
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanochemical synthesis of mononuclear gold(I) halide complexes of diphosphine ligands with tuneable luminescent properties†
Andrea
Deák
 *a,
Csaba
Jobbágy
a,
Attila
Demeter
b,
Ladislav
Čelko
c,
Jaroslav
Cihlář
c,
Pál T.
Szabó
d,
Péter
Ábrányi-Balogh
e,
Deborah E.
Crawford
f,
David
Virieux
g and
Evelina
Colacino
*g
*a,
Csaba
Jobbágy
a,
Attila
Demeter
b,
Ladislav
Čelko
c,
Jaroslav
Cihlář
c,
Pál T.
Szabó
d,
Péter
Ábrányi-Balogh
e,
Deborah E.
Crawford
f,
David
Virieux
g and
Evelina
Colacino
*g
aSupramolecular Chemistry Research Group, Institute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, Eötvös Loránd Research Network (ELKH), Magyar Tudósok körútja 2, 1117 Budapest, Hungary. E-mail: deak.andrea@ttk.hu
bRenewable Energy Research Group, Institute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, Eötvös Loránd Research Network (ELKH), Magyar Tudósok körútja 2, 1117 Budapest, Hungary
cCentral European Institute of Technology, Brno University of Technology, Purkyňova 123, 61200 Brno, Czech Republic
dCentre for Structure Study, Research Centre for Natural Sciences, Eötvös Loránd Research Network (ELKH), 1117 Budapest, Magyar Tudósok körútja 2, Hungary
eMedicinal Chemistry Research Group, Institute of Organic Chemistry, Research Centre for Natural Sciences, Eötvös Loránd Research Network (ELKH), 1117 Budapest, Magyar Tudósok körútja 2, Hungary
fSchool of Chemistry and Biosciences, University of Bradford Richmond Road, BD7 1DP, Bradford, UK
gICGM, Univ Montpellier CNRS, ENSCM, Montpellier, France. E-mail: evelina.colacino@umontpellier.fr
First published on 23rd August 2021
Abstract
A mechanochemical method is reported for the synthesis of Au(diphos)X complexes of diphosphine (diphos = XantPhos and N-XantPhos) ligands and halide ions (X = Cl and I). The Au(XantPhos)X (1: X = Cl; 2: X = I) and Au(N-XantPhos)Cl (3) complexes exhibited either yellowish green (1) or bluish green (2) emission, whereas 3 was seemingly non-emissive in the solid state at room temperature. Blue- (2B) and bluish green (2G) luminescent concomitant solvates of 2 were obtained by recrystallization. Luminescent colour changes from blue (2B) or bluish green (2G) to yellow were observed when these forms were subjected to mechanical stimulus, while the original emission colour can be recovered in the presence of solvent vapours. Moreover, the luminescence of 2B can be reversibly altered between blue and yellow by heating/cooling-cycles. These results demonstrate the power of mechanochemistry in the rapid (4 min reaction time), efficient (up to 98% yield) and greener synthesis of luminescent and stimuli-responsive gold(I) complexes.
Introduction
Molecular functional materials based on gold(I) complexes have gained increasing attention for their numerous promising applications in new optoelectronic devices,1–3 sensing,4,5 anticancer therapeutics,6 bioimaging,7 catalysis,8 gold nanoparticles and nanocluster synthesis.9 The gold(I) diphosphine family10 is one of the most attractive classes of gold(I) compounds as they exhibit various possible coordination modes,11–17 distinct luminescence,11–14,18 are stimuli-responsive photophysically,14,18–20 catalytic15,16 and have promising anticancer properties.21 Moreover, mono- and dinuclear gold(I) phosphines have also been used in the preparation of electroluminescent (LED) devices,22–24 for biochemical25 and biomedical applications26,27 and for bioimaging.28Indeed, the increasing demand for clean processes and eco-conscious reaction conditions has fostered the application of mechanochemistry in organic29,30 and organometallic syntheses,31 in metal catalysis,32 for the preparation of co-crystals,33 metal clusters,34–36 metal–organic frameworks (MOFs)37 and coordination polymers (CPs),38,39 pharmaceutical materials40–42 and ingredients (APIs and metallo-drugs).43–45 In the last few decades, this technology has provided access to an impressive number of discrete metal complexes,31,46,47 as well as infinite CPs48,49 and MOFs,49 in a rapid and environmentally-sustainable fashion. Furthermore, it should be noted that there is the possibility to translate mechanochemical batch protocols at laboratory scale into continuous manufacturing, large-scale processes by twin screw extrusion (TSE),50 not only for the preparation of APIs,51 but also to access OLED materials.52 Indeed, very recently, the International Union of Pure and Applied Chemistry (IUPAC) has acknowledged TSE as one of the ‘top ten emerging technologies in chemistry’.53 While there are extensive studies on the conventional solution-based synthesis of gold(I) complexes,54,55 the use of mechanochemical methods remains limited despite their unique properties. Friščić et al. reported the mechanochemical oxidation of gold into simple water-soluble KAuCl4 and NH4AuCl4 salts, followed by reduction with Ph3P into the monomeric (Ph3P)AuCl complex.56 Pilarski et al. demonstrated the usefulness of mechanochemistry in the synthesis of (hetero)arylAu(I) complexes.57 We demonstrated the effectiveness of mechanochemistry in the synthesis of dinuclear [Au2(diphos)2](X)2 complexes of diverse diphosphine (diphos) ligands, which exhibit unique luminescent properties.13,18 Moreover, we also prepared, by mechanochemical anion-exchange, a set of stimuli-responsive luminescent crystalline and amorphous [Au2(diphos)2](X)2 complexes.58 The striking stimuli-responsive luminescence of these gold(I) compounds has been correlated with external stimuli induced changes in the intra- and/or intermolecular noncovalent interactions such as aurophilic, hydrogen bonding and π⋯π stacking.19,20 Although, the usefulness of mechanochemistry for the generation of the above mentioned materials has been demonstrated, the mechanochemical synthesis of mononuclear gold(I) halide complexes of diphosphine ligands, Au(diphos)X, has never been reported.
Encouraged by these results, and the growing need for a rapid, simple and efficient approach to high value products or functional metal complexes, we were interested in using mechanochemical methods as a versatile tool to build a more sustainable, rapid and simple approach for the preparation of Au(diphos)X complexes. Three-coordinate gold(I) complexes of diphos ligands and halide ions are rare, and only the solution-based preparation of Au(Xantphos)X (XantPhos = 9,9-dimethyl-4,5-bis(diphenylphosphino)-xanthene; X = Cl15,16 and I,59 complexes 1 and 2 respectively in Scheme 1), Au(Rphos)Cl (Rphos = 1,2-bis[bis(2-methylphenyl)phosphino]benzene or 1,2-bis[bis(2-ethylphenyl)phosphino]benzene)12 and solvothermal synthesis of Au(N-XantPhos)I (N-XantPhos = 4,6-bis(diphenylphosphino)phenoxazine)59 have been reported so far. The solvent-based synthesis of these Xantphos- and N-XantPhos-derived gold(I) compounds required longer reaction times (up to 120 minutes),15,16,59 whereas the use of ball-milling conditions could provide a general environmentally friendly approach to obtain gold(I) complexes with dramatically reduced solvent use, reaction times and improved yields, compared to solution-based procedures.
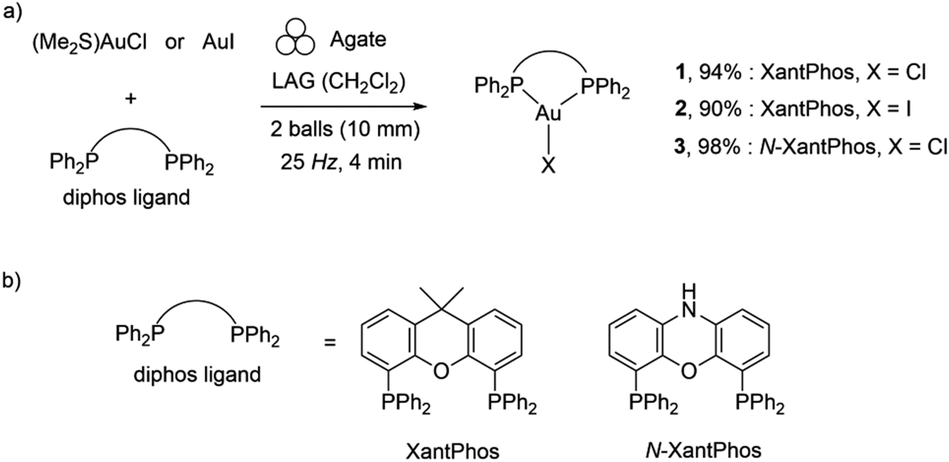 | ||
| Scheme 1 (a) Mononuclear three-coordinate 1–3 complexes prepared by mechanochemistry; (b) ligands. Mechanochemical activation was represented with the formalism recently introduced.47 | ||
As an extension of our studies on mechanochemical synthesis of dinuclear gold(I) complexes, we now employ minimal-solvent conditions and dramatically reduced reaction times for the mechanochemical synthesis of Au(diphos)X complexes. Moreover, these three-coordinate gold(I) complexes are expected to luminesce, thus, systematic variation in the nature of diphosphines and halide ions is also expected to result in an efficient tuning of the solid state luminescence and stimuli-responsive properties of these complexes.19,20,26 Moreover, due the unique properties of gold(I) complexes and their applications for biomedical purposes,25–27 their preparation by mechanochemistry will contribute to a flourishing area of investigation referred to as ‘medicinal mechanochemistry’.41 Indeed, gold(I) complexes have been FDA approved as oral drug for the treatment of rheumatoid arthritis, selectively targetting cysteine sites in glutathione, proteins and enzymes upon rapid ligand displacement in vivo.25
Results and discussion
Mononuclear, three-coordinate Au(diphos)X (1–3) complexes of chelating bidentate diphosphine ligands (Scheme 1) were synthesised mechanochemically (see ESI, Table S1†).In all cases, ball milling stoichiometric amounts of the gold(I) precursors, (Me2S)AuCl or AuI, with the corresponding diphosphine ligands in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio under liquid-assisted grinding conditions (LAG; liquid-to-reactant ratio η = 0.25 μL mg−1)60 gave the expected mononuclear complexes 1–3, as determined by HRMS and other analytical techniques (Scheme 1 and ESI†).
:1 stoichiometric ratio under liquid-assisted grinding conditions (LAG; liquid-to-reactant ratio η = 0.25 μL mg−1)60 gave the expected mononuclear complexes 1–3, as determined by HRMS and other analytical techniques (Scheme 1 and ESI†).
As shown in Fig. 1, product formation is also easily observed by the appearance of a luminescence colour of the barely emitting reaction mixture, as complexes 1 and 2 exposed to 365 nm UV light illumination emit intense yellowish green and bluish green light. Complex 3, however, is non-emissive upon exposure to 365 nm UV light illumination.
 | ||
| Fig. 1 As-prepared 1–3 complexes under 365 nm UV light exposure. | ||
Even though the yields obtained from the solution synthesis (87% for both complexes 1 and 2 and 98% for complex 3) were comparable with those obtained by mechanochemistry (94%, 90% and 98% respectively for complexes 1–3, see ESI, Table S1†) the reaction time (4 minutes) needed for the mechanochemical synthesis of complexes 1–3 was significantly shorter compared to previously reported solution-based procedures (60 and 30 minutes).15,59 Moreover, these reactions proceed in the presence of 105–115 μL quantities of solvent (LAG), compared to 15 mL quantities of dichloromethane in the conventional synthesis, representing a greener (and less toxic) route to the synthesis of gold(I)-based materials. Additionally, for complexes 1–3 prepared mechanochemically, the work-up was simplified and the products can be recovered as crystalline compounds directly from the ball mill, as indicated by the PXRD patterns (Fig. S11†).
The products of LAG reactions were characterized by high resolution mass spectrometry (HRMS), 1H and 31P nuclear magnetic resonance (NMR) spectroscopy, powder (PXRD) and single-crystal X-ray diffraction (2), thermogravimetric analysis coupled with mass spectroscopy (TGA/MS) and fluorescence spectroscopy (see ESI†). Formation of the mononuclear three-coordinate 1–3 complexes was confirmed by HR-MS analyses of the mechanochemically prepared crude products. The HR-MS spectra of the crude products 1–3 (see ESI, Fig. S1, S3 and S5†) show the peaks of the corresponding dehalogenated cationic fragments [Au(XantPhos)]+ and [Au(N-XantPhos)]+. The 31P NMR spectra of 1 and 2 also display the characteristic singlet resonances of Au(diphos)X species at 28.1 and 26.0 ppm, respectively. However, as it was previously reported by Ito and co-workers,15,16 partial disproportionation of Au(XantPhos)Cl (1) to Au2(XantPhos)Cl2 and [Au(XantPhos)2]Cl occurred immediately after dissolution in CH2Cl2. The poor solubility of 3 in common organic deuterated solvents hampered its characterisation by solution-based NMR spectroscopy, as previously reported for similar gold(I) complexes.59
Solid state photophysical properties of 1–3 complexes were evaluated by measuring their emission and excitation spectra as well as their respective fluorescence lifetimes. Upon excitation at 355 nm, the emission maximum of 1 and 2 is centred at 520 and 511 nm (Fig. S16†), respectively, while 3 is practically non-emissive at room temperature. The emission quantum yields (Φem) are 0.81 (1) and 0.33 (2), respectively. The quantum yield of 2 is comparable to that prepared by solution-based method (Φem = 0.28).59 Both luminescent compounds display biexponential emission decay with two lifetimes of 19.7 ± 0.3 (85%) and 3.8 ± 0.2 μs (15%) for 1, as well as, 7.0 ± 0.9 (76%) and 1.66 ± 0.04 μs (24%) for 2. These results demonstrate that the luminescence properties of Au(diphos)X complexes 1 and 2 can be tuned by changing the nature of the halide ions.
Moreover, as shown in Fig. 2, crystallization of 2 by slow evaporation from dichloromethane solution layered with n-hexane or n-heptane provided blue (2B) and bluish green (2G) luminescent crystals concomitantly. The crystals were separated manually and both were characterized by single crystal X-ray diffraction. This analysis reveals that the 2B and 2G crystals are both dichloromethane solvates of 2 (see ESI, Table S2†). The experimental PXRD patterns of mechanochemically prepared bluish green-emitting 2 resemble with the simulated patterns derived from the single crystal data of 2G form (see ESI, Fig. S17†), however there are additional reflections present only in the diffractogram of 2.
 | ||
| Fig. 2 Optical micrographs showing the sorted concomitant solvates of blue 2B and bluish green 2G luminescent crystals under 365 nm UV-light exposure. | ||
Polymorph and solvate formation of gold(I) compounds frequently resulted in marked changes in the optical properties, such as luminescence.61–67 Three-coordinate gold(I) complexes that lack aurophilic interactions such as (Ph3P)2AuX (X = halide) often display polymorphism and exhibit luminescence both in the solid and liquid states.61,62 Photocrystallographic studies revealed that photoexcitation-induced structural changes of (Ph3P)2AuCl molecules caused alterations in the molecular arrangement of the green- and blue-emitting crystals.61,62
Moreover, dinuclear three-coordinate gold(I) diphosphine halides, Au2(dppe)2X2 (dppe = bis-(diphenylphosphino)ethane; X = Br, I), alter their emission colour from green to orange upon exposure to solvent vapours.66–68 This has been attributed to marked flexibility of Au2(dppe)2X2 molecules and has been associated with the variations in the Au–X and intramolecular Au⋯Au distances, as well as in the P–Au–P angles.66,67 Therefore, the polymorphism-dependent emission of 2 allowed us to explore the correlation between the molecular structures or arrangements and photoluminescent behaviour. As shown in Fig. 3, the molecular structure of 2B and 2G reveals a three-coordinate gold(I) centre with a chelating XantPhos (P–Au–P angle of 116.64(6)° in 2B and 116.11(4)° in 2G) and coordinated iodide ligand (Au–I bond distance of 2.6883(4) Å in 2B and 2.7004(3) Å in 2G). In contrast to centrosymmetric 2B structure with equivalent Au–P distance of 2.3403(10) Å, the acentric 2G structure features two Au–P distances of 2.3355(12) and 2.3678(11) Å, respectively. The most notable conformational difference between these structures (see Fig. 3a) is in the I–Au–P–CPh torsion angles (θ1 = −1.14°, θ2 = 1.14°, θ3 = −110.87° and θ4 = 110.87° in 2B; θ1 = −12.91°, θ2 = 10.56°, θ3 = −136.54° and θ4 = 133.74° in 2G). Additionally, the structural analysis also indicates the distortion of the XantPhos ligand backbone in 2G polymorph (P–C–C–P torsion angle (θ3) is −2.90° versus 0.0° in 2B). Moreover, the conformational flexibility is closely related to the molecular arrangements (Fig. 3c and d): the columnar stacks of 2B molecules are linearly arranged (I–Au⋯I–Au torsion angle is ±180.0°), while the 2G molecules show a bent arrangement (I–Au⋯I–Au torsion angle is 158.3° and −90.5°) along the c-axis. There are no intermolecular aurophilic bonds in the crystal structure of these polymorphs, as the shortest distance between the Au atoms is 8.658 Å (2B) and 10.008 Å (2G), respectively. The disordered dichloromethane molecule forms C–H⋯Cl interactions (H⋯Cl distance of 2.020(11) and 2.450(11) Å, C⋯Cl distance of 2.655(12) and 2.859(11) Å, and C–H⋯Cl angle of 122.0° and 106.0°) with the gold(I) complex in 2B, while there are no hydrogen bonding interactions in 2G. Thus, these polymorphs mainly differ in the orientation of the monomers with respect to one another, and in the nature of the C–H⋯Cl interactions between the three-coordinated complexes and solvent molecules. In each polymorph, the molecular structure is remarkably similar to that found in the acetonitrile solvate of 2,59 wherein the molecular complex exhibit a somewhat distorted conformation (θ1, θ2 and θ3 torsion angles of −14.06, 10.29 and −4.62°, respectively), which is also reflected in the bent arrangement of the molecules (I–Au⋯I–Au torsion angle is 108.8° and −165.3°) along the c-axis. As shown in Fig. 4ii, the crystals of 2B and 2G emit with λmax = 489 nm (blue) and 502 nm (bluish green) at room temperature (see ESI, Table S3†), respectively. The emission lifetimes for 2B and 2G crystals (see ESI, Table S3†) are indicative of the phosphorescent character of the emission. The photoemission characteristics of 2B and 2G polymorphs, however, are in good agreement with that of acetonitrile solvate of 2 (λmax = 486 nm; τav = 9.1 μs)59 prepared by solution-based procedures.
 | ||
| Fig. 3 Molecular structure and molecular arrangement (viewed along the a-axis) of blue 2B (a and c) and bluish green luminescent 2G (b and d) polymorphs, respectively. Disordered parts of 2B are not shown, the hydrogen atoms and the dichloromethane molecules are omitted for the clarity in the molecular packing diagrams. Colour scheme: gold, yellow; phosphorous, orange; carbon, grey; oxygen, red; iodide, violet; hydrogen, dark brown. | ||
 | ||
| Fig. 4 (i) Photographs showing the (a) amorphous 2Y phase (obtained by ball-milling of 2B or 2G) and (b) crystalline 2BY and (c) 2GY phases (obtained upon solvent vapour exposure of 2Y) under 365 nm UV light illumination; (ii) Normalised excitation and emission spectra of crystalline and amorphous forms of 2. | ||
It was observed that a number of gold(I) compounds not only exhibited polymorphism-dependent luminescence properties but also displayed changes in the luminescence in response to diverse external-stimuli.19,63–67 Thus, the existence of these crystalline phases with distinct multicolour luminescence foreseeds the stimuli-responsive luminescent behaviour of 2. Both crystalline phases 2B and 2G exhibit mechanochromic luminescence upon milling and give a yellow-emitting 2Y phase (Fig. 4i) as a consequence of a crystalline-to-amorphous (CTA) phase transformation (see ESI, Fig. S18 and S19†). This amorphous 2Y phase exhibits a broad emission spectrum (Fig. 4ii) with λmax at 588 nm (see ESI, Table S3†). Exposure of 2Y to dichloromethane vapours afforded a bluish green luminescent 2GY phase, while a blue luminescent 2BY phase was formed in the presence of ethanol vapours (Fig. 4i and ESI, Table S3†). As indicated by the PXRD data (see ESI, Fig. S19†), the solvent vapours induce the amorphous-to-crystalline (ATC) phase transition and this leads to vapoluminescent responses. The emission maxima of these 2GY and 2BY phases is at 501.5 and 487 nm, respectively (Fig. 4ii, Table S3†). These luminescence switchings induced by mechanical and subsequent chemical stimuli occurred as a consequence of dynamic CTA and ATC phase transitions.
Having identified the mechanochromic and vapochromic luminescence of the polymorphs of 2, we also explored their thermochromic luminescence properties. The bluish green emission of 2G faded upon thermal treatment, while reversible luminescence alterations of 2B was observed upon heating/cooling cycles. As shown in Fig. 5, a yellow emission appeared when 2B was heated to 300 °C, which reverted back to an intense blue emission upon cooling back to room temperature. A movie taken under 365 nm UV light illumination shows the reversible switching between blue and yellow emission during heating and cooling cycles (Movie S1†). Thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), and coupled mass spectrometry (MS) were carried out simultaneously on 2B (see the ESI, Table S4†). The results showed that 2B is thermally stable and no decomposition occurred during heating to 350 °C. The sharp DTG (derivative thermogravimetry) peak was related to the removal of the dichloromethane solvate molecules (Fig. S21†). The DSC curve showed that 2B melted at 305.9 °C and this solid-to-liquid phase transformation begins at 289.6 °C and ends at 315.6 °C (Fig. S21†). This indicates that at least a part of 2B converted into an amorphous phase (2Yt) upon heating to 300 °C during the thermochromic luminescence experiment, and the observed yellow luminescence originated from the as-resulting amorphous phase. The 2Bt phase obtained upon cooling back to room temperature also has some amorphous character as evidenced by the presence of broadened peaks in the PXRD patterns (Fig. S19†) and in agreement with the results of the emission spectroscopy data. As shown in Fig. 4ii, there are two well-resolved emission bands with 480 and 600 nm maxima in the emission spectrum of this new 2Bt phase (Table S3†). The blue emitting band (at around 480 nm) could be described with an 8 nm blue shift of the emission spectrum of 2B. However, the other band is much more similar than that of the emission of amorphous 2Y. It is observable on the time-resolved transients that the blue emitting state excites the red emitting one (at least partly), indicating by that there is a quick (1 μs) decay in the blue band while in the other transient, a rise of fluorescence intensity may be observed with almost the same time parameter (Fig. S26†). Furthermore, very similar excitation spectra were measured at the 460 and 630 nm wavelengths (Fig. S26†). Almost all crystalline and amorphous phases of 2 show two or three exponential luminescence decay character (Table S3†), which used to be explained by the local disorder in the molecular arrangement due to the presence of residual solvent or multiple molecular conformations in the solid state.69–71 Additional experiments aimed to explore the details of this thermally-induced solid state transformation.
 | ||
| Fig. 5 Reversible thermochromic luminescence of 2B (under 365 nm UV illumination) upon heating/cooling cycles. | ||
Experimental
All chemicals and solvents used for the syntheses were of reagent grade. The solvents for synthesis were used without further purification. All manipulations were performed in air. Mechanochemical synthesis were performed by a Retsch MM400 shaker mill in a 10 mL agate jar with two 10 mm diameter agate balls (weight of each ball m = 1.5 g, mtot = 2 × 1.5 g) operating at 25 Hz.In a general procedure, stoichiometric amounts of (Me2S)AuCl (147 mg, 0.50 mmol) or AuI (162 mg, 0.50 mmol) with bidentate phosphine ligands (Table S1†) were added into a 10 mL agate milling jar with two 10 mm diameter agate balls (weight of each ball m = 1.5 g, mtot = 2 × 1.5 g). The reaction mixture was then ball milled at 25 Hz in the presence of few drops (105–115 μL, η = 0.25 μL mg−1, Table S1†) of dichloromethane for 4 minutes. The crude product of LAG reaction (1–3) can be directly recovered as crystalline material and without any work-up.
High resolution mass spectrometric measurements were performed on a Sciex 5600+ QTof mass spectrometer (Sciex, USA) equipped with DuoSpray ion source operated in electrospray ionization mode. The resolution of the instrument was >30000 over the entire mass range. Samples were dissolved in acetonitrile and flow injected into the mass spectrometer. Analyst TF 1.7. software was used for the instrument control and data processing as well. The commonly accepted criteria level of 5 ppm was used during the high-resolution measurements. 1H and 31P NMR spectra were recorded in CD2Cl2 solution at room temperature, on a Varian Unity Inova 500 spectrometer (500 and 202 MHz for 1H and 31P NMR spectra, respectively), with the residual solvent signal as the lock and TMS as the internal standard. Chemical shifts (δ) and coupling constants (J) are given in ppm and Hz, respectively. The corrected luminescence spectra were measured with a JASCO FP-8300 spectrofluorometer with 2.5 nm resolution using 370 nm light for excitation, the phosphorescence decays with a 25 MHz Hitachi VC-6041 (Z) oscilloscope (with typically 5 nm resolution) exciting with the third harmonic (355 nm) excitation beam of a Nd-YAG laser (Continuum Surelite) with very low light intensity weakened by neutral filters (less than 0.03 mJ per flash). The averaged 50–500 flashes were fitted with 1–2–3 exponentials (OriginPro 2018) with background optimization. Furthermore, for 2G crystals the single exponential decay was checked by a picosecond single photon counting (SPC) equipment (Edinburg Instrument FLSP920 spectrofluorimeter, EPL-378 nm diode laser) producing practically the same result. The absolute emission quantum yields of the solid samples 1 and 2 were determined on a Jobin–Yvon Fluoromax-4 spectrofluorometer equipped with a Ø2′′ integrating sphere (Thorlabs). The powder X-Ray diffraction analysis of 1–3 (Fig. S19†) was performed on the multifunctional powder diffractometer PANalytical Empyrean using Cu anode and Kα radiation source, λ = 1.540598 Å for Kα1, accelerating voltage 45 kV, beam current 40 mA, diffraction angle 2θ in the range from 5° to 35° with a step scan of 0.04°. Collected data were evaluated using the PANalytical X'Pert HighScore Plus software and ICSD 2012, COD 2011 and PDF2 databases. Powder diffractograms (Fig. S11, S12 and S17†) were produced with Cu–Kα radiation on a vertical high-angle Philips PW1050 powder diffractometer. Single crystals of 2B and 2G were mounted in Paratone-N oil within a conventional cryo-loop, and intensity data were collected on a Rigaku R-AXIS RAPID image plate diffractometer (λ(Mo-Kα radiation) = 0.71075 Å, fitted with an X-stream low temperature attachment (see the ESI, Table S2†). Optical micrographs were recorded with a Linkam Imaging Station microscope equipped with Nikon DS-Fi1c digital camera and 365 nm hand-held UV lamp. Linkam DSC600 stage has been used to study the reversible thermochromic luminescence of 2B. Thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), and coupled mass spectrometry (MS) were carried out simultaneously (TGA-DSC-MS) in a Netzsch STA 409c/CD apparatus. Analyses were performed in an argon atmosphere using Al2O3 crucibles. Simultaneous thermal analysis (STA) of the 2B was performed according to the program: first heating from 25 °C to 350 °C at a rate of 5 °C min−1, cooling from 350 °C to 80 °C at a rate of 10 °C min−1 and second heating from 80 °C to 450 °C at a rate of 5 °C min−1 (Fig. S21†).
Conclusions
In summary, we have demonstrated the power of mechanochemistry in the simple and rapid synthesis of highly luminescent mononuclear, three-coordinate Au(diphos)X complexes. To the best of our knowledge, this is the first report on the mechanochemical synthesis of gold(I) halide complexes of diphosphines ligands, wherein the systematic variation in the nature of diphosphine ligands and halide ions coupled with the phase change properties allows luminescences colour tuning from blue to green and to yellow. In the case of gold(I) complexes 1 and 2, solution-based procedures and mechanochemical syntheses were comparable in terms of yields.15,59 However, mechanochemistry outperformed in comparison to previously reported solution-based procedures15,59 as the reaction times were significantly shorter (only four minutes instead of hours), the method is practically solvent-free, the experimental set-up is simple (no need of dry solvents or washing/precipitation operations during the work-up), the reactions proceeding with an overall reduced ecological footprint. Scheme 2 summarizes the stimuli-induced transformations among different forms of 2 that we have observed. Thus, it was also possible to discover two different solvates of 2 with distinct blue and bluish green luminescence that also displayed stimuli-responsive luminescent behaviour. Upon mechanical grinding both polymorphs alter their luminescence into yellow, which converted back into original emission colours in the presence of different solvent vapours. A reversible blue-to-yellow emission change of 2B was observed during consecutive heating and cooling cycles over a temperature range of 25–300 °C. Such multistimuli-responsive materials, which show luminescent colour changes in response to mechanical grinding, in the presence of volatile chemical compounds or temperature alteration are of great promise owing to their potential applications in sensing-, switching- and optical-recording systems. | ||
| Scheme 2 Stimuli-induced transformations of 2 forms. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This article is based upon work from COST Action CA18112,72,73 supported by COST (European Cooperation in Science and Technology).‡§ The authors are indebted to Dr Tamás Holczbauer, Dr Nóra May and Laura Nagyné Dr Bereczki for single crystal X-ray diffraction (Research Centre for Natural Sciences, Eötvös Loránd Research Network, Budapest, Hungary) and Mariano Casas Luna (Central European Institute of Technology, Brno University of Technology, Brno, Czech Republic) for the PXRD measurements.Notes and references
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- E. Y. Hong and V. W.-W. Yam, ACS Appl. Mater. Interfaces, 2017, 9, 2616–2624 CrossRef CAS PubMed.
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. Rivett, S. Jones, T. H. Thomas, M. Abdi Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, Science, 2017, 356, 159–163 CrossRef CAS PubMed.
- C. Y. Liu, H. F. Wang, Z. G. Ren, P. Braunstein and J. P. Lang, Inorg. Chem., 2019, 58, 8533–8540 CrossRef CAS PubMed.
- J. X. Chen, W. H. Zhang, X. Y. Tang, Z. G. Ren, H. X. Li, Y. Zhang and J. P. Lang, Inorg. Chem., 2006, 45, 7671–7680 CrossRef CAS PubMed.
- T. Zou, C. T. Lum, C. N. Lok, J. J. Zhang and C. M. Che, Chem. Soc. Rev., 2015, 44, 8786–8801 RSC.
- C. Y. Liu, X. R. Wei, Y. Chen, H. F. Wang, J. F. Ge, Y. J. Xu, Z. G. Ren, P. Braunstein and J. P. Lang, Inorg. Chem., 2019, 58, 3690–3697 CrossRef CAS PubMed.
- R. Ciriminna, E. Falletta, C. Della Pina, J. H. Teles and M. Pagliaro, Angew. Chem., Int. Ed., 2016, 55, 14210–14217 CrossRef CAS PubMed.
- R. Jin, Y. Zhu and H. Qian, Chem. – Eur. J., 2011, 17, 6584–6593 CrossRef CAS PubMed.
- A. V. Paderina, I. O. Koshevoy and E. V. Grachova, Dalton Trans., 2021, 50, 6003–6033 RSC.
- A. Pintado-Alba, H. de la Riva, M. Nieuwhuyzen, D. Bautista, P. R. Raithby, H. A. Sparkes, S. J. Teat, J. M. Lopez-de-Luzuriaga and M. C. Lagunas, Dalton Trans., 2004, 3459–3467 RSC.
- M. Osawa, M. A. Aino, T. Nagakura, M. Hoshino, Y. Tanaka and M. Akita, Dalton Trans., 2018, 47, 8229–8239 RSC.
- C. Jobbágy, P. Baranyai, G. Marsi, B. Rácz, L. Li, P. Naumov and A. Deák, J. Mater. Chem. C, 2016, 4, 10253–10264 RSC.
- P. Baranyai, G. Marsi, C. Jobbágy, A. Domján, L. Oláh and A. Deák, Dalton Trans., 2015, 44, 13455–13459 RSC.
- H. Ito, K. Takagi, T. Miyahara and M. Sawamura, Org. Lett., 2005, 7, 3001–3004 CrossRef CAS PubMed.
- H. Ito, T. Saito, T. Miyahara, C. Zhong and M. Sawamura, Organometallics, 2009, 28, 4829–4840 CrossRef CAS.
- A. X. Zheng, Z. G. Ren, L. L. Li, H. Shang, H. X. Li and J. P. Lang, Dalton Trans., 2011, 40, 589–596 RSC.
- A. Deák, C. Jobbágy, G. Marsi, M. Molnár, Z. Szakács and P. Baranyai, Chem. – Eur. J., 2015, 21, 11495–11508 CrossRef PubMed.
- A. Deák, in Nanomaterials Design for Sensing Applications, ed. O. V. Zenkina, Elsevier Inc., 2019, ch. 9, pp. 281–324 Search PubMed.
- C. Jobbágy and A. Deák, Eur. J. Inorg. Chem., 2014, 4434–4449 CrossRef.
- N. Mirzadeh, T. S. Reddy and S. K. Bhargava, Coord. Chem. Rev., 2019, 388, 343–359 CrossRef CAS.
- V. W.-W. Yam, C.-L. Chan, S. W.-K. Choi, K. M.-C. Wong, E. C.-C. Cheng, S.-C. Yu, P.-K. Ng, W.-K. Chan and K.-K. Cheung, Chem. Commun., 2000, 53–54 Search PubMed.
- Y. Ma, X. Zhou, J. Shen, H.-Y. Chao and C.-M. Che, Appl. Phys. Lett., 1999, 74, 1361–1363 CrossRef CAS.
- Y. Ma, C.-M. Che, H.-Y. Chao, X. Zhou, W.-H. Chan and J. Shen, Adv. Mater., 1999, 11, 852–857 CrossRef CAS.
- H. E. Abdou, A. A. Mohamed, J. P. Fackler, A. Burini, R. Galassi, J. M. López-de-Luzuriaga and M. E. Olmos, Coord. Chem. Rev., 2009, 253, 1661–1669 CrossRef CAS.
- A. A. Karasik, E. I. Musina, I. D. Strelnik, I. R. Dayanova, J. G. Elistratova, A. R. Mustafina and O. G. Sinyashin, Pure Appl. Chem., 2019, 91, 839–849 CrossRef CAS.
- B. Dominelli, J. D. G. Correia and F. E. Kühn, J. Organomet. Chem., 2018, 866, 153–164 CrossRef CAS.
- E. E. Langdon-Jones and S. J. Pope, Chem. Commun., 2014, 50, 10343–10354 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., 2020, 59, 1018–1029 CrossRef PubMed.
- D. Tan and F. Garcia, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC.
- A. Porcheddu, E. Colacino, L. De Luca and F. Delogu, ACS Catal., 2020, 10, 8344–8394 CrossRef CAS.
- D. Braga, L. Maini and F. Grepioni, Chem. Soc. Rev., 2013, 42, 7638–7648 RSC.
- J. X. Chen, Q. F. Xu, Y. Xu, Y. Zhang, Z. N. Chen and J. P. Lang, Eur. J. Inorg. Chem., 2004, 2004, 4247–4252 CrossRef.
- H. Yu, Q. F. Xu, Z. R. Sun, S. J. Ji, J. X. Chen, Q. Liu, J. P. Lang and K. Tatsumi, Chem. Commun., 2001, 2614–2615 RSC.
- J. P. Lang and K. Tatsumi, Inorg. Chem., 1998, 37, 6308–6316 CrossRef CAS.
- A. Lazuen-Garay, A. Pichon and S. L. James, Chem. Soc. Rev., 2007, 36, 846–855 RSC.
- V. Cabras, M. Pilloni, A. Scano, R. Lai, M. C. Aragoni, S. J. Coles and G. Ennas, Eur. J. Inorg. Chem., 2017, 1908–1914 CrossRef CAS.
- F. Wang, C. Y. Ni, Q. Liu, F. L. Li, J. Shi, H. X. Li and J. P. Lang, Chem. Commun., 2013, 49, 9248–9250 RSC.
- D. Hasa and W. Jones, Adv. Drug Delivery Rev., 2017, 117, 147–161 CrossRef CAS PubMed.
- D. Tan, L. Loots and T. Friščić, Chem. Commun., 2016, 52, 7760–7781 RSC.
- S. Quaresma, V. André, A. Fernandes and M. T. Duarte, Inorg. Chim. Acta, 2017, 455, 309–318 CrossRef CAS.
- M. Pérez-Venegas and E. Juaristi, ACS Sustainable Chem. Eng., 2020, 8, 8881–8893 CrossRef.
- E. Colacino, A. Porcheddu, C. Charnay and F. Delogu, React. Chem. Eng., 2019, 4, 1179–1188 RSC.
- A. S. McCalmont, A. Ruiz, M. C. Lagunas, W. T. Al-Jamal and D. E. Crawford, ACS Sustainable Chem. Eng., 2020, 8, 15243–15249 CrossRef CAS.
- A. Beillard, X. Bantreil, T. X. Metro, J. Martinez and F. Lamaty, Chem. Rev., 2019, 119, 7529–7609 CrossRef CAS PubMed.
- N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352–2362 RSC.
- C. Jobbágy, T. Tunyogi, G. Pálinkas and A. Deák, Inorg. Chem., 2011, 50, 7301–7308 CrossRef PubMed.
- T. Friščić, Chem. Soc. Rev., 2012, 41, 3493–3510 RSC.
- E. Colacino, V. Isoni, D. Crawford and F. Garcia, Trends Chem., 2021, 3, 335–339 CrossRef CAS.
- D. E. Crawford, A. Porcheddu, A. S. McCalmont, F. Delogu, S. L. James and E. Colacino, ACS Sustainable Chem. Eng., 2020, 8, 12230–12238 CrossRef CAS.
- D. E. Crawford, S. L. James and T. McNally, ACS Sustainable Chem. Eng., 2017, 6, 193–201 CrossRef.
- F. Gomollón-Bel, Chem. Int., 2019, 41, 12–17 Search PubMed.
- M. C. Gimeno and A. Laguna, Chem. Rev., 1997, 97, 511–522 CrossRef CAS PubMed.
- M. C. Gimeno, in Modern Supramolecular Gold Chemistry, ed. A. Laguna, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, pp. 1–63 Search PubMed.
- J. L. Do, D. Tan and T. Friščić, Angew. Chem., Int. Ed., 2018, 57, 2667–2671 CrossRef CAS PubMed.
- F. J. L. Ingner, Z. X. Giustra, S. Novosedlik, A. Orthaber, P. J. Gates, C. Dyrager and L. T. Pilarski, Green Chem., 2020, 22, 5648–5655 RSC.
- C. Jobbágy, M. Molnár, P. Baranyai and A. Deák, Dalton Trans., 2014, 43, 11807–11810 RSC.
- N. Glebko, T. M. Dau, A. S. Melnikov, E. V. Grachova, I. V. Solovyev, A. Belyaev, A. J. Karttunen and I. O. Koshevoy, Chem. – Eur. J., 2018, 24, 3021–3029 CrossRef CAS PubMed.
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418–426 RSC.
- M. Hoshino, H. Uekusa, S. Sonoda, T. Otsuka and Y. Kaizu, Dalton Trans., 2009, 3085–3091 RSC.
- M. Hoshino, H. Uekusa, S. Ishii, T. Otsuka, Y. Kaizu, Y. Ozawa and K. Toriumi, Inorg. Chem., 2010, 49, 7257–7265 CrossRef CAS PubMed.
- T. Seki, S. Kurenuma and H. Ito, Chem. – Eur. J., 2013, 19, 16214–16220 CrossRef CAS PubMed.
- T. Seki, T. Ozaki, T. Okura, K. Asakura, A. Sakon, H. Uekusa and H. Ito, Chem. Sci., 2015, 6, 2187–2195 RSC.
- M. A. Malwitz, S. H. Lim, R. L. White-Morris, D. M. Pham, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2012, 134, 10885–10893 CrossRef CAS PubMed.
- S. H. Lim, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2011, 133, 10229–10238 CrossRef CAS PubMed.
- S. H. Lim, M. M. Olmstead and A. L. Balch, Chem. Sci., 2013, 4, 311–318 RSC.
- O. S. Wenger, Chem. Rev., 2013, 113, 3686–3733 CrossRef CAS PubMed.
- M. T. Dau, J. R. Shakirova, A. J. Karttunen, E. V. Grachova, S. P. Tunik, A. S. Melnikov, T. A. Pakkanen and I. O. Koshevoy, Inorg. Chem., 2014, 53, 4705–4715 CrossRef CAS PubMed.
- A. Demeter and K. A. Zachariasse, Chem. Phys. Lett., 2003, 380, 699–703 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 3rd edn, 2006 Search PubMed.
- M. Baláž, L. Vella-Zarb, J. G. Hernández, I. Halasz, D. E. Crawford, M. Krupička, V. André, A. Niidu, F. García, L. Maini and E. Colacino, Chim. Oggi – Chem. Today, 2019, 37, 32–34 Search PubMed.
- J. G. Hernández, I. Halasz, D. E. Crawford, M. Krupicka, M. Baláž, V. André, L. Vella-Zarb, A. Niidu, F. García, L. Maini and E. Colacino, Eur. J. Org. Chem., 2020, 8–9 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental section, HR-MS, 1H and 31P NMR spectra, PXRD patterns, solid state photophysical data, STA analysis. Crystal data, details of data collections and refinements. CCDC 2074756 (2B) and 2074757 (2G). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt01751a |
| ‡ For more information on COST Action CA18112 ‘Mechanochemistry for Sustainable Industry’ (MechSustInd): http://www.mechsustind.eu/. |
| § For more information on COST Association: http://www.cost.eu/. |
| This journal is © The Royal Society of Chemistry 2021 |