle and H.-J. Wester, Nucl. Med. Biol., 2013, 40, 33–41 CrossRef CAS PubMed.
- A. Frei, E. Fischer, B. C. Childs, J. P. Holland and R. Alberto, Dalton Trans., 2019, 48, 14600–14605 RSC.
- Z. Li, W. Cai, Q. Cao, K. Chen, Z. Wu, L. He and X. Chen, J. Nucl. Med., 2007, 48, 1162–1171 CrossRef CAS PubMed.
- X. Zhang, Z. Xiong, Y. Wu, W. Cai, J. R. Tseng, S. S. Gambhir and X. Chen, J. Nucl. Med., 2006, 47, 113–121 CAS.
- J. K. Bordoloi, D. Berry, I. U. Khan, K. Sunassee, R. T. M. De Rosales, C. Shanahan and P. J. Blower, Dalton Trans., 2015, 44, 4963–4975 RSC.
- A. J. North, J. A. Karas, M. T. Ma, P. J. Blower, U. Ackermann, J. M. White and P. S. Donnelly, Inorg. Chem., 2017, 56, 9725–9741 CrossRef CAS PubMed.
- Y. Mizuno, T. Uehara, H. Hanaoka, Y. Endo, C. W. Jen and Y. Arano, J. Med. Chem., 2016, 59, 3331–3339 CrossRef CAS PubMed.
- A. Davison, C. Orvig, H. S. Trop, M. Sohn, B. V. DePamphilis and A. G. Jones, Inorg. Chem., 2002, 19, 1988–1992 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: ESI-MS data, NMR, data HPLC chromatograms, SPECT/CT and biodistribution data. See DOI: 10.1039/d1dt03177e |
| This journal is © The Royal Society of Chemistry 2021 |