
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of diacylgermanes: persistent derivatives with superior photoreactivity†
Sabrina D.
Püschmann
a,
Philipp
Frühwirt
b,
Stefanie M.
Müller
b,
Stefan H.
Wagner
c,
Ana
Torvisco
a,
Roland C.
Fischer
 a,
Anne-Marie
Kelterer
b,
Thomas
Griesser
c,
Georg
Gescheidt
b and
Michael
Haas
*a
a,
Anne-Marie
Kelterer
b,
Thomas
Griesser
c,
Georg
Gescheidt
b and
Michael
Haas
*a
aInstitute of Inorganic Chemistry, Technical University Graz, Stremayrgasse 9/IV, 8010 Graz, Austria. E-mail: michael.haas@tugraz.at
bInstitute of Physical and Theoretical Chemistry, Technical University Graz, Stremayrgasse 9/II, 8010 Graz, Austria
cInstitute of Chemistry of Polymeric Materials, Montanuniversitaet Leoben, Otto-Gloeckelstrasse 2, A-8700 Leoben, Austria
First published on 5th August 2021
Abstract
Acylgermanes are known as highly efficient photoinitiators. In this contribution, we present the synthesis of new diacylgermanes 4a–evia a multiple silyl abstraction methodology. The method outperforms the state-of-the-art approach (Corey–Seebach reaction) towards diacylgermanes in terms of group tolerance and toxicity of reagents. Moreover, these compounds are decorated with bulky mesityl groups in order to improve their storage stability. The isolated diacylgermanes were characterized by multinuclear NMR-, UV-Vis spectroscopy and X-ray crystallography, as well as photolysis experiments (photobleaching) and photo-DSC measurements (photopolymerization behavior). Upon irradiation with an LED emitting at 385 nm, all compounds except for 4a and 4c bleach efficiently with quantum yields above 0.6. Due to their broad absorption bands, the compounds can be also bleached with blue light (470 nm), where especially 4e bleaches more efficiently than Ivocerin®.
Introduction
State-of-the-art visible light initiators for photo-induced free radical polymerization are used for a variety of different applications such as 3D-printing,1 tissue engineering,2 coatings3 and dental applications.4 The vast majority of these state-of-the-art initiators decompose into radical species via Norrish type I (α-cleavage). Modern Norrish type I initiators absorb light above 400 nm e.g. acylphosphines,5 acylstannanes6 and acylgermanes.7–10Especially the latter were intensively investigated in the last two decades, which boosted the number of available synthetic pathways towards acylgermanes significantly. However, di(4-methoxybenzoyl)diethylgermane (Ivocerin®), a commercially available diacylgermane, is synthesized via a complex procedure, which relies on a Corey–Seebach reaction followed by column chromatography and consequently results in the high costs of this PI (Chart 1).11 Another disadvantage is the inefficient curing depth at wavelengths above 500 nm.7 Our group has introduced a new one-pot synthetic protocol providing tetraacylgermanes Ge[C(O)R]4 (R = aryl) in high yields (Chart 1).8 However, most implemented acylgermanes as initiators have stability issues in various solvents limiting the field of applications.
 | ||
| Chart 1 The state-of-the-art germanium-based photoinitiators. | ||
Here, we introduce an easy to perform synthetic pathway towards diacylgermanes 4a–f with bulky mesityl groups, avoiding the Corey–Seebach reaction. The introduction of these mesityl groups leads to an increased stability in comparison to the state-of-the-art germanium based photoinitiators. Moreover, the broad absorption band of these compounds lead to a good applicability in the field of photopolymerization.
Results and discussion
Synthetic procedures
The entry into this chemistry is provided by the easy to perform synthetic protocol towards dimesityldi(trimethylsilyl)germane 2. Subsequently 2 was reacted with equimolar amounts of KOtBu and 18-crown-6 generating the germanide 3 as crucial intermediate. On the one hand the addition of 18-crown-6 is necessary to stabilize the anion and on the other hand the usage of crown ether circumvents the introduction of sulfur protecting groups and therefore is more sustainable. Analytical and spectroscopic data clearly support the structure of 3 (for details consult the Experimental section and the ESI†). This germanide was added to the respective twofold excess of acid fluoride in situ yielding the desired diacylgermanes 4a–e in good yields (see Scheme 1). Analytical and spectroscopic data that support the structural assignment are given in the Experimental section, together with experimental details. Particularly striking is the possible synthesis and isolation of compound 4e, where a benzothiophene group is substituted at the carbonyl moiety, as the introduction of heterocyclic groups is not possible with the Corey–Seebach reaction. Compound 4f was also formed with this protocol, unfortunately due to the instability with silica gel it was not isolable. | ||
Scheme 1 Synthetic protocol towards diacylgermanes 4a–f. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Yield determined by 1H NMR analysis. bIsolated yield. Yield determined by 1H NMR analysis. bIsolated yield. | ||
UV-Vis spectroscopy
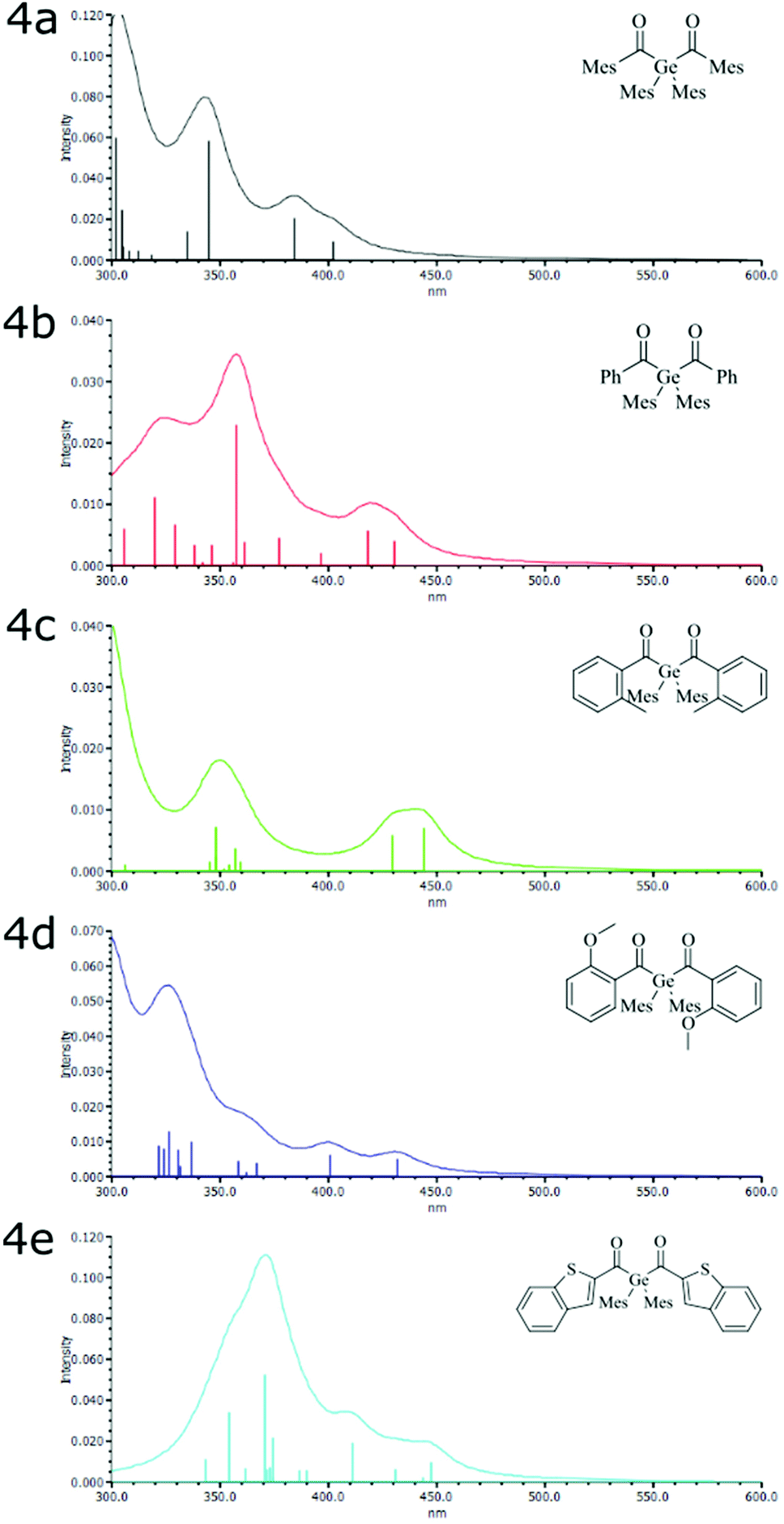
The broad absorption band of compounds 4a–e are centered at around 410 nm (see Fig. 1), which can be allocated to the n–π* transition. This band is responsible for the photo-induced cleavage of the Ge–C bond. In comparison to Ivocerin® these new diacylgermanes show broader absorption bands, which results in a bathochromic shift of their absorption edge. Consequently, 4b–e absorb light above 450 nm whereas the commercial PI, Ivocerin®, has a weak performance at this wavelength. In comparison to tetra(o-toluoyl)germane 5 and tetra(mesitoyl)germane 6 as representative examples of tetraacylgermanes, 4a–e have similar absorption edges, however lower extinction coefficients, which is due to the presence of only two chromophore groups at the diacylgermanes, compared to the four chromophores in the case of tetraacylgermanes. | ||
| Fig. 1 Absorption spectrum of synthesized compounds 4a–e compared with the commercially available Ivocerin®, tetra(o-toluoyl)germane (5) and tetra(mesitoyl)-germane (6) with a concentration of 1 × 10−3 M in chloroform. | ||
DFT calculation
Based on the X-ray data, we have optimized the geometries of 4a–4e by DFT. Basically, the calculated geometries correspond with the experimental data. For 4a, an additional, more stable conformer (Erel = 8.2 kJ mol−1) was found. However, the electronic transitions calculated for the latter isomer were essentially identical with those computed for that corresponding to the X-ray structure. The vertical excitation energies (TDDFT) and are presented in the ESI (Tables S3 and S4†).The TDDFT calculated excitation spectra of 4a–e agree well with the experimental UV-Vis spectra (Fig. 2). The first absorption band at ca. 420 nm consists of two vertical excitations, HOMO → LUMO and HOMO → LUMO+1. Both transitions are n–π* transitions. The HOMO is localized on both oxygen lone pairs of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups. LUMO and LUMO+1 are both linear combinations of the CO π-orbitals with contributions of the neighboring aromatic system. The influence of substitution on orbital energies and localization can be explained by considering the electronic donating (EDG) effects of the substituents. The methyl group is inductive electronic donating and the methoxy group is mesomeric electronic donating. Consequently, the presence of EDG groups (Me in 4a and 4c and OMe in 4d) increase the electron density of the HOMO orbital on the aromatic systems. Additionally, both EDG groups reduce the HOMO–LUMO-gap slightly compared to the unsubstituted 4b (Table S4†), and, thus, cause a bathochromic shift (see Table S3† and Fig. 2).
O groups. LUMO and LUMO+1 are both linear combinations of the CO π-orbitals with contributions of the neighboring aromatic system. The influence of substitution on orbital energies and localization can be explained by considering the electronic donating (EDG) effects of the substituents. The methyl group is inductive electronic donating and the methoxy group is mesomeric electronic donating. Consequently, the presence of EDG groups (Me in 4a and 4c and OMe in 4d) increase the electron density of the HOMO orbital on the aromatic systems. Additionally, both EDG groups reduce the HOMO–LUMO-gap slightly compared to the unsubstituted 4b (Table S4†), and, thus, cause a bathochromic shift (see Table S3† and Fig. 2).
 | ||
| Fig. 2 Simulated spectrum of 4a–e together with the vertical transitions (vertical lines). | ||
The extension of the π system in 4e by the annelated thiophene also leads to a red-shift compared with 4a–d. For 4e the four calculated transitions between 411 and 447 nm are in good consent with the rather broad absorptions between ca. 400 and 470 nm. Whereas the lines at 411, 431, and 447 nm possess two dominating components contributions, that at 443 is dominated by the HOMO–LUMO transition (Table S3†).
Fig. 3 shows the relevant orbitals for the first two vertical excitations of 4b and 4e. For all other compounds the relevant orbitals can be found in the ESI (Fig. S34†).
 | ||
| Fig. 3 (a) Relevant orbitals for the first two vertical excitations of 4b. (b) Relevant orbitals for the first two vertical excitations of 4e. | ||
In particular, the dihedral angle between the aromatic substituent and the adjacent acyl group directs the electronic properties of the diacylgermanes. The bulky mesityl group in 4a causes an almost perpendicular arrangement of the CO group and the π-plane of the mesityl substituent (dihedral angle = 75.8°) whereas the π-system and the CO group are almost coplanar in 4e (3.28°) and 4c (21.3°) > 4d (16.36°) > 4b (12.4°) being “intermediate” cases.
Consequently, in 4a, the electron density is almost entirely localized at the acyl group (Fig. S34†). Solely, HOMO−1 indicates distinguishable coefficients at one mesityl group. For 4c–4e with smaller dihedral angles delocalization is more pronounced rationalizing the red shifts in the UV-VIS spectra.
Photobleaching
Efficient curing of photoinitiator/monomer mixtures is crucial for applications like dental restoration, therefore steady-state photolysis experiments were performed to assess the photobleaching behavior of the compounds. To that end, degassed solutions of 4a–e in a 1/1 (v/v) mixture of toluene/methyl methacrylate (with absorbance of about 0.7 at 385 nm) were irradiated with two different low power LEDs with emission maxima at about 385 and 470 nm (LED385 and LED470; described in the Experimental section of the ESI† in more detail) – emission wavelengths, which are also found in commercial dental lamps.12Fig. 4 shows the time traces of the normalized absorbances when irradiating with LED385 or LED470. It can be seen that 4a and 4c bleach least efficiently, whereas 4d is almost comparable to Ivocerin® or to 6. From an exponential fit of the concentration traces, the quantum yields of decomposition can be determined using the procedure by Stadler et al.13 The compounds 4a and 4c feature the smallest quantum yields (0.20 and 0.49, respectively), while the quantum yields of the other compounds are above 0.62 (see Table 1). For compound 4d a higher quantum yield comparable to the one of Ivocerin® (ϕ = 0.8314) is obtained. We ascribe this to the electronic properties of the methoxy groups having a compatible effect when residing in either o- or p-position of the benzoyl substituents. Despite the fact, that 4c and 4e show nearly identical extinctions above 450 nm (see Fig. 1), photobleaching of 4e with the blue LED emitting at 470 nm is more efficient due to its higher quantum yield of decomposition and even faster than Ivocerin® (see Fig. 4b).
 | ||
| Fig. 4 Steady-state photolysis of 4a–e with (a) LED385, (b) LED470 in toluene/MMA (1/1 v/v). The absorbance traces are normalized to the initial absorptions at the observation wavelengths (maxima of n/σ–π* transitions; 4a: 387 nm, 4b: 413 nm, 4c: 413 nm, 4d: 399 nm, 4e: 419 nm). Data for Ivocerin® and tetramesitoylgermane (6) were taken from ref. 10. | ||
| Compound | λ max,exp [nm] | ε [M−1 cm−1] at λmax,exp | Φ (385 mm) |
|---|---|---|---|
| 4a | 387 | 903 | 0.20 ± 0.01 |
| 4b | 413 | 510 | 0.68 ± 0.02 |
| 4c | 413 | 624 | 0.49 ± 0.02 |
| 4d | 399 | 495 | 0.83 ± 0.02 |
| 4e | 419 | 606 | 0.62 ± 0.01 |
NMR spectroscopy
NMR spectra and detailed characterization of 4a–f are provided in the ESI.† All compounds show very similar 13C chemical shifts for the carbonyl carbon between 219.47 ppm and 240.67 ppm, which is characteristic for the carbonyl groups directly linked to the germanium atom.Storage stability tests
Low long-term stability in various solvents (such as chloroform, benzene or toluene) is a drawback of Ivocerin® and teraacylgermanes limiting the field of applications. By introducing bulky mesityl groups at the germanium atom the storage stability was drastically improved. UV-Vis spectroscopy as well as NMR spectroscopy were used to determine the storage stability of Ivocerin®, tetra(o-toluoyl)germane 5 and compounds 4a–e. For the UV-Vis method, all acylgermanes were measured in chloroform with a concentration of 1 × 10−3 M and the spectra were recorded once a week over a period of 21 days. To confirm the stability in MMA, UV-Vis spectra of Ivocerin®, reference compound 5, compound 4c and 4d were recorded once a week over a period of 21 days with a concentration of 1 × 10−3 M. In case of the NMR method, the measurements were performed in degassed CDCl3 and C6D6 (0.01 M in 0.5 mL). Once a week over a period of 21 days a spectrum of those were recorded. During these long-term tests, the NMR tubes, as well as the brown glass volumetric flask for the UV-Vis measurements were sealed with parafilm and stored in the dark. As representative example, compound 4e, as well as the spectra of Ivocerin® and tetra(o-toluoyl)germane for comparison, are shown in Fig. 5 (UV-Vis data) and Fig. 8 (NMR data, left: in C6D6, right: in CDCl3). The measurements in MMA are shown in Fig. 6. All other spectra can be found in the ESI.† | ||
| Fig. 5 Comparison of the long-term stability in solution (1 × 10−3 M in chloroform) detected by UV-Vis spectroscopy. Zoomed in part of the UV-Vis spectra of (a) compound 4e, (b) Ivocerin® and (c) 5. | ||
 | ||
| Fig. 6 Comparison of the long-term stability in solution (1 × 10−3 M in MMA) detected by UV-Vis spectroscopy. (a) 4c, (b) 4d and (c) Ivocerin®. | ||
As shown in Fig. 5, the newly synthesized compound 4e is stable and even after 21 days no degradation is observed. In contrast to that the absorption of Ivocerin® and 5 slightly decrease (see Fig. 5b and c). Moreover, we could confirm by our stability tests that all investigated diacylgermanes are stable in MMA (Fig. 6a–c).
To conclude, the commercial PIs degrade in solution, in particular in chloroform as well as in benzene, whereas the new synthesized compounds 4a–e are significantly more stable and show no degradation. In monomers, like in MMA, diacylgermanes are stable over time.
The same statement can be made after measuring the NMR spectra of all compounds. The newly synthesized compounds (e.g.4e) do not show any degradation. Even after 21 days, the spectra remain unaffected (see Fig. 8a and b). On the opposite, Ivocerin® and 5 already show degradation after 7 days. During the course of this experiments the degradation product, the aldehyde, get more and more dominant, until only the degradation product is present (Table 2 and Fig. 8). Although the acute toxicity of aldehydes is low, the exposure to aldehydes causes irritation of the skin, eyes and mucous membranes of the respiratory passage. As our new presented initiators 4a–e do not show any formation of these degradation product, we think that this is an important step forward in this research field.
| Compound | Solvent | Days | Aldehyde formation [%] |
|---|---|---|---|
| Ivocerin® | C6D6 | 7 | 15 |
| 14 | 28 | ||
| 21 | 50 | ||
| CDCl3 | 7 | 18 | |
| 14 | 32 | ||
| 21 | 100 | ||
| 5 | C6D6 | 7 | 25 |
| 14 | 62 | ||
| 21 | 100 | ||
| CDCl3 | 7 | 44 | |
| 14 | 73 | ||
| 21 | 100 | ||
X-Ray crystallography
Crystals suitable for single-crystal XRD were obtained for compounds 4a–e. Compound 4e is shown as representative example (Fig. 7), the other structures can be found in the ESI.† The torsion angle between the CO bond and the aromatic plane vary a lot among the different substituents. The evaluated data of the bond lengths are slightly elongated compared to the average Ge–C bond (1.97 Å)15 and the average CO bond (1.19 Å) (Table 3).16
 | ||
| Fig. 7 ORTEP representation of 4e. Thermal ellipsoids are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. The torsion angle (mean value) between the CO group and the aromatic ring plane of the thiofuran group is 1.72°. | ||
O group and the aromatic ring plane [°] of compounds 4a–e
| Compound | d Ge–C |
d
CO
|
∠OC–R |
|---|---|---|---|
| 4a | 2.060 | 1.213 | 68.45 |
| 4b | 2.041 | 1.219 | 7.95 |
| 4c | 2.044 | 1.217 | 25.54 |
| 4d | 2.033 | 1.217 | 13.19 |
| 4e | 2.032 | 1.223 | 1.72 |
Photo-DSC measurements
Photo-DSC is a versatile method to evaluate the performance of PIs in polymerizable resins. One single measurement can give information about the reaction kinetics (time to reach the maximum heat flow (tmax), maximum rate of polymerization (Rp,max), time to reach 95% of final conversion (t95%)) and the double bond conversion (DBC, calculated from the overall reaction enthalpy ΔH (peak area) and the theoretical heat of polymerization (ΔH0,p)).The photopolymerization experiments were conducted in 1,6-hexanediol diacrylate (HDDA) as model monomer system (for further details consult the ESI†). Besides the synthesized PIs (i.e.4a–4e), the polymerization behavior of Ivocerin® and 5 was determined as reference.
The PI performance was analyzed at (1) equal molar PI (0.30 mol%) as well as (2) equal photo-cleavable group (PCG) concentration (0.15 mol% for 5; 0.30 mol% for 4a–4e and Ivocerin®), respectively. In general, the synthesized PIs provide kinetics and a DBC comparable to the reference compounds. In more detail, 4b and 4d show a slightly faster polymerization initiation than the other synthesized PIs and can be compared in their reactivity with 5 at the same PI concentration. However, measured at similar PCG concentration, they show a slightly slower turnover than Ivocerin®, but higher double bond conversion than both Ivocerin® and 5. This correlates to the quantum yields measured at 385 nm, where 4b and 4d show the highest values (Fig. 9).
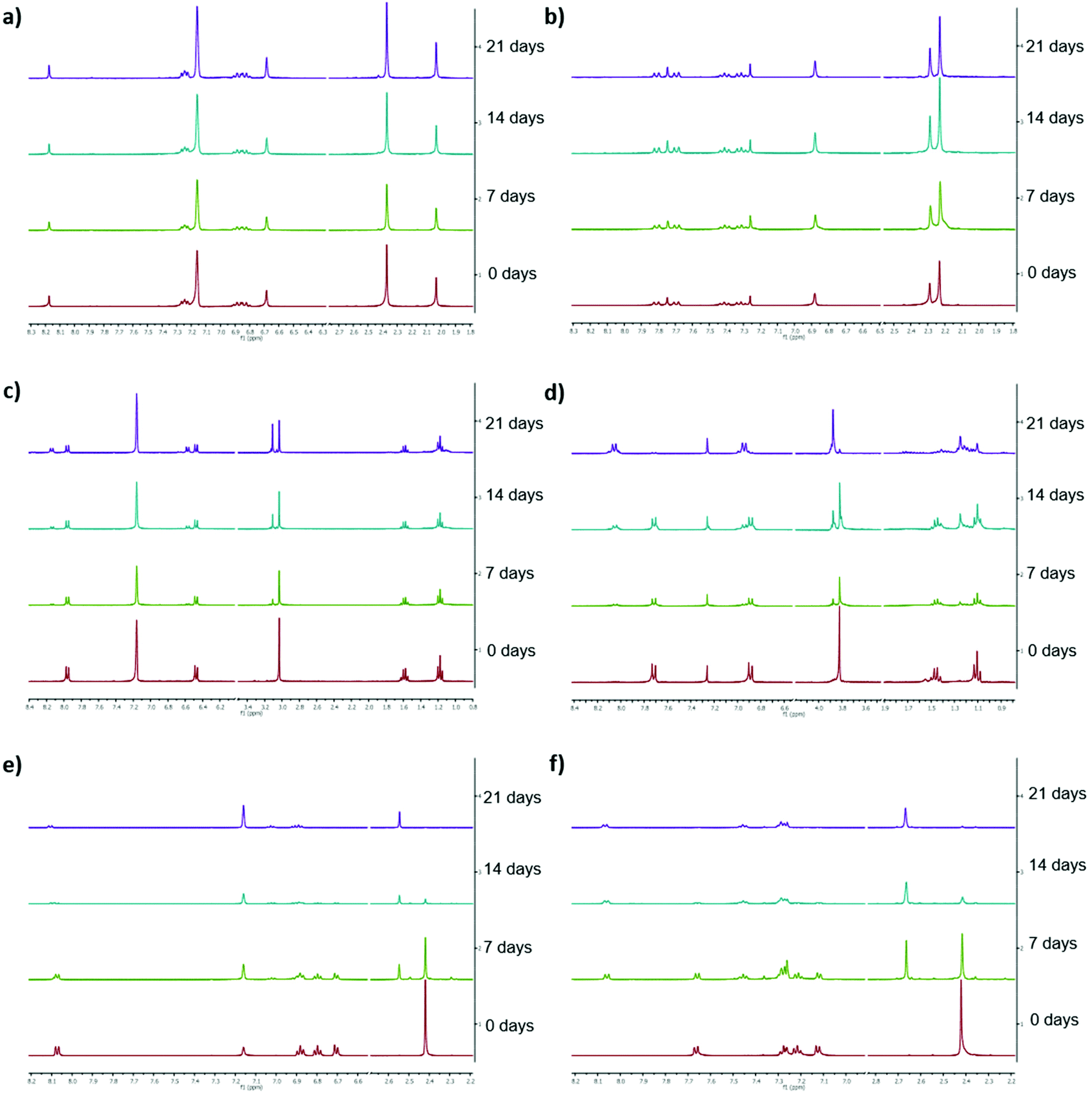 | ||
| Fig. 8 Representation of the stacked 1H NMR spectra (300 MHz) of the respective compound (0.01 M in 0.5 mL) in the corresponding degassed solvent, left: in C6D6 (61.7 ppm H2O) right: in CDCl3 (26 ppm H2O). (a) shows compound 4e in C6D6, (b) compound 4e in CDCl3, (c) Ivocerin® in C6D6, (d) Ivocerin® in CDCl3, (e) 5 in C6D6 and (f) 5 in CDCl3. | ||
 | ||
| Fig. 9 Photo-DSC (left) and conversion plots (right) for the photopolymerization of HDDA with 0.3 mol% PI (top) and equal PCG concentration (bottom). | ||
Experimental
General considerations
All synthetic steps were performed under inert conditions using standard Schlenk techniques. Solvents were dried with a column purification system.17 Commercial tetrachlorogermane (GeCl4), tert-butyllithium (t-BuLi), mesitylbromide (MesBr), chloro(trimethyl)silane (SiMe3Cl), and KOtBu were used without further purification. The used acid fluorides were produced according to the corresponding literature.181H (299.95 MHz) and 13C (75.43 MHz) NMR spectra were recorded with a Varian INOVA 300 spectrometer in CDCl3 or C6D6 solution and were referenced versus TMS by using the internal 2H lock signal of the solvent. UV-Vis spectra were recorded with an Agilent Cary 60 UV-Vis spectrometer. Mass spectra were acquired with a Q-TOF Premier from Waters, Manchester, England. The original ESI source of the instrument was replaced by a standard LIFDI source from Linden CMS, Weyhe, Germany. IR spectra were recorded with a Brucker ALPHA. Melting points were determined by a Stuart automatic melting point (SMP50).Synthesis of dichlorodimesitylgermane (1)
A 3.0 L flask was charged with MesBr (52 mL, 339 mmol, 2.00 eq.) and THF (800 mL). The solution was cooled to −78 °C. 400 mL of t-Buli (1.70 M, 678 mmol, 4.00 eq.) were added with a dropping funnel and stirred for additional 30 minutes. Afterwards GeCl4 (36.4 g, 170 mmol, 1.00 eq.) was dissolved in small amounts of THF and were added to the reaction. After warming up to room temperature, the reaction was stirred overnight. The reaction mixture was transferred into a flask and the solvent was removed under reduced pressure. The solid was extracted with pentane and the solvent was again removed under reduced pressure leading to white crystals (24.4 g, 38%).1 H NMR (300 MHz, CDCl3, ppm): δ 6.81 (s, 4H, Mes–H), 2.28 (s, 12H, Mes–oCH3), 2.26 (s, 6H, Mes–pCH3).
Synthesis of dimesityldi(trimethylsilyl)germane (2)
A 500 mL 3-neck flask with a dropping funnel and a reflux condenser was charged with THF (90 mL) and Li (1.70 g, 249 mmol, 4.00 eq.), which was cooled to −70 °C. Trimethylchlorosilane (13.3 mL, 104 mmol, 3.35 eq.) was added to the solution and afterwards compound 1 (11.9 g, 31.1 mmol, 1.00 eq.) was dissolved in THF (90 mL) and was added over one hour. The reaction was stirred for further 2 h at −70 °C. The cooling was removed and the reaction was stirred over night at room temperature. After aqueous workup with saturated NH4Cl, the phases were separated and the aqueous phase was extracted three times with diethyl ether. The organic layer was dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The crude product was recrystallized from acetone at −30 °C to yield 11.5 g (81%).1 H NMR (300 MHz, CDCl3, ppm): δ 6.76 (s, 4H, Mes–H), 2.23 (s, 6H, Mes–pCH3), 2.17 (s, 12H, Mes–oCH3), 0.21 (s, 18H, Si–(CH3)3).
Synthesis of dimesityldimesitoylgermane (4a)
Mes2Ge(SiMe3)22 (2.00 g, 4.37 mmol, 1.00 eq.), 18-crown-6 (1.27 g, 4.81 mmol, 1.10 eq.) and KOtBu (0.54 g, 4.81 mmol, 1.10 eq.) were dissolved 25 mL benzene and stirred for 1 h at room temperature. A second flask was charged with 2,4,6-trimethylbenzoyl fluoride (1.45 g, 8.75 mmol, 2.00 eq.) in toluene (10 mL) and cooled to 0 °C. The germanide 3 was added dropwise to the fluoride solution. The cooling was removed, and the reaction mixture was allowed to warm to room temperature and stirred overnight. After aqueous workup with saturated NH4Cl, the phases were separated and the aqueous phase was extracted three times with DCM. The organic layer was dried over Na2SO4, filtered with silica gel to remove the crown ether, and the solvent was removed under reduced pressure. The crude product was recrystallized from pentane at −70 °C to yield 1.51 g (57%) of pure slightly yellow crystals.
M.p. 183–186 °C; UV-Vis (chloroform): λ = 385 and 399 nm, ε = 924 and 902 L mol−1 cm−1; IR: v[cm−1] = 1636, 1603 (m, νCO); elemental analysis (%) calcd for C38H44GeO2: C 75.39, H 7.33; found: C 75.31, H 7.34; 1H NMR (300 MHz, CDCl3, ppm): δ 6.59 (s, 4H, C(O)Mes–H), 6.51 (s, 4H, Mes–H), 2.12 (s, 6H, C(O)Mes–pCH3), 2.12 (s, 6H, Mes–pCH3), 1.98 (s, 12H, C(O)Mes–oCH3), 1.91 (s, 12H, Mes–oCH3). 13C NMR (75 MHz, CDCl3, ppm): δ 240.67 (GeCOMes), 143.99, 142.35, 138.88, 138.73, 135.25, 134.15, 129.32, 128.75, 128.69 (Aryl–C), 25.14 (Mes–oCH3), 21.17 (Mes–pCH3), 21.05 (Mes–pCH3), 19.68 (Mes–oCH3). HRMS: calcd for [C38H44GeO2]+ (M+): 606.2562. Found: 606.3292.
Synthesis of dimesityldibenzoylgermane (4b)
Compound 2 (4.00 g, 8.75 mmol, 1.00 eq.), 18-crown-6 (2.54 g, 9.62 mmol, 1.10 eq.) and KOtBu (1.08 g, 9.62 mmol, 1.10 eq.) were dissolved in benzene (60 mL) and stirred for 1 h at room temperature. The germanide 3 was added dropwise to a solution of benzoyl fluoride (1.90 mL, 17.5 mmol, 2.00 eq.) in toluene (10 mL) at 0 °C. After heating up to room temperature, the orange-red reaction solution was stirred overnight. Followed by aqueous workup with NH4Cl solution, extraction with DCM, drying over Na2SO4 and filtration over silica gel to remove the excess of crown ether. The solvent was removed under reduced pressure. The crude product was further purified by column chromatography (pentane/toluene 1:2) and recrystallized from pentane at −70 °C, yielding 2.50 g (55%) of yellow crystals.
M.p. 155–157 °C; UV-Vis (chloroform): λ = 411 and 437 (sh) nm, ε = 369 and 241 (sh) L mol−1 cm−1; IR: v[cm−1] = 1620, 1590, 1574 (m, νCO); elemental analysis (%) calcd for C32H32GeO2: C 73.74, H 6.19; found: C 73.31, H 6.17; 1H NMR (300 MHz, CDCl3, ppm): δ 7.80–7.77 (d, J = 7.3 Hz, 4H, Ph–H), 7.44–7.39 (t, J = 7.3 Hz, 2H, Ph–H), 7.31–7.28 (d, J = 7.7 Hz, 4H, Ph–H), 6.87 (s, 4H, Mes–H), 2.28 (s, 6H, Mes–pCH3), 2.20 (s, 12H, Mes–oCH3). 13C NMR (76 MHz, CDCl3, ppm): δ 228.15 (GeCOPh), 143.59, 140.61, 139.45, 134.12, 133.04, 129.64, 128.75, 128.35 (Aryl–C), 25.11 (Mes–oCH3), 21.15 (Mes–pCH3). HRMS: calcd for [C32H32GeO2]+ (M+): 522.1622. Found: 522.2216.
Synthesis of dimesityldi(o-toluoyl)germane (4c)
Compound 2 (4.00 g, 8.75 mmol, 1.00 eq.), 18-crown-6 (2.54 g, 9.62 mmol, 1.10 eq.) and KOtBu (1.08 g, 9.62 mmol, 1.10 eq.) were dissolved in benzene (60 mL) and stirred for 1 h at room temperature. A second flask was charged with o-toluoyl fluoride (2.43 g, 17.5 mmol, 2.00 eq.) and dissolved in 10 mL toluene. The solution was cooled to 0 °C. After complete addition of the germanide 3 to this solution, aqueous workup with 10% H2SO4 followed. The aqueous phase was extracted three times with DCM, dried over Na2SO4, filtrated over silica gel and the solvent was removed under reduced pressure. The crude product was further purified by column chromatography (pentane/toluene 2:1) and recrystallization from pentane at −70 °C, pure yellow crystals were isolated (2.59 g, 54%).
M.p. 142 °C; UV-Vis (chloroform): λ = 415 nm, ε = 658 L mol−1 cm−1; IR: v[cm−1] = 1630, 1598, 1563 (m, νCO); elemental analysis (%) calcd for C34H36GeO2: C 74.35, H 6.61; found: C 74.19, H 6.45; 1H NMR (300 MHz, CDCl3, ppm): δ 7.68–7.66 (d, J = 7.6 Hz, 2H, Ph–H), 7.20–7.15 (t, J = 7.4 Hz, 2H, Ph–H), 7.06–7.01 (t, J = 7.1 Hz, 4H, Ph–H), 6.86 (s, 4H, Mes–H), 2.31 (s, 6H, Ph–oCH3), 2.27 (s, 6H, Mes–pCH3), 2.25 (s, 12H, Mes–oCH3). 13C NMR (76 MHz, CDCl3, ppm): δ 231.25 (GeCO), 143.66, 140.04, 139.22, 137.19, 134.59, 132.60, 132.44, 131.99, 129.61, 129.57, 125.20 (Aryl–C), 25.15 (Mes–oCH3), 25.02 (oTol–CH3), 21.49 (Mes–pCH3). HRMS: calcd for [C34H36GeO2]+ (M+): 550.1927. Found: 550.2034.
Synthesis of dimesityldi(o-methoxy)germane (4d)
Compound 2 (4.00 g, 8.75 mmol, 1.00 eq.), 18-crown-6 (2.54 g, 9.62 mmol, 1.10 eq.) and KOtBu (1.08 g, 9.62 mmol, 1.10 eq.) were dissolved in benzene (60 mL) and stirred for 1 h at room temperature. In a second flask, o-methoxybenzoyl fluoride (2.71 g, 17.5 mmol, 2.00 eq.) was dissolved in toluene (10 mL) and cooled to 0 °C. The germanide 3 was added dropwise to the fluoride and after warming up to room temperature stirred overnight. Followed by aqueous workup with 10% H2SO4, extraction with dichloromethane, drying over Na2SO4, filtration over silica gel and removal of the solvent under reduced pressure. The crude product was further purified by column chromatography (pentane/toluene 1:3) and recrystallization from pentane at −70 °C, 4d was isolated as yellow crystals (2.64 g, 52%).
M.p. 201–203 °C; UV-Vis (chloroform): λ = 396, 418 (sh) and 440 (sh) nm, ε = 516, 429 (sh) and 265 (sh) L mol−1 cm−1; IR: v[cm−1] = 1617, 1590 (m, νCO); elemental analysis (%) calcd for C34H36GeO4: C 70.25, H 6.24; found: C 70.02, H 6.10; 1H NMR (300 MHz, CDCl3, ppm): δ 7.58–7.55 (d, J = 7.7 Hz, 2H, Ph–H), 7.15–7.10 (t, J = 7.8 Hz, 2H, Ph–H), 6.82 (s, 4H, Mes–H), 6.79 (d, J = 7.7 Hz, 2H, Ph–H), 6.35–6.62 (d, J = 8.3 Hz, 2H, Ph–H), 3.16 (s, 6H, OCH3), 2.26 (s, 12H, Mes–oCH3), 2.24 (s, 6H, Mes–pCH3). 13C NMR (76 MHz, CDCl3): δ 222.52 (GeCO), 158.49, 144.05, 137.81, 135.71, 134.04, 128.72, 126.96, 120.43, 110.29 (Aryl–C), 52.25 (OCH3), 24.28 (Mes–oCH3), 21.09 (Mes–pCH3). HRMS: calcd for [C34H36GeO4]+ (M+): 582.1833. Found: 582.2579.
Synthesis of dimesityldibenzothiophenegermane (4e)
A flask was charged with Mes2Ge(SiMe3)22 (1.00 g, 2.18 mmol, 1.00 eq.), 18-crown-6 (0.64 g, 2.40 mmol, 1.10 eq.) and KOtBu (0.27 g, 2.40 mmol, 1.10 eq.). The compounds were dissolved in benzene (20 mL) and stirred for 1 h at room temperature. In a second flask, thiofuran fluoride (0.99 g, 5.47 mmol, 2.50 eq.) was dissolved in 10 mL toluene and cooled to 0 °C. The germanide 3 was added dropwise to the acid fluoride and was allowed to warm up to room temperature. At this temperature, the reaction was stirred overnight. Followed by aqueous workup with saturated NH4Cl solution and the aqueous layer was extracted with DCM. The combined organic phases were dried over Na2SO4, filtered over silica gel and the solvent was removed under reduced pressure. The crude product was further purified by column chromatography (pentane/toluene 1 + 2) and recrystallization from pentane at −70 °C, yielding in 0.25 g (23%) of yellow crystals.
M.p. 171–174 °C; UV-Vis (chloroform): λ = 371 (sh), 414 (sh), 441 (sh) nm, ε = 1990 (sh), 636 (sh) and 403 (sh) L mol−1 cm−1; IR: v[cm−1] = 1602, 1591 (m, νCO); elemental analysis (%) calcd for C36H32GeO2S2: C 68.27, H 5.09, S 10.12; found: C 68.31, H 5.08, S 10.09; 1H NMR (300 MHz, CDCl3) δ 7.82–7.80 (d, J = 8.1 Hz, 2H, Aryl–H), 7.75 (s, 2H, Aryl–H), 7.71–7.68 (d, J = 7.8 Hz, 2H, Aryl–H), 7.44–7.38 (t, J = 7.6 Hz, 2H, Aryl–H), 7.34–7.29 (t, J = 7.5 Hz, 2H, Aryl–H), 6.88 (s, 4H, Mes–H), 2.29 (s, 6H, Mes–pCH3), 2.23 (s, 12H, Mes–oCH3). 13C NMR (76 MHz, CDCl3) δ 219.47 (GeCO), 148.34, 143.98, 142.22, 140.05, 139.37, 133.13, 129.89, 127.84, 126.89, 124.78, 123.14 (Aryl–C), 25.21 (Mes–oCH3), 21.22 (Mes–pCH3). HRMS: calcd for [C34H36GeO4]+ (M+): 634.1062. Found: 634.1927.
Synthesis of dimesityldibenzofurangermane (4f)
Compound 2 (0.50 g, 1.09 mmol, 1.00 eq.) was dissolved in benzene (15 mL). KOtBu (0.14 g, 1.20 mmol, 1.10 eq.) and 18-crown-6 (0.32 g, 1.20 mmol, 1.10 eq.) were added and the reaction was stirred for 1 h at room temperature. A second flask was charged with benzofuran fluoride (0.45 g, 2.73 mmol, 2.50 eq.) and was dissolved in toluene (9 mL). The germanide 3 was added to the fluoride solution at 0 °C and after full addition the solution was allowed to warm up to room temperature. At this temperature, the reaction was stirred overnight. A portion was separated and the solvent was removed, which was then dissolved in CDCl3. A NMR spectrum was measured. Followed by aqueous workup with saturated NH4Cl solution and the aqueous layer was extracted with DCM. The combined organic phases were dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The purification by either crystallization (normal atmosphere and inert atmosphere) or by column chromatography was not successful. Therefore, clean 4f could not be isolated.1 H NMR (300 MHz, CDCl3) δ1H NMR (300 MHz, CDCl3) δ 7.57–7.50 (dd, J = 17.7, 7.2 Hz, 4H, Aryl–H), 7.34–7.33 (m, 4H, Aryl–H), 7.23–7.21 (d, J = 5.9 Hz, 2H, Aryl–H), 6.86 (s, 4H, Mes–H), 2.34 (s, 6H, Mes–pCH3), 2.25 (s, 12H, Mes–oCH3).
Conclusions
To conclude, we were able to synthesize a variety of substituted diacylgermanes 4a–e with bulky mesityl groups by the multiple silyl abstraction methodology, avoiding the Corey–Seebach reaction. Moreover, these compounds show excellent stabilities in solvents, which was determined by long-term stability tests in chloroform, benzene and MMA via UV-Vis spectroscopy and in chloroform and benzene via NMR spectroscopy. The new compounds show a broadening in their absorption bands with absorption above 450 nm, which makes photobleaching with blue light (470 nm) quite efficient. With 4e a tailing up to 490 nm was observed. All compounds show high quantum yields above 0.5, the only exception is compound 4a. DSC-measurements show good photopolymerization behavior for all synthesized compounds with double bond conversions comparable to or higher than Ivocerin®. Therefore, these new derivatives can be implemented in a broad field of applications. Further studies to probe the scope of these initiators are currently in progress.Author contributions
S. D. P. performed the synthesis towards the diacylgermanes. P. F. performed the Steady-State Photolysis and Determination of Quantum Yields. S. M. M. did the Photo-DSC measurements. S. H. W. and A-M. K. performed the DFT computations and analysed the results. A. T. and R. C. F. measured the X-ray structures. S. D. P., S. M. M. and P. F. jointly wrote the manuscript with help from T. G., G. G. and M. H. All authors discussed the results and commented on the manuscript. M. H. provided supervision and wrote the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from NAWI Graz and FWF (Vienna, Austria) (project number P 32606-N). The authors thank Linden CMS GmbH, Germany for measuring the mass spectra.Notes and references
-
(a) A. Bagheri and J. Jin, ACS Appl. Polym. Mater., 2019, 1, 593–611 CrossRef CAS
; (b) H.-B. Sun and S. Kawata, in NMR • 3D Analysis • Photopolymerization, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, vol. 170, pp. 169–273 Search PubMed
-
(a) T. Billiet, M. Vandenhaute, J. Schelfhout, S. van Vlierberghe and P. Dubruel, Biomaterials, 2012, 33, 6020 CrossRef CAS PubMed
-
I. V. Khudyakov, M. B. Purvis and N. J. Turro, in Photoinitiated Polymerization, Am. Chem. Soc, 2003, vol. 847, pp. 113–126 Search PubMed
-
(a) N. Moszner and U. Salz, Prog. Polym. Sci., 2001, 26, 535–576 CrossRef CAS
-
(a) H. Grützmacher, J. Geier, D. Stein, T. Ott, H. Schönberg, R. H. Sommerlade, S. Boulmaaz, J.-P. Wolf, P. Murer and T. Ulrich, Chimia, 2008, 62, 18–22 CrossRef
-
(a) M. Haas, J. Radebner, A. Eibel, G. Gescheidt and H. Stueger, Chem. – Eur. J., 2018, 24, 8258–8267 CrossRef CAS PubMed
- B. Ganster, U. K. Fischer, N. Moszner and R. Liska, Macromolecules, 2008, 41, 2394 CrossRef CAS
- J. Radebner, A. Eibel, M. Leypold, C. Gorsche, L. Schuh, R. Fischer, A. Torvisco, D. Neshchadin, R. Geier, N. Moszner, R. Liska, G. Gescheidt, M. Haas and H. Stueger, Angew. Chem., Int. Ed., 2017, 56, 3103 CrossRef CAS PubMed
-
(a) J. Radebner, M. Leypold, A. Eibel, J. Maier, L. Schuh, A. Torvisco, R. Fischer, N. Moszner, G. Gescheidt, H. Stueger and M. Haas, Organometallics, 2017, 36, 3624 CrossRef CAS
- P. Frühwirt, A. Knoechl, M. Pillinger, S. M. Müller, P. T. Wasdin, R. C. Fischer, J. Radebner, A. Torvisco, N. Moszner, A.-M. Kelterer, T. Griesser, G. Gescheidt and M. Haas, Inorg. Chem., 2020, 59, 15204–15217 CrossRef
- N. Moszner, F. Zeuner, I. Lamparth and U. K. Fischer, Macromol. Mater. Eng., 2009, 294, 877 CrossRef CAS
-
T. Völkel, Scientific Documentation Bluephase® Family – LED for Every Use, Ivoclar Vivadent AG, Schaan, Liechtenstein, 2009 Search PubMed
- E. Stadler, A. Eibel, D. Fast, H. Freißmuth, C. Holly, M. Wiech, N. Moszner and G. Gescheidt, Photochem. Photobiol. Sci., 2018, 17, 660–669 CrossRef CAS PubMed
- A. Eibel, J. Radebner, M. Haas, D. E. Fast, H. Freißmuth, E. Stadler, P. Faschauner, A. Torvisco, I. Lamparth, N. Moszner, H. Stueger and G. Gescheidt, Polym. Chem., 2018, 9, 38–47 RSC
- K. M. Baines and W. G. Stibbs, Coord. Chem. Rev., 1995, 145, 157–200 CrossRef CAS
- M. Palusiak, S. Simon and M. Solà, J. Org. Chem., 2006, 71, 5241–5248 CrossRef CAS PubMed
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS
-
Methoden der organischen Chemie, ed. B. Baasner, J. L. Adcock, J. Houben, E. Müller, T. Weyl, H. Kropf, K. H. Büchel and O. Bayer, Thieme, Stuttgart, 4th edn, 1998, vol 10a, pp. 552–554 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all new compounds, full details of computational studies. Crystal data, details of data collections and refinements. CCDC 2078935–2078939. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt02091a |
| This journal is © The Royal Society of Chemistry 2021 |