
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReduced quenching effect of pyridine ligands in highly luminescent Ln(III) complexes: the role of tertiary amide linkers†
Daniel
Kocsi
,
Daniel
Kovacs
,
Jordann A. L.
Wells
 and
K. Eszter
Borbas
*
and
K. Eszter
Borbas
*
Department of Chemistry, Ångström Laboratory, Box 523, Uppsala University, 75120, Uppsala, Sweden. E-mail: eszter.borbas@kemi.uu.se
First published on 26th October 2021
Abstract
Luminescent Eu(III) and Tb(III) complexes were synthesised from octadentate ligands carrying various carbostyril sensitizing antennae and two bidentate picolinate donors. Antennae were connected to the metal binding site via tertiary amide linkers. Antennae and donors were assembled on a 1,4,7-triazacyclononane (tacn) platform. Solution- and solid-state structures were comparable to those of previously reported complexes with tacn architectures, with nine-coordinate distorted tricapped trigonal prismatic Ln(III) centres, and distinct from those based on 1,4,7,10-tetraazacyclododecane (cyclen) macrocycles. In contrast, the photophysical properties of these tertiary amide tacn-based complexes were more comparable to those of previously reported systems with cyclen ligands, showing efficient Eu(III) and Tb(III) luminescence. This represents an improvement over secondary amide-linked analogues, and is due to a greatly increased sensitization efficiency in the tertiary amide-linked complexes. Tertiary amide-linked Eu(III) and Tb(III) emitters were more photostable than their secondary amide-linked analogues due to the suppression of photoinduced electron transfer and back energy transfer.
Introduction
The luminescent properties of trivalent lanthanide ions (Ln) have long attracted attention due to their applications in lasers and lighting, encryption, barcoding, and as biological probes. Unlike organic fluorophores that often suffer from short emission lifetimes, broad emission bands, and small Stokes shifts, Ln luminescence is due to f–f transitions, and is therefore atomic-like, unique to the metal, and long-lived.1,2 The inefficient absorption of Ln(III) centres can be overcome by allowing a light-harvesting antenna to absorb the excitation energy and transfer it to the metal ion. In addition to a high apparent absorption coefficient, such a strategy also yields a large apparent Stokes shift. A dazzling variety of antennae have been reported that encompass amino acids,3,4 both small and complex heterocycles5–21 transition metal complexes,22–29 and even other Lns.3,30A crucial component of an emissive Ln coordination compound is the ligand. Stable complexes require the saturation of most of the 8–10 coordination sites of the Ln(III).31 Hepta-, octa-, and nonadentate ligands can be constructed from tetra- and triazamacrocyclic (cyclen and tacn, respectively, Fig. 1) building blocks upon the addition of mono- and bidentate donors, e.g. carboxylates, carbamides, or picolinates.32 While the primary role of the ligand is to create a well-defined, stable emitter, it has additional functions. Ln(III) luminescence is sensitive to nearby X–H oscillators.3,33 While X can be O, N or, in the case of near infrared emitters, even C, the largest quenching effect is due to inner-sphere O–H-containing solvent molecules, such as water or MeOH.34,35 A high-denticity ligand can displace most inner-sphere solvent molecules, and thus improve Ln(III) luminescence.
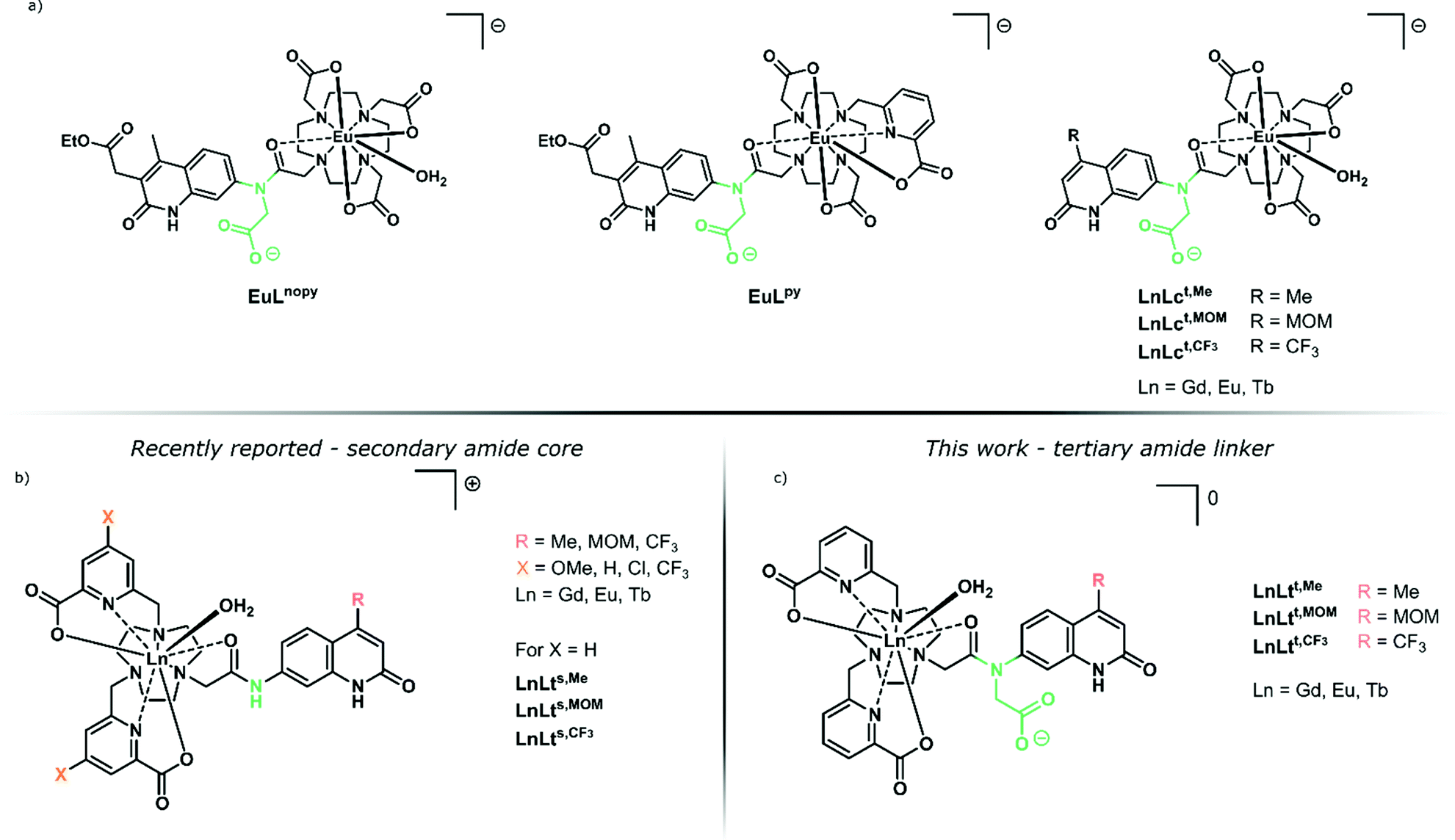 | ||
| Fig. 1 (a) Previously reported tertiary amide-linked complexes with carbostyril antennae,39 including cyclen-based benchmarks LnLct,R.42 (b) Secondary amide-linked carbostyril-sensitised LnLts,R consisting of a tacn ligand framework equipped with substituted picolinic acid donors.38 (c) LnLtt,R complexes studied here. | ||
A further role of the ligand is to impose a coordination geometry. The shielded 4f-orbitals do not participate in directional bonding, and Lns bind preferentially hard donor atoms (such as O, F) through coulombic interactions. The ligand can thus determine the symmetry around the Ln(III), which in turn will govern the radiative lifetime (τrad) of the ion. The overall luminescence quantum yield (ΦLn) is the product of the efficiency with which the Ln(III) excited state is populated (ηsens) via absorption, intersystem crossing, energy transfer, etc., and the intrinsic quantum yield (ΦLnLn) of the ion (eqn (1)). The latter is equal to the proportion of observed (τobs) and radiative lifetimes.36,37 All else being equal, shorter τrad yields higher ΦLn.
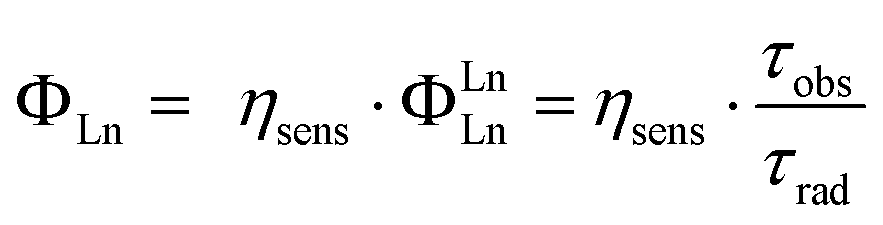 | (1) |
We have previously prepared both tacn and cyclen-based complexes with a variety of carbostyril and coumarin antennae (Fig. 1). Notably, the Eu(III) complexes of the octadentate tacn-based ligands (LnLts,R) had much shorter τrad (∼3 ms)38 than those of their similarly octadentate cyclen-based analogues (e.g.LnLcs,R, τrad ∼ 5 ms).39–42
Ligands can control quenching processes other than X–H quenching. Photoinduced electron transfer (PeT) whereby the excited antenna reduces Ln(III) to Ln(II) is common in emitters with reducible Ln(III), such as Eu.43,44 The re-oxidation of Eu(II) usually yields a quenched complex, although exceptions are known.45,46 The detrimental effects of PeT in some systems is comparable to that of X–H oscillators.40 Ligands that stabilise the Eu(III) oxidation state can increase the luminescence quantum yield.40 The ligand may also directly quench the excited antenna.39 This is the case in the picolinate-equipped LnLts,R (Fig. 1), where the electron-poor pyridines are reduced alongside Eu(III) by the photoexcited antenna. PeT to the pyridines was possible even in the non redox-active Tb(III) and Gd(III), and was more prominent for electron-poor pyridines.38
Here, we report a series of tacn-based ligands and their Ln(III) complexes equipped with the same picolinate ligands that were previously shown to be detrimental to Ln(III) luminescence (Fig. 1). We show that changing the linker that connects the carbostyril antenna to the tacn fragment from a secondary (LnLts,R) to a tertiary amide (LnLtt,R) can result in the recovery of Ln(III) luminescence, and afford emitters that are comparable to what was seen in cyclen-based systems. The complexes were structurally characterised by paramagnetic 1H NMR spectroscopy and X-ray crystallography to enable comparison of the Ln(III) coordination environments in the secondary and tertiary amide-linked complexes. Steady-state and time-resolved luminescence spectroscopy was used to understand the sensitization and quenching pathways in these emitters, and revealed a significantly improved sensitization in the new set of picolinate-containing species compared to the previously reported ones. These results indicate that even efficient quenching processes can be interrupted by judiciously chosen structural changes.
Results and discussion
Synthetic procedures
The ligands Ltt,R were synthesised from 4-methyl-, 4-methoxymethyl- or 4-trifluoromethyl-substituted 7-aminocarbostyrils 1a, 1b and 1c, via the tertiary chloroacetylamides 2a, 2b, and 2c, respectively (Scheme 1). The latter were prepared similarly to their tert-butyl ester analogues.38 Di-substituted triazanonane 3 was monoalkylated with 2a–c to yield protected ligands 4a–c, which were deprotected under basic conditions. Heating Ltt,R with LnCl3 in a warm EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O mixture yielded LnLtt,R after column chromatography on silica gel using iPrOH:H2O as the eluent in quantitative yield as white solids. Detailed experimental procedures and full chemical characterization for all new compounds are given in the ESI.†
:H2O mixture yielded LnLtt,R after column chromatography on silica gel using iPrOH:H2O as the eluent in quantitative yield as white solids. Detailed experimental procedures and full chemical characterization for all new compounds are given in the ESI.†
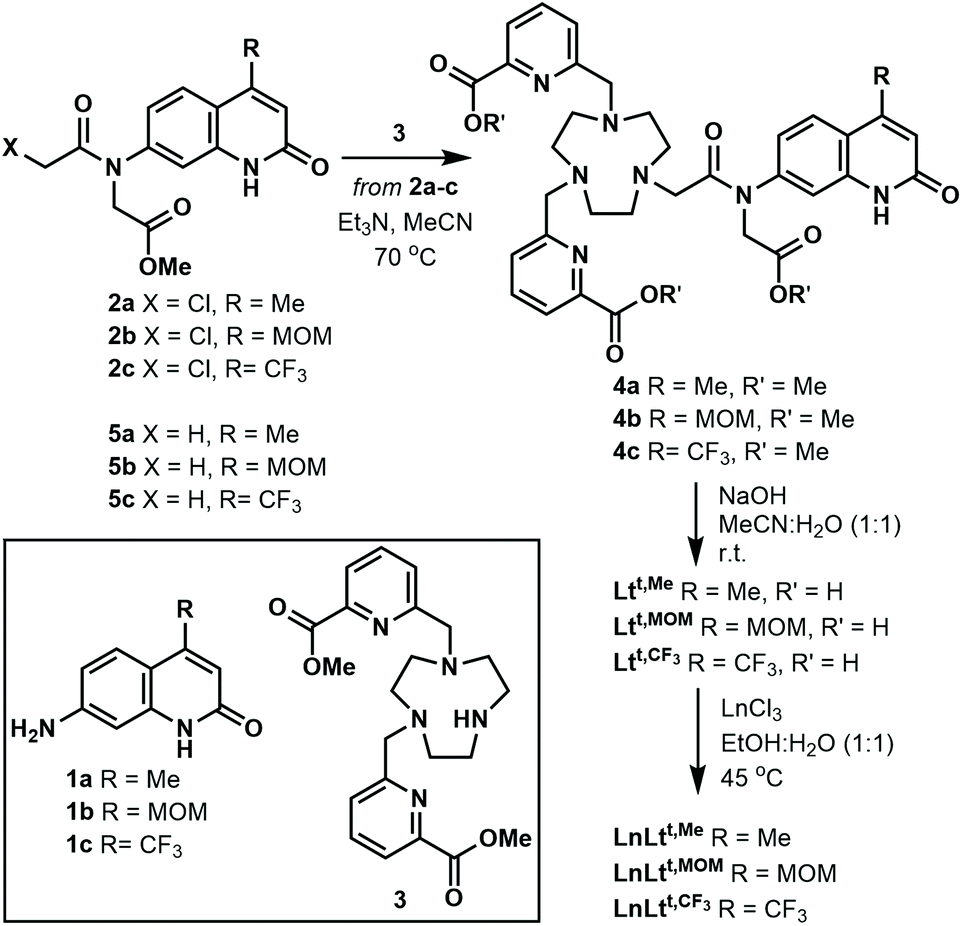 | ||
| Scheme 1 Preparation of LnLtt,R and model compounds 5a–c. | ||
X-ray crystallography
Single crystals suitable for X-ray diffraction analysis were obtained by vapor diffusion of glyme into concentrated aqueous solutions of GdLtt,Me and TbLtt,Me. The presence of the carboxylate group on the amide functionality of the ligand results in a coordination motif not seen for related complexes containing secondary amides.38 The carboxylate group acts as a bridging ligand between LnLt molecules, forming 1D-polymeric chains. In addition, a secondary Ln centre is coordinated to a pyridine carboxylate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, acting as a bridge between 1D-polymeric chains resulting in a 2D-polymeric network (Fig. 2).
O bond, acting as a bridge between 1D-polymeric chains resulting in a 2D-polymeric network (Fig. 2).
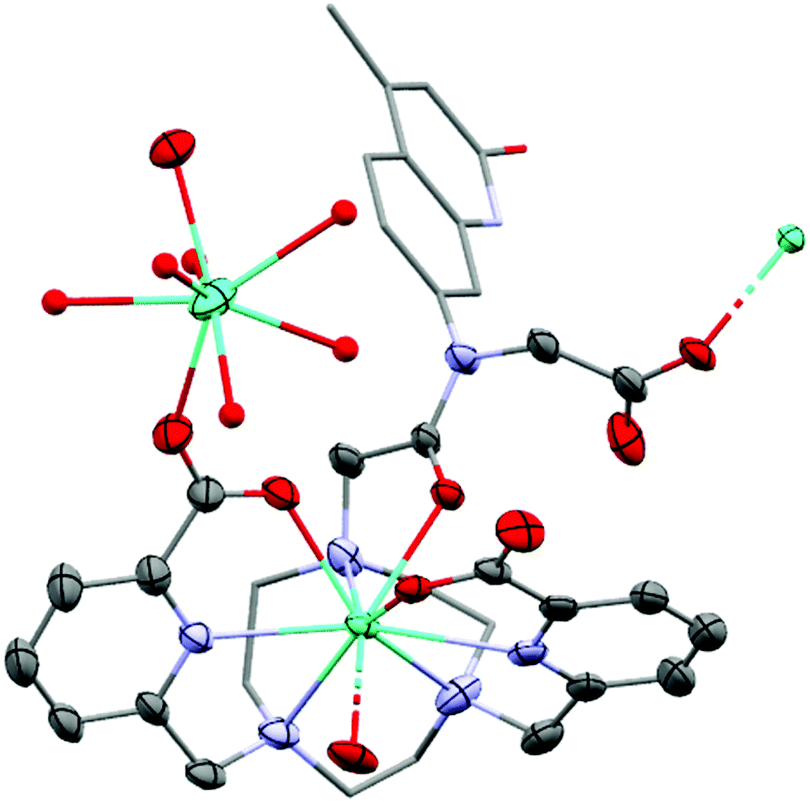 | ||
| Fig. 2 Solid-state structures of TbLtt,Me. H atoms, non-coordinating Cl− counterions and water molecules omitted, triazacyclonane C atoms and antenna displayed as capped sticks and Ln2 co-ligands displayed as ball and stick for clarity. Ellipsoids displayed at 35% probability. Only major component of disordered sites displayed. | ||
The Ln centre within the macrocylic cavity exhibits a nine-coordinate distorted tricapped trigonal prismatic geometry, comparable to that observed in related complexes. The trigonal prism is represented by three tacn N-donor atoms (N3PL), and two pyridine N- and the antenna amide O-atoms (NNOPL). The pyridine carboxylate groups and the bridging carboxylate arm of the tertiary amide serve as the capping ligands of the trigonal prism. The angle between the two planes is 115.1(3)° in Gd and 116.8(6)° in Tb, which compares well to those of related complexes. The deviation of the angle from 180° can be ascribed to the highly distorted geometry of the metal centre. The distance between the Ln centres and the NNOPL are comparable to those of the corresponding secondary amide complexes (0.312(3) and 0.328(6) Å for Gd and Tb, respectively, versus average 0.312 Å), as are the distances to the N3PL (Gd: 2.044(3) Å; Tb: 2.024(8) Å; versus 2.046 Å average). These metrics suggest that the tertiary amide group and the carboxylate bridging ligand have no significant impact on the direct coordination sphere of carbostyril-substituted tacn systems. The complexes are racemic in the solid-state with both Δ and Λ isomers present in the unit cell. The Ln–O (Gd: 2.388(4) Å; Tb: 2.376(6) Å), tacn Ln–N (Gd: 2.646(4) Å; Tb: 2.64(1) Å) and pyridine Ln–N (Gd: 2.548(6) Å; Tb: 2.538(8) Å) bond distances compare well to those of related complexes.
The secondary Ln centre is eight-coordinate and exhibits a square antiprismatic arrangement. Two picolinate carbonyl groups occupy flanking positions, while the remaining six sites are occupied by water ligands, and the charge balancing chloride counter-ions are non-coordinated and reside in the crystal lattice. The metal centre at this position is partially occupied (75%), which results in considerably disordered lanthanide bound ligands and imprecise bond lengths, which are not discussed here (see ESI† for further information).
NMR studies
Paramagnetic 1H and 19F NMR spectroscopic analysis of EuLts and EuLtt was performed in D2O. Complexes with identical metal-binding sites had similar spectra (Fig. 3). At r.t. the signals were broad and indicative of the presence of several species that made the comparison of the data difficult. However, good quality spectra were obtained in all cases upon heating the samples at 80 °C. At high temperature the 1H NMR spectra of the EuLts species contained ∼15 signals, consistent with the presence of a single species or of several species rapidly interconverting on the NMR timescale (Fig. S3–18†). The spectra of EuLtt were much sharper than those of the corresponding EuLts even at room temperature, and further improved upon heating. The 80 °C-spectra of EuLtt contained two sets of signals, which suggests that there are two stable conformers in the solution even at high temperatures. These conclusions were supported by the 19F NMR analysis of EuLts,CF3 and EuLtt,CF3. At r.t. a large number of signals were observed for EuLts,CF3, and a broadened peak consisting of several peaks for EuLtt,CF3. In line with the 1H NMR data, at 80 °C the signals collapsed into a single peak (−61.9 ppm), while that of EuLtt,CF3 into one major and two minor ones (around −62.7 ppm) (Fig. S11–18†). | ||
| Fig. 3 Variable temperature 1H NMR spectra recorded in D2O. (a) EuLts,CF3 at 353 K. (b) EuLts,CF3 at 298 K. (c) EuLtt,CF3 at 353 K. (d) EuLtt,CF3 at 298 K. | ||
Photophysical studies
The photophysical properties of LnLtt,R were measured under the same conditions as those of LnLts,R and LnLct,R,38,42 in PIPES-buffered pH = 6.5 aqueous solution at r. t. The LnLtt,R absorption spectra contained peaks in the 250–310 nm region that were absent from the spectra of LnLct,R, but were present in LnLts,R (Fig. 4, S22–27†). These peaks were assigned to pyridine π–π* transitions. The longest-wavelength absorption was determined by the antenna (Table 1), and the position of the maxima increased in the order LnLtt,Me (λabs = 325 nm) < LnLtt,MOM (λabs = 328 nm) < LnLtt,CF3 (λabs = 338 nm). Upon antenna excitation weak antenna fluorescence was observed at λem = 366–391 nm; longer emission wavelengths were seen for the more red-absorbing antennae. While λabs for LnLtt,R were at wavelengths that were 3–4 nm shorter than seen for the corresponding LnLts,R, this small difference all but disappeared in the emission spectra.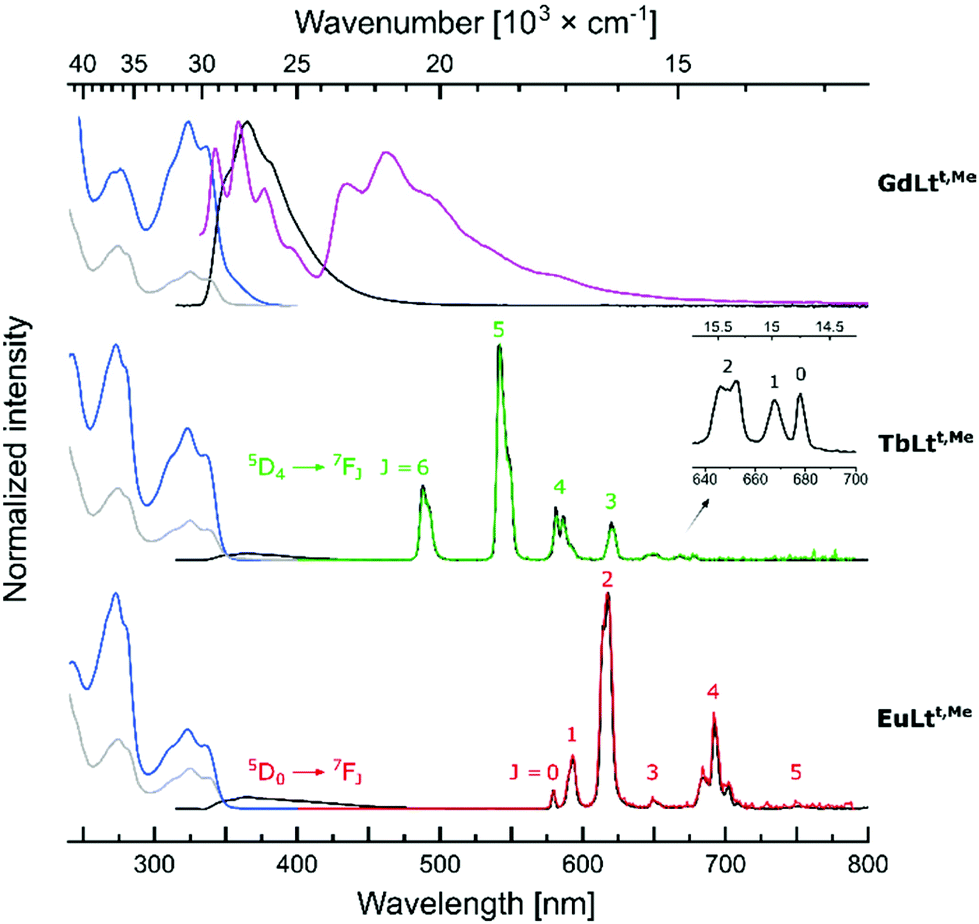 | ||
| Fig. 4 Normalised absorption (left, grey). Excitation [left, blue, λem = 405 nm (Gd), λem = 542 nm (Tb), λem = 618 nm (Eu), 298 K], steady-state emission (middle, black, λex = 325 nm, 298 K), steady-state emission [middle, purple, λex = 325 nm (Gd), 77 K] and time-resolved emission (right, green (Tb), red (Eu), λex = 325 nm) spectra of LnLtt,Me complexes. | ||
| Compound | λ max [nm] | λ em [nm] | E 00(S1)c [cm−1] | E 00(T1)c [cm−1] |
|---|---|---|---|---|
| a In aqueous PIPES buffer (10 mM), pH 6.5, at 10 μM complex concentrations. b λ ex = 329 nm (GdLts,Me), 325 nm (GdLtt,Me), 335 nm (GdLts,MOM), 328 nm (GdLtt,MOM), 331 nm (GdLts,CF3), 330 nm (GdLtt,CF3). c Calculated from the 0–0 transitions of the Gd-complexes recorded at 77 K. | ||||
| GdLts,Me | 329 | 366 | 28900 |
22900 |
| GdLtt,Me | 325 | 365 | 29200 |
23000 |
| GdLts,MOM | 331 | 376 | 28500 |
22500 |
| GdLtt,MOM | 328 | 375 | 28900 |
22800 |
| GdLts,CF3 | 342 | 391 | 27400 |
21700 |
| GdLtt,CF3 | 338 | 391 | 27500 |
22000 |
The antenna first triplet excited states (T1) were determined from the 0–0 transitions of the low-temperature fluorescence spectra (Table 1). The differences between the secondary and tertiary amide-linked antennae are small. The former are lower in energy, but only by 100–300 cm−1. Oxygen sensitivity due to back energy transfer (BET) was previously seen in LnLts,CF3.42 To avoid BET the antenna T1 should be at least 2000 cm−1 above the Tb(III) excited state (20400 cm−1).32,47 The 300 cm−1 increase in T1 energy to 22000 cm−1 in LnLtt,CF3 is probably not sufficient to prevent BET.
Emission spectra were collected with λex > 325 nm to avoid excitation of the pyridines. Antenna excitation yielded Tb(III) and Eu(III) emission from all the complexes. Spectral shapes and the ratios of peak intensities were similar in LnLts,R and LnLtt,R, but differed from those observed in cyclen-based LnLct,R (Fig. S29–40†). Thus, LnLts,R and LnLtt,R have similar coordination environments, which differ from that created by the DO3A-type ligands. As in the cyclen-based systems,42 the influence of the linkers on the coordination geometry is negligible. The Tb(III) and Eu(III) spectra consisted of 6 and 5 major signals corresponding to the 5D4 → 7FJ (J = 6–0) the 5D0 → 7FJ (J = 0–5) transitions, located at 488, 543, 582, 620, 652, 668, and 678 nm, and at 579, 593, 614, 649, 693, and 751 nm, respectively (Fig. 4).
The antenna and Ln(III)-based luminescence quantum yields (ΦL and ΦLn, respectively) were determined by the optically dilute method using quinine sulfate (QS, Φ = 0.59)48 as the external standard (Table 2). Gratifyingly, ΦLn obtained for the current series of Eu(III) and Tb(III) emitters (LnLtt,R) were comparable to the high values previously obtained for the tert-amide-linked DO3A-based species (LnLct,R), a major improvement over the weak emission of LnLts,R. The highest ΦTb and ΦEu were measured for TbLtt,MOM (42%) and EuLtt,CF3 (13%), respectively. As before, ΦL were low, below 7% in all cases. Intriguingly, there was no discernable pattern as to the order of ΦL in the different ligand frameworks. In the non-photoactive and non-redox-active Gd(III) species, ΦL decreases as follows: Lct,Me > Ltt,Me > Lts,Me, Lts,MOM > Lct,MOM ≈ Ltt,MOM, and Lts,CF3 > Ltt,CF3 ≈ Lct,CF3.
| Complex | Φ L [%]a | Φ Ln [%]a |
|---|---|---|
| a Relative to QS (Φ = 0.59) in H2SO4 (0.05 M).48 b Mean ± standard deviation for two independent measurements. c Mean ± standard deviation for three independent measurements. | ||
|
GdLts,Meb |
4.40 ± 0.10 (63%) | — |
|
GdLtt,Meb |
5.47 ± 0.07 (79%) | — |
| GdLct,Me | 6.8 (100%) | — |
GdLts,MOM
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif)
|
6.42 ± 0.28 (131%) | — |
|
GdLtt,MOM
|
4.94 ± 0.06 (98%) | — |
| GdLct,MOM | 5.1 (100%) | — |
|
GdLts,CF3
|
4.53 ± 0.17 (147%) | — |
|
GdLtt,CF3
|
3.62 ± 0.02 (113%) | — |
| GdLct,CF3 | 3.2 (100%) | — |
|
TbLts,Me
|
3.50 ± 0.10 (58%) | 27.05 ± 1.05 (60%) |
|
TbLtt,Me
|
4.59 ± 0.10 (76%) | 40.4 ± 0.40 (93%) |
| TbLct,Me | 5.9 (100%) | 43.4 (100%) |
|
TbLts,MOM
|
4.92 ± 0.01 (109%) | 28.05 ± 0.95 (64%) |
|
TbLtt,MOM
|
4.28 ± 0.03 (96%) | 41.5 ± 0.50 (93%) |
| TbLct,MOM | 4.5 (100%) | 45.1 (100%) |
|
TbLts,CF3
|
4.16 ± 0.24 (142%) | 3.24 ± 0.14 (19%) |
|
TbLtt,CF3
|
3.33 ± 0.28 (116%) | 19.0 ± 1.05 (113%) |
| TbLct,MF3 | 3.1 (100%) | 15.9 (100%) |
|
EuLts,Me
|
0.42 ± 0.03 (29%) | 0.83 ± 0.02 (14%) |
|
EuLtt,Me
|
0.96 ± 0.24 (87%) | 3.45 ± 1.10 (83%) |
| EuLct,Me | 1.5 (100%) | 6.0 (100%) |
|
EuLts,MOM
|
0.16 ± 0.01 (6%) | 2.44 ± 0.07 (28%) |
|
EuLtt,MOM
|
1.32 ± 0.06 (52%) | 5.22 ± 0.15 (61%) |
| EuLct,MOM | 2.5 (100%) | 8.9 (100%) |
|
EuLts,CF3
|
0.61 ± 0.02 (23%) | 7.95 ± 0.42 (68%) |
|
EuLtt,CF3
|
2.81 ± 0.08 (100%) | 13.0 ± 0.10 (108%) |
| EuLct,CF3 | 2.7 (100%) | 11.6 (100%) |
PeT from the antenna to the pyridines and to Eu(III) was calculated to be thermodynamically feasible (eqn (2)). Cyclic voltammetry yielded Eox, the electron donor oxidation potential, as +1.91, +1.94, and +2.20 V (vs. NHE) for the antenna models 5a–c (Scheme 1, Fig. S19–21†), respectively. These models were prepared to enable cyclic voltammetric analysis of the antennae without interference from the other redox-active components of the complexes. Ered is the electron acceptor reduction potential. Pyridine Ered (−1.29 V vs. NHE) has been reported;38Ered of Eu(III) was approximated with values found for a +1 charged cyclen-based complex (−0.80 V vs. NHE for a MOM-substituted complex).40 This was necessary as the presence of the pyridines, which are reduced at similar potentials make Eu(III) Ered determination unreliable in this ligand framework. ES is the antenna excited state energy, determined from the first vibronic band of the 77 K spectra as 3.60, 3.57, and 3.39 eV for GdLtt,Me, GdLtt,MOM, and GdLtt,CF3 respectively. The last term is the coulombic stabilization of the charge-separated system, usually taken as ∼0.15 eV.49
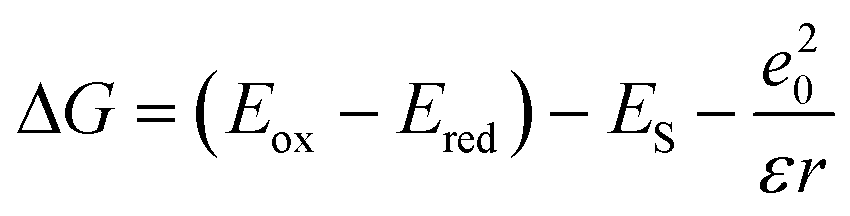 | (2) |
ΔG for PeT from the CF3-substituted antenna to the pyridine was −0.54 eV. Values for the more electron-rich antennae were more negative, −0.98 eV and −1.04 eV for the MOM- and Me-substituted ones, respectively. Eu(III) reduction was even more favorable, with ΔG values of −0.95, −0.87, and −0.45 eV, for Me-, MOM- and CF3-substituted complexes, respectively. In DO3A-complexes, sec-amide-linked antennae are more fluorescent than the tert-amide-linked ones.42 The overall ΦL order is the result of the combination of these effects. In the Tb(III) and Eu(III) complexes the situation is further complicated by the photo- and, in the case of Eu(III), redox-activity of the metals.
The number of metal-coordinated water molecules (q) were determined from τobs of Eu(III) and Tb(III) (Table 3).3,35 The luminescence decays were monoexponential for these complexes, with τobs = ∼0.52 ms and ∼1.34 ms for EuL and TbL, respectively. In all cases q was ∼1, which together with the octadentate ligand gives the expected nine-coordinate complexes. BET in TbLtt,CF3 did not allow for the determination of q, however, it is most likely 1 by analogy with the other complexes.
| Complex | τ H2O [ms] | τ D2O [ms] | q |
|---|---|---|---|
| a Recorded in PIPES-buffered H2O or D2O solutions at pH = 6.5 at room temperature. b Calculated using q = (5 ms) (1/τH2O − 1/τD2O − 0.06 ms−1) for Tb, and q = (1.2 ms) (1/τH2O − 1/τD2O − 0.25 ms−1 − m 0.075 ms−1) for Eu; m = number of nearby amide N–H oscillators.3,35 c Real q value could not be determined. | |||
| TbLtt,Me | 1.37 | 2.13 | 0.99 |
| EuLtt,Me | 0.51 | 1.43 | 1.18 |
| TbLtt,MOM | 1.30 | 2.01 | 1.06 |
| EuLtt,MOM | 0.52 | 1.43 | 1.15 |
| TbLtt,CF3 | 0.62 | 1.01 | 2.71c |
| EuLtt,CF3 | 0.52 | 1.43 | 1.15 |
The similar ΦL and ΦLn values of pyridine-equipped LnLtt,R and DO3A-based LnLct,R suggested that either PeT to the pyridines was less efficient in LnLtt,R than in LnLts,R, or PET was successfully outcompeted by energy transfer to the Ln(III). The efficiency with which the antenna excited state populates the Ln(III) excited state (ηsens) is readily calculated for Eu(III) emitters using eqn (1) and (3) (Table 4).36,37 Antenna excited state quenching affects ηsens. If complexes with identical antennae and similar geometries have different ηsens, this difference may be due to differences in PeT. ΦLnLn in eqn (1) can be calculated36 from τrad of Eu(III), which is obtained from the luminescence spectrum using eqn (3). Itot is the integrated full spectrum (530–800 nm), IMD is the integrated 5D0 → 7F1 transition (582–603 nm), AMD,0 is the spontaneous emission probability (14.65 s−1), n is the refractive index of the aqueous medium (approximated with water), and τobs equals τH2O.
 | (3) |
| Complex | τ rad [ms] | Φ Eu Eu [%] | η sens [%] |
|---|---|---|---|
| a Calculated using eqn (3).36 b Calculated using eqn (1) and (3).36 | |||
| EuLts,Me | 2.87 | 17.4 | 5 |
| EuLtt,Me | 2.94 | 17.7 | 28 |
| EuLct,Me | 5.41 | 12.0 | 50 |
| EuLts,MOM | 2.86 | 17.5 | 14 |
| EuLtt,MOM | 2.96 | 17.9 | 30 |
| EuLct,MOM | 5.40 | 12.2 | 73 |
| EuLts,CF3 | 2.87 | 17.8 | 46 |
| EuLtt,CF3 | 2.99 | 17.4 | 75 |
| EuLct,CF3 | 5.40 | 12.2 | 95 |
EuLtt,R and EuLts,R have similar τrad (∼2.9 ms). This is in line with their having similar Ln(III) coordination environments, as suggested by their superimposable luminescence spectra (Fig. S53–55†). The slightly longer τobs of EuLtt,R than of EuLts,R is presumably due to the removal of the N–H group from the amide linker. Overall, the effect of the tertiary amides on ΦLnLn is small compared to EuLts,R, and all complexes have ΦLnLn ∼ 17.5%. Compared with the ΦLnLn ∼ 12% obtained for EuLct,R, this is a significant improvement. The shortening of τrad to increase ΦLnLn is a rarely used strategy for improving ΦLn.50
Although EuLtt,R have higher ΦLnLn than EuLct,R their ΦLn values were similar or lower, due to the lower ηsens. EuLtt,R shows marked improvement in ηsens compared to secondary amide-linked EuLts,R (e.g. 28% and 5% in EuLtt,Me and EuLts,Me, respectively), just not enough to reach the values seen in EuLct,R (50% in EuLct,Me). The better sensitization in EuLtt,R than in EuLts,R may be due to less efficient PeT to the pyridines. This is suggested by the fact that ΦL in GdLtt,Me is somewhat higher than in GdLts,Me even though in DO3A-based complexes secondary amide linked antennae have higher ΦL than their tertiary amide-linked analogues.42 However, the low ΦL values do not allow for a reliable comparison. A second alternative explanation for the observed results may be more efficient energy transfer due to a different antenna orientation. A third possibility is more efficient population of the Ln(III) feeding level, likely the antenna T1, due to more efficient intersystem crossing in LnLtt,R than in LnLts,R. The additional improvement seen in LnLct,R compared to LnLtt,R is likely due to the absence of PeT to pyridines. As above, antenna orientation could also play a role. Both LnLct,R and Lntt,R have tert-amide-linked antennae, and the similar values of ΦL are consistent with comparable levels of antenna S1 quenching. Thus the higher ηsens in cyclen-based LnLct,R indicates more efficient energy transfer than in tacn-based Lntt,R.
The photostabilities of the complexes reported herein (LnLtt,R) were investigated and compared to those of LnLts,R and LnLct,R (Fig. 5). The Eu(III) complexes proved quite robust, and retained at least 90% of their luminescence after 2.5 h of irradiation. Small differences, however, could be noted. Complexes equipped with a CF3-substituted antenna were the most resistant to photodegradation, while the least stable were the ones carrying the most electron-rich Me-substituted antenna. Cyclen-based EuLct,R were in all cases more stable than their tacn-based analogues EuLts,R and EuLtt,R. Tert-amide-linked EuLtt,R were somewhat more stable then sec-amide-linked EuLts,R. These trends are consistent with PeT opening up a degradation pathway for the complex. This pathway is more prominent for picolinate-carrying EuLts,R and EuLtt,R, which contain both Eu(III) and pyridines as potential electron acceptors.
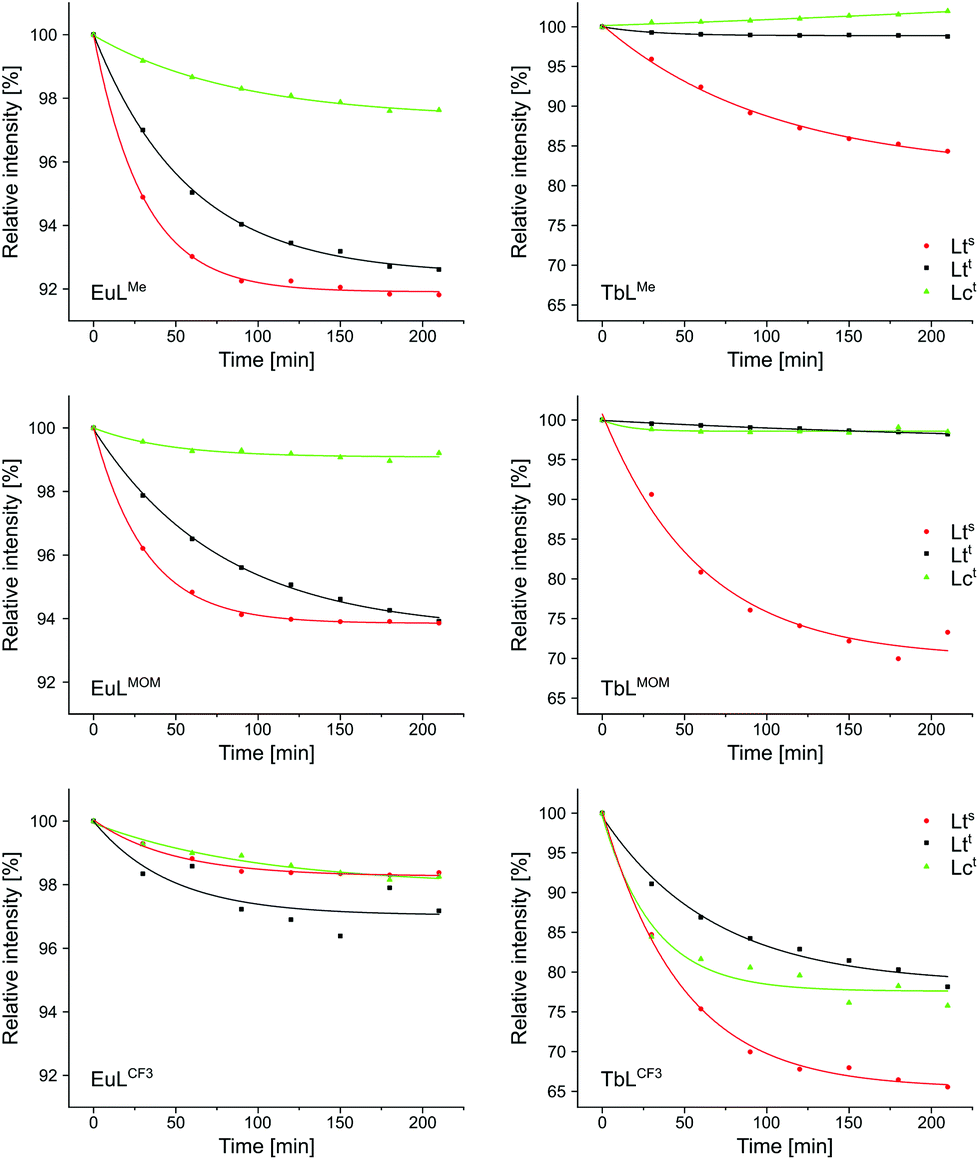 | ||
| Fig. 5 Photostability of Tb and Eu complexes (10 μM) in aqueous PIPES buffer (10 mM), pH = 6.5, using 395 nm filter for Tb and 500 nm filter for Eu complexes, respectively. λex(LnLct,Me) = 324 nm, λex(LnLtt,Me) = 325 nm, λex(LnLct,MOM/CF3) = 327 nm, λex(LnLts,Me/LnLtt,MOM) = 328 nm, λex(LnLts,MOM/(LnLts,CF3/LnLtt,CF3) = 330 nm. Values normalised to integrated emission intensity at t0. | ||
In the case of the Tb(III) complexes fastest degradation was seen for those with the lowest antenna T1. All three CF3-substituted complexes rapidly lost Tb(III) luminescence efficiency. This loss was fastest for TbLts,CF3, with a sec-amide-linked antenna. For the other two antennae attaching them via a tert-amide, and the concomitant increase in T1 was sufficient to protect the integrity of the emitters. These trends indicate that photolability is due to BET populating the antenna T1, followed by T1 reacting with atmospheric oxygen.
Conclusions
We investigated the effect of replacing a secondary amide linker with a methylcarboxylate-substituted tertiary amide one on the photophysical properties of tacn-based luminescent Ln(III) complexes. In solution the amide substituent was non-coordinating, and did not alter the coordination geometry of the complex compared to the parent species. This was indicated by the shapes of the Ln(III) luminescence spectra and the radiative lifetimes of the Eu(III) complexes, and was supported by the results of the paramagnetic NMR spectroscopic and single crystal X-ray crystallographic analyses in the solution and solid states, respectively.Changing the linker from secondary to tertiary amide had a profound effect on the Eu(III) and Tb(III) luminescence. In tacn-based, pyridine-containing Ln(III) complexes with secondary amide linkers pyridine reduction by the photoexcited carbostyril competed efficiently with Eu(III) and Tb(III) sensitization. Upon linker replacement all of the emitters experienced an increase in luminescence quantum yield, with values close to, and in some cases even higher than, those obtained for cyclen-based systems lacking the quenching pyridines. An additional benefit of the tert-amide linkers is higher antenna T1 energy, and thus decreased BET for Tb(III) emitters. Finally, tertiary amide linked complexes based on both tacn and cyclen frameworks showed high photostability, highlighting the importance of closing down PeT and BET quenching pathways to obtain robust emitters.
Author contributions
D. Kocsi and D. Kovacs: conceptualization, resources, investigation, data curation, visualization, formal analysis. J.A.L.W.: investigation, data curation, visualization, formal analysis. K.E.B.: conceptualization, funding acquisition, project administration, resources, formal analysis, visualization, supervision. All authors were involved in the writing of the original draft and took part in the reviewing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Research Council (project grant 2017-04077 for K.E.B.), the Knut och Alice Wallenbergs Foundation (Dnr: 2018.0066).Notes and references
- J.-C. G. Bünzli and S. V. Eliseeva, in Lanthanide Luminescence: Photophysical, Analytical and Biological Aspects, ed. P. Hänninen and H. Härmä, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 1–45 Search PubMed.
- A. de Bettencourt-Dias, in Luminescence of Lanthanide Ions in Coordination Compounds and Nanomaterials, John Wiley & Sons Ltd, 2014, pp. 1–48 Search PubMed.
- W. D. Horrocks and D. R. Sudnick, Acc. Chem. Res., 1981, 14, 384–392 CrossRef CAS.
- W. D. Horrocks, Jr., J. P. Bolender, W. D. Smith and R. M. Supkowski, J. Am. Chem. Soc., 1997, 119, 5972–5973 CrossRef.
- A. S. Chauvin, F. Gumy, D. Imbert and J. C. G. Bünzli, Spectrosc. Lett., 2004, 37, 517–532 CrossRef CAS.
- A. de Bettencourt-Dias, P. S. Barber and S. Bauer, J. Am. Chem. Soc., 2012, 134, 6987–6994 CrossRef CAS PubMed.
- A. de Bettencourt-Dias, S. Viswanathan and A. Rollett, J. Am. Chem. Soc., 2007, 129, 15436–15437 CrossRef CAS PubMed.
- J. H. S. K. Monteiro, A. de Bettencourt-Dias and F. A. Sigoli, Inorg. Chem., 2017, 56, 709–712 CrossRef CAS PubMed.
- R. Xiong, D. Mara, J. Liu, R. Van Deun and K. E. Borbas, J. Am. Chem. Soc., 2018, 140, 10975–10979 CrossRef CAS PubMed.
- J. Laakso, G. A. Rosser, C. Szíjjártó, A. Beeby and K. E. Borbas, Inorg. Chem., 2012, 51, 10366–10374 CrossRef CAS PubMed.
- T. Zhang, X. Zhu, C. C. W. Cheng, W.-M. Kwok, H.-L. Tam, J. Hao, D. W. J. Kwong, W.-K. Wong and K.-L. Wong, J. Am. Chem. Soc., 2011, 133, 20120–20122 CrossRef CAS PubMed.
- Y. Ning, M. Zhu and J.-L. Zhang, Coord. Chem. Rev., 2019, 399, 213028 CrossRef CAS.
- Y. Ning, G.-Q. Jin and J.-L. Zhang, Acc. Chem. Res., 2019, 52, 2620–2633 CrossRef CAS PubMed.
- Y. Ning, J. Tang, Y.-W. Liu, J. Jing, Y. Sun and J.-L. Zhang, Chem. Sci., 2018, 9, 3742–3753 RSC.
- D. Parker, P. K. Senanayake and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1998, 2129–2139 RSC.
- A. Beeby, S. Faulkner, D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 2001, 1268–1273 RSC.
- D. G. Smith, R. Pal and D. Parker, Chem. – Eur. J., 2012, 18, 11604–11613 CrossRef CAS PubMed.
- D. G. Smith, B. K. McMahon, R. Pal and D. Parker, Chem. Commun., 2012, 48, 8520–8522 RSC.
- A. Foucault-Collet, C. M. Shade, I. Nazarenko, S. Petoud and S. V. Eliseeva, Angew. Chem., Int. Ed., 2014, 53, 2927–2930 CrossRef CAS PubMed.
- S. Petoud, G. Muller, E. G. Moore, J. Xu, J. Sokolnicki, J. P. Riehl, U. N. Le, S. M. Cohen and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 77–83 CrossRef CAS PubMed.
- S. Faulkner, M.-C. Carrie, S. J. A. Pope, J. Squire, A. Beeby and P. G. Sammes, Dalton Trans., 2004, 1405–1409 RSC.
- C. Y. Chow, S. V. Eliseeva, E. R. Trivedi, T. N. Nguyen, J. W. Kampf, S. Petoud and V. L. Pecoraro, J. Am. Chem. Soc., 2016, 138, 5100–5109 CrossRef CAS PubMed.
- E. R. Trivedi, S. V. Eliseeva, J. Jankolovits, M. M. Olmstead, S. Petoud and V. L. Pecoraro, J. Am. Chem. Soc., 2014, 136, 1526–1534 CrossRef CAS PubMed.
- T. N. Nguyen, C. Y. Chow, S. V. Eliseeva, E. R. Trivedi, J. W. Kampf, I. Martinic, S. Petoud and V. L. Pecoraro, Chem. – Eur. J., 2018, 24, 1031–1035 CrossRef CAS PubMed.
- T. Lazarides, D. Sykes, S. Faulkner, A. Barbieri and M. D. Ward, Chem. – Eur. J., 2008, 14, 9389–9399 CrossRef CAS PubMed.
- T. Lazarides, N. M. Tart, D. Sykes, S. Faulkner, A. Barbieri and M. D. Ward, Dalton Trans., 2009, 3971–3979 RSC.
- N. M. Shavaleev, L. P. Moorcraft, S. J. A. Pope, Z. R. Bell, S. Faulkner and M. D. Ward, Chem. Commun., 2003, 1134–1135 RSC.
- N. M. Shavaleev, L. P. Moorcraft, S. J. A. Pope, Z. R. Bell, S. Faulkner and M. D. Ward, Chem. – Eur. J., 2003, 9, 5283–5291 CrossRef CAS PubMed.
- S. J. A. Pope, B. J. Coe and S. Faulkner, Chem. Commun., 2004, 1550–1551 RSC.
- S. Faulkner and S. J. A. Pope, J. Am. Chem. Soc., 2003, 125, 10526–10527 CrossRef CAS PubMed.
- S. Cotton, in Lanthanide and Actinide Chemistry, 2006, pp. 35–60 Search PubMed.
- D. Parker, R. S. Dickins, H. Puschmann, C. Crossland and J. A. K. Howard, Chem. Rev., 2002, 102, 1977–2010 CrossRef CAS PubMed.
- W. D. Horrocks, Jr. and D. R. Sudnick, J. Am. Chem. Soc., 1979, 101, 334–340 CrossRef.
- R. S. Dickins, D. Parker, A. S. de Sousa and J. A. G. Williams, Chem. Commun., 1996, 697–698 RSC.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, S. A. S. de, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504 RSC.
- M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542–1548 RSC.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- D. Kovacs, D. Kocsi, J. A. L. Wells, S. R. Kiraev and K. E. Borbas, Dalton Trans., 2021, 50, 4244–4254 RSC.
- D. Kovacs, S. R. Kiraev, D. Phipps, A. Orthaber and K. E. Borbas, Inorg. Chem., 2020, 59, 106–117 CrossRef CAS PubMed.
- D. Kovacs, E. Mathieu, S. R. Kiraev, J. A. L. Wells, E. Demeyere, A. Sipos and K. E. Borbas, J. Am. Chem. Soc., 2020, 142, 13190–13200 CrossRef CAS PubMed.
- S. R. Kiraev, E. Mathieu, F. Siemens, D. Kovacs, E. Demeyere and K. E. Borbas, Molecules, 2020, 25, 5282 CrossRef CAS PubMed.
- D. Kovacs, D. Phipps, A. Orthaber and K. E. Borbas, Dalton Trans., 2018, 47, 10702–10714 RSC.
- D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1996, 1581–1586 RSC.
- D. Kovacs and K. E. Borbas, Coord. Chem. Rev., 2018, 364, 1–9 CrossRef CAS.
- A. K. R. Junker and T. J. Sorensen, Eur. J. Inorg. Chem., 2019, 2019, 1201–1206 CrossRef CAS.
- L.-M. Fu, X.-C. Ai, M.-Y. Li, X.-F. Wen, R. Hao, Y.-S. Wu, Y. Wang and J.-P. Zhang, J. Phys. Chem. A, 2010, 114, 4494–4500 CrossRef CAS PubMed.
- J.-C. G. Bunzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef CAS.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- A. Beeby, S. Faulkner and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 2002, 1918–1922 RSC.
- S. V. Eliseeva, D. N. Pleshkov, K. A. Lyssenko, L. S. Lepnev, J.-C. G. Bunzli and N. P. Kuzmina, Inorg. Chem., 2011, 50, 5137–5144 CrossRef CAS PubMed.
- A. Beeby, D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1996, 1565–1580 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis and chemical characterisation of new compounds, additional photophysical and crystallographic characterization, 1H, 13C, and 19F NMR spectra of new compounds. CCDC 2102702, 2102703. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt02893f |
| This journal is © The Royal Society of Chemistry 2021 |