
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modular synthesis of antimalarial quinoline-based PGM metallarectangles†
Taryn M.
Golding
 ,
Mziyanda
Mbaba
and
Gregory S.
Smith
*
,
Mziyanda
Mbaba
and
Gregory S.
Smith
*
Department of Chemistry, University of Cape Town, Rondebosch, Cape Town, South Africa. E-mail: gregory.smith@uct.ac.za
First published on 4th October 2021
Abstract
A new ditopic, quinoline-based ligand L (7-chloro-4-(pyridin-4-yl)quinoline) was synthesized via a Suzuki cross-coupling reaction. The ligand was utilized to synthesize the corresponding half-sandwich iridium(III) and ruthenium(II) binuclear complexes (1c and 1d) and the subsequent metallarectangles (2c, 2d, 3c, and 3d), via [2 + 2] coordination-driven self-assembly. Single-crystal X-ray diffraction confirmed the proposed molecular structure of the binuclear complex [{IrCl2(Cp*)}2(μ-L)] (1c) and DFT calculations were used to predict the optimized geometry of the rectangular nature of [{Ir(μ-Cl)(Cp*)}4(μ-L)2](CF3SO3)4 (2c). All of the metallarectangles were isolated as their triflate salts and characterized using various spectroscopic (1H, 13C{1H}, DOSY NMR, and IR spectroscopy) and analytical techniques (ESI-MS). The synthesized compounds were screened against the NF54 chloroquine-sensitive (CQS) and K1 chloroquine-resistant (CQR) strains of Plasmodium falciparum. Incorporation of the ubiquitous quinoline core and metal complexation significantly enhanced the in vitro biological activity, with an increase in the nuclearity correlating with an increase in the resultant antiplasmodial activity. This was observed across both parasitic strains, alluding to the potential of supramolecular metallarectangles to act as antiplasmodial agents. Inhibition of haemozoin formation was considered a potential mechanism of action and selected metallarectangles exhibit β-haematin inhibition activity with near comparable activity to chloroquine.
Introduction
Malaria, an infectious mosquito-borne disease caused by a blood parasite of the genus Plasmodium, is associated with high levels of morbidity and mortality in adults and children and occurs throughout tropical and subtropical regions of the world.1–4 Despite the overall number of estimated cases declining between 2010 to 2014, subsequent years have seen a resurgence in malaria cases globally.1 The World Health Organisation (WHO) estimated that in 2019 there were approximately 229 million cases of malaria worldwide.1The eventual recognition of quinoline-based antimalarials, such as chloroquine (CQ),5 provided new hope for the treatment of this disease. CQ quickly became the most effective and widely used antimalarial drug.5 Unfortunately, its efficacy has been greatly compromised by the advent of drug-resistant strains of P. falciparum.2,5 Despite this, the quinoline scaffold continues to be attractive for the design and synthesis of new antimalarial agents. Its limited host toxicity, cost-effective synthesis, ease of use, and excellent clinical efficiency6 have prompted efforts to develop new antimalarial drugs based on this pharmacophoric unit (e.g. amodiaquine, mefloquine, etc.).2 The WHO now recommends artemisinin-based combination therapy (ACT) as the standard first-line therapy for uncomplicated malaria caused by P. falciparum.1 This treatment regimen involves administration of artemisinin, or a derivative thereof, followed by a second antimalarial, preferably with a different mechanism of action, to ensure complete parasite clearance. Despite the proven effectiveness of ACT, disadvantages associated with this treatment regimen include poor patient compliance, increased risk of adverse effects caused by drug–drug interactions, and elevated costs.7–9 Furthermore, even in combination, the individual drugs are also susceptible to the development of resistance, which has been emerging.2,10
The intrinsic and acquired resistance to several chemotherapeutic agents renders drugs that were once highly efficacious, fruitless, hampering the drug development progress made thus far. This has prompted research into alternative approaches, to combat the rising resistance. One approach that is gaining impetus is the incorporation of metals and this has proven to be an invaluable strategy.11–14 This field was stimulated by the success of the metal-based anticancer drug cisplatin.15,16 Since then, organometallic complexes as antimalarial agents have also generated significant scientific interest,17 exemplified by the antimalarial ferroquine (FQ).2,18 Due to the emerging chemoresistance and the limited arsenal of effective antimalarial agents, metal complexes based on novel scaffolds offer an excellent opportunity to find new leads against this infectious disease.
Supramolecular coordination complexes (SCCs), formed via coordination-driven self-assembly, are discrete systems stabilized by non-covalent intermolecular interactions.19,20 The synthetic strategy for the spontaneous assembly of rectangular architectures requires a step-wise synthetic procedure.21–23 The first step toward their synthesis requires synthesizing a stable, pre-organized, bimetallic molecular clip.22–30 Compared to main group metals, transition metals are often utilized due to their well-defined and predictable coordination geometries.31,32 To ensure the formation of a discrete supramolecular architecture, directing ligands such as cyclopentadienyl and arene-based ligands are used to occupy concomitant coordination sites at the octahedral metal centre.19,21,22,32–34 Half-sandwich transition metal complexes are thus promising building blocks to generate metallacycles.24,31,33,35–39 A suitable bifunctional donor, which acts as a bridging ligand, can then coordinate predictably to generate a [2 + 2] rectangular 2-D architecture.22–30,37
Over the years, metallamacrocyclic supramolecular complexes containing organometallic rhodium, ruthenium, and iridium half-sandwich fragments have received a considerable amount of attention due to their myriad of potential applications,40 including as host–guest,41–43 and drug-delivery systems,44–47 and in catalysis.48 Despite extensive research into the pharmacological activity of small metal-based compounds, investigations into the pharmacological activity of metallosupramolecular coordination compounds have primarily centred on their potential as novel anticancer agents (see selected examples in Fig. 1).27,36,49–65 The exploration into the use of such complexes for the treatment of alternative diseases is limited and warrants further exploration.
 | ||
| Fig. 1 Selected examples of metallarectangles displaying potent anticancer cytotoxic activity against selected cancer cell lines.27,53 | ||
Herein we describe the synthesis and characterisation of a series of iridium(III) and ruthenium(II) metallarectangles, containing a new 7-chloro-4-(pyridin-4-yl)quinoline-based ligand. To the best of our knowledge, no studies have explored the antimalarial properties of metallarectangles. Consequently, the metallarectangles prepared in this study were evaluated against the chloroquine-sensitive (CQS) NF54 and chloroquine-resistant (CQR) K1 strains of P. falciparum. This study also explored the β-haematin inhibition ability of selected metallarectangles, as a potential mechanism of action.
Results and discussion
Synthesis and characterisation
The synthesis of the ditopic quinoline-containing ligand L, the corresponding binuclear complexes (1c and 1d) and the subsequent metallarectangles (2c, 2d, 3c, and 3d) is outlined in Scheme 1. The known prototypical complexes (1a, 1b, 2a, 2b, 3a, 3b) containing the bipyridyl ligand were also prepared and evaluated alongside the quinoline-based complexes (vide infra, Table 1). The synthesis of the ligand (L) proceeded via a Suzuki cross-coupling reaction, following a modified literature method by Kudo et al.,66 yielding the desired compound as a white crystalline powder in a moderate yield of 42%. The synthesis of the iridium(III) and ruthenium(II) binuclear complexes (1c and 1d respectively) were achieved via a bridge-splitting reaction of the appropriate dimer, allowing for insertion of the N,N′-ditopic ligand, and subsequent coordination to both nitrogen donor atoms. The resultant complexes (1c and 1d) were isolated in excellent yields (74–76%), as yellow powders. The tetranuclear iridium(III) and ruthenium(II) metallarectangles, [{Ir(μ-Cl)(Cp*)}4(μ-L)2](CF3SO3)4 (2c) and [{Ru(μ-Cl)(p-cymene)}4(μ-L)2](CF3SO3)4 (2d) (L = 7-chloro-4-(pyridin-4-yl)quinoline), were prepared from the binuclear complexes [{IrCl2(Cp*)}2(μ-L)] (1c) and [{RuCl2(p-cymene)}2(μ-L)] (1d), via coordination-driven self-assembly, and isolated as their triflate salts. Metallarectangles 3c and 3d, containing chelating oxalato-ligands, were synthesized by reacting either [{IrCl(Cp*)}2(μ-η2-η2-C2O4)] or [{RuCl(p-cymene)}2(μ-η2-η2-C2O4)] with silver trifluoromethane-sulfonate, ensuring complete abstraction of the chlorides. Thereafter, the addition of ligand L resulted in coordination at the vacant coordination sites, and the resultant complexes (3c and 3d) formed via coordination-driven self-assembly. All of the metallarectangles displayed excellent solubility in CH2Cl2 and DMSO and were insoluble in a range of non-polar organic solvents, such as Et2O, hexane, pentane, and toluene. The ligand L and all the metal complexes were characterized using 1H NMR and 13C{1H} NMR spectroscopy, IR spectroscopy, and electrospray ionization mass spectrometry (ESI-MS). The metallarectangles (2c, 2d, 3c, and 3d) were further analyzed using Diffusion-Ordered Spectroscopy (DOSY) to confirm the presence of a single entity in solution. | ||
| Scheme 1 Synthesis of ditopic quinoline-containing ligand (L) and the corresponding iridium(III) and ruthenium(II) binuclear complexes (1c and 1d) and metallarectangles (2c, 2d, 3c, and 3d). Reagents and conditions: (i) 4-pyridinylboronic acid (1.1 eq.), Pd(OAc)2 (0.01 eq.), PCy3 (0.024 eq.), K3PO4 (aq) (1.7 eq.), 1,4-dioxane, reflux, 18 h; (ii) [IrCp*(μ-Cl)Cl]2 or [Ru(p-cymene)(μ-Cl)Cl]2 (0.8 eq.), CH2Cl2, r.t., 6 h; (iii) AgCF3SO3 (2.0 eq.) in CH3CN, CH2Cl2, r.t., 24 h; (iv) [{IrCl(Cp*)}2(μ-η2-η2-C2O4)] or [{RuCl(p-cymene)}2(μ-η2-η2-C2O4)] (1 eq.), AgCF3SO3 (2.0 eq.) in CH3CN, CH3OH, r.t., 24 h. | ||
|
|
|||
|---|---|---|---|
| Compound | IC50 (μM) ± SE NF54 | IC50 (μM) ± SE K1 | Resistance index (RI)a |
| a (IC50 K1/IC50 NF54). b ND = not determined. | |||
| Ligands | |||
| 4,4′-Bipyridine | 500.50 ± 35.01 | 778.76 ± 12.68 | 1.6 |
| L | 50.88 ± 7.34 | 118.31 ± 5.97 | 2.3 |
| Binuclear complexes | |||
| 1a | 90.66 ± 5.06 | 211.97 ± 1.99 | 2.3 |
| 1b | 13.33 ± 4.21 | 15.54 ± 0.46 | 1.2 |
| 1c | 46.26 ± 2.82 | 50.24 ± 0.16 | 1.1 |
| 1d | 14.75 ± 1.05 | 17.39 ± 1.92 | 1.2 |
| Metallarectangles | |||
| 2a | 19.40 ± 0.73 | 17.14 ± 7.43 | 0.9 |
| 2b | 9.41 ± 2.81 | 19.76 ± 1.29 | 2.1 |
| 2c | 9.28 ± 0.75 | 15.30 ± 2.60 | 1.6 |
| 2d | NDb | NDb | |
| 3a | 26.25 ± 5.98 | 70.32 ± 1.26 | 2.7 |
| 3b | 1.08 ± 0.53 | 2.25 ± 0.82 | 2.1 |
| 3c | 20.55 ± 5.60 | 22.69 ± 5.38 | 1.1 |
| 3d | 9.82 ± 0.89 | 7.65 ± 0.77 | 0.8 |
| CQDP | 0.0134 ± 0.00096 | 0.296 ± 0.0283 | 22.1 |
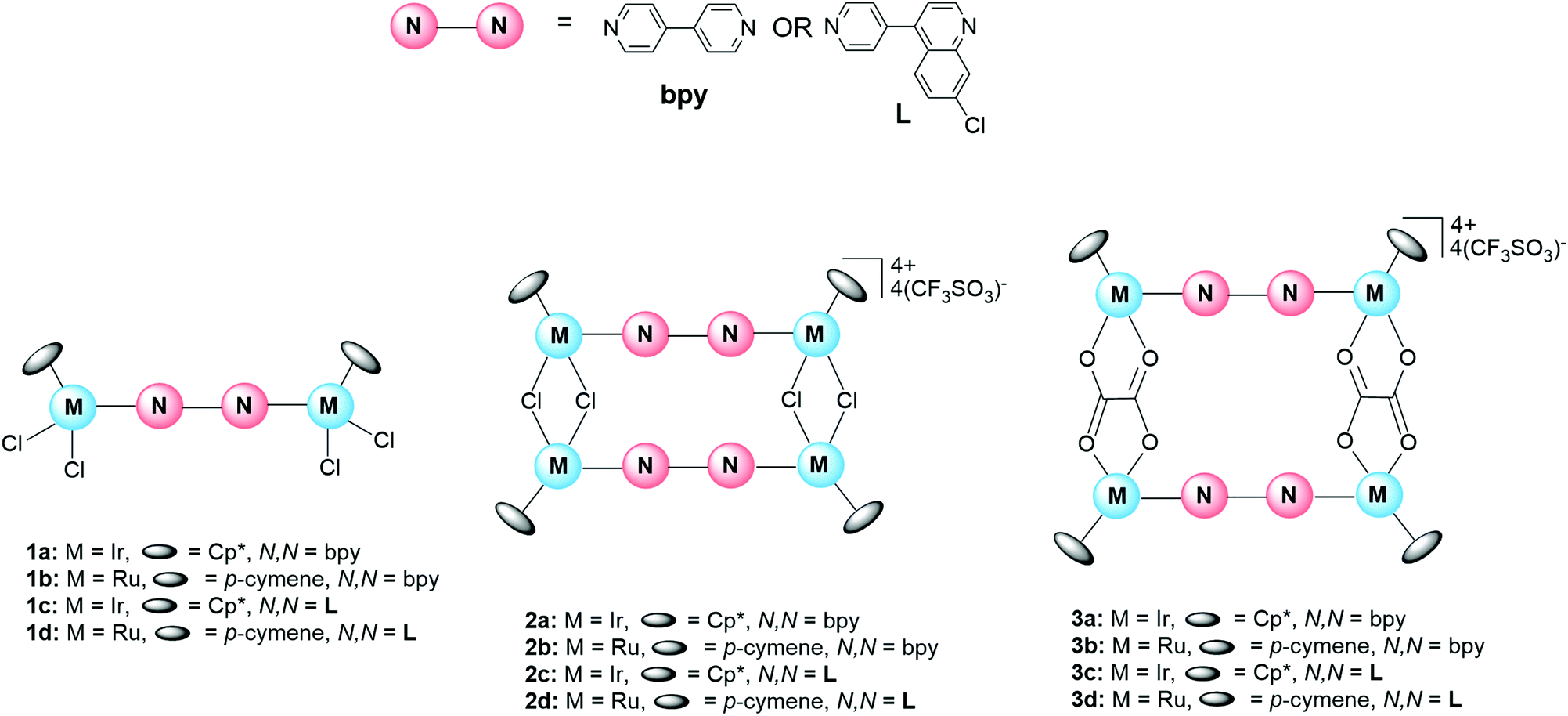
Spectroscopic details of the ligand L and the corresponding binuclear complexes (1c and 1d) are provided in the ESI (Fig. S1–S7†). Analysis of the 1H NMR spectrum of the tetracationic iridium(III) metallarectangle 2c, confirms coordination to both nitrogen atoms of the unsymmetrical ligand L (Fig. S11†). Two singlets at 1.74 and 1.64 ppm are assigned to the methyl protons of the four Cp* ligands, a consequence of the unsymmetrical nature of ligand L. The remaining aromatic proton signals collectively confirm the incorporation of two L ligands per molecule. The NMR spectrum of compound 2c points to the formation of one pure complex. However, it is important to note the existence of one of four possible isomers as illustrated using 2c as an example (Fig. 2). Based on the spectrum of complex 2c (Fig. S11†), one can deduce that a single, symmetrical isomer is favoured, and thus any of the pure compounds, A–D (Fig. 2), could have formed. A mixture of conformational isomers would result in the coalescence of signals, as attested to by variable-temperature (VT) NMR spectroscopic analysis. It is postulated that the absence of observable conformational isomers, or rotamers, may be due to a high barrier to rotation, resulting in restricted rotation. The reason for the selectivity toward a single constitutional isomer, however, is not entirely known, although steric strain may have a role to play.
 | ||
| Fig. 2 Possible isomers of metallarectangle 2c. | ||
In the 1H NMR spectra of metallarectangles 2d (Fig. S12†) and 3c (Fig. S13†), each dominant signal is accompanied by a smaller set of signals. These spectra suggest the presence of conformational isomers, which was confirmed by VT-NMR analysis. The relative integrations confirm the presence of only two ligands (L) and four metal centres, and thus the formation of the desired metallarectangles (2d and 3c). The 1H NMR of metallarectangle 3d (Fig. S14†), however, shows an apparent “doubling-up” of both the ligand L and p-cymene signals. VT-NMR analysis did not result in a coalescence of signals and it is thus speculated that the sample contains a mixture of two constitutional isomers. This is further supported by the presence of two sets of signals corresponding to ligand L in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Moreover, the relative integrations to the p-cymene signals support the presence of two metallarectangles.
:1 ratio. Moreover, the relative integrations to the p-cymene signals support the presence of two metallarectangles.
To further support these assertions, diffusion-ordered NMR (DOSY) spectra of metallarectangles 2c, 2d, 3c, and 3d were also recorded (Fig. S19–S22†) and the presence of a single diffusion line confirms the presence of a single discrete compound in solution. It should also be noted that although the 1H NMR spectra of metallarectangles 2c, 3c, and 3d reveal the presence of either conformational or constitutional isomers, the DOSY spectra still reveals a single vertical trace, as the isomers, despite being chemically different, have the same molecular weight and shape. This excludes the formation of unwanted side-products such as polymers.
For the metallarectangles [{Ir(μ-Cl)(Cp*)}4(μ-L)2](CF3SO3)4 (2c) and [{Ru(μ-Cl)(p-cymene)}4(μ-L)2](CF3SO3)4 (2d), IR spectroscopy reveals ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) stretching vibrations at 1610 and 1589 cm−1 attributed to the two different CN functionalities, CNpy and CNquin., respectively. For the complexes [{Ir(Cp*)}4(μ-η2-η2-C2O4)(μ-L)2](CF3SO3)4 (3c) and [{Ru(p-cymene)}4(μ-η2-η2-C2O4)(μ-L)2](CF3SO3)4 (3d), the characteristic ν(CO) stretching vibration is observed at 1627 cm−1, overlapping with the ν(CN) absorption bands. Furthermore, the infrared spectra of the metallarectangles (2c, 2d, 3c, and 3d) (Fig. S23–S26†) are dominated by strong absorption bands at 1249 cm−1 (CF3 sym.), 1223 cm−1 (SO3 asym.), 1155 cm−1 (CF3 asym.), and 1026 cm−1 (SO3 sym.), due to the stretching vibrations of the triflate anions. These assignments are well documented in literature67–69 and support the formation of the cationic assemblies as their triflate salts. ESI-MS analysis further supports the formation of metallarectangles 2c, 2d, 3c, and 3d. The mass spectral data show tetracationic peaks for iridium metallarectangles 2c (m/z 483.1129) and 3c (m/z 492.0915), corresponding to the loss of four triflate counterions. The presence of these ion peaks, corresponding to the loss of one or more counter ions, is often observed for supramolecular rectangular architectures27,52–54 and correlates with the calculated values. Furthermore, dicationic peaks were observed for all iridium and ruthenium metallarectangles (2c, 2d, 3c, and 3d), which correlate with the loss of two triflate ions, [M − 2CF3SO3]2+.
N) stretching vibrations at 1610 and 1589 cm−1 attributed to the two different CN functionalities, CNpy and CNquin., respectively. For the complexes [{Ir(Cp*)}4(μ-η2-η2-C2O4)(μ-L)2](CF3SO3)4 (3c) and [{Ru(p-cymene)}4(μ-η2-η2-C2O4)(μ-L)2](CF3SO3)4 (3d), the characteristic ν(CO) stretching vibration is observed at 1627 cm−1, overlapping with the ν(CN) absorption bands. Furthermore, the infrared spectra of the metallarectangles (2c, 2d, 3c, and 3d) (Fig. S23–S26†) are dominated by strong absorption bands at 1249 cm−1 (CF3 sym.), 1223 cm−1 (SO3 asym.), 1155 cm−1 (CF3 asym.), and 1026 cm−1 (SO3 sym.), due to the stretching vibrations of the triflate anions. These assignments are well documented in literature67–69 and support the formation of the cationic assemblies as their triflate salts. ESI-MS analysis further supports the formation of metallarectangles 2c, 2d, 3c, and 3d. The mass spectral data show tetracationic peaks for iridium metallarectangles 2c (m/z 483.1129) and 3c (m/z 492.0915), corresponding to the loss of four triflate counterions. The presence of these ion peaks, corresponding to the loss of one or more counter ions, is often observed for supramolecular rectangular architectures27,52–54 and correlates with the calculated values. Furthermore, dicationic peaks were observed for all iridium and ruthenium metallarectangles (2c, 2d, 3c, and 3d), which correlate with the loss of two triflate ions, [M − 2CF3SO3]2+.
Single-crystal X-ray diffraction
Single crystals of the precursor binuclear complex 1c were grown by the slow evaporation of a saturated chloroform solution, and the molecular structure elucidated by single-crystal X-ray diffraction. From the ORTEP diagram of complex 1c (Fig. 3), the ditopic ligand L is observed to bridge two iridium(III) metal centres, with each iridium(III) further coordinated to two chloride ligands and a η5-pentamethylcyclopentadienyl (Cp*) ligand. It is also apparent that one of the Cp* ligands is disordered, with refined site occupancy factors of 0.394(9) and 0.606(9). The complex adopts the well-known “three-legged piano-stool” structure which is commonly observed in many other half-sandwich rhodium, iridium, and ruthenium complexes.70–73 | ||
| Fig. 3 Molecular structure of the iridium(III) binuclear complex 1c. Solvent molecules (5·CHCl3) and hydrogen atoms have been omitted for clarity. Ellipsoids are shown at 30% probability level. | ||
Complex 1c crystallizes in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with a triclinic system. Further crystallographic data and refinement parameters are summarised in Table S1.† Additionally, selected bond lengths and angles are listed in Table S2.† The data in Table S2† suggests that the geometry around the iridium metal centre is pseudo-tetrahedral, as the bond angles around the metal centre range between 83.0° and 90.4°. Furthermore, the Ir1–N1 bond length (2.113(6) Å) is comparable to related iridium(III)-pyridyl complexes,28,30,53,72 however, the Ir2A–N2 and Ir2B–N2 bond lengths are slightly longer.28,30,53 Finally, the quinoline and pyridyl ring systems are not co-planar, as expected, with a torsion angle of 44.3°.
space group with a triclinic system. Further crystallographic data and refinement parameters are summarised in Table S1.† Additionally, selected bond lengths and angles are listed in Table S2.† The data in Table S2† suggests that the geometry around the iridium metal centre is pseudo-tetrahedral, as the bond angles around the metal centre range between 83.0° and 90.4°. Furthermore, the Ir1–N1 bond length (2.113(6) Å) is comparable to related iridium(III)-pyridyl complexes,28,30,53,72 however, the Ir2A–N2 and Ir2B–N2 bond lengths are slightly longer.28,30,53 Finally, the quinoline and pyridyl ring systems are not co-planar, as expected, with a torsion angle of 44.3°.
Density functional theory (DFT) calculations
A theoretical structure indicative of the 2D-rectangular nature of complex 2c is shown in Fig. 4, as determined from a density functional theory (DFT) geometry optimization. Similar to the molecular structure obtained for the binuclear complex 1c (Fig. 3), the optimized structures shown in Fig. 4 also reveal a torsion angle between the pyridyl and quinoline ring systems of ligand L. Within the syn-complex, the torsion angles within the two ligands are 41.48° and 41.43°. Within the anti-complex, these angles are slightly smaller, with values of 33.33° and 34.70°. More notably, however, within a given isomer (syn or anti), the torsion angles within the two coordinated ligands are almost identical, suggesting that if one ring system (e.g. pyridyl or quinoline) rotates, the ring system directly opposite will rotate in a similar manner, possibly to relieve steric strain. This phenomenon is reported in the literature.74 Furthermore, analysis of these structures reveals that the geometry around the iridium(III) metal centre is pseudo-tetrahedral, with bond angles ranging between 81.00° and 94.55°. | ||
| Fig. 4 DFT-optimized computational structure models of the syn- and anti-structures of metallarectangle 2c. | ||
In vitro antiplasmodial assays
4,4′-Bipyridine, the new quinoline-containing ligand L, the corresponding binuclear complexes (1a–1d) and the metallarectangles (2a–2d and 3a–3d) were evaluated for their in vitro antiplasmodial activity against the CQS NF54 and the CQR K1 strains of P. falciparum. The biological results for the ligands and corresponding complexes are summarised in Table 1, with chloroquine diphosphate (CQDP) used as the control drug in this study.Most notably, the antiplasmodial results reveal that the quinoline-containing analogues generally show superior activity in comparison with their corresponding prototypical 4,4′-bipyridyl congeners. The activity of the quinoline-containing ligand L (IC50 value = 50.88 μM) in the NF54 strain, outperforms 4,4′-bipyridine (p = 0.03) by almost 10-fold, pointing to the pharmacological benefits of incorporating a pharmacophoric quinoline scaffold into the framework of a potential drug candidate, as is exemplified in many antimalarial drugs.6,75,76 Furthermore, the activity of both ligands are generally enhanced upon metal complexation (statistically significant, p < 0.05), with most complexes approximately five-fold more potent than their respective ligands. Interestingly, the antiplasmodial activity further increases with an increase in nuclearity. For example, the IC50 value of 4,4′-bipyridine is 500.50 μM, however, upon complexation to form a binuclear complex (e.g.1a), the activity is increased five-fold. Formation of the corresponding metallarectangles (2a or 3a) results in a further increase in activity by approximately four-fold. This trend was similarly observed for the ruthenium-bipyridyl analogues, as well as the quinoline congeners, further underpinning the plethora of studies that emphasize the advantages of metal incorporation on the pharmacological activity of organic compounds.72,77–79 Interestingly, the enhanced activity observed for the supramolecular metallarectangles demonstrates the potential of such multinuclear, highly charged systems, to act as antiplasmodial agents. The increased antiplasmodial activity observed upon complexation suggests that the metal centre may be crucial in reducing the parasitic viability, although the exact mechanism is still unclear. Stability studies demonstrate that most of the complexes are stable as attested to by 1H NMR experiments, except for 3d with small changes observed over the analogous incubation time. The most active complexes containing ligand L are metallarectangles 2c and 3d, with comparable activity (IC50 values of 9.28 and 9.82 μM, respectively), although not as active as the clinical drug CQDP, which has an IC50 value of 0.0134 μM in the NF54 strain.
The inhibitory data of the compounds tested against the multi-drug resistant K1 strain resembles the trend observed for the NF54 strain. The quinoline-containing ligand L was observed to be approximately seven-times more active than 4,4′-bipyridine, reiterating the importance of the pharmacophoric scaffold for antiplasmodial activity. The inhibitory data for the resistant strain similarly reveals that complexation enhances the antiplasmodial activity. Of the synthesized quinoline-containing compounds, metallarectangle 3d showed the greatest activity, with an IC50 value in the low-micromolar range (IC50 = 7.65 μM). Despite the moderate activity of the synthesized compounds, the observed antiplasmodial data is promising, as it points to a potentially new class of metal-based antiplasmodial agents and lends credence to the incorporation of the quinoline scaffold.
The resistance indices (RI) were, in most cases, slightly greater than 1, suggesting that the compounds are likely to experience some cross-resistance, albeit not to the same extent as CQ (RI = 22.1). Notably, the RI values of the quinoline-containing complexes (1c, 1d, 2c, 3c, and 3d), which range between 0.8–1.6, are lower than the RI value observed for the uncoordinated ligand L (RI = 2.3). This suggests that metal incorporation imposes a potentially new mechanism of action, which is not subject to the same resistance mechanism experienced by the uncoordinated ligand, minimizing cross-resistance.
β-Haematin inhibition studies
During the intraerythrocytic stage of the parasites’ life cycle, the parasite degrades haemoglobin (Hb) found within the infected red blood cells of the host.80–82 A side product of this Hb digestion is the production of haem, which is toxic to the parasite. To circumvent these effects, the parasite initiates a detoxification mechanism, converting the free haem into inert crystalline haemozoin, which is non-toxic to the parasite.81 Many researchers have thus exploited this process by developing antimalarials that interfere with this haem detoxification method, the most promising of which has been chloroquine.5,83 Like chloroquine, the target for many aminoquinoline-based antimalarials is the inhibition of haemozoin formation.2,82,84The presence of the quinoline scaffold in ligand L, and the corresponding complexes, prompted investigation into the β-haematin (synthetic haemozoin) inhibition ability of selected compounds, as a potential mechanism of action. The ability of a potential drug candidate to inhibit haemozoin formation can be measured using the NP-40 detergent-mediated β-haematin inhibition assay.85,86 Representative compounds (L, 3c, and 3d) were tested for their ability to inhibit β-haematin formation and the log-based dose–response curves are shown in Fig. 5. The compounds were screened in either duplicate or triplicate and the amount of synthetic haemozoin formed was quantified using the colorimetric pyridine ferrochrome method published by Egan et al.87
 | ||
| Fig. 5 Dose–response curves obtained for metallarectangles 3c and 3d, as well as CQDP, using the NP-40 detergent-mediated β-haematin inhibition assay. | ||
Metallarectangle 3d, which shows the most promising antiplasmodial activity across both strains, metallarectangle 3c, the analogous iridium complex, and ligand L, were tested for their ability to inhibit β-haematin formation. Surprisingly, of the tested compounds, the quinoline-containing ligand L did not show any appreciable β-haematin inhibitory activity. Metallarectangles 3c and 3d, however, were found to inhibit β-haematin formation to almost the same extent as CQ, as indicated by their characteristic log-based sigmoidal curves (Fig. 5). The polyaryl nature of these complexes (3c and 3d) suggests that they are capable of intermolecular π–π interactions and it may be possible that these complexes inhibit β-haematin formation through π–π stacking with haematin. The β-haematin inhibition activity of complexes 3c and 3d were, however, unexpected since ligand L did not demonstrate any β-haematin inhibitory activity. These results should not be read in isolation. Neither complexes 3c nor 3d show antiplasmodial activity comparable with CQ. However, the IC50 value of complex 3c (IC50 = 28.29 μM), obtained from the β-haematin inhibition assay, is fairly comparable with CQ (IC50 = 22.75 μM). This suggests that complex 3c is inhibiting β-haematin to almost the same extent as CQ, however, the observed in vitro antiplasmodial activities do not corroborate this finding. The results of this cell-free assay suggest that these compounds may be likely candidates for haemozoin inhibition in the parasite. However, other factors may be contributing to the limited in vitro antiplasmodial activity. One possibility is that entry into and/or accumulation in the parasitic DV, where haemozoin crystallization takes place, is limited possibly due to the highly charged nature and size of the assembly, as it has a very good cell-free β-haematin inhibitory activity but poor in vitro antiplasmodial activity.
Conclusions
A new quinoline-containing ditopic ligand (L) was prepared, successfully incorporating the ubiquitous quinoline pharmacophore, with the targeted ligand L mimicking the orthodox 4,4′-bipyridine ligand often used in the construction of metallarectangles. The quinoline-based ligand L was reacted with either [IrCl(μ-Cl)(Cp*)]2 or [RuCl(μ-Cl)(p-cymene)]2 to afford the precursor iridium(III) and ruthenium(II) binuclear complexes (1c and 1d), which were further treated with silver triflate and, via coordination-driven self-assembly, formed the concomitant metallarectangles 2c and 2d, containing bridging chlorides. In addition, ligand L was reacted with either [{IrCl(Cp*)}2(μ-η2-η2-C2O4)] or [{RuCl(p-cymene)}2(μ-η2-η2-C2O4)], to form metallarectangles 3c and 3d, containing bridging oxalato ligands, as their triflate salts.Single-crystal X-ray diffraction confirmed the molecular structure of the precursor binuclear complex 2c and inevitably corroborated the structure of the quinoline-containing ligand L. Due to the unsymmetrical nature of the ligand, upon formation of the metallarectangles, there is a possibility of forming constitutional isomers. Interestingly, the 1H NMR spectra of metallarectangles 2c, 2d, and 3c, reveal a single isomer in solution, suggesting increased selectivity toward a preferred configuration. The 1H NMR spectrum of ruthenium metallarectangle 3d, however, suggests a mixture of two metallarectangles (constitutional isomers) in solution.
The new ligand (L), the precursor complexes (1c and 1d), the metallarectangles (2c, 2d, 3c, and 3d), as well as the corresponding 4,4′-bipyridine analogues (1a, 1b, 2a, 2b, 3a, and 3b) were evaluated for their in vitro antiplasmodial activity in the CQS NF54 and CQR K1 strains of P. falciparum. The presence of the pharmacophoric quinoline scaffold significantly enhanced the antiplasmodial activity, emphasising the importance of the quinoline pharmacophore. Metal complexation resulted in a further increase in activity, with enhanced biological activity observed with an increase in the nuclearity. The calculated resistance indices further suggest that the compounds may experience mild cross-resistance, however, not to the extent of CQ. Despite not being very potent antimalarial agents, the metallarectangles do indeed show promising antiplasmodial activity with the potential for further development through fine-tuning and ligand modification. A reason for the limited activity may be due to the size and/or highly charged nature of these SCCs, which may impede their uptake and/or accumulation in the parasite, although further in-depth studies are required to verify this hypothesis.
The presence of the quinoline pharmacophore prompted investigation into the β-haematin inhibition ability of ligand L and selected metallarectangles (3c and 3d). Both metallarectangles show β-haematin inhibitory activity, with the iridium metallarectangle (3c) having an IC50 value (28.29 μM) comparable with CQ (IC50 = 22.75 μM). The superior cell-free β-haematin inhibitory activity of metallarectangle 3c, but limited in vitro antiplasmodial activity, supports the hypothesis that entry of this large, highly charged compound into the digestive vacuole of the parasite is hindered, as entry into the cell will make this compound a likely candidate for haemozoin inhibition, and thus a promising antimalarial agent.
Experimental
General details
All reagents and solvents were purchased from commercial sources (Sigma-Aldrich, Merck, and KIMIX) and were used without further purification. The iridium and ruthenium dimers, [IrCp*(μ-Cl)Cl]288 and [Ru(p-cymene)(μ-Cl)Cl]289 respectively, [{IrCl(Cp*)}2(μ-η2-η2-C2O4)],30 [{RuCl(p-cymene)}2(μ-η2-η2-C2O4)]90 and compounds 1a,341b,912a,342b, 3a,30 and 3b90 were synthesised following literature methods. All reactions were carried out under an inert argon atmosphere using standard Schlenk line techniques unless otherwise stated. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker XR600 MHz spectrometer (1H at 599.95 MHz and 13C{1H} at 151.0 MHz), a Bruker Topspin GmbH (1H at 400.22 MHz and 13C{1H} at 100.65 MHz) or a Varian Mercury 300 (1H at 300.08 MHz) spectrometer. These were equipped with a Bruker Biospin GmbH casing and sample injector at 30 °C and tetramethylsilane (TMS) was used as the internal standard. Infrared (IR) spectroscopy was performed on a PerkinElmer Spectrum 100 FT-IR spectrometer using Attenuated Total Reflectance (ATR) with vibrations measured in units of cm−1. High resolution (HR) electrospray ionisation mass spectrometry (ESI-MS) was performed on a Waters Synapt G2 QTOF mass spectrometer with data recorded using the positive mode. Melting points were obtained using a Büchi Melting Point Apparatus B-540 and are uncorrected.
Synthesis
:ethyl acetate as the eluent to yield compound L as a white crystalline solid. Yield: 42% (1.02 g, 4.25 mmol). Rf: 0.36 (100% EtOAc). LC-MS: (m/z) = 241.0 (100% purity, M + 1). 1H NMR (300 MHz, [D6]-DMSO): δ(ppm) = 9.04 (d, 1H, Ha, 3JH–H = 4.4 Hz); 8.79 (dd, 2H, Hb, J = 4.4, 1.6 Hz); 8.20 (d, 1H, Hc, 4JH–H = 2.2 Hz); 7.84 (d, 1H, Hd, 3JH–H = 9.0 Hz); 7.67 (dd, 1H, He, 3JH–H = 9.0 Hz, 4JH–H = 2.2 Hz); 7.60 (dd, 2H, Hf, J = 4.4, 1.6 Hz); 7.57 (d, 1H, Hg, 3JH–H = 4.4 Hz). 13C{1H} NMR (100 MHz, [D6]DMSO): δ (ppm) = 151.2, 150.0, 148.4, 145.0, 144.3, 134.4, 128.1, 128.0, 127.1, 124.2, 123.8, 121.7; IR (ATR) (vmax/cm−1): 1604, 1580 (CN); M.P. (°C): 156.5–157.1.
[{IrCl2(Cp*)}2(μ-L)], (1c). [IrCp*(μ-Cl)Cl]2 (0.100 g, 0.126 mmol) was reacted with L (0.0350 g, 0.146 mmol), yielding 1c as a bright yellow powder. Yield: 76% (0.0985 g, 0.0949 mmol); 1H NMR (300 MHz, CDCl3): δ(ppm) = 9.65 (br, 1H, Ha), 9.15 (d, 2H, Hb, 3JH–H = 6.6 Hz), 9.01 (br, 1H, Hc), 7.60 (d, 1H, Hd, 3JH–H = 9.0 Hz), 7.52 (dd, 1H, He, 3JH–H = 9.0 Hz, 4JH–H = 2.0 Hz), 7.40 (d, 2H, Hf, 3JH–H = 6.6 Hz), 7.35 (d, 1H, Hg, 3JH–H = 5.2 Hz), 1.59 (s, 15H, Hh′/h), 1.55 (s, 15H, Hh′/h′); 13C{1H} NMR (100 MHz, CDCl3): δ(ppm) = 154.1, 148.5, 146.6, 145.6, 137.2, 131.2, 129.5, 126.5, 125.9, 125.0, 121.8, 86.5, 86.2, 9.2, 8.8; IR (ATR) (vmax/cm−1): 1607 (C
Npy), 1587 (CNquin); M.P. (°C): onset of decomp. with melting = 286.4; HR-MS (ESI (+), m/z): 483.1130 (17%, [M − 2Cl]2+), calculated 483.2480, 603.0919 (49%, [IrC24H24Cl2N2]+), calculated 603.5960.
[{RuCl2(p-cymene)}2(μ-L)], (1d). Ligand L (0.0449 g, 0.187 mmol) and [Ru(p-cymene)(μ-Cl)Cl]2 (0.102 g, 0.167 mmol) yielded 1d as a mustard powder. Yield: 74% (0.105 g, 0.123 mmol); 1H NMR (300 MHz, CDCl3): δ(ppm) = 9.24 (d, 2H, Hb, 3JH–H = 6.2 Hz), 9.05 (br, 1H, Ha), 8.25 (br, 1H, Hc), 7.66 (d, 1H, Hd, 3JH–H = 8.6 Hz), 7.52 (dd, 1H, He, 3JH–H = 9.0 Hz, 4JH–H = 1.7 Hz), 7.42 (br, 2H, Hf), 7.32 (d, 1H, Hg, 3JH–H = 4.5 Hz), 5.54 (d, 2H, Hh/h′, 3JH–H = 5.7 Hz), 5.49 (d, 2H, Hh/h′, 3JH–H = 5.5 Hz), 5.35 (d, 4H, Hi,i′, 3JH–H = 5.5 Hz), 3.18–3.02 (m, 1H, Hj/j′), 3.01–2.85 (m, 1H Hj/j′), 2.22 (s, 3H, Hk/k′), 2.17 (s, 3H, Hk/k′), 1.37 (d, 6H, Hl/l′, 3JH–H = 6.9 Hz), 1.29 (d, 6H, Hl/l′, 3JH–H = 6.9 Hz); 13C{1H} NMR (100 MHz, CDCl3): 155.4, 151.0, 149.0, 147.2, 144.0, 136.4, 129.2, 128.9, 126.2, 124.9, 123.8, 121.2, 104.2, 101.4, 97.4, 96.8, 82.8, 82.6, 81.4, 80.7, 30.9, 30.8, 22.5, 22.3, 19.0, 18.5; IR (ATR) (vmax/cm−1): 1608 (C
Npy), 1587 (CNquin); M.P. (°C): onset of decomp. with melting = 155.5; HR-MS (ESI (+), m/z): 390.9359 (19%, [M − 2Cl]2+), calculated 391.0900, 324.0395 (17%, [M − (p-cymene) − 2Cl]2+), calculated 323.9790, 511.0275 (55%, [RuC24H23Cl2N2]+), calculated 511.4380.
[{Ir(μ-Cl)(Cp*)}4(μ-L)2](CF3SO3)4, (2c). Ag(CF3SO3) (0.0219 g, 0.0852 mmol) in anhydrous acetonitrile (10.0 mL) was added to a solution of 1c (0.0419 g, 0.0404 mmol) in dry CH2Cl2 (10.0 mL), yielding 2c as a pale yellow powder. Yield: 71% (0.0365 g, 0.0144 mmol); 1H NMR (300 MHz, [D6]-DMSO): δ(ppm) = 9.12 (d, 2H, Ha, 3JH–H = 4.4 Hz), 8.94 (d, 4H, Hb, 3JH–H = 6.4 Hz), 8.25 (d, 2H, Hc, 4JH–H = 2.1 Hz), 8.01 (d, 4H, Hf, 3JH–H = 6.7 Hz), 7.97 (d, 2H, Hd, 3JH–H = 9.1 Hz), 7.72 (dd, 2H, He, 3JH–H = 9.0 Hz, 4JH–H = 2.2 Hz), 7.69 (d, 2H, Hg, 3JH–H = 4.4 Hz), 1.75 (s, 20H, Hh/h′), 1.64 (s, 40H, Hh/h′); 13C{1H} NMR (100 MHz, [D6]-DMSO): δ (ppm) = 153.9, 151.6, 148.4, 142.8, 134.7, 128.6, 128.3, 127.1, 123.1, 122.2, 121.7, 119.6, 100.4, 94.5, 8.6, 8.0; IR (ATR) (vmax/cm−1): 1609 (C
Npy), 1589 (CNquin), 1252 (CF3 sym), 1222 (SO3 asym), 1159 (CF3 asym), 1029 (SO3 sym); M.P. (°C): onset of decomp. without melting = 242.5; HR-MS (ESI (+), m/z): 483.2129 (48%, [M − 4OTf]4+), calculated 483.2490, 603.0921 (100%, [Ir2C48H48N4Cl4]2+), calculated 603.596.
[{Ru(μ-Cl)(p-cymene)}4(μ-L)2](CF3SO3)4, (2d). Ag(CF3SO3) (0.0500 g, 0.195 mmol) in anhydrous acetonitrile (3.00 mL) was added to a solution of 1d (0.0721 g, 0.0845 mmol) in dry CH2Cl2 (5.00 mL), yielding 2d as an orange powder. Yield: 24% (0.0218 g, 0.0101 mmol); 1H NMR (300 MHz, [D6]-DMSO): δ(ppm) = 9.10 (d, 2H, Ha, 3JH–H = 4.3 Hz), 8.97 (d, 4H, Hb, 3JH–H = 6.4 Hz), 8.24 (d, 2H, Hc, 4JH–H = 2.0 Hz), 7.94 (d, 4H, Hf, 3JH–H = 7.0 Hz), 7.89–7.79 (m, 2H, Hd), 7.73 (dd, 2H, He, 3JH–H = 9.0 Hz, 4JH–H = 1.9 Hz,), 7.76 (d, 2H, Hg, 3JH–H = 4.3 Hz); 13C{1H} NMR (100 MHz, [D6]-DMSO): δ (ppm) = 153.9, 151.5, 148.4, 148.4, 142.8, 134.7, 128.5, 128.3, 127.1, 123.1, 122.3, 122.2, 119.1, 100.4, 94.5, 40.4, 8.6, 8.3, 7.9; IR (ATR) (vmax/cm−1): 1611 (C
Npy), 1589 (CNquin), 1244 (CF3 sym), 1223 (SO3 asym), 1156 (CF3 asym), 1027 (SO3 sym); M.P. (°C): onset of decomp. with melting = 120.4; HR-MS (ESI (+), m/z): 932.2568 (28%, [M − 2OTf]2+), calculated 932.2460, 1321.9711 (100%, [Ru2C48H46Cl4N4]2+ (CF3SO3)2), calculated 1321.0280.
[{Ir(Cp*)}4(μ-η2-η2-C2O4)(μ-L)2](CF3SO3)4, (3c). [{IrCl(Cp*)}2(μ-η2-η2-C2O4)] (0.0500 g, 0.0614 mmol) was reacted with Ag(CF3SO3) (0.0387 g, 0.151 mmol), followed by L (0.0148 g, 0.0614 mmol), yielding 3c as a mustard-yellow solid. Yield: 74% (0.0586 g, 0.0229 mmol); 1H NMR (300 MHz, [D6]-DMSO): δ(ppm) = 9.19–8.99 (m, 2H, Ha), 8.8–8.53 (m, 4H, Hb), 8.45–8.16 (m, 2H, Hc), 8.12–7.79 (m, 6H, Hf,d), 7.78–7.45 (m, 4H, He,g), 1.91–1.05 (m, 60H, Hh,h′); 13C{1H} NMR (100 MHz, [D6]-DMSO): δ (ppm) = 165.0, 164.1, 152.2, 151.6, 148.4, 142.7, 134.8, 128.4, 127.0, 123.2, 122.3, 119.1, 115.9, 94.5, 93.9, 93.5, 90.9, 83.9, 8.3, 7.9; IR (ATR) (vmax/cm−1): 1627 (C
O), 1250 (CF3 sym), 1222 (SO3 asym), 1155 (CF3 asym), 1019 (SO3 sym); M.P. (°C): onset of decomp. without melting = 271.8; HR-MS (ESI (+), m/z): 492.0915 (32%, [M − 4OTf]4+), calculated 491.8040; 1133.0420 (53%, [M − 2OTf]2+), calculated 1132.6900, 717.0756 (95%, [IrC24H24N2Cl]+(CF3SO3)−), calculated 717.2220.
[{Ru(p-cymene)}4(μ-η2-η2-C2O4)(μ-L)2](CF3SO3)4, (3d). AgOTf (0.0685 g, 0.267 mmol), [{RuCl(p-cymene)}2(μ-η2-η2-C2O4)] (0.0700 g, 0.111 mmol) and L (0.0268 g, 0.111 mmol) yielded 3d as a yellow solid. Yield: 71% (0.0867 g, 0.0395 mmol); 1H NMR (300 MHz, [D6]-DMSO): δ(ppm) = 9.11 (d, 2H, Ha/a′, 3JH–H = 4.4 Hz), 9.05 (d, 2H, Ha/a′, 3JH–H = 4.3 Hz), 8.81 (d, 4H, Hb/b′, 3JH–H = 6.3 Hz), 8.66 (d, 4H, Hb/b′, 3JH–H = 6.3 Hz), 8.26 (d, 2H, Hc/c′, 3JH–H = 1.8 Hz), 8.22 (d, 2H, Hc/c′, 3JH–H = 2.0 Hz), 7.90 (d, 4H, Hf/f′, 3JH–H = 6.4 Hz), 7.83 (d, 4H, Hf/f′, 3JH–H = 6.5 Hz), 7.77–7.67 (m, 4H, He,e′), 7.67–7.28 (m, 8H, Hd,d′, g,g′), 6.12 (d, 8H, Hh/h′, 3JH–H = 6.1 Hz), 5.98 (d, 8H, Hh/h′, 3JH–H = 6.1 Hz), 5.85 (d, 8H, Hi/i′, 3JH–H = 5.9 Hz), 5.63 (d, 8H, Hi/i′, 3JH–H = 6.0 Hz), 2.92–2.69 (m, 8H, Hj,j′), 2.30–1.99 (m, 24H, Hk,k′), 1.44–1.09 (m, 48H, Hl,l′); 13C{1H} NMR (100 MHz, [D6]-DMSO): δ (ppm) = 172.4, 166.3, 164.4, 153.0, 151.6, 150.1, 148.4, 145.3, 145.0, 144.5, 143.1, 134.5, 128.8, 128.2, 128.0, 127.2, 126.1, 124.3, 123.8, 123.3, 121.8, 119.1, 102.7, 96.8, 80.4, 80.0, 33.0 24.0, 21.9, 20.6; IR (ATR) (vmax/cm−1): 1627 (C
O), 1251 (CF3 sym), 1224 (SO3 asym), 1152 (CF3 asym), 1028 (SO3 sym); M.P. (°C): onset of decomp. without melting = 188.0; HR-MS (ESI (+), m/z): 949.0110 (43%, [M − 2OTf]2+), calculated 948.3640, 425.9915 (57%, [Ru3C60H60N4O4Cl2]3+, calculated 425.0967.
X-ray crystallography
Suitable single crystals of the iridium binuclear complex 1c were grown by the slow evaporation of a saturated chloroform solution, at room temperature. Single-crystal X-ray diffraction data were collected on a Bruker KAPPA APEX II DUO diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Data collection was carried out at 100(2) K. The temperature was controlled by an Oxford Cryostream cooling system (Oxford Cryostat). Cell refinement and data reduction were performed using the program SAINT.92 The data were scaled and absorption correction performed using SADABS.93 The structure was solved by direct methods using SHELXS-9793 and refined by full-matrix least-squares methods based on F2 using SHELXL-201893 and using the graphics interface program X-Seed.94,95 The programs X-Seed and POV-Ray were used to prepare molecular graphic images. All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were placed in idealised positions and refined in riding models with Uiso assigned 1.2 or 1.5 times Ueq of their parent atoms and the bond distances were constrained to 0.95 Å, 0.98 Å, and 1.00 Å for different types of C–H.DFT calculations
The structure of the model of the metallarectangle was predicted by density functional theory (DFT) calculations, using the DMol3 interface in BIOVIA Materials Studio 2017 (v. 17.1.0.48, 2016, Dassault Systèmes, Vélizy-Villacoublay Cedex, France).96–98 The molecular structure was optimized at the DFT level of theory, using the GGA-PBE density functional (i.e. generalized gradient approximation with Perdew–Burke–Ernzerhof exchange energies) and DND basis set (i.e. double numerical basis set with d-polarization functions (DND), file 4.4).99 The quantum contributions of all electrons in the system were taken into consideration in the calculation. For quality of the calculation, the integration accuracy was set to coarse, with a SCF tolerance of 1.0 × 10−4 and a maximum number of 50 cycles. The spin polarization was restricted and a global orbital cut off set at 3.40 Å, with no solvation model, as the calculation was done in vacuo.Biological methods
:2:1 was added to all wells in columns 1–11. MilliQ water (140 μL) and NP-40 (305.5 μM, 40.0 μL) was added to column 12. NP-40 is a detergent used to mediate the formation of β-haematin. The respective test compounds (20.0 μL) were then added to column 12 and serially diluted two-fold to give a total of 11 concentrations. Column 1 thus served as a blank containing no test compound. Since the compounds were coloured, the plates were pre-read at 405 nm. A 25 mM stock solution of haematin was then prepared by dissolving haemin in DMSO and sonicating for 1 minute. A 178.8 μL aliquot of this haematin stock solution was suspended in acetate buffer (20.0 mL, 1 M, pH 4.8) and 100 μL of this suspension added to each well. The plates were covered and incubated at 37 °C for 6 hours in an incubator. The assay was analysed using the pyridine-ferrochrome method developed by Egan and co-workers.87 A solution of pyridine/MilliQ water/acetone/HEPES (2 M, pH 7.4) in a v/v ratio of 5:2:2:1 respectively, was prepared, and 32.0 μL of this solution added to each well, followed by acetone (60.0 μL) and mixed. The absorbance was recorded using a Thermo Scientific Multiscan GO plate reader at 405 nm. The IC50 values were obtained using a sigmoidal dose–response curves generated using Graph Pad Prism v.5.0 software.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the University of Cape Town and the National Research Foundation of South Africa (UID: 129288) is gratefully acknowledged. Dr Dale Taylor (H3D), Mr Virgil Verhoog and Mrs Sumaya Salie from the Department of Clinical Pharmacology (University of Cape Town) are gratefully acknowledged for their assistance with the pLDH assays. Professor Timothy Egan and Dr Roxanne Mohunlal from the Department of Chemistry (University of Cape Town) are also gratefully acknowledged for their assistance with the β-haematin inhibition studies.References
- https://www.who.int/news-room/feature-stories/detail/world-malaria-report-2019 .
- P. F. Salas, C. Herrmann and C. Orvig, Chem. Rev., 2013, 113, 3450–3492 CrossRef CAS PubMed.
- R. Carter and K. N. Mendis, Clin. Microbiol. Rev., 2002, 15, 564–594 CrossRef PubMed.
- M. Navarro, C. Gabbiani, L. Messori and D. Gambino, Drug Discovery Today, 2010, 15, 1070–1078 CrossRef CAS PubMed.
- L. Tilley, P. Loria and M. Foley, Antimalarial Chemotherapy, Humana Press Inc., Totowa, New Jersey, 2001 Search PubMed.
- R. A. Jones, S. S. Panda and C. D. Hall, Eur. J. Med. Chem., 2015, 97, 335–355 CrossRef CAS PubMed.
- Guidelines for the treatment of malaria, WHO report, http://helid.digicollection.org/pdf/s13418e/s13418e.pdf, 2006 Search PubMed.
- Guidelines for the treatment of malaria, WHO report, https://www.who.int/docs/default-source/documents/publications/gmp/guidelines-for-the-treatment-of-malaria-eng.pdf?sfvrsn=a0138b77_2, 2015 Search PubMed.
- Y. Bansal and O. Silakari, Eur. J. Med. Chem., 2014, 76, 31–42 CrossRef CAS PubMed.
- E. A. Ashley, M. Dhorda, R. M. Fairhurst, C. Amaratunga, P. Lim, S. Suon, S. Sreng, J. M. Anderson, S. Mao, B. Sam, C. Sopha, C. M. Chuor, C. Nguon, S. Sovannaroth, S. Pukrittayakamee, P. Jittamala, K. Chotivanich, K. Chutasmit, C. Suchatsoonthorn, R. Runcharoen, T. T. Hien, N. T. Thuy-Nhien, N. V. Thanh, N. H. Phu, Y. Htut, K.-T. Han, K. H. Aye, O. A. Mokuolu, R. R. Olaosebikan, O. O. Folaranmi, M. Mayxay, M. Khanthavong, B. Hongvanthong, P. N. Newton, M. A. Onyamboko, C. I. Fanello, A. K. Tshefu, N. Mishra, N. Valecha, A. P. Phyo, F. Nosten, P. Yi, R. Tripura, S. Borrmann, M. Bashraheil, J. Peshu, M. A. Faiz, A. Ghose, M. A. Hossain, R. Samad, M. R. Rahman, M. M. Hasan, A. Islam, O. Miotto, R. Amato, B. MacInnis, J. Stalker, D. P. Kwiatkowski, Z. Bozdech, A. Jeeyapant, P. Y. Cheah, T. Sakulthaew, J. Chalk, B. Intharabut, K. Silamut, S. J. Lee, B. Vihokhern, C. Kunasol, M. Imwong, J. Tarning, W. J. Taylor, S. Yeung, C. J. Woodrow, J. A. Flegg, D. Das, J. Smith, M. Venkatesan, C. V. Plowe, K. Stepniewska, P. J. Guerin, A. M. Dondorp, N. P. Day and N. J. White, N. Engl. J. Med., 2014, 371, 411–423 CrossRef PubMed.
- W. Sim, R. T. Barnard, M. A. T. Blaskovich and Z. M. Ziora, Antibiotics, 2018, 7, 1–15 CrossRef PubMed.
- S. Medici, M. Peana, G. Crisponi, V. M. Nurchi, J. I. Lachowicz, M. Remelli and M. A. Zoroddu, Coord. Chem. Rev., 2016, 327–328, 349–359 CrossRef CAS.
- G. Faa, C. Gerosa, D. Fanni, J. I. Lachowicz and V. M. Nurchi, Curr. Med. Chem., 2018, 25, 75–84 CrossRef CAS PubMed.
- S. Dilruba and G. V. Kalayda, Cancer Chemother. Pharmacol., 2016, 77, 1103–1124 CrossRef CAS PubMed.
- Z. Guo and P. J. Sadler, Angew. Chem., Int. Ed., 1999, 38, 1512–1531 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- U. Schatzschneider, in Advances in Bioorganometallic Chemistry, ed. T. Hirao and T. Moriuchi, Elsevier Inc., 1st edn, 2019, pp. 173–192 Search PubMed.
- J. Held, C. Supan, C. L. O. Salazar, H. Tinto, L. N. Bonkian, A. Nahum, B. Moulero, A. Sié, B. Coulibaly, S. B. Sirima, M. Siribie, N. Otsyula, L. Otieno, A. M. Abdallah, R. Kimutai, M. Bouyou-Akotet, M. Kombila, K. Koiwai, C. Cantalloube, C. Din-Bell, E. Djeriou, J. Waitumbi, B. Mordmüller, D. Ter-Minassian, B. Lell and P. G. Kremsner, Lancet Infect. Dis., 2015, 15, 1409–1419 CrossRef CAS PubMed.
- T. R. Cook, Y. R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- W. X. Gao, H. J. Feng, B. B. Guo, Y. Lu and G. X. Jin, Chem. Rev., 2020, 120, 6288–6325 CrossRef CAS PubMed.
- J.-Q. Wang, C.-X. Ren and G.-X. Jin, Organometallics, 2006, 25, 74–81 CrossRef CAS.
- Y. F. Han, W. G. Jia, W. B. Yu and G. X. Jin, Chem. Soc. Rev., 2009, 38, 3419–3434 RSC.
- Y. F. Han, W. G. Jia, Y. J. Lin and G. X. Jin, Organometallics, 2008, 27, 5002–5008 CrossRef CAS.
- Y. Han and G. Jin, Acc. Chem. Res., 2014, 47, 3571–3579 CrossRef CAS PubMed.
- L. L. Ma, J. Q. Han, W. G. Jia and Y. F. Han, Beilstein J. Org. Chem., 2018, 14, 2027–2034 CrossRef CAS PubMed.
- J. Mattsson, P. Govindaswamy, A. K. Renfrew, P. J. Dyson, P. Stepnicka, B. Suss-Fink and B. Therrien, Organometallics, 2009, 28, 4350–4357 CrossRef CAS.
- A. Dubey, J. W. Min, H. J. Koo, H. Kim, T. R. Cook, S. C. Kang, P. J. Stang and K. W. Chi, Chem. – Eur. J., 2013, 19, 11622–11628 CrossRef CAS PubMed.
- G. L. Wang, Y. J. Lin and G. X. Jin, J. Organomet. Chem., 2010, 695, 1225–1230 CrossRef CAS.
- Y. F. Han, Y. J. Lin, W. G. Jia and G. X. Jin, Organometallics, 2008, 27, 4088–4097 CrossRef CAS.
- Y.-F. Han, Y.-J. Lin, W.-G. Jia, L.-H. Weng and G.-X. Jin, Organometallics, 2007, 26, 5848–5853 CrossRef CAS.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef CAS PubMed.
- B. Therrien, Eur. J. Inorg. Chem., 2009, 2009, 2445–2453 CrossRef.
- Y. Yamamotoa, H. Suzuki, N. Tajima and K. Tatsumi, Chem. – Eur. J., 2002, 8, 372–379 CrossRef CAS.
- K. Severin, Chem. Commun., 2006, 3859–3867 RSC.
- T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K. Chi, Acc. Chem. Res., 2013, 46, 2464–2474 CrossRef CAS PubMed.
- H. N. Zhang, Y. J. Lin and G. X. Jin, J. Am. Chem. Soc., 2021, 143, 1119–1125 CrossRef CAS PubMed.
- P. F. Cui, Y. J. Lin, Z. H. Li and G. X. Jin, J. Am. Chem. Soc., 2020, 142, 8532–8538 CrossRef CAS PubMed.
- Y. Lu, D. Liu, Y.-J. Lin, Z.-H. Li and G.-X. Jin, Natl. Sci. Rev., 2020, 7, 1548–1556 CrossRef CAS.
- C. Y. Zhu, M. Pan and C. Y. Su, Isr. J. Chem., 2018, 59, 209–219 CrossRef.
- Y. F. Han, H. Li and G. X. Jin, Chem. Commun., 2010, 46, 6879–6890 RSC.
- J. Mattsson, P. Govindaswamy, J. Furrer, Y. Sei, K. Yamaguchi, B. Suss-Fink and B. Therrien, Organometallics, 2008, 27, 4346–4356 CrossRef CAS.
- N. P. E. Barry and B. Therrien, Eur. J. Inorg. Chem., 2009, 2009, 4695–4700 CrossRef.
- B. Therrien, G. Süss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem., Int. Ed., 2008, 47, 3773–3776 CrossRef CAS PubMed.
- J. Mattsson, O. Zava, A. K. Renfrew, Y. Sei, K. Yamaguchi, P. J. Dyson and B. Therrien, Dalton Trans., 2010, 39, 8248–8255 RSC.
- N. P. E. Barry, O. Zava, P. J. Dyson and B. Therrien, Chem. – Eur. J., 2011, 17, 9669–9677 CrossRef CAS PubMed.
- A. Casini, B. Woods and M. Wenzel, Inorg. Chem., 2017, 56, 14715–14729 CrossRef CAS PubMed.
- S. L. Huang, Y. J. Lin, T. S. Hor and G. X. Jin, J. Am. Chem. Soc., 2013, 135, 8125–8128 CrossRef CAS PubMed.
- N. P. Barry, F. Edafe and B. Therrien, Dalton Trans., 2011, 40, 7172–7180 RSC.
- V. Vajpayee, Y. J. Yang, S. C. Kang, H. Kim, I. S. Kim, W. Ming, P. J. Stang and K. W. Chi, Chem. Commun., 2011, 47, 5184–5186 RSC.
- H. S. Song, Y. H. Song, N. Singh, H. Kim, H. Jeon, I. Kim, S. C. Kang and K.-W. Chi, Sci. Rep., 2019, 9, 1–13 Search PubMed.
- H. Vardhan, A. Nafady, A. M. Al-Enizi, K. Khandker, H. M. El-Sagher, G. Verma, M. Acevedo-Duncan, T. M. Alotaibi and S. Ma, Molecules, 2019, 24, 2284 CrossRef CAS PubMed.
- G. Gupta, A. Das, S. W. Lee, J. Y. Ryu, J. Lee, N. Nagesh, N. Mandal and C. Y. Lee, J. Organomet. Chem., 2018, 868, 86–94 CrossRef CAS.
- G. Gupta, A. Das, J. Lee, N. Mandal and C. Y. Lee, ChemPlusChem, 2018, 83, 339–347 CrossRef CAS PubMed.
- G. Gupta, B. S. Murray, P. J. Dyson and B. Therrien, Materials, 2013, 6, 5352–5366 CrossRef PubMed.
- G. Gupta, J. M. Kumar, A. Garci, N. Nagesh and B. Therrien, Molecules, 2014, 19, 6031–6046 CrossRef PubMed.
- A. Pothig and A. Casini, Theranostics, 2019, 9, 3150–3169 CrossRef PubMed.
- Y. Zhao, L. Zhang, X. Li, Y. Shi, R. Ding, M. Teng, P. Zhang, C. Cao and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4090–4098 CrossRef CAS PubMed.
- A. Mishra, S. C. Lee, N. Kaushik, T. R. Cook, E. H. Choi, N. K. Kaushik, P. J. Stang and K. W. Chi, Chem. – Eur. J., 2014, 20, 14410–14420 CrossRef CAS PubMed.
- Y. R. Zheng, K. Suntharalingam, P. M. Bruno, W. Lin, W. Wang, M. T. Hemann and S. J. Lippard, Inorg. Chim. Acta, 2016, 452, 125–129 CrossRef CAS PubMed.
- S. M. McNeill, D. Preston, J. E. Lewis, A. Robert, K. Knerr-Rupp, D. O. Graham, J. R. Wright, G. I. Giles and J. D. Crowley, Dalton Trans., 2015, 44, 11129–11136 RSC.
- Z. Yue, H. Wang, Y. Li, Y. Qin, L. Xu, D. J. Bowers, M. Gangoda, X. Li, H. B. Yang and Y. R. Zheng, Chem. Commun., 2018, 54, 731–734 RSC.
- N. P. Barry, F. Edafe, P. J. Dyson and B. Therrien, Dalton Trans., 2010, 39, 2816–2820 RSC.
- A. Casini and J. D. Crowley, Front. Chem., 2019, 7, 293 CrossRef PubMed.
- H. Sepehrpour, W. Fu, Y. Sun and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14005–14020 CrossRef CAS PubMed.
- N. Kudo, M. Perseghini and G. C. Fu, Angew. Chem., Int. Ed., 2006, 45, 1282–1284 CrossRef CAS PubMed.
- P. Govindaswamy, G. Süss-Fink and B. Therrien, Organometallics, 2007, 26, 915–924 CrossRef CAS.
- E. H. Wi, J. Y. Ryu, S. G. Lee, U. Farwa, M. Pait, S. Lee, S. Cho and J. Lee, Inorg. Chem., 2019, 58, 11493–11499 CrossRef CAS PubMed.
- P. Govindaswamy, G. Süss-Fink and B. Therrien, Inorg. Chem. Commun., 2007, 10, 1489–1492 CrossRef CAS.
- B. Therrien, Coord. Chem. Rev., 2009, 253, 493–519 CrossRef CAS.
- E. Ekengard, K. Kumar, T. Fogeron, C. de Kock, P. J. Smith, M. Haukka, M. Monarid and E. Nordlander, Dalton Trans., 2016, 45, 3905–3917 RSC.
- P. Chellan, K. M. Land, A. Shokar, A. Au, S. H. An, D. Taylor, P. J. Smith, K. Chibale and G. S. Smith, Organometallics, 2013, 32, 4793–4804 CrossRef CAS.
- K.-G. Liu, X.-Q. Cai, X.-C. Li, D.-A. Qin and M.-L. Hu, Inorg. Chim. Acta, 2012, 388, 78–83 CrossRef CAS.
- D. C. Caskey, T. Yamamoto, C. Addicott, R. K. Shoemaker, J. Vacek, A. M. Hawkridge, D. C. Muddiman, G. S. Kottas, J. Michl and P. J. Stang, J. Am. Chem. Soc., 2008, 130, 7620–7628 CrossRef CAS PubMed.
- L. Tilley, P. Loria and M. Foley, in Antimalarial Chemotherapy, ed. P. J. Rosenthal, Humana Press Inc., Totowa, NJ, 2001, pp. 87–121 Search PubMed.
- S. K. Singh and S. Singh, Int. J. Pharm. Sci. Rev. Res., 2014, 25, 295–302 Search PubMed.
- R. A. Sánchez-Delgado, M. Navarro, H. Pérez and J. A. Urbina, J. Med. Chem., 1996, 39, 1095–1099 CrossRef PubMed.
- C. S. K. Rajapakse, A. Martínez, B. Naoulou, A. A. Jarzecki, L. Suárez, C. Deregnaucourt, V. Sinou, J. Schrével, E. Musi, G. Ambrosini, G. K. Schwartz and R. A. Sánchez-Delgado, Inorg. Chem., 2009, 48, 1122–1131 CrossRef CAS PubMed.
- D. R. Melis, C. B. Barnett, L. Wiesner, E. Nordlander and G. S. Smith, Dalton Trans., 2020, 49, 11543–11555 RSC.
- D. E. Goldberg, A. F. G. Slater, A. Cerami and G. B. Henderson, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 2931–2935 CrossRef CAS PubMed.
- S. E. Francis, J. David, J. Sullivan and D. E. Goldberg, Annu. Rev. Microbiol., 1997, 51, 97–123 CrossRef CAS PubMed.
- M. Navarro, W. Castro and C. Biot, Organometallics, 2012, 31, 5715–5727 CrossRef CAS.
- M. Mushtaque and Shahjahan, Eur. J. Med. Chem., 2015, 90, 280–295 CrossRef CAS PubMed.
- M. Foley and L. Tilley, Pharmacol. Ther., 1998, 79, 55–87 CrossRef CAS PubMed.
- R. D. Sandlin, M. D. Carter, P. J. Lee, J. M. Auschwitz, S. E. Leed, J. D. Johnson and D. W. Wright, Antimicrob. Agents Chemother., 2011, 55, 3363–3369 CrossRef CAS PubMed.
- M. D. Carter, V. V. Phelan, R. D. Sandlin, B. O. Bachmann and D. W. Wright, Comb. Chem. High Throughput Screening, 2010, 13, 285–292 CrossRef CAS PubMed.
- K. K. Ncokazi and T. J. Egan, Anal. Biochem., 2005, 338, 306–319 CrossRef CAS PubMed.
- C. White, A. Yates and P. M. Maitlis, in Inorganic Syntheses, ed. D. M. Heinekey, John Wiley & Sons Inc., 1992, vol. 29, ch. 53, pp. 228–234 Search PubMed.
- M. A. Bennet, T.-N. Huang, T. W. Matheson and A. K. Smith, in Inorganic Syntheses, ed. S. Ittel and W. Nickerson, John Wiley & Sons Inc., 1982, vol. XXI, ch. 16, pp. 75–78 Search PubMed.
- H. Yan, G. Süss-Fink, A. Neels and H. Stoeckli-Evans, Dalton Trans., 1997, 4345–4350 RSC.
- A. Bacchi, G. Cantoni, P. Pelagatti and S. Rizzato, J. Organomet. Chem., 2012, 714, 81–87 CrossRef CAS.
- SAINT, Version 7.60a, Bruker AXS Inc., Madison, WI, USA, 2006 Search PubMed.
- G. M. Sheldrick, SHELXS-97, SHELXL-2018/3 and SADABS version 2.05, University of Göttingen, Germany, 1997 Search PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- J. L. Atwood and L. J. Barbour, Cryst. Growth Des., 2003, 3, 3–8 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- BIOVIA Materials Studio, https://www.materials-studio.com/products/collaborative-science/biovia-materials-studio, accessed: 07 Dec 2020.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- W. Trager and J. Jensen, Science, 1976, 193, 673–675 CrossRef CAS PubMed.
- M. T. Makler and D. J. Hinrichs, Am. J. Trop. Med. Hyg., 1993, 48, 205–210 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2089889 (1c). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt02842a |
| This journal is © The Royal Society of Chemistry 2021 |