
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMn(I) phosphine-amino-phosphinites: a highly modular class of pincer complexes for enantioselective transfer hydrogenation of aryl-alkyl ketones†
Harikrishnan
Jayaprakash

Department of Chemistry and Applied Biosciences, ETH Zürich, Wolfgang-Pauli-Str. 10, CH-8093 Zürich, Switzerland. E-mail: hari@inorg.chem.ethz.ch
First published on 15th September 2021
Abstract
A series of Mn(I) catalysts with readily accessible and more π-accepting phosphine-amino-phosphinite (P′(O)N(H)P) pincer ligands have been explored for the asymmetric transfer hydrogenation of aryl-alkyl ketones which led to good to high enantioselectivities (up to 98%) compared to other reported Mn-based catalysts for such reactions. The easy tunability of the chiral backbone and the phosphine moieties makes P′(O)N(H)P an alternative ligand framework to the well-known PNP-type pincers.
The combination of chiral pincer ligands with base metals has been established to be effective in replacing precious noble metal catalysts in asymmetric reactions,1–8 especially for hydrogenation of ketones.9–17 Despite base metal catalysts being efficient, high synthetic costs and tedious synthetic protocols triggered the search for simpler and cheaper alternative chiral ligand frameworks with desired chirality.
Though the chiral base metal hydrogenation catalysts are known for decades, Mn(I) catalysts were left unexplored until 2016. Moreover, the Mn(I) complexes with classical chiral phosphines of the types PNP, PN(H)P, and PNN are commonly used for such reactions.18–28 No efforts have been directed towards replacing phosphine pincers with electron-deficient29 chiral bis-phosphinite (P(O)N(H)P(O)) or unsymmetrical phosphine-phosphinite pincer ligands (P′(O)N(H)P). Also, it must be noted that Mn(I) catalyzed asymmetric transfer hydrogenation (ATH) of ketones18–23 is less explored compared to the asymmetric direct hydrogenation (AH).24–28 Moreover, the observed enantioselectivities for ATH with well-defined Mn(I) catalysts A, B and C (Chart 1, 20%–85% ee) and further Mn(I) catalytic systems21,23 (1%–90 ee%) are significantly lower when compared to those for AH (7%–99% ee).
 | ||
| Chart 1 Examples of chiral Mn(I) catalysts for the asymmetric transfer (ATH) and direct hydrogenation (AH) of ketones. | ||
The ligand precursors 1a and 1b were prepared by the condensation of the corresponding aldehydes 5a and 5b with relatively cheap and readily available (1S,2S)-2-aminocyclohexan-1-ol in methanol at room temperature, followed by their in situ reduction with sodium tetrahydridoborate. Deprotonation of 1a and 1b, followed by the addition of PR′2Cl, resulted in the formation of ligands 2a and 2b with 95–96% purity as determined by 31P{1H} NMR.19,29 The desired [Mn(CO)2(2a–2b)]Br (3a and 3b) were then obtained as a yellow solid upon reacting 2a and 2b with [MnBr(CO)5] in toluene (Scheme 1, see the ESI†).
 | ||
| Scheme 1 Synthesis of the ligands (2a and 2b) and complexes (3a and 3b). | ||
The 31P{1H} NMR spectra of 3a and 3b exhibit an AX system that suggests mutually trans positioned nuclei.19,29 The IR spectra of complexes 3a (1931 cm−1, 1851 cm−1, Fig. 2) and 3b (1930 cm−1, 1850 cm−1) show two strong CO bands of similar intensity which further support the presence of mutually cis-oriented CO ligands which are in agreement with the previously reported Mn(I) carbonyl pincer complexes.19,29 The spectroscopic evidence taken together points to the formation of cis, mer-[MnBr(CO)2(2a–2c)] (3a and 3b). In fact, such a structure was also confirmed by X-ray crystallography in the case of 3a (Fig. 1, see the ESI† for more details).
 | ||
| Fig. 1 ORTEP plot of 3a (ellipsoid sets at 40% probability). Selected bond lengths [Å] and angles [°]: P(1)–Mn(1) 2.3083(19), P(2)–Mn(1) 2.2274(19), C(1)–Mn(1) 1.758(8), C(2)–Mn(1) 1.779(7), P(2)–Mn(1)–C(2) 88.7(2) and P(2)–Mn(1)–Br(1) 89.74(6). | ||
 | ||
| Fig. 2 Comparison of the IR stretching frequency of Mn(I) phosphine and phosphinite pincer complexes. | ||
Notably, the Mn1–P2 bond length of Mn1 with phosphorus atom P2 (2.227 Å) is shorter than that for Mn1–P1 (2.308 Å). Moreover, a comparison of the IR stretching frequency of 3a with that of its homologous trans bis-phosphine pincer complex 11 (1921 cm−1, 1842 cm−1, Fig. 2; see the ESI† for the synthesis) suggests that phosphinite (P2) is a weaker electron donor and possesses better dπ–pπ back bonding (M → L back donation) compared to phosphine P1. The observed CO stretching frequencies are analogous to those of the previously reported Mn(I) bis-phosphine (12) and bis-phophinite complex (13).29,30
The activity of 3a was next investigated in ATH with 2-acetonaphthone (8a) as a prototypical substrate31 under a broad range of conditions. The reaction temperature and nature of the base and catalyst have the largest effect on the enantioselectivity and activity of the catalytic system. A strong base is necessary to achieve high activity of catalyst 3a and notably, catalyst 3b (82% ee) has shown better enantioselectivity in comparison with 3a (79% ee, see the ESI†). The activity of the complex 3a was found to be diminishing with increasing temperature from 40 °C to 80 °C (see the ESI†). It is noteworthy that the tendency of Mn(I) complexes to form a metal-aziridine intermediate at high temperature and an amido species at lower temperature has been previously reported.32 Therefore, it is expected that 4a and 4b are formed at 40 °C and 80 °C, respectively, leading to different reactivities at the corresponding temperatures (see Scheme 4 for the proposed mechanism). The greater acidity of the benzylic proton and better dπ–pπ back bonding of the phosphinite ligand possibly lead to the easier formation of metal-aziridine intermediate 4b that remains as the resting species. This could prevent the formation of 5a which could be a possible explanation for the lower activity of 3a at higher temperatures (Scheme 2). A poisoning test with trimethyl phosphine (30 mol% vs.3a) and the fact that the enantioselectivity remains the same throughout the reaction indicate the presence of a homogeneous catalytic system.33,34
 | ||
Scheme 2 Asymmetric transfer hydrogenation with catalyst 3b: isolated yields are given and ee values were determined by GC and HPLC, respectively. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Optimized conditions C1 (catalytic loading: 2 mol%, 4 mol% tBuOK, 40 °C, 18 h, 0.2 M). bRe-optimized conditions C2 (catalytic loading: 1 mol%, 2 mol% tBuOK, RT, 24 h). Optimized conditions C1 (catalytic loading: 2 mol%, 4 mol% tBuOK, 40 °C, 18 h, 0.2 M). bRe-optimized conditions C2 (catalytic loading: 1 mol%, 2 mol% tBuOK, RT, 24 h). | ||
Hence, with the identified optimal reaction conditions C1 (catalytic loading (3b) 2 mol%, 4 mol% tBuOK (1 M in THF), 40 °C, 18 h, 0.2 M, Scheme 2), the ATH of a large scope of aryl-alkyl ketones was investigated (see Schemes 2 and 3). The corresponding secondary alcohols were obtained in good yields with high enantioselectivities (80%–98% ee). With acetophenones, 3b tolerates ortho, meta and para substituents, with enantioselectivities in the order of ortho (91%–98% ee) > meta (90%–92% ee) > para (82%–87% ee) substituents and yields in the order of meta, para (68%–99%) > ortho (21%–55%) substituents.
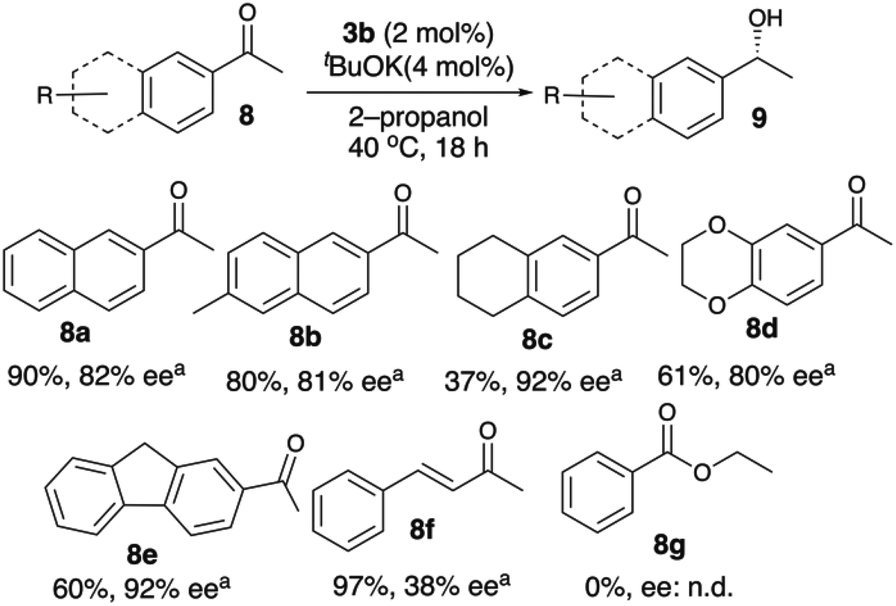 | ||
| Scheme 3 Asymmetric transfer hydrogenation with catalyst 3b. Reaction conditions C1 (catalytic loading: 2 mol%, 4 mol% tBuOK, 40 °C, 18 h, 0.2 M). aIsolated yields are given and ee values were determined by GC and HPLC, respectively. | ||
Meta-substituted acetophenones (6e–6g) including 3,5 di-substituted acetophenone (6m) give high enantioselectivities (up to 94% ee, Scheme 2) and quantitative yields. Catalyst 3b gives the trifluoromethyl-substituted alcohols 7h and 7l which are important synthons for fungicides35 and NK1 antagonists35 in quantitative yields and with 90% and 94% ee, respectively. It is to be noted that these are the best enantioselectivities observed with meta-chloro-acetophenone (6e, 90% ee) and 3,5-bis-trifluoromethyl-acetophenone (6l, 94% ee) for reported manganese complexes for ATH (24–85% ee, 6–85% ee, respectively).18,19,21–23
Challenging substrates like ortho-substituted acetophenones (6i–6k) displayed high enantioselectivity (91–98% ee), though with lower yield, likely due to the greater steric hindrance of the system (Scheme 2). Despite such shortcomings, the observed enantioselectivities are the highest for ortho-methoxyacetophenone (6k, 91% ee) in comparison with other reported manganese catalysts for ATH (61–90% ee).18,19,21–23
Para-substituted acetophenones (6o–6p) were reduced by 3b with quantitative yields, albeit with significantly lower enantioselectivities (82–87%). The use of lower temperature, i.e., room temperature and a catalytic loading of 1 mol% (re-optimized conditions C2: catalytic loading 1 mol%, 2 mol% tBuOK, RT, 24 h) allows the improvement of enantioselectivity while maintaining good yields. The highest difference in enantioselectivity was observed with the electron-rich para-substituted acetophenone (6q, by 4%). Moreover, these conditions improved the observed enantioselectivities for electron-deficient meta acetophenones (6e and 6h by 3% and 4%, respectively) or 3,5 disubstituted acetophenone (6l by 2%). It is again noteworthy that catalyst 3b provides high enantiomeric excess (83% ee) with para-trifluoromethyl-acetophenone (6o) when comparing with other Mn-based catalysts for ATH (55–76% ee).18,19,21–23 The heterocycle 6r was also reduced by 3b with high enantioselectivity (97% ee) but with lower yield (25% and 41%, respectively).
Alkyl-phenyl ketones with various alkyl substituents were also investigated for their reactivity with catalyst 3b. Ethyl phenyl ketone (6b) has shown a higher ee (90%) and a lower isolated yield (49%) compared to acetophenone (6a, 86% ee and 84% yield) and the trend remains the same with further increase in the alkyl chain length (6c, Scheme 2). Since these catalysts are highly sensitive towards steric hindrance, their activity with 2° alkyl-aryl ketone 6d (91% ee and 10% yield) is greater than that with 3° alkyl-aryl ketones. Substrates with high steric bulk such as cyclohexyl phenyl ketone, 2-methoxy-benzophenone and tertiary butyl phenyl ketone do not get reduced with the catalyst 3b.
The extended aromatic ketones 8a and 8b were reduced with 82% ee (Scheme 3). Both 2-acetyl-fluorene (8e) and tetralone (8c) showed a higher ee of 92% when compared with 8a; however, 8d gave a lower ee of 80%. It is noteworthy that α,β-unsaturated ketones (8f) have shown low enantioselectivity (38% ee) since the carbonyl group is placed remotely from the aryl ring. The enantioselectivity observed with 8a (82% ee) is the highest on comparing with other Mn(I) catalysts (up to 76% ee) for ATH.18,19,21–23 The “R” isomer is formed as the major enantiomer during the reduction of ketones with 3b. The aromatic ester 8g was also not reduced with complex 3b (Scheme 3).
Based on the experimental results obtained by the screening of the catalysts 3a and 3b and the reported Mn(I) intermediates,32 a possible reaction mechanism in which temperature is the key parameter is proposed (Scheme 4). As discussed earlier, the higher activity of the catalyst at 40 °C could be due to the possible formation of the Mn(I) amido species 4a. The Mn(I) amido species 4a formed at 40 °C in the presence of tBuOK leads to the possible formation of hydride 5avia oxidation of 2-propanol. An Re or Si selective hydride attack on acetophenone leads to the formation of 4c. The intermediate 4c then regenerates 4a by eliminating the enantioenriched alcohol.
 | ||
| Scheme 4 Proposed mechanism for asymmetric transfer hydrogenation of acetophenone with 3a. | ||
In conclusion, readily accessible chiral pincer ligands (2a and 2b) and their corresponding well-defined Mn(I) complexes (3a and 3b) were developed. The complex 3b shows high enantioselectivities (80%–97% ee) for the ATH of ketones, thus outperforming other Mn(I) catalysts. This demonstrates that the ligand scaffolds like 2 are a valuable alternative to the conventional chiral phosphine pincers (PN(H)P) as they are cheap and highly modular. Most importantly this class of ligands can be even extrapolated to various other asymmetric catalytic applications. Further studies on the catalytic system will be carried out in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
I thank the Swiss National Science Foundation for financial support (grant no. 200020_184606) and Prof. Antonio Mezzetti for his contribution in the initial phase of the project. I also thank Dr Jordan Meyet and Dr Michael. D. Wörle for the single-crystal X-ray analysis. I sincerely thank Profs. Antonio Togni, Christophe Copéret and Hansjörg Grützmacher for their most valuable assistance in finalizing the manuscript and for their general support.References
- S. Murugesan and K. Kirchner, Dalton Trans., 2016, 45, 416–439 RSC.
- D. Benito-Garagorri and K. Kirchner, Acc. Chem. Res., 2008, 41, 201–213 CrossRef CAS PubMed.
- S. K. Gibbons, Z. Xu, R. P. Hughes, D. S. Glueck and A. L. Rheingold, Organometallics, 2018, 37, 2159–2166 CrossRef CAS.
- H. Valdés, M. A. García-Eleno, D. Canseco-Gonzalez and D. Morales-Morales, ChemCatChem, 2018, 10, 3136–3172 CrossRef.
- K. Junge, V. Papa and M. Beller, Chem. – Eur. J., 2019, 25, 122–143 CrossRef CAS PubMed.
- G. Bauer and X. Hu, Inorg. Chem. Front., 2016, 3, 741–765 RSC.
- Y. Xiang, Q. Ge, S. Wu, X. Zheng and Z. Yang, RSC Adv., 2020, 10, 9563–9578 RSC.
- A. Casnati, M. Lanzi and G. Cera, Molecules, 2020, 25, 3889 CrossRef CAS PubMed.
- M. Garbe, Z. Wei, B. Tannert, A. Spannenberg, H. Jiao, S. Bachmann, M. Scalone, K. Junge and M. Beller, Adv. Synth. Catal., 2019, 361, 1913–1920 CrossRef CAS.
- (a) F. Agbossou-Niedercorn and C. Michon, Coord. Chem. Rev., 2020, 425, 213523 CrossRef CAS; (b) J. Wen, F. Wang and X. Zhang, Chem. Soc. Rev., 2021, 50, 3211–3237 RSC; (c) Y. Wang, M. Wang, Y. Li and Q. Liu, Chem, 2021, 7, 1180–1223 CrossRef CAS.
- A. Passera and A. Mezzetti, Adv. Synth. Catal., 2019, 361, 4691–4706 CrossRef CAS.
- J. F. Sonnenberg, A. J. Lough and R. H. Morris, Organometallics, 2014, 33, 6452–6465 CrossRef CAS.
- A. Zirakzadeh, K. Kirchner, A. Roller, B. Stöger, M. Widhalm and R. H. Morris, Organometallics, 2016, 35, 3781–3787 CrossRef CAS.
- S. A. M. Smith, P. O. Lagaditis, A. Lüpke, A. J. Lough and R. H. Morris, Chem. – Eur. J., 2017, 23, 7212–7216 CrossRef CAS PubMed.
- R. Huber, A. Passera and A. Mezzetti, Organometallics, 2018, 37, 396–405 CrossRef CAS.
- P. O. Lagaditis, P. E. Sues, J. F. Sonnenberg, K. Y. Wan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2014, 136, 1367–1380 CrossRef CAS PubMed.
- R. Huber, A. Passera and A. Mezzetti, Chem. Commun., 2019, 55, 9251–9266 RSC.
- A. Zirakzadeh, S. R. M. M. de Aguiar, B. Stöger, M. Widhalm and K. Kirchner, ChemCatChem, 2017, 9, 1744–1748 CrossRef CAS.
- K. Z. Demmans, M. E. Olson and R. H. Morris, Organometallics, 2018, 37, 4608–4618 CrossRef CAS.
- C. S. G. Seo, B. T. H. Tsui, M. V. Gradiski, S. A. M. Smith and R. H. Morris, Catal. Sci. Technol., 2021, 3153–3163 RSC.
- K. Azouzi, A. Bruneau-Voisine, L. Vendier, J. B. Sortais and S. Bastin, Catal. Commun., 2020, 142, 106040 CrossRef CAS.
- R. Van Putten, G. A. Filonenko, A. Gonzalez De Castro, C. Liu, M. Weber, C. Müller, L. Lefort and E. Pidko, Organometallics, 2019, 38, 3187–3196 CrossRef CAS PubMed.
- J. Schneekönig, K. Junge and M. Beller, Synlett, 2019, 503–507 Search PubMed.
- M. B. Widegren, G. J. Harkness, A. M. Z. Slawin, D. B. Cordes and M. L. Clarke, Angew. Chem., Int. Ed., 2017, 56, 5825–5828 CrossRef CAS PubMed.
- M. B. Widegren and M. L. Clarke, Catal. Sci. Technol., 2019, 9, 6047–6058 RSC.
- L. Zhang, Y. Tang, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2019, 58, 4973–4977 CrossRef CAS PubMed.
- (a) M. Garbe, K. Junge, S. Walker, Z. Wei, H. Jiao, A. Spannenberg, S. Bachmann, M. Scalone and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 11237–11241 CrossRef CAS PubMed; (b) F. Ling, H. Hou, J. Chen, S. Nian, X. Yi, Z. Wang, Di. Song and W. Zhong, Org. Lett., 2019, 21, 3937–3941 CrossRef CAS PubMed.
- L. Zeng, H. Yang, M. Zhao, J. Wen, J. H. R. Tucker and X. Zhang, ACS Catal., 2020, 10, 13794–13799 CrossRef CAS.
- H. Li, D. Wei, A. Bruneau-Voisine, M. Ducamp, M. Henrion, T. Roisnel, V. Dorcet, C. Darcel, J. F. Carpentier, J. F. Soulé and J. B. Sortais, Organometallics, 2018, 37, 1271–1279 CrossRef CAS.
- S. Fu, Z. Shao, Y. Wang and Q. Liu, J. Am. Chem. Soc., 2017, 139, 11941–11948 CrossRef CAS PubMed.
- According to the preliminary DFT studies, the π–π interaction between the aryl rings on the ligand phosphine atom (not the aryl rings on the phosphinite donor) and the incoming ketones plays the key role in the enantioselectivity of our reaction. Hence 2-acetonaphthone was chosen as the prototypical substrate over acetophenone (unpublished results).
- S. Chakraborty, U. Gellrich, Y. Diskin-Posner, G. Leitus, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2017, 56, 4229–4233 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS PubMed.
- Y. Li, S. Yu, X. Wu, J. Xiao, W. Shen, Z. Dong and J. Gao, J. Am. Chem. Soc., 2014, 136, 4031–4039 CrossRef CAS PubMed.
- (a) K. Tanaka, M. Katsurada, F. Ohno, Y. Shiga, M. Oda, M. Miyagi, J. Takehara and K. Okano, J. Org. Chem., 2000, 65, 432–437 CrossRef CAS PubMed; (b) K. M. J. Brands, J. F. Payack, J. D. Rosen, T. D. Nelson, A. Candelario, M. A. Huffman, M. M. Zhao, J. Li, B. Craig, Z. J. Song, D. M. Tschaen, K. Hansen, P. N. Devine, P. J. Pye, K. Rossen, P. G. Dormer, R. A. Reamer, C. J. Welch, D. J. Mathre, N. N. Tsou, J. M. McNamara and P. J. Reider, J. Am. Chem. Soc., 2003, 125, 2129–2135 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2083796. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt02257a |
| This journal is © The Royal Society of Chemistry 2021 |