
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Steric effects on acetate-assisted cyclometallation of meta-substituted N-phenyl and N-benzyl imidazolium salts at [MCl2Cp*]2 (M = Ir, Rh)†
David L.
Davies
 *,
Kuldip
Singh
and
Neringa
Tamosiunaite
*,
Kuldip
Singh
and
Neringa
Tamosiunaite
Department of Chemistry, University of Leicester, Leicester LE1 7RH, UK. E-mail: dld3@le.ac.uk
First published on 31st August 2021
Abstract
meta-Substituted N-phenyl,N′-methyl and N-benzyl,N′-methyl imidazolium salts undergo acetate-assisted cyclometallation to provide mixtures of ortho and para substituted cyclometallated complexes. The effect of the substituents on the isomer ratios is discussed; steric effects are more important in the 6-membered rings derived from the N-benzyl imidazolium salts than 5-membered rings from the N-phenyl salts. Comparisons are made to steric effects with some other common directing groups.
Introduction
Carboxylate-assisted cyclometallation is now a very well-established reaction both stoichiometrically and in catalysis.1 Whilst there are now hundreds of examples in catalysis there are still relatively few publications that focus on a detailed understanding of the steric and electronic influences on the cyclometallation step. Acetate assisted cyclometallation at Cp*M (M = Ir, Rh) centres proceeds from [M(OAc)2Cp*] which can be accessed by the reaction of [MCl2Cp*]2 with NaOAc.2 Cyclometallation consists of a number of steps (i) coordination of the directing group, (ii) possible anion loss, (iii) proton transfer to form coordinated carboxylic acid, (iv) substitution of carboxylic acid either by halide if stoichiometric, or by another substrate in catalysis. Recently, we described steric and electronic effects on acetate assisted cyclometallation of phenyl pyrazoles at Cp*M (M = Ir, Rh)3 and (arene)Ru centres.4 We showed that cyclometallation is kinetically favoured at electron rich phenyl groups but thermodynamically at electron poor ones. For meta substituted substrates steric factors were particularly important in controlling the ortho/para selectivity with para isomers being favoured thermodynamically for all substituents studied except fluorine. DFT studies surprisingly showed that initial proton transfer to form ortho isomers could actually be favoured over the para isomers, even for sterically bulky substituents. However, in those cases loss of coordinated acetic acid from the ortho isomer was significantly more endergonic leading to fast reverse proton transfer, which could only be detected by H/D exchange. The only exceptions to this were meta-fluorinated phenyl rings which always favoured the ortho-fluorine substituted products; a preference that is well precedented in other systems and is known as the “ortho effect”.5Jones et al. compared the effect of different directing groups phenylimines L1-R and phenylpyridines L2-R with [MCl2Cp*]2 (M = Ir, Rh) on the regioselectivity of cyclometallation of differently meta-substituted phenyls (Scheme 1).6 We have subsequently examined related reactions with phenylpyrazoles L3-R3 and re-examined some phenylpyridines.7 In making comparisons between different directing groups it is important to bear in mind that the ortho![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : para ratios of the products can vary over time. Therefore, ideally, final ratios corresponding to thermodynamic ratios should be compared. Jones et al. left their reactions a set amount of time and there is no mention of whether the ratios changed over time. It should be noted that reactions at Cp*Ir are faster and less easily reversible than those at Cp*Rh and those with electron donating substituents equilibrate faster than those with electron withdrawing substituents.3,6,8
: para ratios of the products can vary over time. Therefore, ideally, final ratios corresponding to thermodynamic ratios should be compared. Jones et al. left their reactions a set amount of time and there is no mention of whether the ratios changed over time. It should be noted that reactions at Cp*Ir are faster and less easily reversible than those at Cp*Rh and those with electron donating substituents equilibrate faster than those with electron withdrawing substituents.3,6,8
 | ||
| Scheme 1 Meta-substituted phenylimines L1-R,6 phenylpyridines L2-R,6,7 and phenylpyrazoles L3-R,3 and their products from acetate assisted cyclometallation with [MCl2Cp*]2 (M = Ir, Rh).6 | ||
The ortho:para ratio of products for L1-3 are shown in Table 1. As can be seen for the larger substituents CF3 and Me the para isomer is heavily favoured. With less bulky substituents (R = OMe) two isomers were formed but the para-isomer was still preferred. However, for the F-substituted ligands the ortho isomer was favoured in all cases. Jones et al. suggested that the selectivity of meta-substituted phenylpyridines L2-R was slightly less than with the imines because the phenyl imines are more bulky than the corresponding pyridines. However, this seems to be mainly based on the selectivity with L2-CF3, results which we were unable to reproduce. In our study L2-CF3 gave only the para isomer for both metals.
:para ratios of acetate-assisted cyclometallation of meta-substituted ligands L1-R,6L2-R6,7 and L3-R3
| L1-R | L2-R | L3-R | ||||
|---|---|---|---|---|---|---|
|
a Jones reported ortho:para ratios of 1:1.1 and 1:2.5 for Ir and Rh respectively but we found these changed with further heating.
b Jones reported a small amounts of a second species presumed to be the ortho isomer,6 however the selectivity with Rh is usually higher than with Ir hence it is likely that the minor species is a very small amount of an impurity.
c Jones reported ortho:para ratios of 1:6.4 and 1:8.4 for Ir and Rh respectively. However, we found no evidence for ortho isomers in the 19F or 1H NMR spectra.
|
||||||
| R | Ir, | Rh | Ir | Rh | Ir | Rh |
| OMe | 1:1.7 |
1:1.7 |
1:2.5a |
1:3.0a |
1:1.4 |
1:3.9 |
| Me | para only | para only | para only | para onlyb | 1:10 |
para only |
| CF3 | para only | para only | para only | para only | para only | para only |
| F | 2.3:1 |
8.5:1 |
3.4:1 |
11:1 |
40:1 |
44:1 |
All the ligands mentioned above cyclometallate to form five membered rings. Formation of six-membered rings by acetate assisted cyclometallation is known, though it is less facile than for five-membered rings. For example, cyclometallation of 2-phenylpyridine with [IrCl2Cp*]2 is complete within 4 hours,9 whilst the corresponding reaction of 2-benzylpyridine takes 20 hours.10 In addition, 2-phenylpyridines react with both Ir and Rh,6,9 whilst 2-benzylpyridine was only shown to give a complex with Ir and not Rh.6
Both five and six-membered cyclometallated ring complexes with NHCs are well known11 however in nearly all cases the phenyl that is activated has a para-substituent so only one product can be formed with the substituent meta to the metal. Here we examine acetate assisted cyclometallation of meta-substituted N-phenyl and N-benzyl imidazolium salts to consider the effect of directing group and ring size on steric effects on the cyclometallation.
Results and discussion
To examine NHCs as donor ligands we studied cyclometallation of meta-substituted N-phenyl, N′-methyl imidazolium salts L4-R (R = OMe, F, CF3, CN) which were prepared as shown in Scheme 2 in high yields (72–98%). L4-OMe, L4-F and L4-CN are new compounds, whilst L4-CF3 is known as the iodide salt.12 | ||
| Scheme 2 Preparation and NMR labelling scheme of meta-substituted N-phenyl,N′-methyl limidazolium salts L4-R. | ||
The reactions of L4-R with [MCl2Cp*]2 (M = Ir, Rh) were carried out in the presence of NaOAc at 75 °C in dichloroethane; however very low conversions were observed after heating overnight.13 The reactions were repeated in the presence of Et4NCl14 and proceeded slowly even at room temperature and gave high conversions (Scheme 3) and the products 4a/b-R (R = OMe, CN, CF3, F) were fully characterised.
 | ||
| Scheme 3 Reactions of L4-R with [MCl2Cp*]2 (M = Ir, Rh) and NMR labelling scheme. | ||
Each isomer has a characteristic pattern in the 1H NMR spectrum, for the para isomer H4 is a very narrow doublet and H2 shows an noe to the Cp* signal. The reactions were repeated and monitored at room temperature (ca. 20% conversion) and upon heating (50 °C overnight) to further investigate if the ratios change and so whether selectivity is kinetic or thermodynamic (see below).
All the reactions gave a mixture of two isomers and the ratios are shown in Table 2. In no case was an intermediate non-cyclometallated complex observed. This is consistent with activation of the imidazolium CH bond being relatively slow and the cyclometallation of the phenyl being much faster.15 The reactions of L4-R (R = OMe, CN, CF3) with both Ir and Rh showed that a mixture of the ortho and para-isomers was formed initially with increasing fraction of the para-isomer after heating (entries 1–3 Table 2).
:para ratios of acetate-assisted cyclometallation of meta-Substituted ligands L4-R in DCM/MeOH
| Entry | R | Ir | Rh | ||
|---|---|---|---|---|---|
| a In DCE at 50 °C after 1 hour and then after 6 hours. b Due to the small amount of minor species present it is not possible to unambiguously identify it as the ortho isomer. | |||||
| r.t. | 50 °C | r.t. | 50 °C | ||
| 1 | OMea | 1:1.5a |
1:3.0a |
1:3.0 |
1:3.0 |
| 2 | CN | 1:1.3 |
1:2.2 |
1.2:1 |
1:2.0 |
| 3 | CF3 | 1:3b |
1:>20b | 1:6b |
1:>40b |
| 4 | F | 2.2:1 |
2.2:1 |
6.0:1 |
10:1 |
This indicates that the para-isomer is thermodynamically favoured, whilst kinetically there is no clear preference for either the para or the ortho-isomers, except for R = CF3 which favours the para isomer kinetically and thermodynamically. For the reactions of L4-F both Ir and Rh favour the ortho isomer and with Rh the selectivity increases with heating indicating that the ortho isomer is favoured thermodynamically. This preference for ortho fluorine has been observed previously.3,5,6
Overall, the steric bulk mainly controls the regioselectivities in agreement with the results observed with phenylimines,6 phenylpyridines6,7 and phenylpyrazoles.3 However, in those cases none of the ortho-isomer was observed for the reactions with R = CF3, whilst it was present in substantial quantities for the reactions of L4-CF3 suggesting that there is less steric crowding at the metal centre in 4a/b-R compared to phenylimine,6 phenylpyridine,6,7 and phenylpyrazole complexes.3
To examine the effect of ring size on regioselectivity we examined cyclometallation of N-benzyl,N′-methyl imidazolium salts L5-R (R = OMe, CF3, F). These were prepared by reaction of N-methyl imidazole with an excess of appropriately meta-substituted benzyl chloride for 1–2 days. All three salts L5-R (R = OMe, CF3, F) were obtained in moderate to good yields (55–87%) and have been reported previously.16
The cyclometallated complexes 6a/b-R were prepared in a stepwise manner, transmetallation to form NHC bound complexes 5a/b-R followed by cyclometallation (Scheme 4).11a Thus, L5-R (R = OMe, CF3, F) was stirred with Ag2O in the dark for 1 hour to give a silver NHC complex. The reactions were filtered through Celite to remove the excess of Ag salts, and the resulting filtrate was reacted with [MCl2Cp*]2 (M = Ir, Rh) (Scheme 4) which after work up gave the new complexes 5a/b-R (R = OMe, CF3, F), in moderate to excellent yields (67–92%).
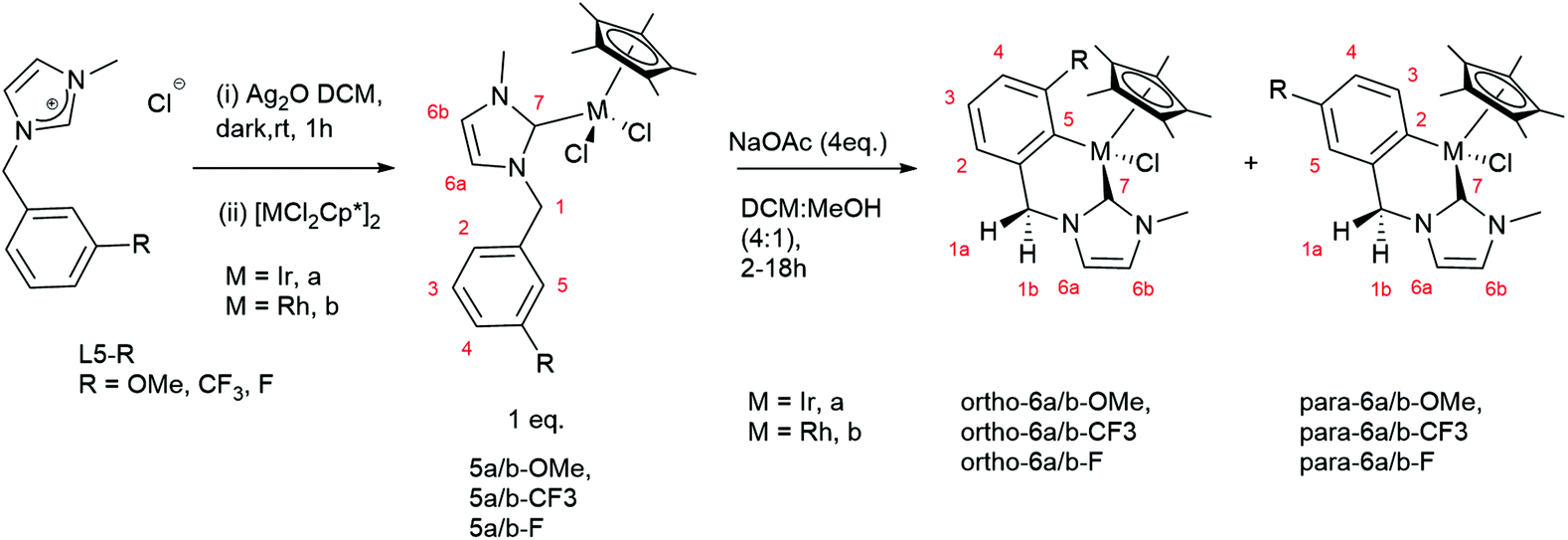 | ||
| Scheme 4 Synthesis and labelling of 6a/b-R. | ||
The 1H NMR spectra of 5a/b-R show two mutually coupled doublets at δ 5–6.5 due to the benzylic protons showing the chiral nature of the complexes with no mirror plane. Complex 5a-OMe gave crystals suitable for X-ray diffraction and the structure is shown in Fig. 1. The orientation of the NHC ligand confirms that the benzyl protons are inequivalent and that the benzyl group has not cyclometallated.
 | ||
| Fig. 1 The molecular structures of 5a-OMe with 50% ellipsoids. H-atoms omitted for clarity. Selected bond distances Å; Ir–C(1), 2.047(9), Ir–Cl(1) 2.417(2), Ir–Cl(2) 2.409(3). | ||
The cyclometallated complexes 6a/b-R were prepared in good to excellent yields (68–96%) by reaction of 5a/b-R (R = OMe, CF3, F) with NaOAc in DCM:MeOH (4:1) at room temperature (Scheme 4). The 1H NMR spectra of 6a/b-R show the mutually coupled benzylic proton doublets are closer together (between δ 4.5 and 5.0) than in complexes 5a/b-R. For both the OMe and CF3 substituted complexes only the para-isomer was observed in each case; for the F-substituted products 6a/b-F a mixture of both isomers (approximately 10:1) was formed favouring the ortho isomer in each case. The reactions were repeated in CDCl3, in which they were considerably slower, to measure the initial product ratios and see if these changed over time and with heating (Table 3).
:para ratios of acetate-assisted cyclometallation of complexes 5a/b-R
For the reactions of 5a/b-OMe (entry 1 Table 3) at low conversions (<20%), the ortho and para-isomers 6a/b-OMe were observed in 1:1 ratio. As the time and conversion increased the ratio between the two isomers changed significantly in favour of the para-isomer for both Ir (ortho:para 1:10) and Rh which showed only traces of the ortho-isomer. These results indicate that the para-isomer is favoured thermodynamically in this case, whilst there is almost no kinetic preference for either the ortho or para isomer.
This is consistent with the isolation of only the para-isomers 6a/b-OMe from the preparative reactions in DCM:MeOH (4:1). In the case of 5a/b-CF3 all the 1H NMR spectra irrespective of conversion only showed the para-isomer (as in DCM:MeOH). Based on our related work with phenylpyrazoles it is likely that formation of the ortho-isomer is significantly endergonic so is not observed. The cyclometallations of 5a/b-F led to the formation of 6a/b-F in 10:1 ortho![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :para ratio for both Ir and Rh, (entries 5 and 6) irrespective of percentage conversion. Approximately the same ratios were formed in DCM:MeOH (4:1) after heating for 50 °C for two days. As the ratios did not change it is likely that the kinetic and thermodynamic selectivity are similar.
:para ratio for both Ir and Rh, (entries 5 and 6) irrespective of percentage conversion. Approximately the same ratios were formed in DCM:MeOH (4:1) after heating for 50 °C for two days. As the ratios did not change it is likely that the kinetic and thermodynamic selectivity are similar.
The reversibility of the cyclometallation of the benzyl complexes was probed by deuteration studies as in similar studies.3–4,7,8b Thus, 5a/b-OMe, and 5b-CF3 were reacted with NaOAc in CD3OD and the percentage D-incorporation was determined by integration and the results are shown in Scheme 5.17 For 5a/b-OMe, a high D-incorporation (>80%) was observed in the para isomer products, para-6a-OMe and para-6b-OMe. The formation of the deuterated products shows that formation of the ortho-isomer had occurred but was easily reversible, ultimately leading to preferential formation of the thermodynamically favoured para-isomer (a more detailed scheme showing how D incorporation occurs is in the SI). This is consistent with entries 1 and 2 in Table 3 discussed above which show that the ortho:para ratio changes over time favouring the para isomer. Note, for Rh complex 5b-OMe the reaction only reached about 55% conversion overnight and the starting complex was deuterated at sites 2 and 5 showing that formation of both isomers is reversible under these conditions. No D-incorporation was detected for the cyclometallation of 5b-CF3. This result shows that either the formation of the ortho-isomer has a significantly higher activation barrier than that of the para-isomer so the para isomer is kinetically preferred and/or formation of the ortho-isomer is so easily reversible that there is no time for H/D-exchange. In addition, the lack of observation of deuterated starting material means that formation of para-6b-CF3 is exergonic so not easily reversible.
 | ||
| Scheme 5 Deuterium incorporation experiments with 5a/b-OMe and 5b-CF3. | ||
Comparing the regioselectivity of the phenyl and benzyl complexes the phenyl-NHC complexes 4a/b-R even for the largest substituent (R = CF3) show some ortho-isomer (25 and 14% for Ir and Rh respectively) and about 30% ortho-isomer was seen for R = OMe with both metals after heating. Whereas, with the benzyl complexes 6a/b-R for R = CF3 only the para isomer is observed and for R = OMe less than 10% of the ortho isomer is observed after heating. These results suggest a more sterically hindered metal centre for the six-membered rings leading to less of the ortho isomer. Steric distortion of bidentate NHC containing ligands has been analysed previously in terms of a “yaw”-distortion (see Fig. 2).18 A number of structures of Ir complexes have been reported for phenyl complexes (n = 0) the yaw angle varies from 9.2 to 10.2° whilst the benzyl complexes (n = 1) the yaw angles are much less at 2.1 to 3.4°.18b Interestingly the lower distortion in NHC coordination in the benzyl complexes leads the ortho H to be closer to the metal M⋯H distances 3.02 to 3.07 Å for benzyl complexes compared to 3.21 to 3.25 Å for the phenyl complexes. There are much smaller differences between 5-membered rings with different directing groups. The observation of a second species with L4-CF3 with an NHC directing group suggests the steric hindrance is slightly less in this case than with L1-3 however it should be borne in mind that the NHC has an NMe substituent on the non-cyclometallated side, compared to a CH for the other ligands, and this may impact the overall geometry at the metal.
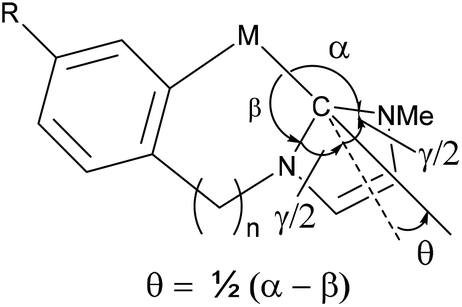 | ||
| Fig. 2 Yaw distortion of chelating NHC complexes. | ||
Conclusions
Steric effects on C–H activation were assessed using meta-substituted N-phenyl and N-benzylimidazolium salts L4-R and L5-R respectively. N-Phenyl limidazolium salts L4-R (R = OMe, CN, CF3, F) underwent cyclometallation easily in the presence of NaOAc14 and no intermediate non-cyclometallated NHC bound complexes were observed. The para-isomers were favoured thermodynamically over the ortho with both metals for R = OMe, CN, and particularly for the more bulky CF3, whilst at shorter reaction times the selectivity was less. For L4-F cyclometallation at both metals favoured the ortho isomer as has been observed in other systems.Cyclometallation to form 6-membered rings is less favourable than 5-membered ones hence for N-benzylimidazolium salts L5-R(R = OMe, CF3, F) intermediate non-cyclometallated NHC bound complexes 5a/b-OMe could be prepared by transmetallation and the cyclometallation studied as a separate step. Treatment of 5a/b-OMe with NaOAc resulted in formation of the ortho and para isomers of 6a/b-OMe initially (<20% conversion) in equal quantities. At high conversions the proportion of the ortho-isomers diminished to <10% for Ir and none for Rh indicating a thermodynamic preference for the para-isomer. For cyclometallation of 5a/b-CF3 none of the ortho-isomer could be detected even at low conversions likely due to the bulkier CF3 group. Therefore, steric effects control regioselectivity with the para-isomer being the major one for cyclometallation of 5a/b-R (R = OMe, CF3). The selectivity for the para-isomer observed in six membered ring complexes 6a/b-R is larger compared to the formation of the five-membered ring complexes 4a/b-R consistent with a more sterically hindered metal centre for the six-membered rings.
Experimental
meta-Substituted N-phenylimidazoles were prepared according to a modified literature procedure.19meta-Substituted arylhalide (1 eq.), imidazole (1.5 eq.), Cs2CO3 (2 eq.), CuO2 (10 mol%) and MeCN or DMF (5–10 mL) were added to a Schlenk flask, sealed with a screw-cap, placed under N2 atmosphere, partially evacuated, transferred to an oil bath and stirred at 100–120 °C for 1–5 days behind a blast shield. Afterwards the reaction mixture was cooled to rt, followed by filtration through Celite. The solvent was removed by rotary evaporation and pure phenylimidazole was obtained by column chromatography. Data were in agreement with the literature.20General procedure for preparation of meta-substituted phenylimidazolium salts L4-R
A nitrogen flushed Schlenk flask was charged with magnetic stirrer, meta-substituted phenylimidazole (1 eq.), dry DCM (4–6 mL), methyl trifluoromethanesulfonate (1.1 eq.), capped and stirred at rt for 2–4 h. The solvent was removed by rotary evaporation and the product was either precipitated from DCM/Et2O mixture or washed with Et2O to yield imidazolium salt L4-R.General procedure for cyclometallation of meta-substituted phenylimidazolium salts L4-R with [MCl2Cp*]2 (M = Ir, Rh) in DCE
[MCl2Cp*]2 (M = Ir, Rh) (1 eq.), NaOAc (8 eq.) were placed in an oven-dried Schlenk flask, sealed with a screw-cap, evacuated for 15 min and placed under N2 atmosphere. DCE (2 mL) was added and mixture stirred for another 15 min. The appropriate imidazolium salt L4-R (2.1 eq.) and Et4NCl (2 eq.) was added, and the Schlenk flask transferred to a preheated oil bath and stirred at 50 °C for 1–2 h, then at 70 °C for 1–6 h. The reaction mixture was cooled to rt with continuous stirring, diluted with DCM (10 mL), filtered through Celite and solvent removed by rotary evaporation. The pure products were isolated by several precipitations from DCM/hexane.General procedure for cyclometallation of meta-substituted phenylimidazolium salts L4-R with [MCl2Cp*]2 (M = Ir, Rh) in DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :MeOH
:MeOH
[MCl2Cp*]2 (M = Ir, Rh) (1 eq., 0.0251 mmol), NaOAc (8 eq.) were placed in an oven-dried Schlenk flask, sealed with a screw-cap, evacuated for 15 min and placed under N2 atmosphere. Dry DCM (1.6 mL) and MeOH (0.4 mL) were added and the mixture stirred for another 15 min. The appropriate imidazolium salt (2.1 eq.) and Et4NCl (2 eq.) was added and the mixture stirred at rt overnight and then heated to 50 °C overnight. The reactions were monitored by 1H NMR spectroscopy by comparing the relative integrations of the appropriate signals (H3 of the ortho-isomers compared to the H3 of the para-isomers).
:para ratio 1:2.2) as a yellow powder (29.9 mg, 94%). ortho-4a-OMe. 1H NMR (400 MHz, CDCl3): δ 1.85 (s, 15H, C5Me5), 3.82 (s, 3H, OMe), 3.97 (s, 3H, Me), 6.54 (dd, J = 7.9, 1.1 Hz, 1H, H3), 6.83 (dd, J = 7.7, 1.1 Hz, 1H, H1), 6.93 (t, J = 8.3 Hz, 1H, H2), 6.95 (d, J = 2.2 Hz, 1H, H5b), 7.31 (d, J = 2.2 Hz, 1H, H5a). ESIMS: m/z 513 [M − Cl]+. HRMS (ESI): Calcd for C21H26N2O193Ir [M − Cl]+ 515.1674, found 515.1675. para-4a-OMe. 1H NMR (400 MHz, CDCl3): δ 1.79 (s, 15H, C5Me5), 3.80 (s, 3H, OMe), 3.98 (s, 3H, Me), 6.65 (dd, J = 8.2, 2.6 Hz, 1H, H3), 6.76 (d, J = 2.4 Hz, 1H, H4), 6.97 (d, J = 2.2 Hz, 1H, H5b), 7.29 (d, J = 2.2 Hz, 1H, H5a), 7.60 (d, J = 8.3 Hz, 1H, H2), 13C{1H} NMR (101 MHz, CDCl3): δ 9.74 (C5Me5), 37.0 (Me), 55.5 (OMe), 90.6 (C5Me5), 98.6 (C4), 110.8 (C3), 114.8 (C5a), 121.3 (C5b), 131.2, 136.1 (C2), 146.9 (C1), 156.3, 166.6 (C6). ESIMS: m/z 513 [M − Cl]+. HRMS (ESI): Calcd for C21H26N2O193Ir [M − Cl]+ 515.1674, found 515.1675.
:para 1:1.9 ratio) as an orange-yellow powder (14 mg, 61%). ortho-4b-OMe. 1H NMR (400 MHz, CDCl3): δ 1.68 (s, 15H, C5Me5), 3.83 (s, 3H, OMe), 3.97 (s, 3 H, Me), 6.58 (dd, J = 8.0, 0.9 Hz, 1H, H3), 6.78 (dd, J = 7.7, 1.0 Hz, 1H, H1), 6.98 (d, J = 2.0 Hz, 1H, H5b), 6.99 (t, J = 7.8 Hz, 1H, H2), 7.37 (d, J = 2.0 Hz, 1H, H5a), 13C{1H} NMR (101 MHz, CDCl3): δ 9.9 (C5Me5), 37.0 (Me), 56.3 (OMe), 97.9 (d, 1JC–Rh = 5.2 Hz, C5Me5), 104.9 (C1), 108.9 (C3), 115.9 (C5a), 121.9 (C5b), 124.0 (C2), 146.1, 145.6 (d, 1JC–Rh = 42.1 Hz, C4), 156.7, 184.2 (d, 1JC–Rh = 55.5 Hz, C6). ESIMS: m/z 425 [M − Cl]+. HRMS (ESI): Calcd for C21H26N2O103Rh [M − Cl]+ 425.1100, found 425.1100. para-4b-OMe. 1H NMR (400 MHz, CDCl3): δ 1.70 (s, 15H, C5Me5), 3.78 (s, 3H, OMe), 4.01 (s, 3H, Me), 6.66 (dd, J = 8.3, 2.6 Hz, 1H, H3), 6.71 (d, J = 2.5 Hz, 1H, H4), 6.98 (d, J = 2.1 Hz, 1H, H5b), 7.35 (d, J = 2.0 Hz, 1H, H5a), 7.63 (d, J = 8.3 Hz, 1H, H2), 13C{1H} NMR (101 MHz, CDCl3): δ 9.9 (C5Me5), 37.0 (Me), 55.5 (OMe), 97.2 (d, 1JC–Rh = 5.0 Hz, C5Me5), 99.0 (C4), 110.5 (C3), 115.1 (C5a), 122.2 (C5b), 137.2 (C2), 146.1, 146.6 (d, 1JC–Rh = 35.7 Hz, C1), 165.0, 184.2 (d, JC–Rh = 55.5 Hz, C6). ESIMS: m/z 425 [M−Cl]+. HRMS (ESI): Calcd for C21H26N2O103Rh [M−Cl]+ 425.1100, found 425.1100.
:para ratio 1:2.0) as an orange-yellow powder (24 mg, 90%). ortho-4a-CN. 1H NMR (400 MHz, CDCl3): δ 1.83 (s, 15H, C5Me5), 3.98 (s, 3H, Me), 7.00 (t, J = 7.8 Hz, 1H, H2), 7.01 (d, J = 2.6 Hz, 1H, H5b), 7.21 (dd, J = 7.8, 1.2 Hz, 1H, H1), 7.33 (d, J = 2.2 Hz, 1H, H5a), 7.37 (dd, J = 7.7, 1.2 Hz, 1H, H3), 13C{1H} NMR (101 MHz, CDCl3): δ 9.6 (C5Me5), 36.8 (Me), 92.6 (C5Me5), 94.8, 112.5 (C1), 115.6 (C5a), 120.3, 121.6 (C2), 122.0 (C5b), 131.2 (C3), 148.0, 151.1 (C4), 167.9 (C6). ESIMS: m/z 510 [M − Cl]+. HRMS (ESI): Calcd for C21H23193IrN3 [M − Cl]+ 510.1521, found 510.1523. ESIMS: m/z 551 [M − Cl + MeCN]+. HRMS (ESI): Calcd for C23H26N4193Ir [M − Cl + MeCN]+ 551.1787, found 551.1790. para-4a-CN. 1H NMR (400 MHz, CDCl3): δ 1.79 (s, 15H, C5Me5), 3.99 (s, 3H, Me), 7.03 (d, J = 2.2 Hz, 1H, H5b), 7.21 (dd, J = 7.7 Hz, 1.7, 1H, H3), 7.32 (d, J = 1.7 Hz, 1H, H4), 7.34 (d, J = 2.1 Hz, 1H, H5a), 7.88 (d, J = 7.7 Hz, 1H, H2), 13C{1H} NMR (101 MHz, CDCl3): δ 9.7 (C5Me5), 37.0 (Me), 91.8 (C5Me5), 104.6, 112.6 (C4), 114.9 (C5a), 122.2 (C5b), 122.6, 129.0 (C3), 137.2 (C2), 147.3, 153.4 (C1), 166.2 (C6). ESIMS: m/z 510 [M − Cl]+. HRMS (ESI): Calcd for C21H23193IrN3 [M − Cl]+ 510.1521, found 510.1523. ESIMS: m/z 551 [M − Cl + MeCN]+. HRMS (ESI): Calcd for C23H26N4193Ir [M − Cl + MeCN]+ 551.1787, found 551.1790.
:para ratio 1:1.9) as an orange-yellow powder (13 mg, 58%). ortho-4b-CN. 1H NMR (400 MHz, CDCl3): δ 1.75 (s, 15H, C5Me5), 3.99 (s, 3H, Me), 7.01 (d, J = 2.2 Hz, 1H, H5b), 7.05 (t, J = 7.7 Hz, 1H, H2), 7.17 (dd, J = 7.8, 1.3 Hz, 1H, H1), 7.40 (m, 1H, H3), 7.41 (d, J = 2.0 Hz, 1H, H5a), 13C{1H} NMR (101 MHz, CDCl3): δ 9.7 (C5Me5), 37.1 (Me), 94.0, 98.8 (d, 1JC–Rh = 4.8 Hz, C5Me5), 113.1 (C1), 116.1 (C5a), 122.0, 123.1 (C2), 123.3 (C5b), 130.7 (C3), 147.3, 167.4 (d, 1JC–Rh = 38.2 Hz, C4), 185.1 (d, 1JC–Rh = 54.8 Hz, C6). ESIMS: m/z 420 [M − Cl]+. HRMS (ESI): Calcd for C21H23N3103Rh [M − Cl]+ 420.0947, found 420.0947. ESIMS: m/z 461 [M − Cl + MeCN]+. HRMS (ESI): Calcd for C23H26N4103Rh [M − Cl + MeCN]+ 461.1213, found 461.1213. para-4b-CN. 1H NMR (400 MHz, CDCl3): δ 1.71 (s, 15H, C5Me5), 4.00 (s, 3H, Me), 7.03 (d, J = 2.2 Hz, 1H, H5b), 7.27 (m, 1H, H3), 7.27 (br.s, 1H, H4), 7.41 (d, J = 2.1 Hz, 1H, H5a), 7.94 (dd, J = 8.1, 0.9 Hz, 1H, H2), 13C{1H} NMR (101 MHz, CDCl3): δ 9.8 (C5Me5), 37.1 (Me), 98.2 (d, 1JC–Rh = 5.6 Hz, C5Me5), 105.5, 112.8 (C4), 115.4 (C5a), 120.1, 123.1 (C5b), 128.0 (C3), 138.4 (C2), 146.6, 170.3 (d, 1JC–Rh = 35.8 Hz, C1), 183.7 (d, 1JC–Rh = 55.6 Hz, C6). Calcd for C21H23N3103Rh [M − Cl]+ 420.0947, found 420.0947. ESIMS: m/z 461 [M − Cl + MeCN]+. HRMS (ESI): Calcd for C23H26N4103Rh [M − Cl + MeCN]+ 461.1213, found 461.1213.
:para ratio 2.1:1) as a yellow powder (18 mg, 67%). ortho-4a-F. 1H NMR (400 MHz, CDCl3): δ 1.80 (d, 15 H, C5Me5), 3.97 (s, 3H, Me), 6.72 (m, 1H, H3), 6.94 (m, 2H, H1,H2), 6.97 (d, J = 2.1 Hz, 1H, H5b), 7.32 (d, J = 2.1 Hz, 1H, H5a), 13C{1H} NMR (101 MHz, CDCl3): δ 9.9 (d, JC–F = 2.7 Hz, C5Me5), 36.9 (Me), 91.6 (C5Me5), 106.6 (d, 4JC–F = 2.7 Hz, C1), 112.1 (d, 2JC–F = 29.4 Hz, C3), 115.5 (C5a), 121.4 (C5b), 123.8 (d, 3JC–F = 8.2 Hz, C2), 126.3 (d, 2JC–F = 44.6 Hz, C4), 148.5 (d, 3JC–F = 18.3 Hz), 166.5 (C6), 167.8 (d, 1JC–F = 235.0 Hz, C–F), 19F{1H} NMR (376 MHz, CDCl3): δ −94.9 (F). ESIMS: m/z 503 [M − Cl]+. HRMS (ESI): Calcd for C20H23N2F193Ir [M − Cl]+ 503.1674, found 503.1676. ESIMS: m/z 544 [M − Cl + MeCN]+. HRMS (ESI): Calcd for C22H26N3F193Ir [M − Cl + MeCN]+ 544.1740, found 544.1744. para-4a-F. 1H NMR (400 MHz, CDCl3): δ 1.79 (s, 15H, C5Me5), 3.99 (s, 3H, Me), 6.78 (ddd, J = 9.8, 8.3, 2.7 Hz, 1H, H3), 6.87 (dd, J = 9.5, 2.6 Hz, 1H, H4), 6.97 (d, J = 2.1 Hz, 1H, H5b), 7.27 (d, J = 2.1 Hz, 1H, H5a), 7.64 (dd, J = 8.3, 6.5 Hz, 1H, H2), 19F{1H} NMR (376 MHz, CDCl3): δ −122.9 (F). ESIMS: m/z 503 [M − Cl]+. HRMS (ESI): Calcd for C20H23193IrN2F [M − Cl]+ 503.1674, found 503.1676. ESIMS: m/z 544 [M − Cl + MeCN]+. HRMS (ESI): Calcd for C22H26N3F193Ir [M − Cl + MeCN]+ 544.1740, found 544.1744.
:para ratio 10:1) as an orange-yellow powder (13 mg, 59%).
General procedure for preparation of meta-substituted benzylimidazolium salts L5-R
A Schlenk flask was charged with N-methylimidazole (1 eq.), meta-substituted benzyl chloride (1–2 eq.) and MeCN (5 mL) and stirred at 55 °C for 1 day. The resulting mixture was concentrated in vacuo, the residue dissolved in DCM and washed with hexane, then the solvent removed by rotary evaporation giving pure imidazolium salt as an oil or a sticky solid.General procedure for complexation of meta-substituted benzylimidazolium salts L5-R with [MCl2Cp*]2 (M = Ir, Rh)
An aluminium foil wrapped Schlenk flask was charged with a magnetic stirrer bar, the appropriate meta-substituted benzylimidazolium salt L5-R (2.1 eq.) and Ag2O (2.2 eq.), capped, purged with N2. Dry DCM (2.5 mL) was added and the mixture stirred at rt for 1–2 h. Then the reaction mixture was filtered through Celite and the solvent removed by rotary evaporation. The residue was re-dissolved in dry DCM (2.5 mL) and added to an N2 purged Schlenk flask wrapped in aluminium foil, followed by addition of [MCl2Cp*]2 (M = Ir, Rh) (1 eq.). The reaction mixture was stirred at rt for 1–2 h, filtered through Celite, the solvent removed by rotary evaporation and the final product 5a/b-R obtained by precipitation/crystallisation from DCM/Et2O or DCM/hexane.General procedure for cyclometallation of 5a/b-R
An oven-dried and N2 purged Schlenk flask was charged with a magnetic stirrer bar, 5a/b-R (1 eq.), NaOAc (5 eq.) and dry DCM (2 mL) and MeOH (0.5 mL) and stirred at rt for indicated time. The reaction mixture was filtered through Celite, which was washed with additional DCM (5–10 mL), the solvent removed by rotary evaporation and the residue purified by precipitation from DCM/hexane.:para ratio 10:1) as a yellow powder (27 mg, 96%). ortho-6a-F. 1H NMR (400 MHz, CDCl3): δ 1.73 (s, 15H, C5Me5), 3.89 (s, 3H, Me), 4.71 (dd, J = 13.9, 1.3 Hz, 1H, H1b), 4.88 (d, J = 13.9 Hz, 1H, H1a), 6.76 (m, 3H, H2, H3, H4), 6.90 (d, J = 1.9 Hz, 1H, H6b), 6.94 (d, J = 2.0 Hz, 1H, H6a), 13C{1H} NMR (101 MHz, CDCl3): δ 9.4 (C5Me5), 37.0 (Me), 57.0 (C1), 90.04 (C5Me5), 114.2 (d, 2JC–F = 31.0 Hz, C4), 120.3 (C6a), 120.6 (d, 4JC–F = 1.6 Hz, C2), 121.6 (C6b), 123.8 (d, 3JC–F = 8.7 Hz, C3), 128.0 (d, 2JC–F = 38.9 Hz, C5), 141.0 (d, 3JC–F = 14.3 Hz, C), 156.4 (C7), 167.2 (d, 1JC–F = 232.9 Hz, C–F), 19F{1H} NMR (376 MHz, CDCl3): δ −87.8 (F). ESIMS: m/z 517 [M − Cl]+. HRMS (ESI): Calcd for C21H25N2F193Ir [M − Cl]+ 517.1631, found 517.1631. para-6a-F.1H NMR (400 MHz, CDCl3): δ 1.66 (s, 15H, C5Me5), 3.91 (s, 3H, Me), 4.6 (d, J = 14.1 Hz, 1H, H1a), 4.81 (d, J = 13.9 Hz, 1H, H1b), 6.71–6.74 (m, 2H, H4, H5), 6.89 (d, J = 1.9 Hz, 1H, H6b), 6.93 (d, J = 1.9 Hz, 1H, H6a), 7.51 (dd, J = 8.3, 7.0 Hz, 1H, H3), 19F{1H} NMR (376 MHz, CDCl3): δ −125.3 (F). ESIMS: m/z 427 [M − Cl]+. HRMS (ESI): ESIMS: m/z 517 [M − Cl]+. HRMS (ESI): Calcd for C21H25N2F193Ir [M − Cl]+ 517.1631, found 517.1631.
:para ratio 10:1) as a yellow powder (22.5 mg, 96%). ortho-6b-F. 1H NMR (400 MHz, CDCl3): δ 1.66 (s, 15H, C5Me5), 3.99 (s, 3H, Me), 4.71 (dd, J = 14.2, 1.0 Hz, 1H, H1a), 4.97 (d, J = 14.2 Hz, 1H, H1b), 6.76 (m, 2 H, H2, H4), 6.82 (m, 1H, H3), 6.97 (d, J = 1.9 Hz, 1H, H6b), 7.02 (d, J = 1.9 Hz, 1H, H6a), 13C{1H} NMR (101 MHz, CDCl3): δ 9.7 (C5Me5), 37.8 (Me), 56.4 (C1), 97.5 (d, 1JC–Rh = 5.6 Hz, C5Me5), 114.4 (d, 2JC–F = 31.0 Hz, C4), 120.9 (C6a), 121.3 (C2), 122.6 (C6b), 124.1 (d, 3JC–F = 8.7 Hz, C3), 141.60 (d, 3JC–F = 15.1 Hz, C), 142.8 (m, C5), 166.9 (d, 1JC–F = 232.1 Hz, C–F), 174.2 (d, 1JC–Rh = 54.8 Hz, C7), 19F{1H} NMR (376 MHz, CDCl3): δ −84.7 (F). m/z 427 [M − Cl]+. HRMS (ESI): Calcd for C21H25N2F103Rh [M − Cl]+ 427.1057, found 427.1049. para-6b-F. 1H NMR (400 MHz, CDCl3): δ 1.73 (s, 15H, C5Me5), 3.99 (s, 3H, Me), 4.68 (d, J = 14.2, 1.0 Hz, 1H, H1a), 4.92 (d, J = 14.2 Hz, 1H, H1b), 6.73 (dd, J = 10.0, 3.0 Hz, 1H, H5), 6.76 (td, J = 9.0, 2.8 Hz, 1H, H4), 6.97 (d, J = 1.9 Hz, 1H, H6b), 7.02 (d, J = 1.9 Hz, 1H, H6a), 7.61 (dd, J = 8.3, 7.0 Hz, 1H, H3), 19F{1H} NMR (376 MHz, CDCl3): δ −125.0 (F). ESIMS: m/z 427 [M − Cl]+. HRMS (ESI): Calcd for C21H25N2F103Rh [M − Cl]+ 427.1057, found 427.1049.
Deuteration studies
An NMR tube was charged with 5a/b-R (5 mg) and CD3OD (0.5 mL). The 1H NMR spectrum was recorded and then NaOAc (4 eq.) was added. The reactions were allowed to sit at rt overnight. The spectra in CD3OD were broad so the samples were evaporated and redissolved in CDCl3. The percentage deuteration was determined by 1H NMR spectroscopy by comparing the relative integrations for H3 and H5 for the para-isomers in the 1H NMR spectra.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for financial support through awards EP/J002917/1 and EP/J021709/1 (D.L.D.) and EUCOST Action CA15106 “C−H Activation in Organic Synthesis (CHAOS)”. NT thanks the Joy and George Fraser Bursary Fund for support.Notes and references
- (a) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (b) M. Albrecht, Chem. Rev., 2010, 110, 576–623 CrossRef CAS PubMed; (c) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC; (d) Y.-F. Han and G.-X. Jin, Chem. Soc. Rev., 2014, 43, 2799–2823 RSC; (e) A. Peneau, C. Guillou and L. Chabaud, Eur. J. Org. Chem., 2018, 5777–5794 CrossRef CAS.
- (a) D. L. Davies, O. Al-Duaij, J. Fawcett, M. Giardiello, S. T. Hilton and D. R. Russell, Dalton Trans., 2003, 4132–4138 RSC; (b) A. P. Walsh and W. D. Jones, Organometallics, 2015, 34, 3400–3407 CrossRef CAS.
- R. A. Alharis, C. L. McMullin, D. L. Davies, K. Singh and S. A. Macgregor, J. Am. Chem. Soc., 2019, 141, 8896–8906 CrossRef CAS PubMed.
- R. A. Alharis, C. L. McMullin, D. L. Davies, K. Singh and S. A. Macgregor, Faraday Discuss., 2019, 220, 386–403 RSC.
- (a) E. Clot, M. Besora, F. Maseras, C. Mégret, O. Eisenstein, B. Oelckers and R. N. Perutz, Chem. Commun., 2003, 490–491 RSC; (b) E. Clot, C. Mégret, O. Eisenstein and R. N. Perutz, J. Am. Chem. Soc., 2009, 131, 7817–7827 CrossRef CAS PubMed; (c) A. D. Selmeczy, W. D. Jones, M. G. Partridge and R. N. Perutz, Organometallics, 1994, 13, 522–532 CrossRef CAS.
- L. Li, W. W. Brennessel and W. D. Jones, Organometallics, 2009, 28, 3492–3500 CrossRef CAS.
- D. L. Davies, K. Singh and N. Tamosiunaite, Dalton Trans., 2020, 49, 2680–2686 RSC.
- (a) A. I. VanderWeide, W. W. Brennessel and W. D. Jones, J. Org. Chem., 2019, 84, 12960–12965 CrossRef CAS PubMed; (b) K. J. T. Carr, D. L. Davies, S. A. Macgregor, K. Singh and B. Villa-Marcos, Chem. Sci., 2014, 5, 2340–2346 RSC.
- Y. Boutadla, O. Al-Duaij, D. L. Davies, G. A. Griffith and K. Singh, Organometallics, 2009, 28, 433–440 CrossRef CAS.
- Y. Boutadla, D. L. Davies, R. C. Jones and K. Singh, Chem. – Eur. J., 2011, 17, 3438–3448 CrossRef CAS PubMed.
- (a) R. Corberán, M. Sanaú and E. Peris, J. Am. Chem. Soc., 2006, 128, 3974–3979 CrossRef PubMed; (b) R. Corberán, M. Sanaú and E. Peris, Organometallics, 2006, 25, 4002–4008 CrossRef; (c) A. C. Marr, P. J. Morgan, G. C. Saunders and H. P. Thomas, Dalton Trans., 2019, 48, 1947–1949 RSC; (d) S. Gülcemal, D. Gülcemal, G. F. S. Whitehead and J. Xiao, Chem. – Eur. J., 2016, 22, 10513–10522 CrossRef PubMed.
- T.-Y. Li, X. Liang, L. Zhou, C. Wu, S. Zhang, X. Liu, G.-Z. Lu, L.-S. Xue, Y.-X. Zheng and J.-L. Zuo, Inorg. Chem., 2015, 54, 161–173 CrossRef CAS PubMed.
- Similar N,N‘-diphenylimidazolium salts cyclometallate much more efficiently under the same conditions.
- We found that addition of Et4NCl was crucial for high conversions, further details of this effect will be published elsewhere.
- R. Thenarukandiyil, S. K. Gupta and J. Choudhury, ACS Catal., 2016, 6, 5132–5137 CrossRef CAS.
- I. Kilpeläinen, H. Xie, A. King, M. Granstrom, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142–9148 CrossRef PubMed.
- Due to the low concentration of the samples attempts to record the 2D NMR spectra were unsuccessful.
- (a) C. H. Leung, C. D. Incarvito and R. H. Crabtree, Organometallics, 2006, 25, 6099–6107 CrossRef CAS; (b) S. Semwal, I. Mukkatt, R. Thenarukandiyil and J. Choudhury, Chem. – Eur. J., 2017, 23, 13051–13057 CrossRef CAS PubMed.
- X. Wang, M. Wang and J. Xie, Synth. Commun., 2017, 47, 1797–1803 CrossRef CAS.
- (a) X. Ge, X. Chen, C. Qian and S. Zhou, RSC Adv., 2016, 6, 58898–58906 RSC; (b) A. V. Nakhate and G. D. Yadav, ChemistrySelect, 2017, 2, 2395–2405 CrossRef CAS; (c) D. P. Phillips, X.-F. Zhu, T. L. Lau, X. He, K. Yang and H. Liu, Tetrahedron Lett., 2009, 50, 7293–7296 CrossRef CAS; (d) Q. Zhang, J. Luo and Y. Wei, Synth. Commun., 2012, 42, 114–121 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data (CIF), and NMR spectra of all new compounds. CCDC 2085491. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt02677a |
| This journal is © The Royal Society of Chemistry 2021 |