
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetic anisotropies of Ho(III) and Dy(III) single-molecule magnets experimentally determined via polarized neutron diffraction†
Emil A.
Klahn
a,
Andreas M.
Thiel
a,
Rasmus B.
Degn
a,
Iurii
Kibalin
b,
Arsen
Gukassov
b,
Claire
Wilson
c,
Angelos B.
Canaj‡
 *c,
Mark
Murrie
*c and
Jacob
Overgaard
*a
*c,
Mark
Murrie
*c and
Jacob
Overgaard
*a
aDepartment of Chemistry, Aarhus University, DK-8000 Aarhus C, Denmark. E-mail: jacobo@chem.au.dk
bLaboratoire Léon Brillouin, CEA-CNRS, CE-Saclay, 91191 Gif-sur-Yvette, France
cSchool of Chemistry, University of Glasgow, University Avenue, Glasgow, G12 8QQ, UK. E-mail: mark.murrie@glasgow.ac.uk
First published on 2nd September 2021
Abstract
We present the magnetic anisotropy of two isostructural pentagonal-bipyramidal complexes, [Ln(H2O)5(HMPA)2]I3·2HMPA (HMPA = hexamethylphosphoramide, Ln = Dy, Ho). Using ac magnetic susceptibility measurements, we find magnetic relaxation barriers of 600 K and 270 K for the Dy- and Ho-compounds, respectively. This difference is supported by polarized neutron diffraction (PND) measured at 5 K and 1 T which provides the first experimental evidence that the transverse elements in the magnetic anisotropy of the Ho-analogue are significant, whereas the Dy-analogue has a near-axial magnetic anisotropy with vanishing transverse contributions. The coordination geometries of the two complexes are highly similar, and we attribute the loss of strong magnetic axiality as expressed in the atomic susceptibility tensors from PND, as well as the smaller relaxation barrier in the Ho-complex compared to the Dy-complex, to the less favorable interaction of the pentagonal bipyramidal crystal field with the characteristics of the Ho(III) 4f-charge distribution.
Introduction
Single-ion magnets (SIMs) have attracted growing scientific attention over the last three decades.1–4 These compounds are organometallic or coordination compounds that show magnetically bistable ground states, thus giving rise to slow relaxation of a molecular magnetic moment, upon the removal of a magnetizing field. The scientific interest in these compounds originates in the possibility of using magnetic molecules as individual bits in high-density memory storage technology, or the promise of using molecules as qubits in quantum computing.5–8Magnetic bistability in SIMs originates in magnetic anisotropy, describing the fact that the magnetic properties of the molecules depend on the direction from which they are measured. Controlling the molecular environment to tailor the optimal conditions for magnetic bistability of a given ion is therefore a central challenge in molecular magnetism. Successes in this regard includes the report of an 80 K blocking temperature for magnetic relaxation in a Dy(III)-containing molecular compound, owing to a ligand field tailored to the Dy(III)-ion.1
Such advancements are made possible by developing sound design criteria. For lanthanide SIMs, the foremost guiding principle was set forward by Rinehart and Long,9 based on the description of the anisotropic valence charge distributions of trivalent lanthanide ions by Sievers.10 These design criteria have not only given rise to the dysprosocenium-type complexes,11 but also to the large and diverse pentagonal-bipyramidal family of lanthanide complexes.12–20 The guiding principle is to design an appropriate ligand field that complements the characteristic lanthanide valence charge distribution of the most magnetic ground mJ-state, which ranges between fully oblate for some ions (Dy(III), Tb(III)) and prolate for others (most notably Yb(III), Tm(III)). Most well-known is perhaps the Dy(III)-ion as an archetypical example of an oblate-shaped electron density.21,22 The pentagonal-bipyramidal geometry has indeed been extensively explored for Dy(III), giving rise to a large variety of complexes, which have, in their own right, shown advancements in molecular magnetic properties with for example, exceedingly large barriers to magnetic relaxation.
While Dy(III)-based compounds have gained a lot of attention, there have been fewer reports of Ho(III)-based molecular magnets.23–29 On one hand, the f10-ion Ho(III) with a free-ion J-value of 8 and a value of gJ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ·J = 10, should be equally valid for molecular magnetic purposes as the f9-ion Dy(III). On the other hand, the Ho(III)-ion is more likely to suffer from fast magnetic relaxation due to quantum tunneling of magnetization (QTM) than Dy(III) because Kramers’ degeneracy, which ensures that there is no mixing of time-reversed states in Dy(III), is absent in Ho(III). However, it has also been shown that QTM can sometimes be suppressed in Ho(III)-based species: Ho has a 100% natural abundance of the isotope with nuclear spin I = 7/2, meaning that coupling to the nuclear spin can give this ion some Kramers-character, and that this effect might actually quench QTM.26
·J = 10, should be equally valid for molecular magnetic purposes as the f9-ion Dy(III). On the other hand, the Ho(III)-ion is more likely to suffer from fast magnetic relaxation due to quantum tunneling of magnetization (QTM) than Dy(III) because Kramers’ degeneracy, which ensures that there is no mixing of time-reversed states in Dy(III), is absent in Ho(III). However, it has also been shown that QTM can sometimes be suppressed in Ho(III)-based species: Ho has a 100% natural abundance of the isotope with nuclear spin I = 7/2, meaning that coupling to the nuclear spin can give this ion some Kramers-character, and that this effect might actually quench QTM.26
Related to the question of suppressing QTM in non-Kramers ions such as Ho(III) is the challenge of designing an optimal crystal field for a Ho(III)-based SMM. Fig. 1 shows Sievers-type representations of the shapes of the Ho(III) and Dy(III) charge distributions. It is evident that Ho(III) does not as easily as Dy(III) fit into the popular dichotomous classification of lanthanide ions as having oblate or prolate 4f-valence charge distributions (4f-CD). This begs the question as to whether we can experimentally quantify this decrease in “oblateness” for Ho(III). Or posed differently: if we place Ho(III)- and Dy(III)-ions in the same pentagonal bi-pyramidal coordination, will we then witness a similar stabilization of the mJ = ±J ground state and the same strongly axial magnetic anisotropy? Obviously, ab initio calculations can provide energy levels and thus quantify the effect but these are complex and challenging calculations. In similar complexes incorporating the Ho(III)-ion,26,27 they do tend to show a stabilization of the mJ = ±8 states. However, our goal is to provide experimental insights into this question.
 | ||
| Fig. 1 Sievers plots for the largest mJ state of (a) the Ho(III)-ion (mJ = 8) and (b) the Dy(III)-ion (mJ = 15/2, right). The Dy(III)-ion is widely recognized as oblate, but the situation is less clear-cut for the Ho(III)-ion, which is more elongated along the axial direction and also has a horizontal “waistband” shape. | ||
To achieve this, we have therefore synthesized two isomorphous examples of the coordination compound [Ln(H2O)5(HMPA)2]I3·2HMPA (HMPA = hexamethyl-phosphoramide) with LnIII = Dy (1) and Ho (2). The magnetic properties of 1 were recently studied by some of us by both magnetic measurements and ab initio calculations.12 In this work, we have studied the magnetic properties of 2 using a combination of DC and AC susceptibility studies. In addition, we have quantified the magnetic anisotropy of both 1 and 2 in the form of atomic magnetic site susceptibility tensors using polarized neutron diffraction (PND). While both AC and DC magnetic measurements are standard techniques in molecular magnetism, the use of PND to study the magnetic anisotropy of molecular magnets is extremely rare, with only a handful of published studies in the literature.30–36 Thus, this work represents the first comparative study of two lanthanide-ions in identical crystal fields using the PND technique.
PND has proved an extremely powerful tool for accessing the individual atomic magnetic susceptibility in crystalline matter, and it can therefore be used to describe the anisotropy of the magnetic parameters of atoms and molecules. The major strength of the technique is that it provides a completely experimental view of the magnitude and direction of the magnetic easy-axis for each magnetic site.37
Alternative techniques that can give related information about molecular magnetic susceptibilities are angular resolved magnetometry (ARM) and torque magnetometry (TM).38,39 These techniques, however, are not generally applicable for all space groups and site symmetries, and so the retrieval of atomic magnetic anisotropy using PND presents the only completely general approach to measuring the directional dependence of the magnetic properties of molecules embedded in crystals. Our earlier results showed how ARM-measurements and the results of PND measurements are in very good agreement.34
Very recently, it was shown by some of us40 that it is possible to refine the atomic susceptibility tensor using powder PND data, instead of relying on the large single crystals that have previously been required. In combination with the new ESS facility under construction in Sweden, this fundamental development has the potential to change this method from a niche technique to a widely used diagnostic tool, of huge importance in single-molecule magnet studies.
The molecular compounds discussed here both show nearly pentagonal bipyramidal coordination (Table S3 and Fig. S1 and S2†) to the trivalent lanthanide ion and both crystallize in the monoclinic space group Cc (Tables S1 and S2†).12 This coordination geometry has, as already mentioned, remarkable variety in terms of the number of published combinations of axial and equatorial ligands.12–18,23–27,29 For the Dy(III)-based analogues, there is significant evidence that the Orbach barrier to thermal relaxation correlates with the strength of the axial ligands: the shorter the average Dy-L-distance (where L refers to any axial ligand), the higher is the measured Ueff.41 A similar correlation of increasing energy barrier with increasing axial ligand strength was found for a series of pentagonal bipyramidal Ho(III)-complexes.24
To consolidate such findings, it is important to determine the magnetic anisotropy, and by extension the magnetic easy-axis direction, of such complexes by experimental means. Single-state easy-axis directions based on g-tensors from ab initio measurements are often used as a simple guide to the possible relaxation pathways of SIMs. For this reason, it is especially important to keep challenging the results of theoretical calculations. In one case of ab initio calculations on a series of Ho(III)-containing pseudo-trigonal bipyramidal complexes, the theoretical easy-axes obtained were tilted and in some cases even perpendicular to the axial direction of the coordination sphere.23 In such a surprising case, PND can provide unparalleled experimental corroboration.
Methods
Synthesis
All manipulations were performed under aerobic conditions, using materials as received. No safety hazards were encountered during the described experimental procedures.:1) and was left for slow evaporation. After ∼3 days, colourless single crystals of [Dy(H2O)5(HMPA)2]I3·2HMPA (1) were isolated from the solution. For [Ho(H2O)5(HMPA)2]I3·2HMPA (2), exactly the same procedure was followed as in the case of 1, with the use of HoI3 (164 mg, 0.3 mmol). Typical yields of compounds 1 and 2 are 30–35%.
Elemental analysis calculated (found) for 2·0.55H2O: C 21.31 (21.16), H 6.11 (6.15), N 12.43 (12.34) %.
X-ray crystallography
The crystal structure of 1 has been determined previously.12 For 2, crystallographic details are available in the ESI and CIF (CCDC 2057601†).Single-crystal polarized neutron diffraction
The magnetic anisotropy of individual atoms in molecules embedded in crystals can be studied using the method of site susceptibilities as developed by Gukasov and Brown in 2002.37 The method works by applying a magnetic field on the sample position, inducing a magnetization in the crystal. This magnetization can be picked up by a detecting coil. More importantly, however, is the fact that the crystal magnetization is the vector sum of individual magnetic moments on each magnetic atom in the crystal. Such magnetic moments can be studied by using PND. Polarized neutrons enhance the sensitivity to the magnetic scattering, which for molecular crystals is generally weak compared to the nuclear scattering that is the primary contribution to the diffraction pattern.42 The intensity of magnetic scattering observed in a Bragg peak is proportional to the magnetic structure factor, which can be written as37
In this expression the underlying assumption is that the magnetic moments of individual magnetic ions in the structure can be parametrized by their susceptibility tensor χ, such that the magnetic moment of the j′th atom is given by the vector equation mJ = χjH. H is the vector describing the magnetic field applied to the crystal, and fj(Q) is the magnetic form factor, which describes the spatial extent of spin around the atom. To emphasize the fact that the susceptibility tensors of all molecules in the crystal are the same, related by symmetry, the sum over j is taken over all magnetic atoms in the asymmetric unit of the crystal structure, and the sum over i is then over all symmetry operators of the space group, with rotational and translational parts being Ri and ti respectively. This technique can then be used, by mapping the magnetic susceptibility tensor of individual atoms in a molecule, to determine the magnetic anisotropy of the molecule directly from an experiment.30–33
Flipping ratio data, which suppress the need for deuterated samples, were collected on the 6T2-diffractometer of the Laboratorie Léon Brillouin (LLB) on the Orpheé reactor.43 Diffraction data was collected for three (1) and four (2) crystal orientations. The crystals were glued onto an aluminium pin, which was then mounted in a cradle, attached to the end of a stick, and lowered into a cryomagnet. The cradle allows for an easy change of the crystal orientation with respect to the field of the cryomagnet. This orientation stays the same throughout the measurement. Each orientation then gives a new direction of the magnetic field with respect to the crystal. From these data, a full magnetic susceptibility tensor can be reconstructed for each ion – a maximum of 6 susceptibility parameters for a C1 site symmetry. The data for both crystals were collected at a temperature of 5 K and under an applied magnetic field of 1 T. Flipping ratios for the PND refinement were then extracted for each orientation using an in-house suite of programs from the LLB.
Susceptibility tensors were refined with the Python-script RHOCHI,40 using the published12 100 K X-ray structure for 1 and the 150 K X-ray structure for 2. It is a known challenge that hydrogen positions determined from X-ray diffraction data gives bond lengths to hydrogen that are smaller than the results from complementary methods, such as neutron diffraction. To control this effect, positions of hydrogens in the crystal structures were changed such that bond lengths to hydrogen atoms match what has been observed for similar atom groups in neutron diffraction experiments.44 Site susceptibilities refined against structures with artificially elongated bonds to H robustly reproduce the site susceptibilities found by using a nuclear structure refined directly from neutron diffraction data (for details, see ESI†).
One caveat to the susceptibility tensor refinement presented here is the fact that the two structures used to simulate the structure factors for the refinement of the PND data were measured at different temperatures. As mentioned, the structure for 1 was measured at 100 K, while the structure for 2 was measured at 150 K. This is also seen in the thermal parameters, which are larger for 2 than for 1 (Fig. 4). We are convinced that this is not the primary cause for the difference in the susceptibility tensors. Firstly, a study on a Dy(III) SIM showed that a change in temperature from 20 K to 100 K in the absence of a phase transition resulted only in minor changes to the molecular structure, that turned out to be insignificant to the magnetic anisotropy.45 We believe that those results are applicable here as well. Furthermore, we have seen here how the crystal fields in 1 and 2, determined at 100 K and 150 K respectively, are very similar, as described by the Ln–O-distances (Table 1).
| 1 | 2 | |
|---|---|---|
| Axial O | 2.202(4) | 2.203(6) |
| Axial O | 2.208(4) | 2.204(8) |
| Equatorial O | 2.375(4) | 2.349(8) |
| Equatorial O | 2.357(4) | 2.321(7) |
| Equatorial O | 2.343(4) | 2.329(7) |
| Equatorial O | 2.364(4) | 2.323(7) |
| Equatorial O | 2.359(4) | 2.343 (7) |
We note that in both compounds we find one of the eigenvalues of the magnetic susceptibility tensor to be negative. For 1, the negative value is not so different from zero to be a cause for concern, considering its associated esd. On the other hand, there is a noticeable deviation from zero for 2. The magnetic properties of both compounds must originate in the unpaired electrons of the paramagnetic lanthanide ions, so a “diamagnetic direction” of susceptibility is unreasonable. Based on the direction of the susceptibility tensor of 2 with respect to the magnetic field orientations during the measurements on 2, what we are seeing in this case is a correlation between the two smallest eigenvalues. The data measured on 2 can roughly be collected in two groups of magnetic field orientations (see ESI†), with the group probing the transverse elements of the susceptibility tensor containing only few flipping ratios. It has not been possible to separate the two smallest eigenvalues, but notice, however, that their mean is positive with a value of 0.6 μBT−1, in agreement with the paramagnetic nature of the compound.
Results
Direct-current (DC) magnetic susceptibility measurements
Direct-current (DC) magnetization studies were performed on 2. The magnetic susceptibility χMT product of 2, shown in Fig. 2, is 14.0 cm3 mol−1 K at 290 K. Upon cooling, χMT decreases slowly to 12.6 cm3 mol−1 K at 20 K due to thermal depopulation of excited states, followed by a near plateau and then a sharper decrease observed below 10 K where χMT = 12.0 cm3 mol−1 K at 2 K. The high temperature χMT value of 2 is in close agreement with the theoretical value of 14.1 cm3 mol−1 K expected for a free Ho(III) ion (5I8, S = 2, L = 6, g = 5/4). The general χMT profile is in good agreement with the ab initio calculated susceptibility (Fig. 2, left). In addition, the slow decrease of χMT upon cooling with the sharp drop at low temperatures indicates strong crystal field splitting. Moreover, the overlapping reduced magnetization data at low temperatures (Fig. S3†) suggest the presence of well-separated excited energy states. The field dependence of the magnetization studies were performed at 2, 4 and 6 K (Fig. 2, right) with the magnetization curves at 4 K and 6 K following an approximately linear trend up to 1 T before reaching 5.04 μB and 4.96 μB at 5 T. Since the PND (carried out at 5 K, 1 T) approach assumes a linear relationship between magnetization and field, this means that the retrieved atomic susceptibility tensor values will be correct on an absolute scale. | ||
| Fig. 2 (Left) Temperature dependence of the χMT product under an applied dc field of 1000 Oe from 290-2 K for 2 (yellow circles). The red line is the ab initio computed temperature dependence. (Right) Variable-field magnetization data at 2, 4 and 6 K for 2 from 0.04–5 T. | ||
Alternating-current (AC) magnetic susceptibility measurements
Compound 1 has a reported energy barrier of 600 K under zero applied dc field.12Ac susceptibility measurements, between 1–940 Hz in zero dc field and a small oscillating ac field of Hac = 3 Oe, were performed for 2 in order to investigate the magnetic relaxation dynamics (Fig. 3 and S4–S7, ESI†). Under zero external dc field the out-of-phase ac susceptibility data exhibit well defined maxima in χ′′M (T) and χ′′M (v) (Fig. 3 and S4 and S5†), indicative of slow relaxation of magnetization. The χ′′M(v) out-of-phase ac susceptibility data exhibit peaks clearly observable in the range of 6–23 K (Fig. 3 and S5†). The relaxation times, τ, were extracted by fitting the χM′′ vs. χM′ Argand plots using a generalised Debye model (Fig. S6†).46 The α parameters extracted are in the range of 0.06–0.298 (6–23 K) for 2. The τ−1(T) data were fitted using the equation τ−1 = τ0−1exp(−Ueff/T) + CTn, in which C and n are the Raman process parameters (Fig. 3 and S7†). The best fit gives a magnetization reversal barrier of Ueff = 270 K, τ0 = 4.77 × 10−9 s, n = 4.11 and C = 0.008 K−n s−1 under zero applied dc field (Fig. 3). The values of τ0, n and C are in agreement with the observed range for HoIII SIMs.23,24,26,27,47 Demagnetization through high excited mJ-states is not so common in HoIII SIMs because of their non-Kramers nature, which normally promotes fast quantum tunneling of magnetization. However, in the case of 2, the compressed pentagonal bipyramidal geometry stabilizes an anisotropic ground state, suggesting that Ho(III) does indeed benefit from a compressed and hence significantly axial ligand field, and that the 4f-valence charge distribution of the ground mJ-state, to some extent, may be classified as oblate.
 | ||
| Fig. 3 (Left)Frequency dependence of the out-of-phase, χ′′M ac susceptibility, in zero dc field, for 2. The solid lines correspond to the best fit to Debye's law.48 (Right) Log–Log plot of the relaxation times, τ−1versus T for 2 (see main text for details). | ||
This is supported by ab initio calculations on 2, which reveal the pseudo doublet of the ground state (purely mJ = ±8) shows strong axiality (gzz = 19.96) with a negligible tunnel splitting value of 0.0016 cm−1 (see Table S4†). In addition, the principal anisotropy axis is found nearly collinear with the shortest PO–Ho–OP bonds (Fig. S8†). The first excited pseudo doublet is located at ∼260 K and shows a mixture of wavefunctions with a tunnel splitting value of 0.90 cm−1, large enough to result in relaxation via this state. The maximum calculated relaxation barrier, Ucal, is estimated at 261 K which is in excellent agreement with the experimentally determined magnetization reversal barrier of 270 K, found in zero applied dc field.
X-ray crystallography on [Ho(H2O)5(HMPA)2]I3·2HMPA (2)
To justify the comparison of the susceptibility tensors of the two compounds measured here, we first revisit the structures of the two molecules. Our initial assumption is that the lanthanide ions in 1 and 2 experience identical crystal fields, such that differences in the magnetic anisotropy can be attributed mainly to the difference in the metal ions and the respective interactions between the 4f-valence charge distribution with the surrounding ligand field. The distances between Ln and the nearest seven oxygen atoms are shown in Table 1 (and Table S2†).As can be seen in Table 1, the axial Ln–O distances are shorter than the equatorial Ln–O distances and are very similar for the two complexes. The equatorial Ln–O-distances, which are between Ln and the oxygen-atoms of the water molecules around the “waist” of the molecules, are also very similar. The mean values of the equatorial distances are 2.36(1) Å and 2.33(1) Å in 1 and 2, respectively. This confirms the assertion that the two crystal fields are indeed very similar.
PND: magnetic susceptibility tensors
The refined magnetic susceptibility tensor of Dy in 1 can be diagonalized to give the following eigenvalues (λ) and eigenvectors (v), the latter given in the crystallographic basis.| λ1 = 10.1(1)μBT−1,λ2 = 0.2(1)μBT−1,λ3 = 0.4(1)μBT−1 |

The anisotropy ellipsoid that graphically represents these values is shown in Fig. 4. As the eigenvalues give the magnitudes of the ellipsoid along its primary axes, they are showing the degree of anisotropy of the magnetic susceptibility of 1. It is evident that 1 exhibits extreme axial magnetic anisotropy with the easy-axis value being 10.1 μBT−1 and the transverse susceptibilities negligible: |χx|,|χx|<0.4 μBT−1. Within the effective spin ½-model, the relationship between susceptibility and g-value is g = (4kBχTμB−2)1/2, and this easy-axis magnetic susceptibility value then amounts to a g|| = 17.3(1).
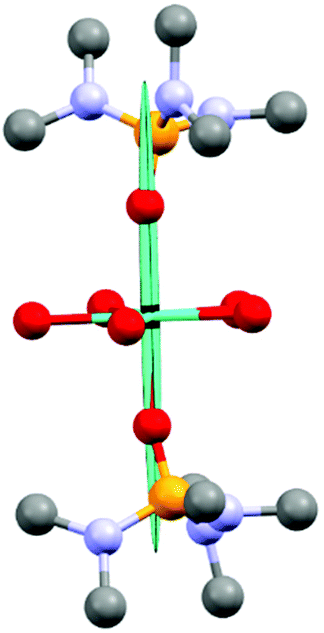 | ||
| Fig. 4 Magnetic susceptibility tensor of 1. The magnetic susceptibility tensor has been visualized as an ellipsoid and scaled arbitrarily to fit the molecular structure. Negative eigenvalues are here shown as positive for the purpose of visualization. The units on the tensor axes are μBT−1. Hydrogen atoms, HMPA co-crystalized ligands and counter-ions have been omitted. | ||
The refinement of the magnetic susceptibility tensor for 2 presents more of a challenge than for 1, due to a smaller dataset, particularly for the orientations perpendicular to the easy axis. Although four magnetic field orientations were measured for 2, orientations 3 and 4 (see ESI†) gave only 7 and 6 discernable flipping ratios, respectively. In order to ensure that the global minimum was reached, we compared five different models for the magnetic susceptibility tensor against the data. Two of these were simulated models, based partly on the results obtained for 1 (see the ESI† for more details). This in-depth investigation resulted in the best model for 2 being one in which the magnetic easy axis is tilted with respect to the molecular axis, with eigenvalues (λ) and eigenvectors (v), given in the crystallographic basis, of
| λ1 = 10.7(2)μBT−1,λ2 = 2.9(1)μBT−1,λ3 = −2.5(1)μBT−1 |

In the same way as with 1, the susceptibility tensor of 2 is represented as an ellipsoid on top of the molecular structure in Fig. 5. Recalculating this result in terms of an effective spin ½-model gives an easy-axis g-value (g||) of 17.8(2), which is still significantly less than the expected value of 20, and in the same range as that obtained experimentally for 1. For 2 we obtain a value of gz = 19.96 from the ab initio calculations with negligible values of gx and gy. Agreement statistics between the models obtained from site susceptibility modelling of the diffraction data and the measured PND data is shown in Tables S10 and S11.†
 | ||
| Fig. 5 Magnetic susceptibility tensor of 2 within the model that best fits the measured data. The magnetic susceptibility tensor has been visualized as an ellipsoid and scaled arbitrarily to fit the molecular structure. Negative eigenvalues are here shown as positive for the purpose of visualization. The units on the tensor axes are μBT−1. Hydrogen atoms, HMPA co-crystalized ligands and counter-ions have been omitted. | ||
Discussion
The best fits of the atomic susceptibility tensors for the central Ln(III)-ion in 1 and 2 are noticeably different, both in terms of their directions and anisotropies, as shown in Fig. 4 and 5, and also in terms of the model quality (χ2(1) = 2.5; χ2(2) = 15.7).Let us first examine compound 1; here, we obtain a susceptibility tensor that is almost completely axial with vanishing transverse values, as outlined above. The experimental g||-value of 17.3(1) contrasts with the ab initio calculations from the recent paper on 1 that found the g-tensor of the computed ground doublet of 1 to be strictly axial with a value of g|| = 19.96.12 The experimental and theoretical easy axis directions are identical, and the discrepancy between the experimental g|| and the computational result is similar to what has been seen in earlier investigations of the anisotropy of the magnetic susceptibility, which all show a smaller experimental g|| than the calculated value based on the magnetic ground state.49,50
Let us now address the differences in the orientation of the easy axis and in the anisotropy between the experimental and theoretical magnetic anisotropy for 2. We consider first the orientation. The best model for 2 shows a pronounced tilting (25(1)°) of the experimental magnetic easy-axis away from the PO–Ho–OP-axis. If we impose an axial susceptibility tensor oriented along this axis, with the same eigenvalues as obtained for 1 (model 5 in ESI†), the result is a significantly worse agreement with the data. We note that this large tilt is in stark contrast to the ab initio result, which shows the easy-axis direction for 2 to be parallel to the PO–Ho–OP direction. There is a relatively small error on the angular discrepancy. This error, however, is purely a least-squares derived error and since most of the data for 2 has been measured with a magnetic field direction that induces a response along the transverse plane of the molecule (see Fig. S12†), the true experimental error on the easy axis direction is probably substantially larger. In addition, although the value of χ2 increases substantially when different models are tested, the graphical representation of the discrepancies between observed and calculated flipping ratios shows that the differences are more subtle. Thus, despite the final model being robust, we cannot conclude that the magnetic easy axis is tilted significantly away from the PO–Ho–OP direction. The main reasons for this are (1) the large goodness of fit-values; (2) the accidental near-overlap of crystal orientations during flipping ratio measurement, and (3) the much lower number of flipping ratios for 2 relative to 1; (4) combined with the fact that no ab initio calculations, neither on this nor on similar compounds,26,27 lead to deviations from axiality of the magnetic easy axis.
This now leads us to a comparison of the difference in anisotropy in 1 and 2. The magnetic anisotropy barrier, Ueff, which is 600 K for 112 and 270 K for 2 shows that slow magnetic relaxation is better supported for Dy(III) than for Ho(III) in these compounds. Considering this in relation to the Rinehart-Long-model of complementarity between 4f-CD and ligand charges, this finding reiterates that the axial crystal field in 1 and 2 provides an environment more suitable for stabilizing axial anisotropy with the Dy(III) charge distribution than with that of Ho(III). The degree of anisotropy (here evaluated as the ratio of transverse to axial susceptibility) is one of several important parameters for the determination of the magnetic properties of a SIM, and the prevailing opinion is that a reduced degree of anisotropy leads to a lower barrier to magnetization reversal. Since all used magnetic field orientations during flipping ratio measurements for 2 involved the transverse plane of the molecule (Fig. S12†), we believe we can trust a comparison of the easy-axis susceptibility to the transverse susceptibilities for both compounds. Thus, the transverse elements of the susceptibility tensors for 1 and 2 obtained with PND give magnitudes in 1 that are only 2% and 4% of the easy axis magnetic susceptibility, whereas they make up 23% and 27% in 2, supporting the decrease of magnetic anisotropy from 1 to 2.
Finally, we return to the question posed in the introduction, of whether we can quantify the decrease of “oblateness” in Ho(III) compared to Dy(III). Importantly, the decrease of magnetic anisotropy in 2 compared to 1, seen both in the experimentally determined effective barriers from magnetometry and the magnetic anisotropies from PND, is not captured in the comparison of pseudo g-tensors from ab initio calculations, which show a ground state for 2 with an almost identical value of g|| to that of 1. This highlights the fact that experimental measurement of magnetic anisotropy is an important technique for complementing theoretical calculations, and with the advent of next-generation neutron sources there is a huge potential for further quantitative insights from PND experiments.
Conclusion
We have presented the new compound [Ho(H2O)5(HMPA)2]I3·2HMPA (2), measured its magnetic properties and compared these to the known magnetic properties of 1. We have measured PND data on both the Dy and Ho analogues to make an experimental determination of the atomic magnetic anisotropy of the two compounds and thereby undertaken the first site susceptibility measurement on a Ho(III)-containing molecular species. The trends observed in the PND results correlate well with those measured by DC magnetization measurements.PND allows for the retrieval of the atomic magnetic anisotropy of complexes embedded in crystals, where other methods are unable to provide an unambiguous answer. It is common in the literature to find reports of the magnetic anisotropy of molecular complexes, but we stress the fact that the easy-axes and magnetic anisotropies presented here are purely experimental results and show not only the magnitude of the magnetic susceptibility, but also the primary axes of the magnetic environment. Our results show that there is easy-axis character of both 1 and 2. Thus, we reiterate the preference for the axial geometry of the Dy(III)-ion, which has been shown here to be experimentally verifiable for 1 in close agreement with the theoretical results. The subtle differences between Ho(III) and Dy(III) mean that the optimal crystal field for the former might not be strictly linear. We have shown here by experimental means using PND that in the example of 2, the magnetic anisotropy of a Ho(III)-ion embedded in an axial environment decreases compared to the Dy(III)-analogue.
Author contributions
The project was conceived by JO, MM and ABC. ABC carried out the synthesis. CW collected and analysed the single crystal X-ray data of compound 2. RBD, AMT and EAK collected and analysed the PND data. IK and AG collected PND data and developed software for analysis. EAK carried out the theoretical calculations. ABC and MM collected and analysed the magnetization data. The manuscript was written by ABC, MM, EAK and JO, with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to the staff of the Orphée-reactor and the LLB for their assistance with this work. Affiliation with the Center for Integrated Materials Research (iMAT) at Aarhus University is gratefully acknowledged. Funding from the Danish Ministry of Higher Education and Science through the SMART Lighthouse is gratefully acknowledged. The computational results were obtained at the Centre for Scientific Computing at Aarhus University. This research was funded by the Villum Foundation, the Danish National Research Foundation (DNRF-93) and Danscatt. MM thanks the UK Engineering and Physical Sciences Research Council for financial support (EP/N01331X/1).Notes and references
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Magnetic hysteresis up to 80 kelvin in a dysprosium metallocene single-molecule magnet, Science, 2018, 362, 1400 CrossRef CAS PubMed.
- A. K. Bar, P. Kalita, M. K. Singh, G. Rajaraman and V. Chandrasekhar, Low-coordinate mononuclear lanthanide complexes as molecular nanomagnets, Coord. Chem. Rev., 2018, 367, 163–216 CrossRef CAS.
- S. G. McAdams, A.-M. Ariciu, A. K. Kostopoulos, J. P. S. Walsh and F. Tuna, Molecular single-ion magnets based on lanthanides and actinides: Design considerations and new advances in the context of quantum technologies, Coord. Chem. Rev., 2017, 346, 216–239 CrossRef CAS.
- G. A. Craig and M. Murrie, 3d single-ion magnets, Chem. Soc. Rev., 2015, 44, 2135–2147 RSC.
- M. N. Leuenberger and D. Loss, Quantum computing in molecular magnets, Nature, 2001, 410, 789–793 CrossRef CAS PubMed.
- A. Gaita-Ariño, F. Luis, S. Hill and E. Coronado, Molecular spins for quantum computation, Nat. Chem., 2019, 11, 301–309 CrossRef PubMed.
- J. Ferrando-Soria, E. Moreno Pineda, A. Chiesa, A. Fernandez, S. A. Magee, S. Carretta, P. Santini, I. J. Vitorica-Yrezabal, F. Tuna, G. A. Timco, E. J. McInnes and R. E. Winpenny, A modular design of molecular qubits to implement universal quantum gates, Nat. Commun., 2016, 7, 11377 CrossRef CAS PubMed.
- R. E. P. Winpenny, Quantum Information Processing Using Molecular Nanomagnets As Qubits, Angew. Chem., Int. Ed., 2008, 47, 7992–7994 CrossRef CAS PubMed.
- J. D. Rinehart and J. R. Long, Exploiting single-ion anisotropy in the design of f-element single-molecule magnets, Chem. Sci., 2011, 2, 2078–2085 RSC.
- J. Sievers, Asphericity of 4f-Shells in Their Hund Rule Ground-States, ZPhys-e.B: Condens. Matter, 1982, 45, 289–296 CAS.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Molecular magnetic hysteresis at 60 kelvin in dysprosocenium, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- A. B. Canaj, M. K. Singh, C. Wilson, G. Rajaraman and M. Murrie, Chemical and in silico tuning of the magnetisation reversal barrier in pentagonal bipyramidal Dy(iii) single-ion magnets, Chem. Commun., 2018, 54, 8273–8276 RSC.
- A. B. Canaj, S. Dey, C. Wilson, O. Cespedes, G. Rajaraman and M. Murrie, Engineering macrocyclic high performance pentagonal bipyramidal Dy(iii) single-ion magnets, Chem. Commun., 2020, 56, 12037–12040 RSC.
- Y.-C. Chen, J.-L. Liu, L. Ungur, J. Liu, Q.-W. Li, L.-F. Wang, Z.-P. Ni, L. F. Chibotaru, X.-M. Chen and M.-L. Tong, Symmetry-Supported Magnetic Blocking at 20 K in Pentagonal Bipyramidal Dy(III) Single-Ion Magnets, J. Am. Chem. Soc., 2016, 138, 2829–2837 CrossRef CAS PubMed.
- S. K. Gupta, T. Rajeshkumar, G. Rajaraman and R. Murugavel, An air-stable Dy(iii) single-ion magnet with high anisotropy barrier and blocking temperature, Chem. Sci., 2016, 7, 5181–5191 RSC.
- J. Liu, Y. C. Chen, J. L. Liu, V. Vieru, L. Ungur, J. H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X. M. Chen and M. L. Tong, A Stable Pentagonal Bipyramidal Dy(III) Single-Ion Magnet with a Record Magnetization Reversal Barrier over 1000 K, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS PubMed.
- Y.-S. Ding, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, On Approaching the Limit of Molecular Magnetic Anisotropy: A Near-Perfect Pentagonal Bipyramidal Dysprosium(III) Single-Molecule Magnet, Angew. Chem., Int. Ed., 2016, 55, 16071–16074 CrossRef CAS PubMed.
- Y. S. Ding, T. Han, Y. Q. Zhai, D. Reta, N. F. Chilton, R. E. P. Winpenny and Y. Z. Zheng, A Study of Magnetic Relaxation in Dysprosium(III) Single-Molecule Magnets, Chem. – Eur. J., 2020, 26, 5893–5902 CrossRef CAS PubMed.
- P. E. Kazin, M. A. Zykin, V. V. Utochnikova, O. V. Magdysyuk, A. V. Vasiliev, Y. V. Zubavichus, W. Schnelle, C. Felser and M. Jansen, “Isolated” DyO+Embedded in a Ceramic Apatite Matrix Featuring Single-Molecule Magnet Behavior with a High Energy Barrier for Magnetization Relaxation, Angew. Chem., Int. Ed., 2017, 56, 13416–13420 CrossRef CAS PubMed.
- I. F. Díaz-Ortega, J. M. Herrera, S. Dey, H. Nojiri, G. Rajaraman and E. Colacio, The effect of the electronic structure and flexibility of the counteranions on magnetization relaxation in [Dy(L)2(H2O)5]3+(L=phosphine oxide derivative) pentagonal bipyramidal SIMs, Inorg. Chem. Front., 2020, 7, 689–699 RSC.
- N. F. Chilton, D. Collison, E. J. McInnes, R. E. Winpenny and A. Soncini, An electrostatic model for the determination of magnetic anisotropy in dysprosium complexes, Nat. Commun., 2013, 4, 2551 CrossRef PubMed.
- C. Gao, A. Genoni, S. Gao, S. Jiang, A. Soncini and J. Overgaard, Observation of the asphericity of 4f-electron density and its relation to the magnetic anisotropy axis in single-molecule magnets, Nat. Chem., 2020, 12, 213–219 CrossRef CAS PubMed.
- S. M. Chen, Y. Q. Zhang, J. Xiong, B. W. Wang and S. Gao, Adducts of Tris(alkyl) Holmium(III) Showing Magnetic Relaxation, Inorg. Chem., 2020, 59, 5835–5844 CrossRef CAS PubMed.
- Y. Ma, Y.-Q. Zhai, Y.-S. Ding, T. Han and Y.-Z. Zheng, Understanding a pentagonal-bipyramidal holmium(III) complex with a record energy barrier for magnetisation reversal, Chem. Commun., 2020, 56, 3979–3982 RSC.
- T. Kajiwara, A Holmium(III)-Based Single-Molecule Magnet with Pentagonal-Bipyramidal Geometry, Angew. Chem., Int. Ed., 2017, 56, 11306–11308 CrossRef CAS PubMed.
- Y.-C. Chen, J.-L. Liu, W. Wernsdorfer, D. Liu, L. F. Chibotaru, X.-M. Chen and M.-L. Tong, Hyperfine-Interaction-Driven Suppression of Quantum Tunneling at Zero Field in a Holmium(III) Single-Ion Magnet, Angew. Chem., Int. Ed., 2017, 56, 4996–5000 CrossRef CAS PubMed.
- S. K. Gupta, T. Rajeshkumar, G. Rajaraman and R. Murugavel, Is a strong axial crystal-field the only essential condition for a large magnetic anisotropy barrier? The case of non-Kramers Ho(iii) versus Tb(iii), Dalton Trans., 2018, 47, 357–366 RSC.
- M. Ren, S.-S. Bao, B.-W. Wang, R. A. S. Ferreira, L.-M. Zheng and L. D. Carlos, Lanthanide phosphonates with pseudo-D5 h local symmetry exhibiting magnetic and luminescence bifunctional properties, Inorg. Chem. Front., 2015, 2, 558–566 RSC.
- S. K. Gupta and R. Murugavel, Enriching lanthanide single-ion magnetism through symmetry and axiality, Chem. Commun., 2018, 54, 3685–3696 RSC.
- O. Iasco, Y. Chumakov, F. Guégan, B. Gillon, M. Lenertz, A. Bataille, J.-F. Jacquot and D. Luneau, Mapping the Magnetic Anisotropy inside a Ni4 Cubane Spin Cluster Using Polarized Neutron Diffraction, Magnetochemistry, 2017, 3, 25–38 CrossRef.
- K. Ridier, A. Mondal, C. Boilleau, O. Cador, B. Gillon, G. Chaboussant, B. Le Guennic, K. Costuas and R. Lescouëzec, Polarized Neutron Diffraction to Probe Local Magnetic Anisotropy of a Low-Spin Fe(III) Complex, Angew. Chem., Int. Ed., 2016, 55, 3963–3967 CrossRef CAS PubMed.
- K. Ridier, B. Gillon, A. Gukasov, G. Chaboussant, A. Cousson, D. Luneau, A. Borta, J.-F. Jacquot, R. Checa, Y. Chiba, H. Sakiyama and M. Mikuriya, Polarized Neutron Diffraction as a Tool for Mapping Molecular Magnetic Anisotropy: Local Susceptibility Tensors in CoII Complexes, Chem. – Eur. J., 2016, 22, 724–735 CrossRef CAS PubMed.
- A. Borta, B. Gillon, A. Gukasov, A. Cousson, D. Luneau, E. Jeanneau, I. Ciumacov, H. Sakiyama, K. Tone and M. Mikuriya, Local magnetic moments in a dinuclear Co2 + complex as seen by polarized neutron diffraction: Beyond the effective spin-1/2 model, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 184429 CrossRef.
- E. A. Klahn, C. Gao, B. Gillon, A. Gukasov, X. Fabreges, R. O. Piltz, S. D. Jiang and J. Overgaard, Mapping the Magnetic Anisotropy at the Atomic Scale in Dysprosium Single-Molecule Magnets, Chem. – Eur. J., 2018, 24, 16576–16581 CrossRef CAS PubMed.
- F. Guégan, J. Jung, B. Le Guennic, F. Riobé, O. Maury, B. Gillon, J.-F. Jacquot, Y. Guyot, C. Morell and D. Luneau, Evidencing under-barrier phenomena in a Yb(iii) SMM: a joint luminescence/neutron diffraction/SQUID study, Inorg. Chem. Front., 2019, 6, 3152–3157 RSC.
- S. Tripathi, S. Vaidya, N. Ahmed, E. A. Klahn, H. Cao, L. Spillecke, C. Koo, S. Spachmann, R. Klingeler, G. Rajaraman, J. Overgaard and M. Shanmugam, Structure-property correlation in stabilizing axial magnetic anisotropy in octahedral Co(II) complexes, Cell Rep. Phys. Sci., 2021, 2, 100404 CrossRef CAS.
- A. Gukasov and P. J. Brown, Determination of atomic site susceptibility tensors from polarized neutron diffraction data, J. Phys.: Condens. Matter, 2002, 14, 8831 CrossRef CAS.
- M. Perfetti, Cantilever torque magnetometry on coordination compounds: from theory to experiments, Coord. Chem. Rev., 2017, 348, 171–186 CrossRef CAS.
- M. Perfetti, G. Cucinotta, M. E. Boulon, F. El Hallak, S. Gao and R. Sessoli, Angular-resolved magnetometry beyond triclinic crystals part II: torque magnetometry of Cp*ErCOT single-molecule magnets, Chem. – Eur. J., 2014, 20, 14051–14056 CrossRef CAS PubMed.
- I. A. Kibalin and A. Gukasov, Local magnetic anisotropy by polarized neutron powder diffraction: Application of magnetically induced preferred crystallite orientation, Phys. Rev. Res., 2019, 1, 033100 CrossRef CAS.
- P. Kalita, J. Acharya and V. Chandrasekhar, Mononuclear pentagonal bipyramidal Ln(III) complexes: Syntheses and magnetic properties, J. Magn. Magn. Mater., 2020, 498, 166098 CrossRef CAS.
- E. Ressouche, Polarized neutron diffraction, Collection SFN, 2014, 13, 02002 CrossRef.
- A. Gukasov, A. Goujon, J. L. Meuriot, C. Person, G. Exil and G. Koskas, Super-6T2, a new position-sensitive detector polarized neutron diffractometer, Physica B, 2007, 397, 131–134 CrossRef CAS.
- F. H. Allen and I. J. Bruno, Bond lengths in organic and metal-organic compounds revisited: X-H bond lengths from neutron diffraction data, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 380–386 CrossRef CAS PubMed.
- K. Qian, J. J. Baldovi, S.-D. Jiang, A. Gaita-Arino, Y.-Q. Zhang, J. Overgaard, B.-W. Wang, E. Coronado and S. Gao, Does the thermal evolution of molecular structures critically affect the magnetic anisotropy?, Chem. Sci., 2015, 6, 4587–4593 RSC.
- D. Gatteschi, R. Sessoli and J. Villain, Molecular nanomagnets, Oxford University Press, Oxford; New York, 2006 Search PubMed.
- R. J. Blagg, F. Tuna, E. J. McInnes and R. E. Winpenny, Pentametallic lanthanide-alkoxide square-based pyramids: high energy barrier for thermal relaxation in a holmium single molecule magnet, Chem. Commun., 2011, 47, 10587–10589 RSC.
- M. L. Baker and H. Mutka, Neutron spectroscopy of molecular nanomagnets, Eur. Phys. J.: Spec. Top., 2012, 213, 53–68 CAS.
- E. A. Klahn, C. Gao, B. Gillon, A. Gukasov, X. Fabrèges, R. O. Piltz, S.-D. Jiang and J. Overgaard, Mapping the Magnetic Anisotropy at the Atomic Scale in Dysprosium Single-Molecule Magnets, Chem. – Eur. J., 2018, 24, 16576–16581 CrossRef CAS PubMed.
- G. Cucinotta, M. Perfetti, J. Luzon, M. Etienne, P.-E. Car, A. Caneschi, G. Calvez, K. Bernot and R. Sessoli, Magnetic Anisotropy in a Dysprosium/DOTA Single-Molecule Magnet: Beyond Simple Magneto-Structural Correlations, Angew. Chem., Int. Ed., 2012, 51, 1606–1610 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Magnetic studies, crystallographic details, ab initio studies. CCDC 2057601. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt01959g |
| ‡ Current address: Department of Chemistry and Materials Innovation Factory, University of Liverpool, 51 Oxford Street, Liverpool L7 3NY, UK. E-mail: angelos.tsanai@liverpool.ac.uk |
| This journal is © The Royal Society of Chemistry 2021 |