
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and ex vivo biological evaluation of gallium-68 labelled NODAGA chelates assessing cardiac uptake and retention†
Bradley E.
Osborne
 ab,
Thomas T. C.
Yue
a,
Edward C. T.
Waters
b,
Friedrich
Baark
b,
Richard
Southworth
*b and
Nicholas J.
Long
*a
ab,
Thomas T. C.
Yue
a,
Edward C. T.
Waters
b,
Friedrich
Baark
b,
Richard
Southworth
*b and
Nicholas J.
Long
*a
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, London, W12 0BZ, UK. E-mail: n.long@imperial.ac.uk
bSchool of Biomedical Engineering and Imaging Sciences, King's College London, 4th Floor Lambeth Wing, St Thomas’ Hospital, London, SE1 7EH, UK. E-mail: richard.southworth@kcl.ac.uk
First published on 21st September 2021
Abstract
Radiolabelled lipophilic cations can be used to non-invasively report on mitochondrial dysfunction in diseases such as cardiovascular disease, cardiotoxicity and cancer. Several such lipophilic cations are currently used clinically to map myocardial perfusion using SPECT imaging. Since PET offers significant advantages over SPECT in terms of sensitivity, resolution and the capacity for dynamic imaging to allow pharmacokinetic modelling, we have synthesised and radiolabelled a series of NODAGA-based radiotracers, with triarylphosphonium-functionalisation, with gallium-68 to develop PET-compatible cationic complexes. To evaluate their capacity to report upon mitochondrial membrane potential, we assessed their pharmacokinetic profiles in isolated perfused rat hearts before and after mitochondrial depolarisation with the ionophore CCCP. All three tracers radiolabel with over 96% RCY, with log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D7.4 values above −0.4 observed for the most lipophilic example of this family of radiotracers. The candidate tracer [68Ga]Ga4c exhibited non-preferential uptake in healthy cardiac tissue over CCCP-infused cardiac tissue. While this approach does show promise, the lipophilicity of this family of probes needs improving in order for them to be effective cardiac imaging agents.
D7.4 values above −0.4 observed for the most lipophilic example of this family of radiotracers. The candidate tracer [68Ga]Ga4c exhibited non-preferential uptake in healthy cardiac tissue over CCCP-infused cardiac tissue. While this approach does show promise, the lipophilicity of this family of probes needs improving in order for them to be effective cardiac imaging agents.
Introduction
The radiolabelled lipophilic cations [99mTc]Tc-sestaMIBI (Fig. 1) and [99mTc]Tc-tetrofosmin are widely used clinically to visualise defects in myocardial perfusion using single photon emission computed tomography (SPECT).1–4 While the delivery of these imaging agents to tissues is dependent upon perfusion, as cations, their retention within mitochondria is actually determined by mitochondrial membrane potential (ΔΨm)-dependent trapping according to the Nernst equation.5–7 Accordingly, if the retention of such lipophilic cations could be corrected for perfusion, either by pharmacokinetic modelling or by parallel injection of non-mitochondrially dependent perfusion imaging agents, they could be repurposed to provide a non-invasive imaging index on mitochondrial dysfunction in cancer and cardiovascular disease, as we have recently proposed.8 | ||
| Fig. 1 [99mTc]Tc-sestaMIBI and [18F]MitoPhos_07, two examples of lipophilic and cationic radiotracers.1,19 | ||
Positron emission tomography (PET) offers significant advantages over SPECT in terms of spatial resolution, sensitivity, and the capacity for dynamic imaging to allow pharmacokinetic modelling and 3-dimensional image-derived perfusion correction or parameter mapping. Translating this class of perfusion imaging agents to PET is therefore an attractive prospect. Lipophilic triphenylphosphonium (TPP)-functionalised compounds are also capable of targeting the mitochondria due to their delocalised positive charge across the large surface area of the aromatic rings, and TPP-based PET tracers radiolabelled with carbon-11 and fluorine-18 (t½ = 20 and 109.7 min respectively) have been extensively explored for mitochondrial imaging.9–18 In 2014, Haslop et al. synthesised a series of fluorine-18 radiolabelled compounds, [18F]MitoPhos (Fig. 1), consisting of alkyl-functionalised motifs on triarylphosphonium (TAP) moieties, achieved via a copper-catalysed click reaction of an 18F synthon to the TAP cation.19 Functionalisation of the TAP moiety afforded the tuneability of radiotracer lipophilicity which facilitated the modification of its pharmacokinetic properties and capacity to successfully penetrate cell membranes.
The recent development and clinical approval of 68Ge/68Ga generators to produce the positron-emitting radiometal gallium-68, has provided the opportunity for clinical PET imaging without the expensive infrastructure required for cyclotron-produced radioisotopes like fluorine-18 and carbon-11.20,21 Gallium-68 also affords a convenient balance between patient radiation dosage and effective imaging due to its half-life (t½ = 68 min). The first use of gallium radionuclides in lipophilic cations was reported by Sharma et al., in 2014, who developed a Schiff-base phenolic complex, consisting of a gallium-68 metal core surrounded by aromatic and isopropoxide groups.22 In 2017, Kardashinsky et al. synthesised a series of TAP-functionalised DO3A compounds for the gamma-emitting radionuclide gallium-67 (t½ = 78 h), which consisted of methyl and methoxy aryl functionalisation, as well as monoiodo and pyridyl asymmetric variants.23 In 2018, Smith et al. synthesised a family of DO3A-xy-TAP chelates for gallium-68, functionalised with triphenylphosphonium (TPP), tritolylphosphonium (TTP) and trixylylphosphonium (TXP).24 These radiotracers were designed with the idea of combining the tuneable lipophilicity afforded by alkyl-functionalisation of the TAP moiety, and the rationale behind the DO3A-xy-TPP chelators used for gallium-67.23 The most lipophilic tracers of this series, [68Ga]Ga-DO3A-xy-TXP (Fig. 2), was assessed in vitro in cultured tumour cells, and exhibited a three-fold increase in uptake compared to unchelated [68Ga]Ga(III). The capacity of this radiotracer to report on mitochondrial membrane potential was assessed ex vivo in isolated perfused rat hearts, which exhibited less than 1% retention in healthy hearts (compared to approximately 40% uptake typically observed with the “gold standard” [99mTc]Tc-sestaMIBI).8 This was not significantly reduced by mitochondrial depolarisation with the ionophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP), suggesting the lipophilicity of this family (logD7.4 between −3.12 and −1.81) was insufficient to penetrate cell membranes to reach the mitochondria. In 2020, Smith et al. attempted to address this limitation by synthesising and radiolabelling two series of DO2A-based chelates with bisTAP- and bisaryl-functionalisation, resulting in logD7.4 values of −0.3 for the most lipophilic examples, [68Ga]Ga-DO2A-(xy-TXP)2 and [68Ga]Ga-DO2A-Xy2 (Fig. 2).25 Both sets of tracers radiolabelled with over 90% radiochemical purity (RCP) when heated at 100 °C for 30 min, however formation of kinetic/thermodynamic pairs of species were observed upon gallium-68 chelation which were visualised and separated by radioHPLC. These tracers exhibited much improved retention in healthy hearts compared to their respective DO3A analogues, and their cardiac washout following mitochondrial depolarisation with infusion of CCCP suggested a ΔΨm-dependent component in their cardiac uptake and retention. However, these tracers retained significant non-specific cardiac retention, which limits their usefulness.
 | ||
| Fig. 2 [68Ga]Ga-DO3A-xy-TXP, [68Ga]Ga-DO2A-(xy-TXP)2 and [68Ga]Ga-DO2A-Xy2, three examples of gallium-68 lipophilic and cationic radiotracers synthesised in our group.24,25 | ||
The harsh radiolabelling conditions needed for successful 68Ga-labelling of the DO3A- and DO2A-based chelates were a result of the size mismatch of these macrocyclic chelates and Ga(III), and increased the overall hydrophilicity of these complexes, with the unbound carboxylate arm present in the Ga-DO3A complexes exhibiting low logD7.4 values. In order to address these limitations, we have herein developed a series of (R)-NODAGA-based ligands for gallium-68 chelation for the formation of 68Ga-labelled cationic complexes, which are presented in this study (Fig. 3). The chosen chelator for this study, (R)-NODAGA, displays the appropriate hexadentate coordination environment necessary for Ga(III) whilst providing a better size match between NODAGA and Ga(III), leading to significantly improved radiolabelling kinetics and stability of Ga-labelled compounds.26,27 The absence of an unbound carboxylate arm will increase the overall lipophilicity of the final radiotracers.
 | ||
| Fig. 3 The three [68Ga]Ga-(R)-NODAGA-based radiotracers investigated in this study; (R)-NODAGA-xy-TAP, and the phenyl, p-tolyl and m,m′-xylyl motifs used. | ||
By combining the rationales discussed above, we have synthesised three (R)-NODAGA-based chelators with tuneable lipophilicity afforded by alkyl-functionalised TAP moieties in order to form the novel ligands (R)-NODAGA-xy-TPP, (R)-NODAGA-xy-TTP and (R)-NODAGA-xy-TXP. After the synthesis and characterisation of the ligands, their radiolabelling profiles with gallium-68 and logD7.4 values were assessed as an initial indication of their capacity to report on mitochondrial dysfunction. The pharmacokinetics and sensitivity of the tracers to ΔΨm were then evaluated using the ex vivo Langendorff isolated heart model monitored by our triple γ-detection system.24,25,28–31
Results and discussion
Synthesis of (R)-NODAGA-xy-TAP chelators
Building on our previous work on the synthesis of DO3A-xy-TAP, DO2A-(xy-TAP)2 and DO2A-Ar2 chelators, (R)-NODAGA-xy-TAP (4) ligands with triphenylphosphonium (TPP, 4a), tritolylphosphonium (TTP, 4b) and trixylylphosphonium (TXP, 4c) functionalisation were synthesised as illustrated in Scheme 1. | ||
| Scheme 1 Synthesis of (R)-NODAGA-xy-TAP ligands. 1a–c were synthesised according to previously reported procedures.25,32,33 Reaction conditions: (i) NH3(aq), EtOH, rt, 48 h; (ii) (R)-NODAGA(tBu)3, DCC, pyridine, H2O:MeCN (1:1), rt, 48 h; (iii) TFA, rt, 48 h, reverse-phase flash chromatography. | ||
The synthesis of 4a–c combined methodologies used by Eisenwiener et al., Smith et al. and Schirrmacher et al.25,32,33 Firstly the TAP-functionalised bromoxylenes 1a–c, were reacted with aqueous ammonia in ethanol to form the TAP-functionalised aminoxylenes 2a–c. The monocationic aminoxylene phosphonium species 2 was then reacted with tris-tert-butyl-protected (R)-NODAGA to form TAP-functionalised NODAGA derivatives (3). Due to the insolubility of the TAP-functionalised aminoxylenes (2) in organic solvents, we performed the reaction in a 50:50 mixture of water and acetonitrile using N,N′-dicyclohexylcarbodiimide (DCC) as the coupling reagent, as a modification of the procedure used by Schirrmacher et al.33 DCC was dissolved in pyridine and added in a dropwise manner to the reaction mixture, activating the carboxylic acid and providing a base for the reaction. The byproduct of this amide coupling, dicyclohexylurea (DCU), was easily isolated and removed by filtration by dissolving the crude residue in acetonitrile, which DCU is highly insoluble in. The protected intermediates (3a–c) were purified by reverse-phase flash chromatography, followed by deprotection of the tert-butyl protected carboxylates in trifluoroacetic acid (TFA) and purification using reverse-phase flash chromatography to obtain the final ligands (4a–c). The modular approach used in this work allows for a library of compounds to be synthesised and purified in a simple manner, utilising identical reaction protocols.
Radiolabelling studies of (R)-NODAGA-xy-TAP chelates with [68Ga]GaCl3 eluate
All three (R)-NODAGA-xy-TAP compounds were radiolabelled and analysed according to the same protocol, a modification of the methodology previously described by Smith et al.24,25 The ligand was dissolved in clinical grade 3.6 M NaOAc to generate a ligand concentration of 500 μM, before adding generator-produced [68Ga]Ga(III) (in 0.1 M aqueous HCl, 500 μL) and the resulting mixture (pH 5.0) was left at room temperature for 15 minutes. Instant thin-layer chromatography (iTLC) was employed as a quality control (QC) method. Two different mobile phases were employed for iTLC: (1) 0.1 M EDTA – [68Ga]Ga-(R)-NODAGA-xy-TAP Rf < 0.1; unchelated [68Ga]Ga(III) Rf > 0.9. (2) 2.0 M NH4OAc:MeOH (1:1) – unchelated [68Ga]Ga(III) Rf < 0.1; [68Ga]Ga-(R)-NODAGA-xy-TAP Rf = 0.6–0.7. Implementing this method for the QC of 68Ga-labelling with these compounds provides the basis for analysing future gallium-68 labelled lipophilic and cationic radiotracers due to the facile chelation of Ga(III) by EDTA, along with promotion of 68Ga-labelled radiotracers to the solvent front by a 1-to-1 mixture of 2.0 M NH4OAc:MeOH. In order to determine the efficiency of gallium-68 chelation, a series of reactions were undertaken in which the [68Ga]Ga(III) eluate was added to a solution of ligand in 3.6 M NaOAc (42 μL) at a ligand concentration in the range 500 μM to 5 μM. After 15 minutes reaction time and room temperature, radiochemical yields (RCY) were measured using iTLC, with a ligand concentration of 100 μM, RCYs were greater than 97% for [68Ga]Ga-(R)-NODAGA-xy-TPP, TTP and TXP. At a ligand concentration of 50 μM, RCYs were greater than 85% for all radiolabelled compounds, and at a ligand concentration of 5 μM, RCYs of less than 50% were obtained. Due to near quantitative RCYs obtained for all 68Ga-labelled complexes at 100 μM ligand concentration, this ligand concentration was used to perform myocardial uptake and retention experiments due to there being no requirement for post-purification at this concentration.
To assess the purity of the obtained 68Ga-radiolabelled complexes, the reaction mixture was analysed by radioHPLC. This also serves as an initial indication of radiotracer lipophilicity. The traces obtained for all three compounds are shown normalised form in Fig. 4A, with the HPLC traces of their respective proligands shown in Fig. 4B. Unreacted/non-chelated [68Ga]Ga(III) species were identified as a small peak in all HPLC traces, observed at approximately 2 minutes, and the desired complexes corresponded to a single major peak with no evidence of any isomerism. Fig. 4 illustrates that an increase in TAP alkylation results in a longer retention time and indicates superior lipophilicity, with the retention times determined to be 15.8 min, 18.0 min and 19.6 min for 68Ga-labelled NODAGA-xy-TPP, TTP and TXP respectively. This finding is further supported by the HPLC traces of the proligands; retention times found to be 11.8 min, 14.1 min and 15.4 min for NODAGA-xy-TPP, TTP and TXP respectively. The proligands also show shorter retention times by HPLC than their radiolabelled analogues, demonstrating an increase in lipophilicity upon chelation with gallium-68, resulting from a reduction in hydrophilic interactions of the carboxylate arms upon binding with Ga(III).
 | ||
| Fig. 4 (A) RadioHPLC traces of the three [68Ga]Ga-(R)-NODAGA-xy-TAP complexes. (B) HPLC traces of the three respective (R)-NODAGA-xy-TAP proligands (eluent gradient: 5% B for 5 min; 5–95% for 20 min; 95% B for 5 min; flow rate 1 mL min−1). All traces have been normalised such that the maximum activity/absorbance measured for each compound is equal to 1. | ||
Lipophilicity measurements of [68Ga]Ga-NODAGA-xy-TAP radiotracers
Lipophilicities of the radiotracers were measured from their octanol/PBS partition coefficients at pH 7.4 (logD) using a conventional ‘shake-flask’ method. The logD7.4 values, illustrated in Fig. 5, were determined to be −2.43 ± 0.07, −1.00 ± 0.14 and −0.37 ± 0.21 for [68Ga]Ga-NODAGA-xy-TPP, TTP and TXP respectively (n = 6 for all compounds). Increasing the level of alkyl-functionalisation of the TAP moiety causes an increase in logD7.4 value. This trend was studied and shown previously,19,24,25 and was also observed for the [68Ga]Ga4 compounds. The logD7.4 values for [68Ga]Ga4 compounds are greater than those observed for the DO3A analogues studied previously. This provides confirmation that the removal of the unbound carboxylate arm decreases hydrophilic interactions and improves the overall lipophilicity of the [68Ga]Ga4 complexes. An important characteristic exhibited by these complexes is that a small change, such as the addition of three methyl groups to the TPP group, has such a significant effect (1.4 unit difference) on the logD7.4 values, even when attached to the hydrophilic NODAGA macrocyclic ring. This effect was also observed upon the addition of three more methyl groups to the TTP group, resulting in a 0.6 unit difference between the TTP and TXP-functionalised compounds. As with other 68Ga-labelled lipophilic cations studied previously, the logD7.4 values of the compounds are low when compared to the ‘gold-standard’ SPECT-based lipophilic cation, [99mTc]Tc-sestaMIBI, which has a logD7.4 value of 1.29.19,24,25,34 However, since there is precedent of lipophilic cations with low logD7.4 values crossing lipid bilayer membranes, we decided to perform an ex vivo assessment of the candidate radiotracer.15,35
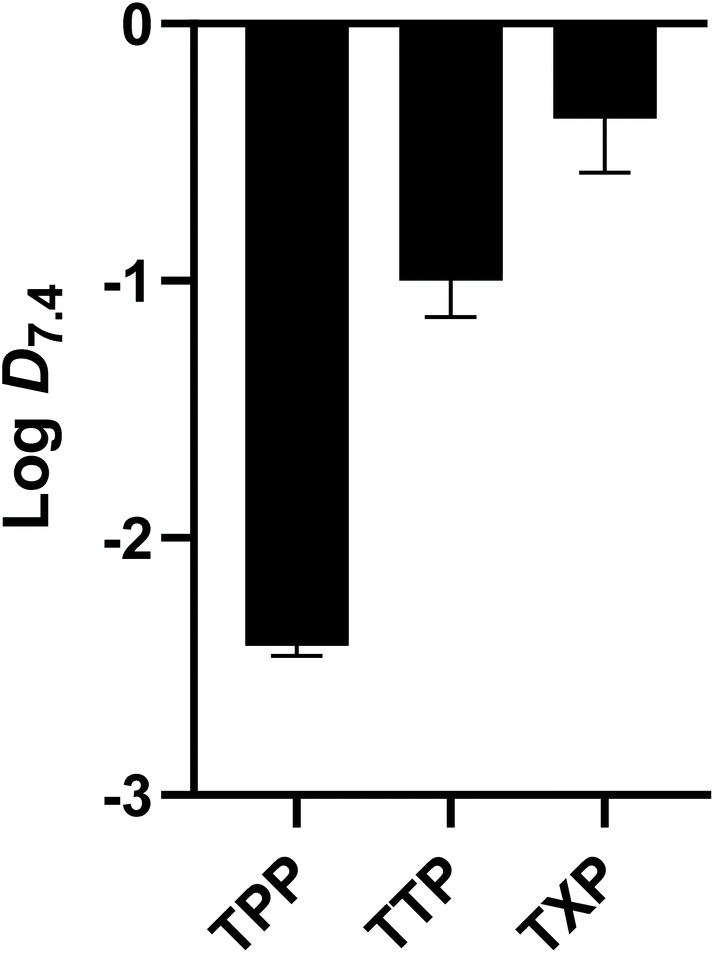 | ||
| Fig. 5 logD7.4 values obtained of the three [68Ga]Ga-NODAGA-xy-TAP compounds. Data are mean ± SD (n = 6). | ||
Ex vivo quantification of [68Ga]Ga-NODAGA-xy-TXP myocardial uptake using the langendorff isolated perfused heart model
In order to determine whether the tracer exhibited ΔΨm-dependent mitochondrial uptake, we employed the Langendorff isolated perfused heart model. This ex vivo model allows accurate and reproducible interventions on intact beating cardiac tissue which would otherwise be lethal to an animal in vivo, whilst also maintaining a high degree of in vivo relevance. Crucially for these experiments it also allows coronary perfusion (and hence radiotracer delivery) to be maintained constant, allowing the separation of the respective effects of ΔΨm and perfusion on radiotracer retention, whilst also simplifying pharmacokinetic modelling.28To determine the dependence of cardiac radiotracer uptake and retention on ΔΨm, cardiac uptake and washout curves were obtained during a control baseline period, and then during the infusion of 600 nM carbonyl cyanide m-chlorophenyl hydrazone (CCCP),36 which we have previously shown to completely depolarise ΔΨm, validated using the clinical SPECT radiopharmaceutical [99mTc]Tc-sestaMIBI (Fig. 1).8,24,25 We confirm this in Fig. 6A, where under baseline conditions, 25.4 ± 1.92% of an injected [99mTc]Tc-sestaMIBI bolus is retained in the heart 15 minutes post-injection when the mitochondria are polarised, while 0.00 ± 0.53% is retained when the mitochondria are depolarised by 600 nM CCCP (n = 3).
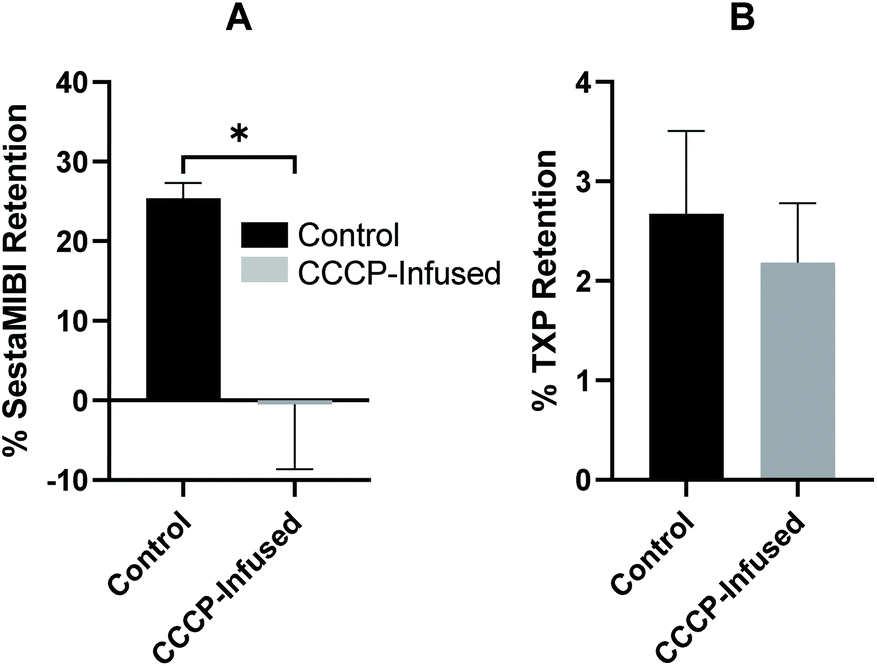 | ||
| Fig. 6 (A) and (B) Cardiac retention values obtained using the isolated perfused heart model and two-injection protocol 15 minutes post-injection for [99mTc]Tc-sestaMIBI and [68Ga]Ga-(R)-NODAGA-xy-TXP respectively. Control data were obtained from the injection before CCCP infusion, whilst CCCP data was obtained during 600 nM CCCP infusion. Data for [99mTc]Tc-sestaMIBI are mean ± SD (n = 3). * = P < 0.05, data for [68Ga]Ga-(R)-NODAGA-xy-TXP are mean ± SD (n = 3). | ||
The myocardial uptake and retention of [68Ga]Ga4c was assessed, since this complex displayed the highest lipophilicity of the three compounds synthesised. Data from the isolated heart characterisation of [68Ga]Ga4c are summarised in Fig. 6B, 2.67 ± 0.83% of injected dose was retained in the heart 15 minutes post-injection when cardiac mitochondria are polarised, and upon 600 nM CCCP infusion, retention of [68Ga]Ga4c was not significantly diminished in depolarised cardiac mitochondria, falling to 2.19 ± 0.60%. This lower cardiac uptake compared [99mTc]Tc-sestaMIBI is likely due to the lower lipophilicity of [68Ga]Ga4c hindering passage through the lipid bilayer membrane. As a result of its increased lipophilicity, [68Ga]Ga4c does demonstrate a near three-fold increase in uptake and retention in healthy cardiac mitochondria compared to DO3A analogues studied previously. However, in depolarised cardiac mitochondria, both [68Ga]Ga4c and its DO3A derivative exhibited no statistically significant difference in retention compared to in healthy cardiac mitochondria. It is also possible that while net positive charge and gross lipophilicity might make the tracers more amenable to crossing cell membranes and being retained within the mitochondria than previous [68Ga]Ga-chelates we have investigated, the distribution of positive charges across the surface area of the complex is uneven, such that there may be a polarity within the molecule which hinders its capacity to enter the cell; it is possible that while the lipophilic “head” of the molecule may penetrate cell membranes, the cationic “tail” cannot. This polarity effect is further supported by the retention of previous DO2A analogues, which demonstrated the importance of charge distribution in terms of ΔΨm-dependent retention. Based on initial analysis of molecular electrostatic potential (MEP) mapping of the DFT optimised structures of [Ga(4c)]+ and its DO2A counterpart, [Ga(DO2A(-xy-TXP)2]3+ shown in Fig. S50,† a more symmetrical charge distribution was observed for the DO2A analogue along with a greater distribution of positive charge across the entire molecule compared to [Ga(4c)]+. A more substantial and symmetrical positive charge distribution would result in greater uptake and retention polarised mitochondria, which was observed for the previous DO2A tracer, and resulted in a significant difference in retention when no membrane potential is present compared to [68Ga]Ga4c. [68Ga]Ga4c may not exhibit superior ΔΨm-dependent uptake and retention in cardiac cells compared to previous [68Ga]Ga-cationic species, nevertheless, this work does expand our understanding of the structure/activity relationship for refining our synthetic strategy, providing a basis for further functionalisation to improve these characteristics.
Conclusions
We have synthesised a series of TAP-functionalised NODAGA chelates for gallium-68 labelling, with a view to synthesising lipophilic and cationic radiotracers. This work represents an entirely modular and effective synthetic strategy, being the basis for synthesising a range of optimal cationic and lipophilic gallium-68 radiotracers with tuneable lipophilicity. These compounds demonstrate rapid radiolabelling with gallium-68 at room temperature, with high radiochemical yields. Cardiac uptake and retention of the most lipophilic radiotracer of this series was assessed using the Langendorff isolated perfused heart model. The results of this work show optimised radiolabelling kinetics compared to the previous gallium-68 tracer families studied in our group, however the logD7.4 values and cationic charge distribution over the molecules surface-area need to be improved. Nevertheless, the results emphasise and encourage the development of future gallium-68-based lipophilic cations to report on mitochondrial function.
Experimental procedures
Materials and instrumentation
Commercially available reagent grade solvents and chemicals were used without further purification. All gas mixtures were purchased from BOC Industrial Gases. Anhydrous solvents were acquired from solvent towers within the department and stored over 3 Å molecular sieves. 1H, 13C{1H}, 31P{1H}, COSY, HSQC, HMBC NMR spectra were recorded on a Bruker AVIII 400 spectrometer. Chemical shifts are reported in ppm and referenced to residual protonated impurities in the solvent for NMR spectra. High Resolution Electrospray Mass Spectrometry was carried out by Dr Lisa Haigh of the mass spectrometry service at Imperial College, or independently on an Aglient 6200 TOF LC-MS instrument. Flash chromatography used silica gel (60 Å pore size). Where specified, automated flash chromatography was performed using a Biotage Isolera Four unit and 10 g, 25 g, 50 g or 100 g SNAP KP-Sil/Sfar duo cartridge. HPLC was performed on an Agilent 1200 Series Liquid Chromatograph with UV and LabLogic Flow-Count detector with a sodium-iodide probe (B-FC-3200). Mobile phase A contained H2O with 0.1% TFA, and mobile phase B contained MeCN with 0.1% TFA. Analytical reverse-phase HPLC was conducted using a ZORBAX Eclipse XDB-C18 column (4.6 × 150 mm, 5 μm) and UV spectroscopic detection at 250 nm. Gallium-68 was eluted as [68Ga]GaCl3 from an Eckert and Ziegler gallium-68 generator using a 0.1 M solution of hydrochloric acid.Synthesis
TAP-functionalised xylyl bromides (1a–c) were synthesised as previously reported.24 (R)-4-(4,7-Bis(2-(tert-butoxy)-2-oxoethyl)-1,4,7-triazacyclononan-1-yl)-5-(tert-butoxy)-5-oxopentanoic acid (NODAGA(tBu)3) was synthesised according to a previously reported literature procedure by Eisenwiener et al.32General procedure 1: synthesis of NH2-xy-TAP compounds (2)
To a solution of 1 (1 equiv.) in EtOH (150 mL), aqueous NH3 (28% wt., 40 mL) was added, and the resulting mixture was stirred at rt for 48 h. Solvent was removed under reduced pressure, before the residue was purified by reverse-phase flash chromatography (C-18 SiO2, 0–100% B in A) to yield the desired product.![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 4), 7.12 (2H, dd, 3JHH = 8.1, 4JHH = 2.6 Hz, C64), 4.92 (2H, d, 2JHP = 15.1 Hz, C2P), 4.13 (2H, s, C2NH2), 2.51 (9H, s, Ar-CH3). 13C{1H}-NMR (101 MHz, MeOD) δC (ppm): 148.0 (p-Ar), 135.3 (d, 2JCP = 9.8 Hz, o-Ar), 135.1 (
4), 7.12 (2H, dd, 3JHH = 8.1, 4JHH = 2.6 Hz, C64), 4.92 (2H, d, 2JHP = 15.1 Hz, C2P), 4.13 (2H, s, C2NH2), 2.51 (9H, s, Ar-CH3). 13C{1H}-NMR (101 MHz, MeOD) δC (ppm): 148.0 (p-Ar), 135.3 (d, 2JCP = 9.8 Hz, o-Ar), 135.1 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) 6H4), 132.9 (d, 3JCP = 5.6 Hz, 6H4), 132.0 (d, 3JCP = 12.9 Hz, m-Ar), 130.7 (6H4), 130.3 (d, 2JCP = 8.1 Hz 6H4), 115.8 (d, 1JCP = 85.3 Hz, i-Ar), 43.7 (H2NH2), 30.7 (d, 1JCP = 49.6 Hz, H2P), 21.8 (Ar-CH3). 31P{1H}-NMR (162 MHz, MeOD) δP (ppm): 22.1. HRMS (ES-TOF+): m/z calcd for C29H31NP ([M − Br]+) 424.2194. Found: 424.2193.
4), 7.23 (6H, d, 3JHH = 13.0 Hz, o-Ar), 7.09 (2H, dd, 3JHH = 8.1, 4JHH = 2.7 Hz, C64), 4.90 (2H, dd, 2JHP = 15.1, 3JHH = 5.9 Hz, C2P), 4.13 (2H, s, C2NH2), 2.38 (18H, s, Ar-CH3). 13C{1H}-NMR (101 MHz, MeOD) δC (ppm): 141.7 (d, 3JCP = 13.9 Hz, m-Ar), 137.9 (p-Ar), 135.1 (6H4), 133.1 (d, 3JCP = 5.7 Hz, 6H4), 132.6 (d, 2JCP = 10.0 Hz, o-Ar), 130.5 (6H4), 118.9 (d, 1JCP = 85.1 Hz, i-Ar), 43.7 (H2NH2), 30.5 (d, 1JCP = 49.3 Hz, H2P), 21.3 (Ar-CH3). 31P{1H}-NMR (162 MHz, MeOD) δP (ppm): 22.2. HRMS (ES-TOF+): m/z calcd for C32H37NP ([M − Br]+) 466.2664. Found: 466.2646.
6H4), 132.9 (d, 3JCP = 5.6 Hz, 6H4), 132.0 (d, 3JCP = 12.9 Hz, m-Ar), 130.7 (6H4), 130.3 (d, 2JCP = 8.1 Hz 6H4), 115.8 (d, 1JCP = 85.3 Hz, i-Ar), 43.7 (H2NH2), 30.7 (d, 1JCP = 49.6 Hz, H2P), 21.8 (Ar-CH3). 31P{1H}-NMR (162 MHz, MeOD) δP (ppm): 22.1. HRMS (ES-TOF+): m/z calcd for C29H31NP ([M − Br]+) 424.2194. Found: 424.2193.
4), 7.23 (6H, d, 3JHH = 13.0 Hz, o-Ar), 7.09 (2H, dd, 3JHH = 8.1, 4JHH = 2.7 Hz, C64), 4.90 (2H, dd, 2JHP = 15.1, 3JHH = 5.9 Hz, C2P), 4.13 (2H, s, C2NH2), 2.38 (18H, s, Ar-CH3). 13C{1H}-NMR (101 MHz, MeOD) δC (ppm): 141.7 (d, 3JCP = 13.9 Hz, m-Ar), 137.9 (p-Ar), 135.1 (6H4), 133.1 (d, 3JCP = 5.7 Hz, 6H4), 132.6 (d, 2JCP = 10.0 Hz, o-Ar), 130.5 (6H4), 118.9 (d, 1JCP = 85.1 Hz, i-Ar), 43.7 (H2NH2), 30.5 (d, 1JCP = 49.3 Hz, H2P), 21.3 (Ar-CH3). 31P{1H}-NMR (162 MHz, MeOD) δP (ppm): 22.2. HRMS (ES-TOF+): m/z calcd for C32H37NP ([M − Br]+) 466.2664. Found: 466.2646.
General procedure 2: synthesis of tert-butyl protected (R)-NODAGA-xy-TAP compounds (3)
This methodology was a modification of the procedure described by Schirrmacher et al.33 (R)-NODAGA(tBu)3 (1 equiv.) and 2 (1.2 equiv.) were dissolved in mixture of H2O and MeCN (1:1). DCC (1.1 equiv.) was dissolved in pyridine and added dropwise to the mixture before being stirred at rt for 48 h. The solvent was removed under reduced pressure, before the crude residue was dissolved in MeCN (8.0 mL) and the urea by-product was removed by filtration. The solvent was removed under reduced pressure and the residue was purified by reverse-phase flash chromatography (C-18 SiO2, 0–100% B in A) to yield the desired product.
:MeCN (1:1, 9.0 mL), DCC (0.04 g, 0.20 mmol) and pyridine (1.15 mL) as a pale yellow solid (0.07 g, 0.07 mmol, 39%). 1H-NMR (400 MHz, MeOD) δH (ppm): 7.88 (3H, t, 3JHH = 7.6 Hz, p-Ph), 7.70 (6H, m, m-Ph), 7.62 (6H, dd, 3JHP = 12.7, 3JHH = 8.0 Hz, o-Ph), 7.14 (2H, d, 3JHH = 7.7 Hz, C64), 6.95 (2H, dd, 3JHH = 7.9, 4JHH = 2.6 Hz, C64), 4.88 (2H, d, 2JHP = 15.1 Hz, C2P), 4.31 (2H, s, C2NH), 3.51 (5H, m, CN/C2COOtBu), 3.09 (12H, m, macrocycle ), 2.41 (2H, t, 3JHH = 7.8 Hz, C2CONH), 2.02 (2H, m, NCHC2), 1.48 (27H, m, COOC(C3)3). 13C{1H}-NMR (101 MHz, MeOD) δC (ppm): 175.0 (ONH), 173.1 (OOtBu), 141.2 (6H4), 136.5 (p-Ph), 135.3 (d, 2JCP = 9.7 Hz, o-Ph), 132.3 (d, 2JCP = 5.7 Hz, 6H4), 131.4 (d, 3JCP = 12.4 Hz, m-Ph), 129.1 (6H4), 127.4 (6H4), 119.1 (d, 1JCP = 86.5 Hz, i-Ph), 84.4, 83.5, 83.3 ((CH3)3), 64.9 (HNH), 56.4 (H2COOtBu), 52.8, 51.5, 46.9, 45.9 (macrocycle ), 43.5 (H2NH), 33.8 (H2CONH), 30.4 (d, 1JCP = 46.9 Hz, H2P), 28.4 (C(H3)3), 27.1 (NCHH2). 31P{1H}-NMR (162 MHz, MeOD) δP (ppm): 22.4. HRMS (ES-TOF+): m/z calcd for C53H72N4O7P ([M − Br]+) 907.5139. Found: 907.5150.
:MeCN (1:1, 9.0 mL), DCC (0.04 g, 0.20 mmol) and pyridine (1.25 mL) as a pale yellow solid (0.09 g, 0.09 mmol, 46%). 1H-NMR (400 MHz, MeOD) δH (ppm): 7.52 (6H, dd, 3JHH = 8.1, 4JHH = 3.4 Hz, m-Ar), 7.45 (6H, dd, 3JHP = 12.3, 3JHH = 8.1 Hz, o-Ar), 7.15 (2H, d, 3JHH = 7.0 Hz, C64), 6.95 (2H, dd, 3JHH = 7.8, 4JHH = 2.7 Hz, C64), 4.77 (2H, d, 2JHP = 14.9 Hz, C2P), 4.33 (2H, s, C2NH), 3.58 (5H, m, C2COOtBu/CN), 3.04 (12H, m, macrocycle ), 2.50 (9H, s, Ar-C3), 2.40 (2H, m, C2CONH), 2.03 (2H, m, NCHC2), 1.49 (27H, m, COOC(C3)3). 13C{1H}-NMR (101 MHz, MeOD) δC (ppm): 175.0 (ONH), 173.1 (OOtBu), 171.2 (OOtBu), 168.5 (OOtBu), 148.0 (p-Ar), 141.0 (6H4), 135.2 (d, 2JCP = 9.7 Hz, o-Ar), 132.3 (d, 2JCP = 6.1 Hz, 6H4), 131.9 (d, 3JCP = 12.9 Hz, m-Ar), 129.0 (6H4), 127.8 (6H4), 115.9 (d, 1JCP = 89.5 Hz, i-Ar), 84.4, 83.4, 83.2 (COO(CH3)3), 64.9 (HN), 56.4 (H2COOtBu), 52.9, 51.3, 46.9, 46.0 (macrocycle ), 43.5 (H2NH), 33.8 (H2CONH), 30.7 (d, 1JCP = 48.1 Hz, H2P), 28.5, 28.4, 28.2 (COOC(H3)3), 27.2 (NCHH2), 21.8 (Ar-H3). 31P{1H}-NMR (162 MHz, MeOD) δP (ppm): 21.8. HRMS (ES-TOF+): m/z calcd for C56H78N4O7P ([M − Br]+) 949.5603. Found: 949.5592.
:MeCN (1:1, 9.0 mL), DCC (0.04 g, 0.20 mmol) and pyridine (1.25 mL) as a pale yellow solid (0.09 g, 0.09 mmol, 46%). 1H-NMR (400 MHz, MeOD) δH (ppm): 7.51 (3H, m, p-Ar), 7.15 (8H, m, o-Ar/C64), 6.91 (2H, dd, 3JHH = 8.2, 4JHH = 2.5 Hz, C64), 4.75 (2H, d, 2JHP = 14.8 Hz, C2P), 4.34 (2H, s, C2NH), 3.79 (4H, m, C2COOtBu), 3.54 (1H, t, 3JHH = 7.3 Hz, CN), 3.11 (12H, m, macrocycle ), 2.48 (2H, m, C2CONH), 2.35 (18H, s, Ar-C3), 2.02 (2H, m, NCHC2), 1.49 (27H, s, COOC(C3)3). 13C{1H}-NMR (101 MHz, MeOD) δC (ppm): 175.0 (ONH) 173.0 (OOtBu), 141.7 (d, 3JCP = 13.1 Hz, m-Ar), 141.1 (d, 5JCP = 3.9 Hz, 6H4), 137.8 (d, 4JCP = 3.3 Hz, p-Ar), 132.6 (d, 2JCP = 9.7 Hz, o-Ar), 132.4 (d, 3JCP = 5.6 Hz, 6H4), 128.9 (d, 4JCP = 3.4 Hz, 6H4), 128.0 (d, 2JCP = 8.6 Hz, 6H4), 119.1 (d, 1JCP = 71.5 Hz, i-Ar), 83.2 (COO(CH3)3), 65.0 (HN), 56.4 (H2COOtBu), 52.8, 51.4, 46.9, 46.0 (macrocycle ), 43.5 (H2NH), 33.8 (H2CONH), 30.5 (d, 1JCP = 48.3 Hz, H2P), 28.5, 28.4, 28.3 (COOC(H3)3), 27.1 (NCHH2), 21.3 (Ar-H3). 31P{1H}-NMR (162 MHz, MeOD) δP (ppm): 22.2. HRMS (ES-TOF+): m/z calcd for C59H84N4O7P ([M − Br]+) 991.6078. Found: 991.6077.
General procedure 3: deprotection of tert-butyl protected (R)-NODAGA-xy-TAP compounds using trifluoroacetic acid (4)
Compounds 3a–c were dissolved in trifluoroacetic acid (reaction mixture concentration 0.1 g mL−1) and stirred at rt under an N2 atmosphere for 48 h. The acid was removed under reduced pressure, before the residue was purified by reverse-phase flash chromatography (C-18 SiO2, 0–100% B in A) to yield the desired product.4), 6.95 (2H, dd, 3JHH = 7.9, 4JHH = 2.7 Hz, C64), 4.88 (2H, d, 2JHP = 14.9 Hz, C2P), 4.33 (2H, s, C2NH), 3.97 (4H, m, C2COOH), 3.61 (1H, t, 3JHH = 7.1 Hz, HN), 3.11 (12H, m, macrocycle ), 2.47 (2H, m, C2CONH), 2.09 (2H, m, NCHC2). 13C{1H}-NMR (101 MHz, MeOD) δC (ppm): 175.1 (ONH), 175.0 (OOH), 141.2 (d, 5JCP = 4.2 Hz, 6H4), 136.5 (d, 4JCP = 3.2 Hz, p-Ph), 135.3 (d, 2JCP = 9.7 Hz, o-Ph), 132.3 (d, 2JCP = 5.5 Hz, 6H4), 131.3 (d, 3JCP = 12.8 Hz, m-Ph), 129.1 (d, 4JCP = 3.5 Hz, 6H4), 127.4 (d, 3JCP = 8.8 Hz, 6H4), 119.1 (d, 1JCP = 86.5 Hz, i-Ph), 64.3 (HN), 55.8 (H2COOH), 52.1, 47.2 (macrocycle ), 43.5 (H2NH), 33.6 (H2CONH), 30.4 (d, 1JCP = 48.4 Hz, H2P), 26.6 (NCHH2). 31P{1H}-NMR (162 MHz, MeOD) δP (ppm): 22.6. 19F{1H}-NMR (377 MHz, MeOD) δF (ppm): −77.1. HRMS (ES-TOF+): m/z calcd for C41H48N4O7P ([M − CF3CO2]+) 739.3261. Found: 739.3265.
4), 6.94 (2H, dd, 3JHH = 8.1, 4JHH = 2.7 Hz, C64), 4.75 (2H, d, 2JHP = 14.9 Hz, C2P), 4.33 (2H, s, C2NH), 3.88 (5H, m, C2COOH/CN), 3.10 (12H, m, macrocycle ), 2.50 (9H, s, Ar-C3), 2.46 (2H, m, C2CONH), 2.08 (2H, m, NCHC2). 13C{1H}-NMR (101 MHz, MeOD) δC (ppm): 175.1 (ONH/OOH), 148.1 (p-Ar), 141.1 (6H4), 135.2 (d, 2JCP = 9.9 Hz, o-Ar), 132.2 (d, 3JCP = 5.2 Hz, 6H4), 131.9 (d, 3JCP = 12.4 Hz, m-Ar), 129.0 (6H4), 127.8 (d, 2JCP = 8.7 Hz, 6H4), 115.9 (d, 1JCP = 88.5 Hz, i-Ar), 64.3 (HN), 55.8 (H2COOH), 52.0, 47.0 (macrocycle ), 43.6 (H2NH), 33.6 (H2CONH), 30.8 (d, 1JCP = 48.8 Hz, H2P), 26.6 (NCHH2), 21.8 (Ar-H3). 31P{1H}-NMR (162 MHz, MeOD) δP (ppm): 21.8. HRMS (ES-TOF+): m/z calcd for C44H54N4O7P ([M − CF3CO2]+) 781.3730. Found: 781.3740.
4), 6.91 (2H, dd, 3JHH = 8.2, 4JHH = 2.5 Hz, C64), 4.74 (2H, d, 2JHP = 14.8 Hz, C2P), 4.33 (2H, s, C2NH), 3.84 (4H, m, C2COOtBu), 3.60 (1H, t, 3JHH = 7.2 Hz, CN), 3.15 (12H, m, macrocycle ), 2.46 (2H, m, C2CONH), 2.35 (18H, s, Ar-C3), 2.06 (2H, m, NCHC2). 13C{1H}-NMR (101 MHz, MeOD) δC (ppm): 175.1 (ONH/OOH), 141.7 (d, 3JCP = 13.0 Hz, m-Ar), 141.2 (6H4), 137.9 (p-Ar), 132.6 (d, 2JCP = 9.7 Hz, o-Ar), 132.4 (6H4), 128.9 (6H4), 128.0 (6H4), 119.0 (d, 1JCP = 85.0 Hz, i-Ar), 64.2 (HN), 55.8 (H2COOH), 52.2, 51.6, 47.3, 46.5 (macrocycle ), 43.5 (H2NH), 33.6 (H2CONH), 30.6 (d, 1JCP = 48.0 Hz, H2P), 26.6 (NCHH2), 21.3 (Ar-H3). 31P{1H}-NMR (162 MHz, MeOD) δP (ppm): 22.2. 19F{1H}-NMR (377 MHz, MeOD) δF (ppm): −77.4. HRMS (ES-TOF+): m/z calcd for C47H60N4O7P ([M − CF3CO2]+) 823.4200. Found: 823.4175.
General procedure 4: complexation of (R)-NODAGA-xy-TAP compounds with natGa3+ ([natGa]Ga4)
To a solution of 4 (1 equiv.) in NaOAc buffer (3.60 M, 5.00 mL, pH 5.00), Ga(NO3)3·xH2O (1.5 equiv.) was added, and the resulting suspension was stirred at rt for 15 min. The solvent was removed under reduced pressure, before the residue was purified by reverse-phase flash chromatography (C-18 SiO2, 0–100% B in A) to yield the desired product.[natGa]Ga-(R)-NODAGA-xy-TPP trifluoroacetate ([natGa]Ga4a)
Following general procedure 4, the title compound was prepared from 4a (0.03 g, 0.04 mmol) and Ga(NO3)3·xH2O (0.01 g, 0.05 mmol) as a white solid (0.03 g, 0.03 mmol, 83%). 1H-NMR (400 MHz, MeOD) δH (ppm): 7.88 (3H, m, p-Ph), 7.71 (6H, td, 3JHH = 7.7, 4JHH = 3.5 Hz, m-Ph), 7.63 (6H, dd, 3JHP = 12.5, 3JHH = 7.7 Hz, o-Ph), 7.14 (2H, d, 3JHH = 7.5 Hz, C64), 6.99 (2H, m, C64), 4.88 (2H, m, C2P), 4.29 (2H, m, C2NH), 3.67 (5H, m, C2COOGa/CN), 3.49 (4H, m, macrocycle ), 3.13 (8H, m, macrocycle ), 2.63 (2H, m, C2CONH), 2.20 (2H, m, NCHC2). 13C{1H}-NMR (101 MHz, MeOD) δC (ppm): 175.7 (ONH), 174.6 (OOGa), 174.5 (OOGa), 174.4 (OOGa), 141.1 (6H4), 136.5 (p-Ph), 135.4 (d, 2JCP = 9.7 Hz, o-Ph), 132.5 (d, 2JCP = 5.5 Hz, 6H4), 131.4 (d, 3JCP = 12.6 Hz, m-Ph), 129.2 (d, 4JCP = 3.3 Hz, 6H4), 127.4 (6H4), 119.1 (d, 1JCP = 86.1 Hz, i-Ph), 68.5 (HN), 63.43 (H2COOGa), 54.8, 54.5, 54.1, 53.8 (macrocycle ), 43.5 (H2NH), 34.04 (H2CONH), 30.4 (d, 1JCP = 48.4 Hz, H2P), 23.7 (NCHH2). 31P{1H}-NMR (162 MHz, MeOD) δP (ppm): 22.6.
HRMS (ES-TOF+): m/z calcd for C41H46N4O7P69Ga ([M − CF3CO2]+) 806.2360. Found: 806.2341.
[natGa]Ga-(R)-NODAGA-xy-TTP trifluoroacetate ([natGa]Ga4b)
Following general procedure 4, the title compound was prepared from 4b (0.03 g, 0.03 mmol) and Ga(NO3)3·xH2O (0.01 g, 0.04 mmol) as a white solid (0.03 g, 0.03 mmol, 97%). 1H-NMR (400 MHz, MeOD) δH (ppm): 7.52 (6H, dd, 3JHH = 8.2, 4JHH = 3.4 Hz, m-Ar), 7.45 (6H, dd, 3JHP = 12.2, 3JHH = 8.1 Hz, o-Ar), 7.14 (2H, d, 3JHH = 7.7 Hz, C64), 6.98 (2H, m, C64), 4.76 (2H, d, 2JHP = 14.9 Hz, C2P), 4.32 (2H, m, C2NH), 3.67 (5H, m, C2COOGa/CN), 3.47 (4H, m, macrocycle ), 3.22 (8H, m, macrocycle ), 2.62 (2H, m, C2CONH), 2.49 (9H, s, Ar-C3), 2.23 (2H, m, NCHC2). 13C{1H}-NMR (101 MHz, MeOD) δC (ppm): 175.7 (ONH), 174.6 (OOGa), 174.5 (OOGa), 174.4 (OOGa), 148.0 (d, 4JCP = 3.2 Hz, p-Ar), 141.0 (d, 4JCP = 4.2 Hz, 6H4), 135.2 (d, 2JCP = 9.8 Hz, o-Ar), 132.5 (d, 3JCP = 5.3 Hz, 6H4), 131.9 (d, 3JCP = 12.9 Hz, m-Ar), 129.1 (d, 5JCP = 3.3 Hz, 6H4), 127.8 (d, 2JCP = 8.7 Hz, 6H4), 115.9 (d, 1JCP = 89.1 Hz, i-Ar), 68.5 (HN), 63.4 (H2COOGa), 54.8, 54.5, 54.1, 53.8 (macrocycle ), 43.6 (H2NH), 34.03 (H2CONH), 30.8 (d, 1JCP = 48.8 Hz, H2P), 22.9 (NCHH2), 21.8 (Ar-H3). 31P{1H}-NMR (162 MHz, MeOD) δP (ppm): 21.8. HRMS (ES-TOF+): m/z calcd for C44H52N4O7P69Ga ([M − CF3CO2]+) 848.2829. Found: 848.2817.
[natGa]Ga-(R)-NODAGA-xy-TXP trifluoroacetate ([natGa]Ga4c)
Following general procedure 4, the title compound was prepared from 4c (0.03 g, 0.03 mmol) and Ga(NO3)3·xH2O (0.01 g, 0.05 mmol) as a white solid (0.03 g, 0.03 mmol, 99%). 1H-NMR (400 MHz, MeOD) δH (ppm): 7.51 (3H, m, p-Ar), 7.14 (8H, m, o-Ar/C64), 6.94 (2H, dd, 3JHH = 8.2, 4JHH = 2.5 Hz, C64), 4.75 (2H, d, 2JHP = 14.7 Hz, C2P), 4.33 (2H, m, C2NH), 3.67 (5H, m, C2COOGa/CN), 3.45 (4H, m, macrocycle ), 3.10 (8H, m, macrocycle ), 2.58 (2H, m, C2CONH), 2.35 (18H, s, Ar-C3), 2.01 (2H, m, NCHC2). 13C{1H}-NMR (101 MHz, MeOD) δC (ppm): 175.4 (ONH), 174.6 (OOGa), 174.5 (OOGa), 174.4 (OOGa), 141.7 (m-Ar), 141.6 (6H4), 137.9 (p-Ar), 132.6 (d, 2JCP = 8.7 Hz, o-Ar), 128.9 (6H4), 128.0 (6H4), 119.0 (d, 1JCP = 84.8 Hz, i-Ar), 63.4 (HN), 54.8 (H2COOGa), 54.1, 53.8 (macrocycle ), 43.5 (H2NH), 34.0 (H2CONH), 30.3 (d, 1JCP = 48.0 Hz, H2P), 22.8 (NCHH2), 21.3 (Ar-H3). 31P{1H}-NMR (162 MHz, MeOD) δP (ppm): 22.2. HRMS (ES-TOF+): m/z calcd for C47H58N4O7P69Ga ([M − CF3CO2]+) 890.3299. Found: 890.3305.
Radiochemistry procedures
:MeOH (1:1), were implemented. Initial QC was also performed using analytical HPLC (Eluent gradient: 95% A for 5 min, 5–95% B in A for 25 min, 5% B in A for 5 min; flow rate 1 mL min−1).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) D7.4 values.
As an index for lipophilicity, octanol/phosphate buffered saline (PBS) partition coefficient (logD7.4) values of the radiolabelled compounds were determined. A general methodology is as follows: aliquots of the radiolabelled compound (5 μL, approx. 200 kBq) were added to vials containing a layered mixture of n-octanol (500 μL) and PBS (pH 7.4, 500 μL). After vortex mixing for 1 min, the mixture was centrifuged to separate the octanol/PBS phases (104 rpm, 2 min), before aliquots of the two phases (100 μL) were taken and transferred into vials for counting. Relative amounts of test compound in each phase were determined by an automatic gamma counter. The logD7.4 value was reported as the mean ± SD of data obtained in 6 independent experiments.
D7.4 values.
As an index for lipophilicity, octanol/phosphate buffered saline (PBS) partition coefficient (logD7.4) values of the radiolabelled compounds were determined. A general methodology is as follows: aliquots of the radiolabelled compound (5 μL, approx. 200 kBq) were added to vials containing a layered mixture of n-octanol (500 μL) and PBS (pH 7.4, 500 μL). After vortex mixing for 1 min, the mixture was centrifuged to separate the octanol/PBS phases (104 rpm, 2 min), before aliquots of the two phases (100 μL) were taken and transferred into vials for counting. Relative amounts of test compound in each phase were determined by an automatic gamma counter. The logD7.4 value was reported as the mean ± SD of data obtained in 6 independent experiments.
Langendorff isolated heart perfusion
After a 10 min stabilization period, a bolus of radiotracer (1 MBq) was injected into the arterial line, and the activity at the arterial, heart and venous detectors was recorded. After 15 minutes, a 6 μM solution of CCCP in mKHB heated to 37 °C was infused via the arterial line at 1.4 mL min−1, whilst the flow rate at the peristaltic pump was reduced by 10%. A further 1 MBq radiotracer bolus was administered 5 minutes after the commencement of CCCP infusion. The radiotracer retained after 15 min in both the healthy and CCCP-infused hearts was reported as the average of the normalized activity between 14–15 minutes after the radiotracer injection.
DFT calculations
All calculations were performed using the Gaussian 16 package (Revision C.01).38 Full geometry optimisations were performed using the ωB97X-D functional39 in aqueous solution using the polarisable continuum model (PCM),40 with the LANL2DZ basis set and effective core potentials for Ga, and 6-311+G** for all other atoms.41 Frequency analysis of the optimised structures confirmed a true minimum energy structure by the absence of imaginary frequencies.Author contributions
N. J. L. and R. S. jointly supervised the work. B. E. O. carried out the synthetic chemistry, radiochemistry and isolated heart perfusion. E. C. T. W. and F. B. performed Langendorff and triple γ-detector training and supervision and assisted with heart perfusion experiments. T. T. C. Y. performed the DFT calculations. All authors contributed to the writing and proofreading of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All experimental procedures were approved by King's College London's local Animal Care and Ethics Committee, and carried out in accordance with Home Office regulations as detailed in the Guidance on the Operation of Animals (Scientific Procedures) Act 1986.We would like to thank the King's College London & Imperial College London EPSRC Centre for Doctoral Training in Medical Imaging (EP/L015226/1) and the EPSRC Programme grants for Next Generation Molecular Imaging and Therapy with Radionuclides (EP/S032789/1 “MITHRAS”, EP/S019901/1 “RedOx_KCL”) funding, and Imperial College London for studentship funding for B. E. O.
Notes and references
- J. Maddahi, H. Kiat, K. F. Van Train, F. Prigent, J. Friedman, E. V. Garcia, N. Alazraki, E. G. DePuey, K. Nichols and D. S. Berman, Am. J. Cardiol., 1990, 66, E55–E62 CrossRef.
- J. D. Kelly, A. M. Forster, B. Higley, C. M. Archer, F. S. Booker, L. R. Canning, K. W. Chiu, B. Edwards, H. K. Gill and M. McPartlin, J. Nucl. Med., 1993, 34, 222–227 CAS.
- M. L. Chiu, J. F. Kronauge and D. Piwnica-Worms, J. Nucl. Med., 1990, 31, 1646–1653 CAS.
- D. Piwnica-Worms, J. F. Kronauge and M. L. Chiu, Circulation, 1990, 82, 1826–1838 CrossRef CAS PubMed.
- M. P. Murphy, Trends Biotechnol., 1997, 15, 326–330 CrossRef CAS PubMed.
- M. P. Murphy, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 1028–1031 CrossRef CAS PubMed.
- B. Kadenbach, R. Ramzan, R. Moosdorf and S. Vogt, Mitochondrion, 2011, 11, 700–706 CrossRef CAS PubMed.
- Z. M. Safee, F. Baark, E. C. T. Waters, M. Veronese, V. R. Pell, J. E. Clark, F. Mota, L. Livieratos, T. R. Eykyn, P. J. Blower and R. Southworth, Sci. Rep., 2019, 9, 216 CrossRef PubMed.
- H. Fukuda, A. Syrota, P. Charbonneau, J. Vallois, M. Crouzel, C. Prenant, J. Sastre and C. Crouzel, Eur. J. Nucl. Med., 1986, 11, 478–483 CAS.
- H. T. Ravert, I. Madar and R. F. Dannals, J. Labelled Compd. Radiopharm., 2004, 47, 469–476 CrossRef CAS.
- M. F. Ross, T. Da Ros, F. H. Blaikie, T. A. Prime, C. M. Porteous, I. I. Severina, V. P. Skulachev, H. G. Kjaergaard, R. A. J. Smith and M. P. Murphy, Biochem. J., 2006, 400, 199–208 CrossRef CAS PubMed.
- D.-Y. Kim, H.-J. Kim, K.-H. Yu and J.-J. Min, Bioorg. Med. Chem. Lett., 2012, 22, 319–322 CrossRef CAS PubMed.
- D.-Y. Kim, H.-J. Kim, K.-H. Yu and J.-J. Min, Bioconjugate Chem., 2012, 23, 431–437 CrossRef CAS PubMed.
- G. Zhao, Y.-M. Yu, T. M. Shoup, D. R. Elmaleh, A. A. Bonab, R. G. Tompkins and A. J. Fischman, J. Surg. Res., 2014, 188, 473–479 CrossRef CAS PubMed.
- T. Tominaga, H. Ito, Y. Ishikawa, R. Iwata, K. Ishiwata and S. Furumoto, J. Labelled Compd. Radiopharm., 2016, 59, 117–123 CrossRef CAS PubMed.
- D.-Y. Kim and J.-J. Min, Nucl. Med. Mol. Imaging, 2016, 50, 185–195 CrossRef CAS PubMed.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- T. Mou and X. Zhang, Molecules, 2017, 22, 562 CrossRef PubMed.
- A. Haslop, L. Wells, A. Gee, C. Plisson and N. Long, Mol. Pharm., 2014, 11, 3818–3822 CrossRef CAS PubMed.
- S. R. Banerjee and M. G. Pomper, Appl. Radiat. Isot., 2013, 76, 2–13 CrossRef CAS PubMed.
- L. Martiniova, L. De Palatis, E. Etchebehere and G. Ravizzini, Curr. Radiopharm., 2016, 9, 187–207 CrossRef CAS PubMed.
- V. Sharma, J. Sivapackiam, S. E. Harpstrite, J. L. Prior, H. Gu, N. P. Rath and D. Piwnica-Worms, PLoS One, 2014, 9, e109361 CrossRef PubMed.
- M. Kardashinsky, N. Lengkeek and L. M. Rendina, J. Labelled Compd. Radiopharm., 2017, 60, 4–11 CrossRef CAS PubMed.
- A. J. Smith, P. J. Gawne, M. T. Ma, P. J. Blower, R. Southworth and N. J. Long, Dalton Trans., 2018, 47, 15448–15457 RSC.
- A. J. Smith, B. E. Osborne, G. P. Keeling, P. J. Blower, R. Southworth and N. J. Long, Dalton Trans., 2020, 49, 1097–1106 RSC.
- E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- M. I. Tsionou, C. E. Knapp, C. A. Foley, C. R. Munteanu, A. Cakebread, C. Imberti, T. R. Eykyn, J. D. Young, B. M. Paterson, P. J. Blower and M. T. Ma, RSC Adv., 2017, 7, 49586–49599 RSC.
- E. Mariotti, M. Veronese, J. T. Dunn, R. A. Medina, P. J. Blower, R. Southworth and T. R. Eykyn, EJNMMI Res., 2013, 3, 74 CrossRef PubMed.
- M. G. Handley, R. A. Medina, E. Mariotti, G. D. Kenny, K. P. Shaw, R. Yan, T. R. Eykyn, P. J. Blower and R. Southworth, J. Nucl. Med., 2014, 55, 488–494 CrossRef CAS PubMed.
- F. Shaughnessy, E. Mariotti, K. P. Shaw, T. R. Eykyn, P. J. Blower, R. Siow and R. Southworth, EJNMMI Res., 2014, 4, 1–10 CrossRef CAS PubMed.
- R. A. Medina, E. Mariotti, D. Pavlovic, K. P. Shaw, T. R. Eykyn, P. J. Blower and R. Southworth, J. Nucl. Med., 2015, 56, 921–926 CrossRef CAS PubMed.
- K.-P. Eisenwiener, M. I. M. Prata, I. Buschmann, H.-W. Zhang, A. C. Santos, S. Wenger, J. C. Reubi and H. R. Mäcke, Bioconjugate Chem., 2002, 13, 530–541 CrossRef CAS PubMed.
- World Intellectual Property Organization, WO2010/066051A1, 2010, 1–35.
- Y.-S. Kim, Z. He, W.-Y. Hsieh and S. Liu, Bioconjugate Chem., 2007, 18, 929–936 CrossRef CAS PubMed.
- S. E. Abu-Gosh, N. Kolvazon, B. Tirosh, I. Ringel and E. Yavin, Mol. Pharm., 2009, 6, 1138–1144 CrossRef CAS PubMed.
- R. L. Gallo, J. N. Finkelstein and R. H. Notter, Biochim. Biophys. Acta, 1984, 771, 217–227 CrossRef CAS.
- R. Southworth and P. B. Garlick, Am. J. Physiol.: Heart Circ. Physiol., 2003, 285, H316–H324 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. a. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. a. Petersson, H. Nakatsuji, X. Li, M. Caricato, a. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, a. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. a. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. a. Keith, R. Kobayashi, J. Normand, K. Raghavachari, a. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallin, 2016 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dt02224e |
| This journal is © The Royal Society of Chemistry 2021 |