
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydroboration of aldehydes, ketones and CO2 under mild conditions mediated by iron(III) salen complexes†
Samantha
Lau
 ,
Cei B.
Provis-Evans
,
Alexander P.
James
and
Ruth L.
Webster
*
,
Cei B.
Provis-Evans
,
Alexander P.
James
and
Ruth L.
Webster
*
Department of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: r.l.webster@bath.ac.uk
First published on 20th July 2021
Abstract
The hydroboration of aldehydes, ketones and CO2 is demonstrated using a cheap and air stable [Fe(salen)]2-μ-oxo pre-catalyst with pinacolborane (HBpin) as the reductant under mild conditions. This catalyst system chemoselectively hydroborates aldehydes over ketones and ketones over alkenes. In addition, the [Fe(salen)2]-μ-oxo pre-catalyst shows good efficacy at reducing “wet” CO2 with HBpin at room temperature.
Introduction
Hydroboration (HB) of aldehydes and ketones is an efficient route to access primary and secondary alcohols respectively through the facile hydrolysis of organoborate intermediates formed in these reactions.1–3 In recent years there has been a focus on using earth abundant base metals, such as iron, to catalyse hydrofunctionalisation reactions of unsaturated moieties (alkenes, alkynes, nitriles)4–10 which have historically been dominated by late transition metal complexes.11–17Relative to noble metals, there are significantly fewer reports on using iron complexes to mediate hydroboration of aldehydes and ketones.18–24 Notably in 2017, Findlater and co-workers reported the simple use of a Fe(acac)3/NaHBEt3 system to hydroborate a range of aldehydes and ketones in moderate to excellent yields in the presence of 10 mol% catalyst.25 Following on, Baker, Hein and co-workers demonstrated the use of a [Fe(N2S2)]2 complex at 0.1 mol% catalyst loading to hydroborate aldehydes selectively with TON ca. 5200 easily achieved in one instance.26 In addition, the first case of an iron(II) coordination polymer and heterogeneous Fe2O3 nanoparticles mediated hydroboration of these carbonyl moieties have been reported by Zhang and co-workers27,28 and Geetharani, Bose and co-workers29 respectively. A more difficult but highly desired transformation is the asymmetric hydroboration of ketones to achieve optically active secondary alcohols,1 with only a handful of examples using iron complexes reported to date.23,30
Beyond HB of carbonyl functionality of simple molecules, there has been a paucity in reports of HB of CO2 mediated by iron complexes.31 In 2015, Sabo-Etienne, Bontemps and co-workers32 reported high conversion and selectivity for the formation of bis(boryl)acetal using 9-borabicyclo[3.3.1]-nonane (9-BBN) as the borane source and [Fe(H)2(dmpe)2] (dmpe = 1,2-bis(dimethylphosphino)ethane) as the catalyst.
Continuing from our previous studies using a simple iron salen complex (1a) to effect the hydrophosphination of alkenes33,34 and the cyclotrimerization of alkynes,35 here we report the hydroboration of both aldehydes and ketones with good chemoselectivity observed for aldehydes over ketones. Furthermore, we disclose modest rate of reduction of CO2 with HBpin (Scheme 1).
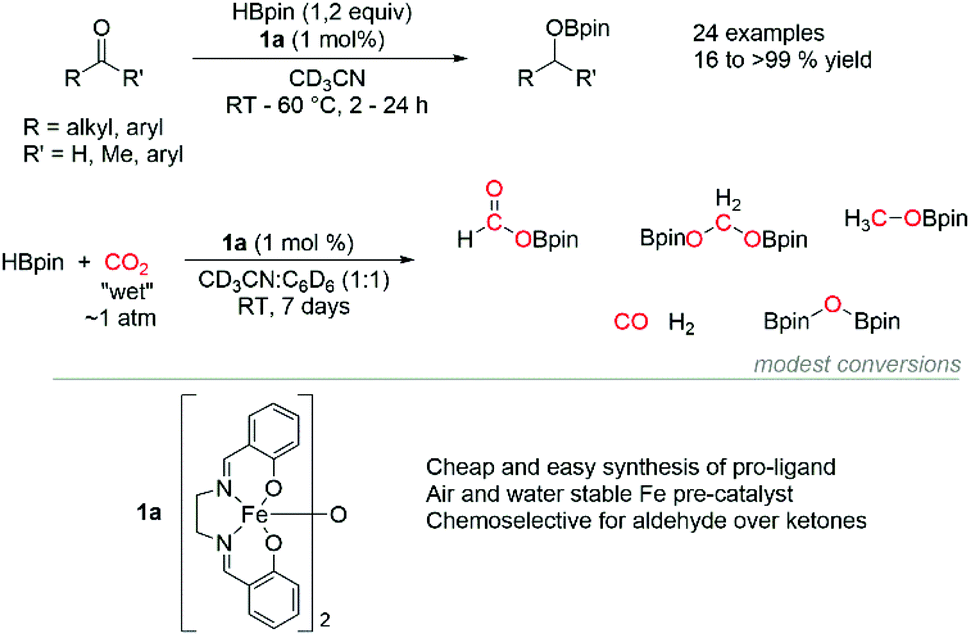 | ||
| Scheme 1 This work using [Fe(salen)]2-μ-oxo pre-catalyst in HB of aldehydes, ketones and CO2. | ||
Results and discussion
Optimisation of reaction conditions using 4-methylbenzaldehyde as the model substrate shows proficiency of the catalyst system to hydroborate the substrate using aromatic solvents (benzene, toluene) and polar solvents (dimethylformamide, methylene chloride, tetrahydrofuran). Acetonitrile was chosen due to higher solubility of 1a and ease of monitoring the reactions in CD3CN. Furthermore, HBpin was found to be much more active than catecholborane (HBcat) under comparable conditions (see ESI†). The reactions are carried out with 1 mol% catalyst loading at room temperature and 60 °C for aldehyde and ketone substrates respectively. Control reactions without any 1a show lower conversion for benzaldehyde (58%) and acetophenone (<5%) under similar reaction conditions (Fig. 1 and Fig. 2). | ||
| Fig. 1 Substrate scope for HB of aldehydes. All reactions were performed using 0.25 mmol aldehyde and 0.30 mmol HBpin in CD3CN (500 μL) unless stated otherwise. Spectroscopic yields reported using 1,2-dichloroethane as the internal standard. a2.4 equiv. of HBpin required for doubly reduced product. bPercentage conversion determined by loss of CHO signal due to 1,2-dichloroethane signal overlapping with characteristic methylene peak from reduction. | ||
 | ||
| Fig. 2 Substrate scope for HB of ketones. All reactions were performed using 0.25 mmol ketone and 0.30 mmol HBpin in CD3CN (500 μL) unless stated otherwise. Spectroscopic yields reported using 1,2-dichloroethane as the internal standard. | ||
Excellent yields are achieved across all the aldehydes tested with almost full conversion to the organoborate product observed spectroscopically. Electronic effects para to the benzaldehyde do not influence yields and the system is proficient for both aryl and alkyl aldehydes. 4-Hydroxybenzaldehyde requires a second equivalent of HBpin due to competing reactivity of the hydroxyl group with the reducing reagent (2f). Chemoselectivity is demonstrated with the alkene functionality left intact for cinnamaldehyde and only the mono hydroboration product observed (2h) by NMR spectroscopy. The organoborate products can be converted to their primary alcohol by quenching the reaction with minimal MeOH and then removal of the iron through a silica plug with methylene chloride as the eluent.
The homogeneity of the HB of aldehydes was tested. Complementary results from parallel PMe3 and Hg dropping test suggest the reaction is homogenous in nature (see ESI†).
Hydroboration of ketones requires a higher temperature and longer reaction time. Standard substrate, acetophenone, gives 3a in good spectroscopic yield on 0.25 mmol scale and 58% (0.59 g) isolated yield of 1-phenyl ethan-1-ol when the reaction is scaled by over 30 times (to 8.3 mmol). Good yields are obtained with electron withdrawing groups (3d–f) in the para position with lower conversions found for electron donating groups (3b, 3g) in the same position. The conversions of the ketones are comparable to the recent literature on iron mediated HB of ketones (vide supra). Aliphatic products 3i and 3j are formed in excellent spectroscopic yield. Competitive alkene HB is not observed, for example 3k is produced in low spectroscopic yield but the alkene is unreacted, while we attribute the modest yield of 3m to the volatility of the starting material. Unfortunately, our catalyst system is unable to hydroborate esters or tertiary amides, with less than 5% conversion of starting material observed for the ester substrates; ethyl benzoate, vinyl benzoate, methyl 2-bromobenzoate, methyl formate, and the amide substrates; N,N-diisopropylbenzamide, N-(tert-butyl)-N-isopropyl-3,5-bis(trifluoromethyl)benzamide and, N-(tert-butyl)-N-ethyl-4-methylbenzamide respectively, even at 80 °C with higher equivalents of HBpin used (Fig. 2).
The chemoselectivity of the catalyst system was further investigated; intermolecular competition reactions show the catalytic system selectively hydroborates aldehydes over ketones and ketones over alkenes (Scheme 2a–c). Due to the high conversions across all benzaldehyde derivatives with different electronic properties in the para position, a qualitative intermolecular competition reaction using 4-bromobenzaldehyde, benzaldehyde and 4-methoxybenzaldehyde was set up (Scheme 2d). The calculated spectroscopic conversions show faster reaction with electron withdrawing substituent –Br at the para position relative to electron donating substituent –OMe.
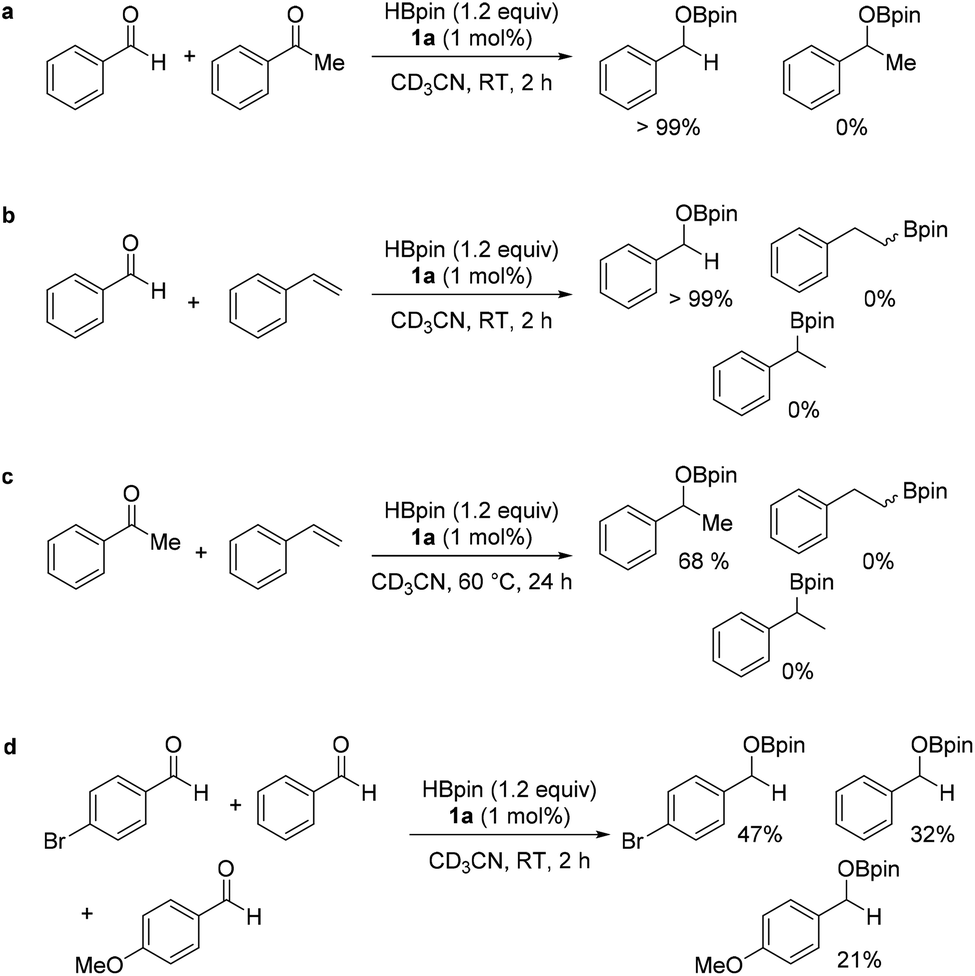 | ||
| Scheme 2 Competition HB reactions: a – intermolecular chemoselectivity between benzaldehyde and acetophenone, b – intermolecular chemoselectivity between benzaldehyde and styrene, c – intermolecular chemoselectivity between acetophenone and styrene and d – intermolecular competition of electronic effects para to benzaldehyde. | ||
To further interrogate the limits of this catalytic system, the HB of acetophenone derivatives using the chiral iron salen complex 1b under the same reaction conditions was carried out to probe if enantioinduction36 can be conferred to the product (Fig. 3, top). The reactions of the substrates are visibly faster; good to excellent conversions are achieved within 2 h at 60 °C compared to 24 h using 1a. Unfortunately, no enantioinduction is observed in the organoborate products after work-up to the secondary alcohols and reacting these with the chiral auxiliary (R)-Mosher's acid37 to form the corresponding diastereoisomeric ester product.38
 | ||
| Fig. 3 (Top) HB of acetophenone derivatives mediated by 1b, unfortunately no enantioinduction was induced to the organoborate product. (Bottom) HB of benzaldehyde (0.25 mmol) and HBpin (0.30 mmol) in CD3CN (500 μL) mediated by 1c. | ||
The different reactivity observed between 1a and 1b on the HB of ketones may allude to the mechanism involved in these reactions. The varying modes of activation that 1a can undergo have been investigated previously by our group showing potential to form a twisted Fe salen complex containing ‘Bpin’ fragment, or alternatively can simply be reduced down to 1c, an Fe(II) air and moisture sensitive analogue of 1a.34,35 Reacting benzaldehyde with 2 mol% 1c, results in 92% conversion to 2a at room temperature after 2 h which is comparable to the efficacy of 1a (Fig. 3, bottom). This data would suggest the mechanism of the HB of carbonyls is likely to undergo a reduction with HBpin to generate the active species 1c that participates in the catalytic cycle. The empirical observation that 1b is a more active pre-catalyst than 1a could be due to the greater electron density on the iron centre from the tert-butyl groups on the phenolate arms as well as the cyclohexyl backbone of the ligand for a faster reduction to the active Fe(II) species. Further detailed study is necessary to determine whether 1c undergoes further activation (i.e. twisting) to generate an on-cycle species, or whether 1c is a discrete on-cycle catalyst in its own right.
We next examined the reduction of CO2 using 1a as the pre-catalyst.‡ The advantage of using boranes for CO2 reduction circumvents harsher reaction conditions normally associated when using dihydrogen gas as the reductant by exploiting the more reactive B–H bond.39–43 Due to the insolubility of bis (pinacolato)diborane (BOB) formed during the reaction in CD3CN, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CD3CN:C6D6 is used for the following studies. Furthermore, “wet” CO2 generated from dry ice/toluene mixture, and without passing through a drying column, is added directly to the reaction vessel. Modest consumption of HBpin is achieved after 7 days at room temperature, with formation of the formoxy (4), acetal (5), methoxy (6) derivatives of HBpin, and undesired BOB confirmed by 1H and 11B NMR spectroscopy.44 Repeating the reaction at 60 °C shows similar conversions to 4, 5, and 6 but with a higher yield of BOB (see ESI†). Although the rate of the reaction is slow and conversions modest, the use of “wet” CO2 demonstrates the air and moisture stability of 1a in this catalytic transformation and the accessibility of this chemistry without the need of specialist equipment. Reaction with “dry” CO2 from gas cylinder shows similar reactivity and confirmed no detrimental effect in using “wet” CO2.
:1 mixture of CD3CN:C6D6 is used for the following studies. Furthermore, “wet” CO2 generated from dry ice/toluene mixture, and without passing through a drying column, is added directly to the reaction vessel. Modest consumption of HBpin is achieved after 7 days at room temperature, with formation of the formoxy (4), acetal (5), methoxy (6) derivatives of HBpin, and undesired BOB confirmed by 1H and 11B NMR spectroscopy.44 Repeating the reaction at 60 °C shows similar conversions to 4, 5, and 6 but with a higher yield of BOB (see ESI†). Although the rate of the reaction is slow and conversions modest, the use of “wet” CO2 demonstrates the air and moisture stability of 1a in this catalytic transformation and the accessibility of this chemistry without the need of specialist equipment. Reaction with “dry” CO2 from gas cylinder shows similar reactivity and confirmed no detrimental effect in using “wet” CO2.
Alternative borane sources 9-BBN and HBcat were also tested but perform poorly in the reaction; differing from trends observed in the reported literature.31,32,45 Furthermore, no advantageous improvement to reduction of CO2 is observed using 1b as the pre-catalyst. However, the potential to optimise the ligand design of these [Fe(salen)]2-μ-oxo complexes provides an attractive opportunity to improve the rate of CO2 reduction but is beyond the scope of this study (Table 1).
|
|
|||||
|---|---|---|---|---|---|
| [Fe] | Borane | 4 | 5 | 6 | COa + H2 + R2BOBR2 |
| a 13CO2 reaction performed to try and detect free 13CO or Fe–13CO species. However, these were not observed, potentially due to the small scale of the reaction and paramagnetic nature of any Fe–CO complex formed. | |||||
| 1a (1 mol%) | HBpin | 5% | 1% | 1% | 8% |
| 1a (1 mol%) | HBcat | — | — | 12% | 44% |
| 1a (1 mol%) | 9-BBN | 6% | — | — | — |
| 1b (1 mol%) | HBpin | — | — | — | 20% |
| 1c (2 mol%) | HBpin | 8% | 2% | 7% | 22% |

Conclusions
The hydroboration of aldehydes and ketones was achieved using an air and moisture stable [Fe(salen)]2-μ-oxo (1a) pre-catalyst. The catalyst system was chemoselective for aldehyde over ketone substrates. Unfortunately, the chemistry could not be expanded to induce enantioinduction using a chiral pre-catalyst, 1b. However, the reduction of CO2 using boranes was investigated with 1a tolerating “wet” CO2, formed from a dry ice/toluene mixture. Although the rate of conversion was slow and conversions modest for the reduction of CO2, future work on ligand design provides an attractive opportunity to improve the reaction whilst still benefitting from the air and moisture stability of these [Fe(salen)]2-μ-oxo complexes.Conflicts of interest
There are no conflicts to declare.Notes and references
- B. T. Cho, Chem. Soc. Rev., 2009, 38, 443–452 RSC.
- J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156–1184 CrossRef CAS.
- C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS.
- D. Wei and C. Darcel, Chem. Rev., 2019, 119, 2550–2610 CrossRef CAS PubMed.
- S. R. Tamang and M. Findlater, Molecules, 2019, 24, 3194–3194 CrossRef CAS PubMed.
- M. L. Shegavi and S. K. Bose, Catal. Sci. Technol., 2019, 9, 3307–3336 RSC.
- J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–15 CrossRef CAS PubMed.
- H. Yoshida, ACS Catal., 2016, 6, 1799–1811 CrossRef CAS.
- Y. Y. Li, S. L. Yu, W. Y. Shen and J. X. Gao, Acc. Chem. Res., 2015, 48, 2587–2598 CrossRef CAS PubMed.
- J. Chen, J. Guo and Z. Lu, Chin. J. Chem., 2018, 36, 1075–1109 CrossRef CAS.
- T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef CAS.
- Z. Xu, W. S. Huang, J. Zhang and L. W. Xu, Synthesis, 2015, 3645–3668 CrossRef CAS.
- Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC.
- C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef CAS PubMed.
- S. Díez-González and S. P. Nolan, Org. Prep. Proced. Int., 2007, 39, 523–559 CrossRef.
- C. Vogels and S. Westcott, Curr. Org. Chem., 2005, 9, 687–699 CrossRef CAS.
- B. Marciniec, Coord. Chem. Rev., 2005, 249, 2374–2390 CrossRef CAS.
- X. Qi, T. Zheng, J. Zhou, Y. Dong, X. Zuo, X. Li, H. Sun, O. Fuhr and D. Fenske, Organometallics, 2019, 38, 268–277 CrossRef CAS.
- J. Liu, A. Zhang, H. Song, Q. Tong, C. H. Tung and W. Wang, Chin. Chem. Lett., 2018, 29, 949–953 CrossRef CAS.
- S. Khoo, J. Cao, F. Ng and C. W. So, Inorg. Chem., 2018, 57, 12452–12455 CrossRef CAS PubMed.
- A. Baishya, S. Baruah and K. Geetharani, Dalton Trans., 2018, 47, 9231–9236 RSC.
- T. Bai, T. Janes and D. Song, Dalton Trans., 2017, 46, 12408–12412 RSC.
- J. Guo, J. Chen and Z. Lu, Chem. Commun., 2015, 51, 5725–5727 RSC.
- T. M. Maier, M. Gawron, P. Coburger, M. Bodensteiner, R. Wolf, N. P. van Leest, B. de Bruin, S. Demeshko and F. Meyer, Inorg. Chem., 2020, 59, 16035–16052 CrossRef CAS PubMed.
- S. R. Tamang and M. Findlater, J. Org. Chem., 2017, 82, 12857–12862 CrossRef CAS PubMed.
- U. K. Das, C. S. Higman, B. Gabidullin, J. E. Hein and R. T. Baker, ACS Catal., 2018, 8, 1076–1081 CrossRef CAS.
- G. Zhang, J. Cheng, K. Davis, M. G. Bonifacio and C. Zajaczkowski, Green Chem., 2019, 21, 1114–1121 RSC.
- L. Li, E. Liu, J. Cheng and G. Zhang, Dalton Trans., 2018, 47, 9579–9584 RSC.
- M. L. Shegavi, A. Baishya, K. Geetharani and S. K. Bose, Org. Chem. Front., 2018, 5, 3520–3525 RSC.
- C. K. Blasius, V. Vasilenko and L. H. Gade, Angew. Chem., Int. Ed., 2018, 57, 10231–10235 CrossRef CAS PubMed.
- A. Aloisi, J. C. Berthet, C. Genre, P. Thuéry and T. Cantat, Dalton Trans., 2016, 45, 14774–14788 RSC.
- G. Jin, C. G. Werncke, Y. Escudié, S. Sabo-Etienne and S. Bontemps, J. Am. Chem. Soc., 2015, 137, 9563–9566 CrossRef CAS PubMed.
- K. J. Gallagher, M. Espinal-Viguri, M. F. Mahon and R. L. Webster, Adv. Synth. Catal., 2016, 358, 2460–2468 CrossRef CAS.
- K. J. Gallagher and R. L. Webster, Chem. Commun., 2014, 50, 12109–12111 RSC.
- C. B. Provis-Evans, S. Lau, V. Krewald and R. L. Webster, ACS Catal., 2020, 10, 10157–10168 CrossRef CAS.
- L. Wenbo and L. Zhan, Chin. J. Org. Chem., 2020, 40, 3596–3604 CrossRef.
- T. R. Hoye, C. S. Jeffrey and F. Shao, Nat. Protoc., 2007, 2, 2451–2458 CrossRef CAS PubMed.
- T. J. Wenzel and C. D. Chisholm, Chirality, 2011, 23, 190–214 CrossRef CAS PubMed.
- Y. Zhang, A. D. MacIntosh, J. L. Wong, E. A. Bielinski, P. G. Williard, B. Q. Mercado, N. Hazari and W. H. Bernskoetter, Chem. Sci., 2015, 6, 4291–4299 RSC.
- A. Pinaka and G. C. Vougioukalakis, Coord. Chem. Rev., 2015, 288, 69–97 CrossRef CAS.
- H. Fong and J. C. Peters, Inorg. Chem., 2015, 54, 5124–5135 CrossRef CAS PubMed.
- C. Ziebart, C. Federsel, P. Anbarasan, R. Jackstell, W. Baumann, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2012, 134, 20701–20704 CrossRef CAS PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- S. Bontemps, L. Vendier and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 51, 1671–1674 CrossRef CAS PubMed.
- S. C. Sau, R. Bhattacharjee, P. K. Vardhanapu, G. Vijaykumar, A. Datta and S. K. Mandal, Angew. Chem., Int. Ed., 2016, 55, 15147–15151 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic methods, analysis data and NMR spectra. See DOI: 10.1039/d1dt02092g |
| ‡ CO2 reactions performed in a standard J. Young NMR tube. Estimated theoretical maximum amount of CO2 in reaction vessel at 1 atm and 25 °C was calculated to be 0.08 mmol. |
| This journal is © The Royal Society of Chemistry 2021 |