
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Colloidal synthesis of metal chalcogenide nanomaterials from metal–organic precursors and capping ligand effect on electrocatalytic performance: progress, challenges and future perspectives
Malik Dilshad
Khan
 *ab,
Marcin
Opallo
*a and
Neerish
Revaprasadu
*b
*ab,
Marcin
Opallo
*a and
Neerish
Revaprasadu
*b
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224, Warsaw, Poland
bDepartment of Chemistry, University of Zululand, Private Bag X1001, Kwa-Dlangezwa 3880, South Africa. E-mail: RevaprasaduN@unizulu.ac.za
First published on 2nd August 2021
Abstract
Renewable and sustainable functional nanomaterials, which can be employed in alternative green energy sources, are highly desirable. Transition metal chalcogenides are potential catalysts for processes resulting in energy generation and storage. In order to optimize their catalytic performance, high phase purity and precise control over shape and size are indispensable. Metal–organic precursors with pre-formed bonds between the metal and the chalcogenide atoms are advantageous in synthesizing phase pure transition metal chalcogenides with controlled shape and sizes. This can be achieved by the decomposition of metal–organic precursors in the presence of suitable surfactants/capping agents. However, the recent studies on electrocatalysis at the nanoscale level reveal that the capping agents attached to their surface have a detrimental effect on their efficiency. The removal of surfactants from active sites to obtain bare surface nanoparticles is necessary to enhance catalytic activity. Herein, we have discussed the properties of different metal–organic precursors and the role of surfactants in the colloidal synthesis of metal chalcogenide nanomaterials. Moreover, the effect of surfactants on their electrocatalytic performance, the commonly used strategies for removing surfactants from the surface of nanomaterials and the future perspectives are reviewed.
Introduction
The rise in population and advancement in technology has resulted in an exponential increase in energy demand. Conventional fossil fuels are non-sustainable and are hazardous for the environment. Therefore sustainable and renewable materials with a lower carbon footprint are highly desirable. The use of hydrogen as a fuel is beneficial as it can be generated from a widely available source i.e. water, and has a low environmental impact. However, water is thermodynamically stable, and current methods of generation of hydrogen are not really green. Therefore efficient, cost-effective, abundant and stable catalytic materials for water splitting are desirable. So far, the maximum efficiency and stability have been shown by precious metals, but their cost and scarcity are a major hurdle in their commercialization. Apart from precious metals, metal chalcogenides have shown some promising catalytic performances. The variable oxidation states can easily promote oxidation/reduction reactions and (electro)catalytic activity. Moreover, they have the potential to be used at a large scale for industrial applications.The principal factors controlling the catalytic activity of the metal chalcogenides are phase purity, size, morphology and the ligands on the surface of the nanocrystals. Over time, synthetic approaches have been well developed to prepare phase pure and morphologically controlled metal chalcogenide nanomaterials. The synthetic routes vary from solution-based synthesis to solvent-free routes. The numerous synthetic techniques include seed-mediated growth,1 hydrothermal,2 microemulsion,3 sol–gel,4 co-precipitation,5 solution combustion synthesis,6 solid state synthesis,7 molten salt synthesis,8 solvent-less pyrolysis,9 heat-up and hot injection routes.10,11 Heat-up and hot injection, though quite different in nature, are the most commonly used methods to obtain uniform monodispersed nanoparticles (Fig. 1). In the hot injection method, the stock solution of a precursor is rapidly injected into the pre-heated surfactants. The elevated temperature facilitates an extremely fast reaction, that results in high supersaturation and formation of nanocrystals. The hot-injection method is particularly effective, because it offers a high level of control over the size of the particles and size distribution by allowing a rapid nucleation separated from the growth stage. The size and shape of the nanoparticles can be controlled by varying the temperature, concentration, reaction time and surfactants. Unlike hot injection method in heat-up method, the reactants are added in the surfactant at room temperature. Then reaction mixture is gradually heated to the desired temperature where formation of nanocrystals take place. The heat up method is a facile approach, as nanocrystals are prepared in a single pot without hot injection step. Reagents/precursors with suitable reactivity are required to achieve better monodispersity. Controlling the reactivity of the precursors allow homogenous nucleation, after reaching a certain temperature, and leads to the formation of monodisperse nanoparticles. Both heat-up and hot injection methods have been successfully employed to prepare wide range of monodisperse metal chalcogenide nanomaterials.
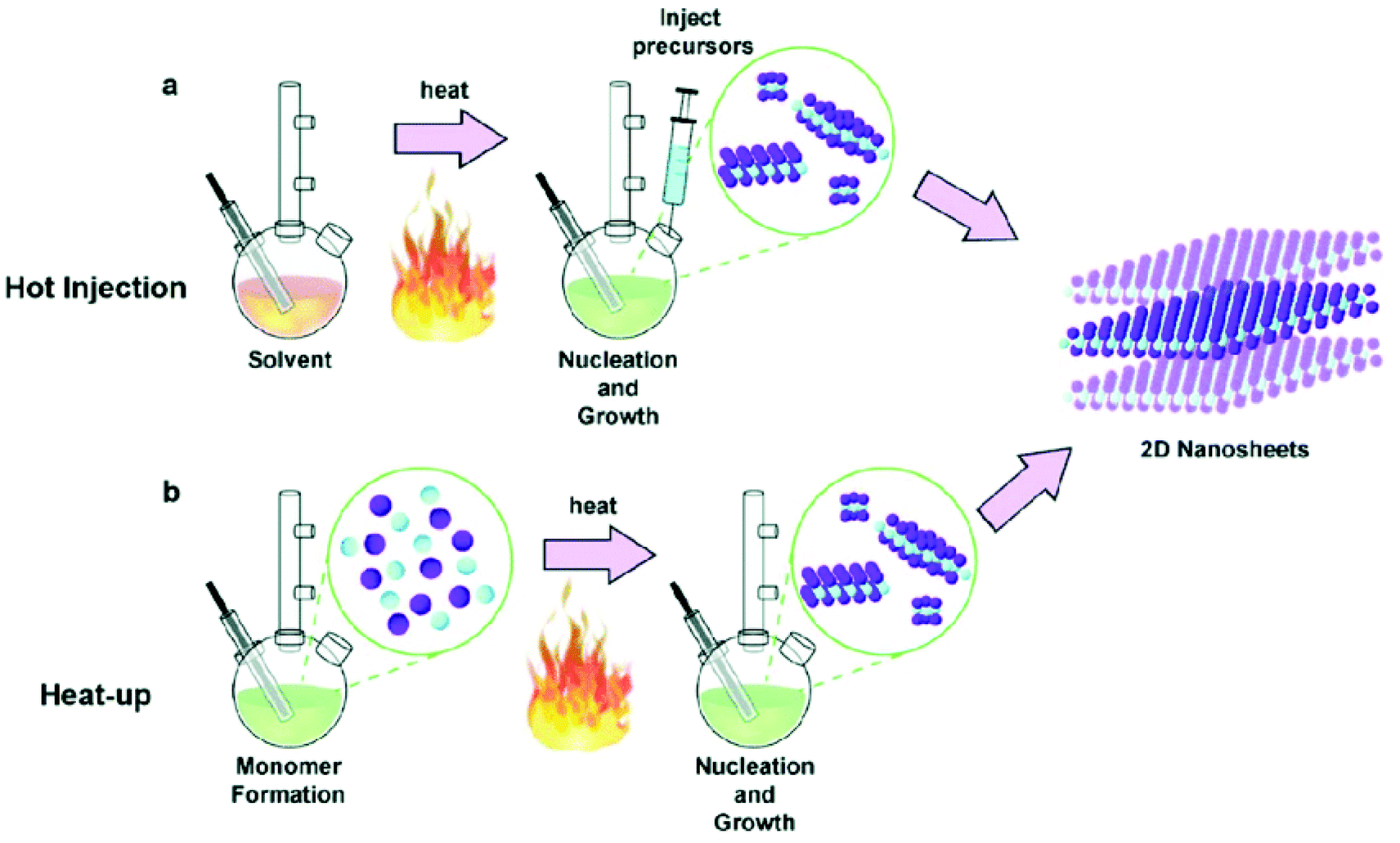 | ||
| Fig. 1 Schematic of solution-based synthesis of nanomaterials by (a) hot injection and (b) heat-up methods. Reproduced from ref. 19, Copyright 2020 with permission from IOP Publishing Ltd. | ||
These developments in colloidal synthesis provide more flexibility in performing reactions under diverse reaction conditions.12 Besides synthetic strategies, other parameters such as temperature, capping agents, solvents and reducing agents play an important role in optimizing the composition and dimensions of the nanomaterials.13,14 Likewise, various studies have explained in detail the role and mechanism of surfactants in controlling the size and morphology of the nanomaterials.15–18
In colloidal synthesis of nanomaterials with required compositions, a variety of reagents can be used as starting materials. Besides multiple source routes,20–22 other starting materials, such as open frameworks,23,24 metal–organic frameworks25,26 and metal–organic precursors27 can be employed for the preparation of desired nanomaterials. Among various available materials, metal–organic precursors stand out as unique and advantageous precursors for preparing metal sulfide/selenide nanomaterials. Metal–organic precursors (also referred to as molecular precursors or single-source precursors) contain pre-formed bonds between metal and the chalcogenide atoms, which provides better control over composition. They are relatively more versatile, as they are not only suitable for the synthesis of nanomaterials by different routes, but they can also be used successfully for the deposition of thin films by various techniques, such as chemical bath deposition, spray pyrolysis, spin coating and aerosol-assisted chemical vapor deposition. They can easily replace moisture-sensitive, hazardous and pyrophoric reagents, such as metal alkyls or phosphines. More importantly, some metal complexes can be pyrolyzed easily at relatively lower temperatures to form metal sulfide/selenide nanomaterials in absence of surfactants. In this way, the activity of a such nanomaterials can be determined without any intervention from the surfactants.
In this Frontier article, we discuss the suitability of the precursors, based on the nature of their backbone, for the synthesis of desired metal chalcogenide nanomaterials. Likewise, instead of the role of surfactants on shape and size, this frontier focuses on the surfactants taking part in the reaction and altering the decomposition path and/or final product. Moreover, the effect of surfactants on electrochemical properties and strategies for their removal from the surface of nanoparticles are discussed.
Selecting a suitable precursor
Among various ligands, only a few simple sulfur-based ones, such as potassium ethyl xanthate, sodium diethyldithiocarbamate, thiobenzoic acid, simple thioethers and thiols, are commercially available. However, these ligands can be synthesized and the reactivity of a ligand can be tuned by the introduction of different substituents (i.e. alkyl or aryl groups) or designing suitable ligand backbone (i.e. symmetrical or unsymmetrical) (Fig. 2), to prepare desired metal chalcogenides in the form of nanomaterials or thin films. Each precursor has its own merits and/or limitations, making the selection of a suitable precursor challenging. Here, we will discuss some aspects of the molecular precursors that may assist in selecting suitable precursors.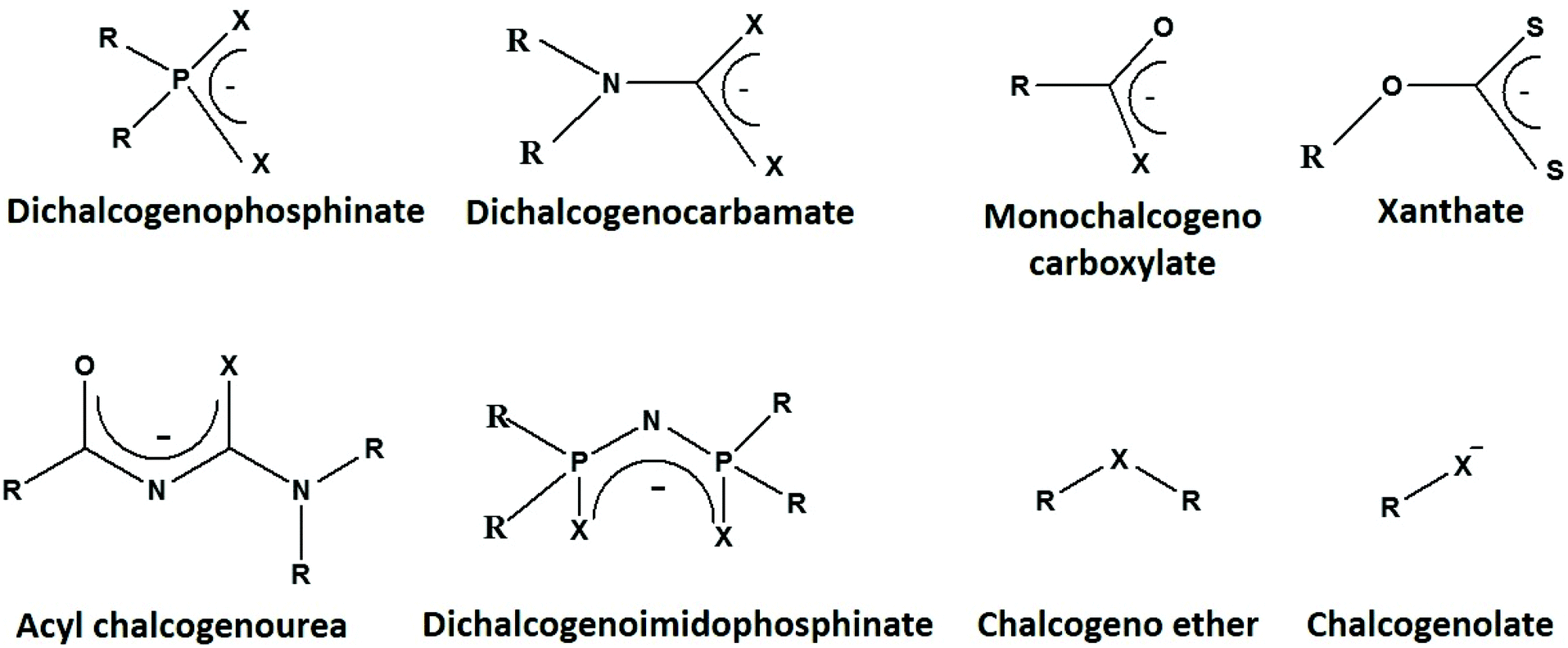 | ||
| Fig. 2 Molecular structures of the ligands (where R = alkyl or aryl and X = S or Se), used for the synthesis of metal–organic precursors. The metal–organic precursors act as single-source precursors for the preparation of metal chalcogenide nanomaterials or thin films. | ||
The first and foremost requirement is the purity of the metal–organic complex, i.e., the precursor can be easily separated/recrystallized from the impurities or by-products. If the precursor is not adequately purified, it may produce metal chalcogenide nanomaterials with undesirable contamination or affect the reproducibility of the reaction. Moreover, the decomposition of the pure metal–organic precursor should lead to the formation of only phase pure metal chalcogenides. The decomposed by-products should not interfere with the purity of the synthesized nanomaterials. For instance, phosphorus-containing precursors may cause phosphorus contamination, depending on the synthetic route. It is known that the deposition of cadmium and zinc chalcogenide thin films from [M((EPiPr2)2N)2] (M = Cd, Zn and E = S, Se) precursors by low-pressure metal–organic chemical vapor deposition LP-MOCVD, resulted in contamination of the films by phosphorus.28 Likewise, decomposition of [Pb((C6H5)2PSSe)2] to deposit Pb chalcogenide films by aerosol-assisted chemical vapor deposition (AACVD) yielded PbSe films contaminated with phosphorus.29 In this respect, the colloidal synthesis of metal chalcogenide nanomaterials is highly advantageous because it yields products with high purity. For example, the decomposition of the Cd[N(SePiPr2)2]2 complex in trioctylphosphineoxide (TOPO), via the hot injection method, yields CdSe quantum dots.30 In contrast, AACVD of the same precursor produces cadmium selenide films contaminated with phosphorus.31 Similarly, molecular precursors with the aromatic or bulky alkyl group result in carbon contamination of the metal chalcogenide films. For the deposition of films by vapour deposition routes, volatility of the precursor or good solubility in a suitable organic solvent is also required.
Toxicity is another primary concern while designing and synthesizing metal–organic precursors. Care should be taken to avoid the use of toxic and pyrophoric reagents where ever possible, and/or in cases where the use of such materials is inevitable, reagents with relatively lower toxicity should be used. Although mercuric compounds are highly toxic, mercuric chalcogenides are indispensable for selected applications. For instance, they are among the few compounds with absorption tunability up to the tetrahertz range.32 Moreover, they are widely used in infra-red (IR) and photovoltaic devices.33,34 Therefore, despite their toxicity, shape and size controlled synthesis of these materials is much required to employ their tunable properties.35,36 So far, due to toxicity concerns, only a few metal–organic precursors of mercury are explored. Diselenoimidodiphosphinate complex of organomercury (where R = methyl, ethyl, thienyl, 2-selenyl and phenyl) derivatives were reported by O'Brien and co-workers for the synthesis of HgSe nanomaterials by in vacuo solid-state decomposition of the complexes at 300 and 350 °C.37 However, the extreme toxicity associated with dimethyl and diethyl mercury rendered only the 2-selenyl and phenyl derivatives effective for the synthesis of mercury-based chalcogenides.
The synthetic conditions also play an important role in determining the applicability of a molecular precursor, and one-step synthesis is preferred. Multi-step reactions are laborious, require more time, and decrease the yield of the end product. By carefully designing and selecting the reactants, the reaction path can be altered significantly. The importance of designing a reaction route was demonstrated in the example of selenobenzoate complexes, which were not well explored, owing to the difficulty in complex preparation. The earlier approach required a reaction between an alkali metal (M = Na, K) and Se to prepare M2Se, in ammonia at −70 °C for 12 h. The selenobenzoate salt was obtained by the reaction of M2Se with benzoyl chloride.38,39 In the modified protocol, NaBH4 was used to prepare NaHSe instead of Na2Se. Then in situ addition of benzoyl chloride and the metal (Bi, Sb and Sn) salts produces selenobenzoate complexes.40–43 Protocol modification allows performing the reaction at room temperature (in ethanol) instead of ammonia at −70 °C. Moreover, highly pyrophoric alkali metals were replaced by a relatively mild reagent, i.e., NaBH4, and the reaction time was decreased from 12 hours to one hour. This is a good example of eliminating toxic and pyrophoric reagents and making the synthesis handier and time and cost-effective.
The stability of the precursors helps to handle the precursors at atmospheric conditions, and the materials can be easily stored for later use. The stability can be tailored by the appropriate selection of the precursor backbone. For instance, metal dithiocarbamate complexes are generally more stable than metal xanthate complexes because the former contains a more substantial electron-donating group (–NR2) than the alkoxy (–OR) group in the latter. The lone pair contribution to the pi-electron system of CS2 in dithiocarbamates provides extra stability to the resulting complex. In contrast, xanthate complexes lack the excess electron density on the sulfur atom arising from the pi donation.44 This effect is evidenced by the stretching frequency of the O–CS2 bond (1020–1280 cm−1) being closer to that of the single O–C bond.45 Thermogravimetric analysis also indicates higher stability for dithiocarbamates compared to xanthates.46,47 The stability of the precursors can be exploited to use them under desired reaction conditions. For example, the stability of xanthate precursors can be enhanced by a change of alkyl chain length, from ethyl to octyl, resulting in an increase in decomposition temperature (from 170 °C to 225 °C) and enhanced solubility in organic solvents.48 The decomposition temperature and the mechanistic route of decomposition, influence the shape and phase of the synthesized nanomaterials. Moreover, it should be kept in mind that the nature of the precursor also affects the atom economy (molecular mass of desired product/molecular mass of reactants) of a reaction. Therefore, the bulky groups on the precursor backbone or long alkyl chains may change the solubility and decomposition temperature as desired, but it will also decrease the atom efficiency. The comparative study concluded that the xanthate complexes were more atom efficient than the dithiocarbamate precursors.49 Metal complexes with low decomposition temperatures, which can be prepared easily in relatively few steps and provide products with high atom efficiency are cost effective and have more potential to be used at the industrial level.
Selecting suitable surfactants
In the colloidal synthesis of metal chalcogenide nanomaterials from metal–organic precursors, the capping agents should be chosen carefully (Fig. 3). One has to bear in mind that apart from controlling shape and size, they may take part in the reaction and alter the decomposition path and/or final product. Typical examples of capping agents acting as decomposition initiators are primary amines. Most metal–organic precursors, such as dithiocarbamate, dithiophosphinate, acyl-thiourea, thiobenzoate and xanthate complexes, contain pi-bonds or electrophilic centers. Strongly nucleophilic primary amines immediately attack the carbonyl carbon, which initiates the decomposition of the precursors. A combined experimental and theoretical study explains the decomposition mechanism of nickel dithiocarbamate in the presence of primary amines.50 It was observed that, through the amide reaction, the primary amine initially replaces the secondary amine of the dithiocarbamate complex and then the deprotonation of the primary amine leads to the decomposition of the product to yield nickel sulfide nanoparticles. It was noticed that, by the addition of a primary amine, the decomposition temperature of the precursor (>300 °C, as indicated by TGA) decreased by almost 200 °C down to 120 °C. A similar mechanistic study was also performed for the dithiocarbamate complex of zinc and the decomposition path analyzed by NMR and LC-MS also indicated the active nature of primary amines.51 Similar influence of primary amines have been observed towards xanthate and chalcogeno-carboxylate precursors as well: they decompose even at room temperature by primary amines.40,41,52–56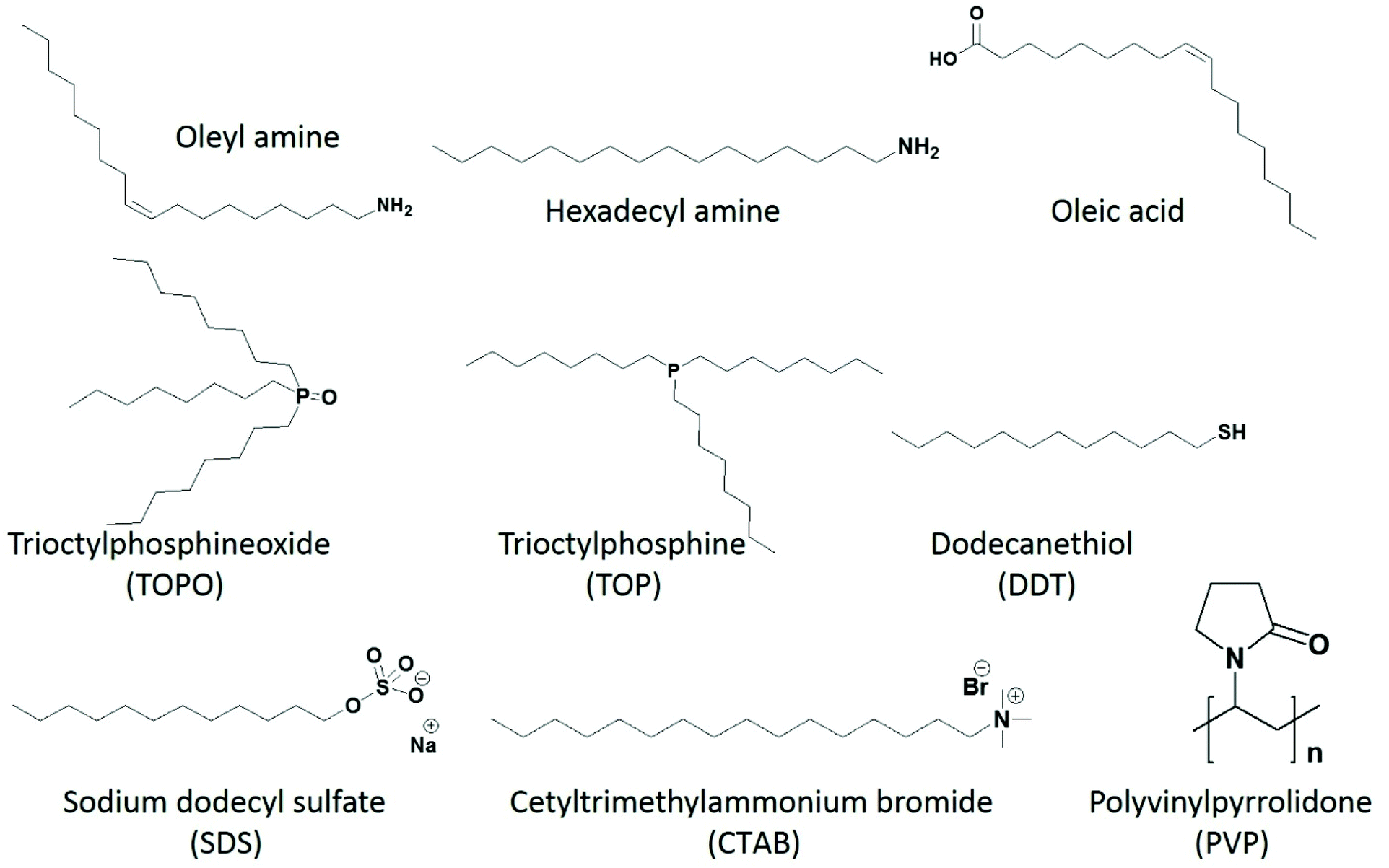 | ||
| Fig. 3 Structures of selected capping agents, commonly used to control the size and morphology of nanoparticles. | ||
Besides catalyzing the decomposition of precursors, the surfactant may act as a reagent as well. It was reported that the decomposition of phosphorus-containing complexes yields either a metal chalcogenide or metal phosphide in the presence of trioctylphosphine (TOP).57–59 Initially, it was thought that intramolecular phosphorus is responsible for the formation of metal phosphide. However, a systematic investigation has concluded that TOP acts as a phosphorus source at elevated temperatures (>300 °C).58 It makes the molecular precursor more versatile and can be selectively used to prepare either metal chalcogenide or phosphide nanomaterials (Fig. 4(a)). Likewise, thiols may also act as either surface capping agents (surface-bound) or as a sulfur source (crystal bound).60 In addition to acting as a phosphorus source, TOP may also act as a reducing agent.61,62 It is interesting to note that TOP can act as a reducing agent in the case of metal–organic precursors as well where metal has pre-formed bonds with the chalcogenide atoms.40 The selenobenzoate complex of bismuth yielded Bi2Se3 nanosheets by AACVD or by the colloidal route in oleylamine and 1-octadecene mixture. However, in the presence of TOP, the decomposition of the complex resulted in the formation of pure bismuth nanocrystals (Fig. 4(b)).
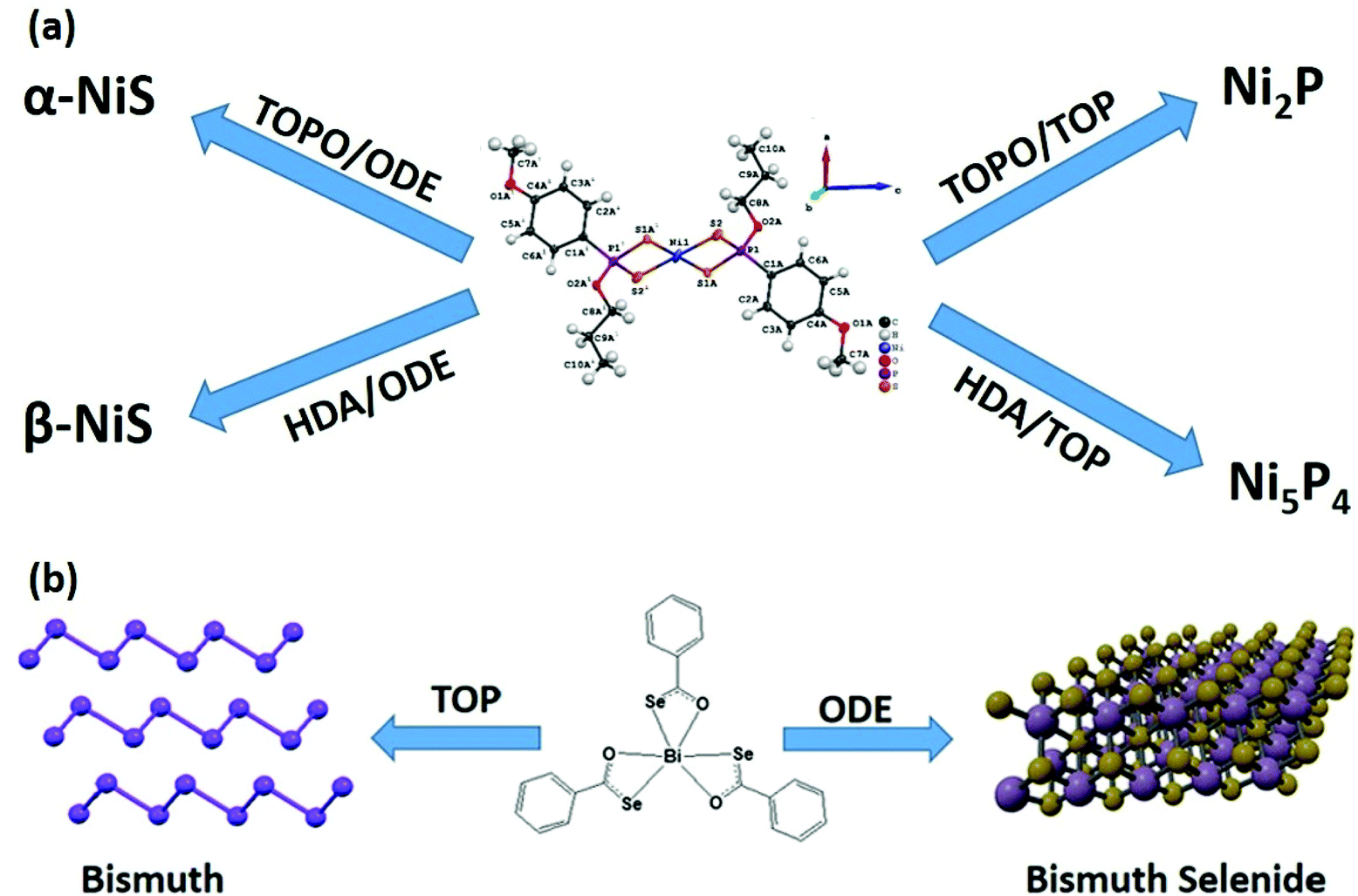 | ||
| Fig. 4 (a) Scheme of phase selective synthesis of nickel sulfide or nickel phosphide, when TOP acts as a phosphorus source.58 (b) Role of TOP as a reducing agent to produce bismuth from metal–organic precursor (reproduced with permission from ref. 40, Copyright 2021, American Chemical Society). | ||
Effect of surfactants on the electrocatalytic activity
Although surfactants are required for better control over size and morphology and to avoid aggregation and agglomeration, they may have a negative impact on the electrocatalytic applications of metal chalcogenides. One has to take into account that the surfactants adhere firmly to the surface of nanocrystals, effectively blocking the active site. Moreover, in the case of conductive nanoobjects, their presence may decrease the probability of electron transport between active sites and electrode support, acting as a tunneling barrier for electrons during the electrode processes. Therefore, it is essential to obtain bare/clean surface nanoparticles to maintain high catalytic efficiency. Investigating the precise effect of surfactants on catalytic activity is highly challenging. This is because, for accurate comparison, the size and morphology of the nanomaterials must not differ significantly. Materials prepared in the absence of surfactants may have the same phase, but control over size and morphology is lost in the absence of surfactants. Likewise, using too harsh conditions for surfactant removal from the surface of already capped nanomaterials may induce size or morphological changes. Therefore, different synthetic techniques with precise control over shape and size and soft methods for removing surfactants without inducing a change in phase, shape or size are highly desirable.Until now, the inhibiting effect of surfactants on catalytic efficiency has been investigated mostly for metallic nanoparticles. Geng et al. prepared surface clean porous Pt nanoparticles via mild seedless method using ascorbic acid as reductant. In electrooxidation of methanol, the surface clean Pt nanoparticles exhibited 2.8 fold greater surface activity and 2.3 fold higher mass activity as compare to commercial Pt/C catalyst. A systematic study on the effect of cationic (cetyltrimethylammonium bromide (CTAB)), anionic (sodium dodecyl sulfate (SDS)) and neutral (polyvinylpyrrolidone (PVP)) surfactants on the electrocatalytic methanol oxidation of porous Pt nanoparticles reveals a notable decrease in catalytic activity and the inhibition effect significantly depends on their nature.63 The inhibition decreases in the order of CTAB > PVP > SDS, and it correlates with the adsorption energy of surfactant and surface environment of the nanoparticles. It may indicate that the use of cationic surfactant is not suitable for catalysts with negatively charged surfaces. The effect of PVP capping was also investigated for ORR at Ag nanorods.64 The PVP capping was removed by dispersing the PVP capped Ag nanorods and 1-hexanethiol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) in ethanol. After 12 hours, a layer of Ag nanorods was floating on the surface of ethanol, suggesting hydrophobic properties of Ag nanorods, acquired by functionalization of 1-hexanethiol. The particles were washed with excess ethanol to remove displaced PVP and excess 1-hexanethiol. The removal of PVP by the ligand exchange process, using 1-hexanethiol, results in a current density increase and a change of ORR mechanism from two-electron to four-electrons. The latter effect may indicate that the ligand environment next to the nanoobject surface may affect the mechanism of the electrode reaction.65,66 Wang and co-workers prepared Pt nanoclusters dispersed on multi-walled carbon nanotubes (MW-CNTs) to avoid agglomeration of Pt nanoclusters without using insulating capping agents.67 CNTs have high electronic conductivity and are resistant to electrochemical oxidation. The composite formation lowered not only the mass loading of Pt, but also multifold enhanced the ORR performance compared to commercial Pt/C catalyst. The Pt nanoparticles prepared in an oleic acid/oleylamine mixture also suffered a decrease in catalytic performance. Therefore the effectiveness of different cleaning processes, such as, washing with acetone, methanol, hexane, and alkaline methanol treatment, was investigated. CO adsorption/oxidation typically used as indicative of catalytic activity was selected to study the effect of these treatments.68 Washing with acetone and methanol were ineffective to remove hydrophobic capping agents, and hexane removed the surfactants to a certain extent, as long alkyl chain acids/amines have relatively better solubility in non-polar solvents. Treating as-synthesized Pt nanoparticles in an alkaline solution of methanol and subsequent washing with acetone/deionized water was more effective. Li et al. also investigated the effectiveness of several cleaning methods (i.e., annealing, washing with acetic acid and UV-ozone treatment) on the performance of oleylamine capped Pt nanoparticles (Fig. 5).69 Their study indicated that for oleylamine capped Pt NPs, simple thermal annealing at 185 °C in the air was more effective than washing with acetic acid and UV-ozone treatment. Importantly, no changes in the size and morphology were observed (Fig. 5(e–g)), therefore, the surfactant was removed without compromising the catalytic activity due to size or morphological changes. However, the technique may be suitable only for limited inert materials that are resistant to surface oxidation.
:3) in ethanol. After 12 hours, a layer of Ag nanorods was floating on the surface of ethanol, suggesting hydrophobic properties of Ag nanorods, acquired by functionalization of 1-hexanethiol. The particles were washed with excess ethanol to remove displaced PVP and excess 1-hexanethiol. The removal of PVP by the ligand exchange process, using 1-hexanethiol, results in a current density increase and a change of ORR mechanism from two-electron to four-electrons. The latter effect may indicate that the ligand environment next to the nanoobject surface may affect the mechanism of the electrode reaction.65,66 Wang and co-workers prepared Pt nanoclusters dispersed on multi-walled carbon nanotubes (MW-CNTs) to avoid agglomeration of Pt nanoclusters without using insulating capping agents.67 CNTs have high electronic conductivity and are resistant to electrochemical oxidation. The composite formation lowered not only the mass loading of Pt, but also multifold enhanced the ORR performance compared to commercial Pt/C catalyst. The Pt nanoparticles prepared in an oleic acid/oleylamine mixture also suffered a decrease in catalytic performance. Therefore the effectiveness of different cleaning processes, such as, washing with acetone, methanol, hexane, and alkaline methanol treatment, was investigated. CO adsorption/oxidation typically used as indicative of catalytic activity was selected to study the effect of these treatments.68 Washing with acetone and methanol were ineffective to remove hydrophobic capping agents, and hexane removed the surfactants to a certain extent, as long alkyl chain acids/amines have relatively better solubility in non-polar solvents. Treating as-synthesized Pt nanoparticles in an alkaline solution of methanol and subsequent washing with acetone/deionized water was more effective. Li et al. also investigated the effectiveness of several cleaning methods (i.e., annealing, washing with acetic acid and UV-ozone treatment) on the performance of oleylamine capped Pt nanoparticles (Fig. 5).69 Their study indicated that for oleylamine capped Pt NPs, simple thermal annealing at 185 °C in the air was more effective than washing with acetic acid and UV-ozone treatment. Importantly, no changes in the size and morphology were observed (Fig. 5(e–g)), therefore, the surfactant was removed without compromising the catalytic activity due to size or morphological changes. However, the technique may be suitable only for limited inert materials that are resistant to surface oxidation.
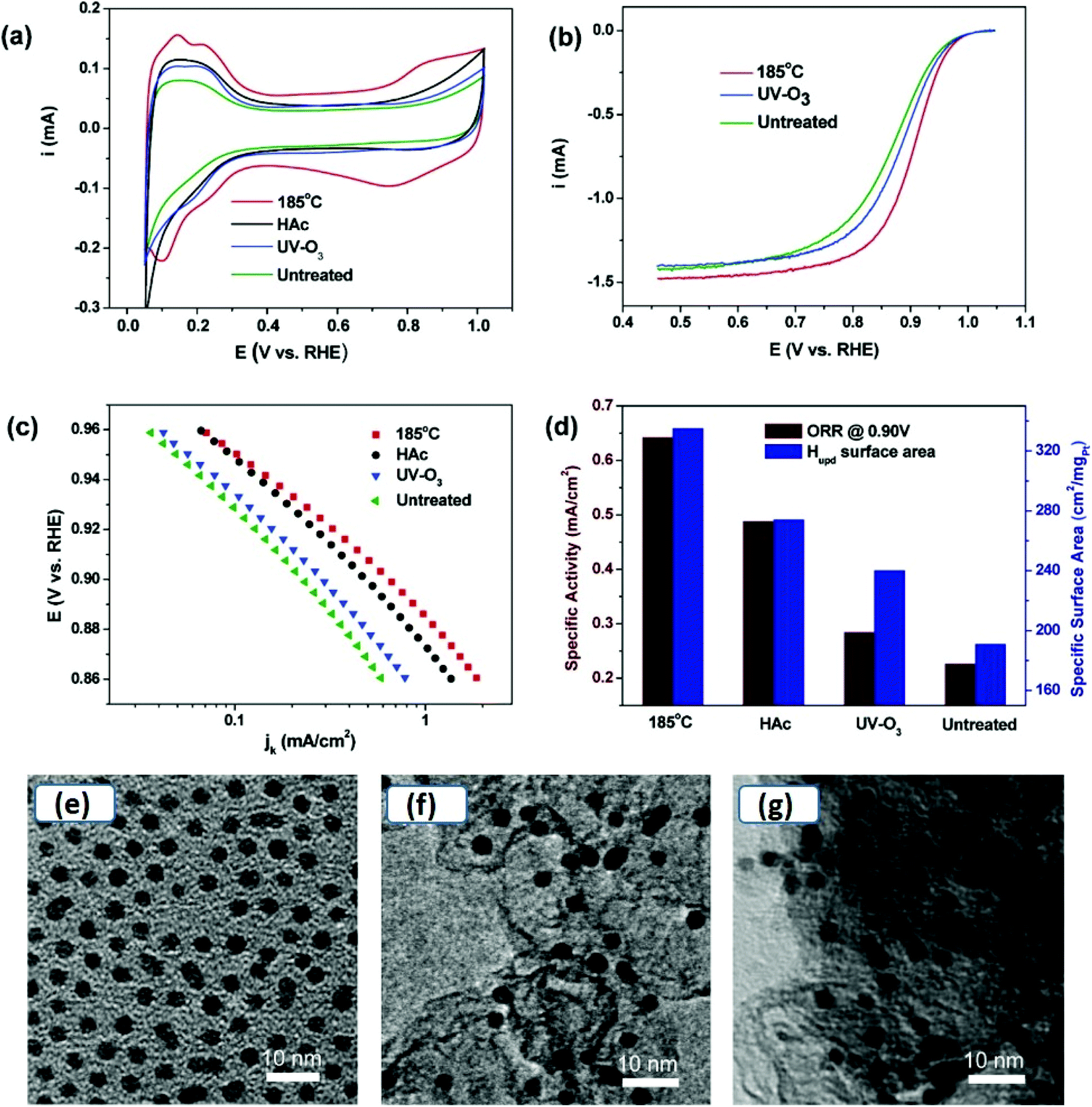 | ||
| Fig. 5 (a) Cyclic voltammograms at a scan rate of 50 mV s−1, (b) ORR polarization curves, (c) Tafel plots obtained from data recorded at rotating disc electrode at 20 mV s−1, 1600 rpm, 20 °C, (d) specific activity at 0.90 V, and specific surface area of annealed, HAc treated, UV-ozone treated and untreated Pt/C catalyst.69 TEM images of (e) as-synthesized Pt NPs, (f) Pt NPs loaded on carbon black, and (g) Pt NPs on carbon black after 185 °C annealing. Reproduced with permission from ref. 69, Copyright 2012, American Chemical Society. | ||
While heating a catalytic material is a simple and useful strategy to strip off surfactants from the surface of the catalyst, it can also alter the surface atomic structure and composition of the crystal facets. Beermann et al. investigated the effect of thermal treatment (at 180 °C, 300 °C and 500 °C), on oxygen reduction reaction activity of Pt–Ni alloyed nanoparticles.70 The alloyed nanoparticles were heated to 180 °C under oxygen, which is then followed by heating in the reduced environment (4% H2/Ar) at 300 °C or 500 °C. They suggested that the thermal treatment in the reductive environment should be carried out carefully to preserve morphology and composition. The correlation between catalytic activity and the annealing temperature shows a 25 times higher ORR activity as compare to commercial Pt/C catalyst at an optimized temperature of 300 °C. At 500 °C, the activity was decreased again as the hydrogen treatment at 500 °C has a destabilizing effect on the shape and the elemental distribution (Fig. 6). Furthermore, the catalytic activity was initially boosted after annealing, but the long-term stability is compromised.
 | ||
| Fig. 6 EDX elemental maps showing the composition of PtNi-raw (a, b, g, h and l), PtNi-300, i.e. annealed at 300 °C, (c, d, i and m), and PtNi-500 i.e. annealed at 500 °C, (e, f, j, k, n and o) in the initial conditions (a–f), after activation (g–k), and after 4k stability test (l–o). If present, also big irregular shaped mostly Ni-rich particles are depicted with their elemental distribution (b and h; d, f, and k; and o). Pt is shown in red and Ni in green.70 Reproduced with permission from ref. 70, Copyright 2017, American Chemical Society. | ||
Liu et al. also reported the removal of oleylamine from the Pt surface by annealing in air at 185 °C and 400 °C under an Ar/H2 environment.71 No oxidation or change in size was observed by annealing in air, whereas particle sintering was observed only in the second case. The carbon-supported as-synthesized Pt nanoparticles exhibited no electrocatalytic activity in H2SO4, as peaks for hydrogen adsorption/desorption were not present. In contrast, the thermal treatment electrochemically activated the Pt nanoparticles and adsorption/desorption of weakly and strongly bound hydrogen was observed. It should also be kept in mind that electrocatalytic activity depends on the environment of annealing (N2, O2 or Ar/H2). For instance, Kramm and co-workers studied the effect of annealing on oleylamine/oleic acid capped IrNi and IrSn catalysts under different environments (N2, O2, Ar/H2) and investigated the catalytic performance of pre- and post-treated bimetallic catalysts for the ethanol oxidation reaction.72 The size of the alloyed nanoparticles was not affected, while heating under N2 and N2/O2, whereas annealing under a reducing environment resulted in an increase in crystallite size (Fig. 7(a and b)). The surface analysis of the catalysts indicates that removal of capping layer was initiated by the N2 annealing step and completed under N2/O2 annealing step. The thermal treatment maximized the performance of IrNi catalyst for ethanol oxidation reaction in the order (Ar/H2 > O2 > N2), but barely affected the IrSn performance. On the other hand, electrochemical cleaning in NaOH was more effective for IrSn catalyst (Fig. 7(c and d)).
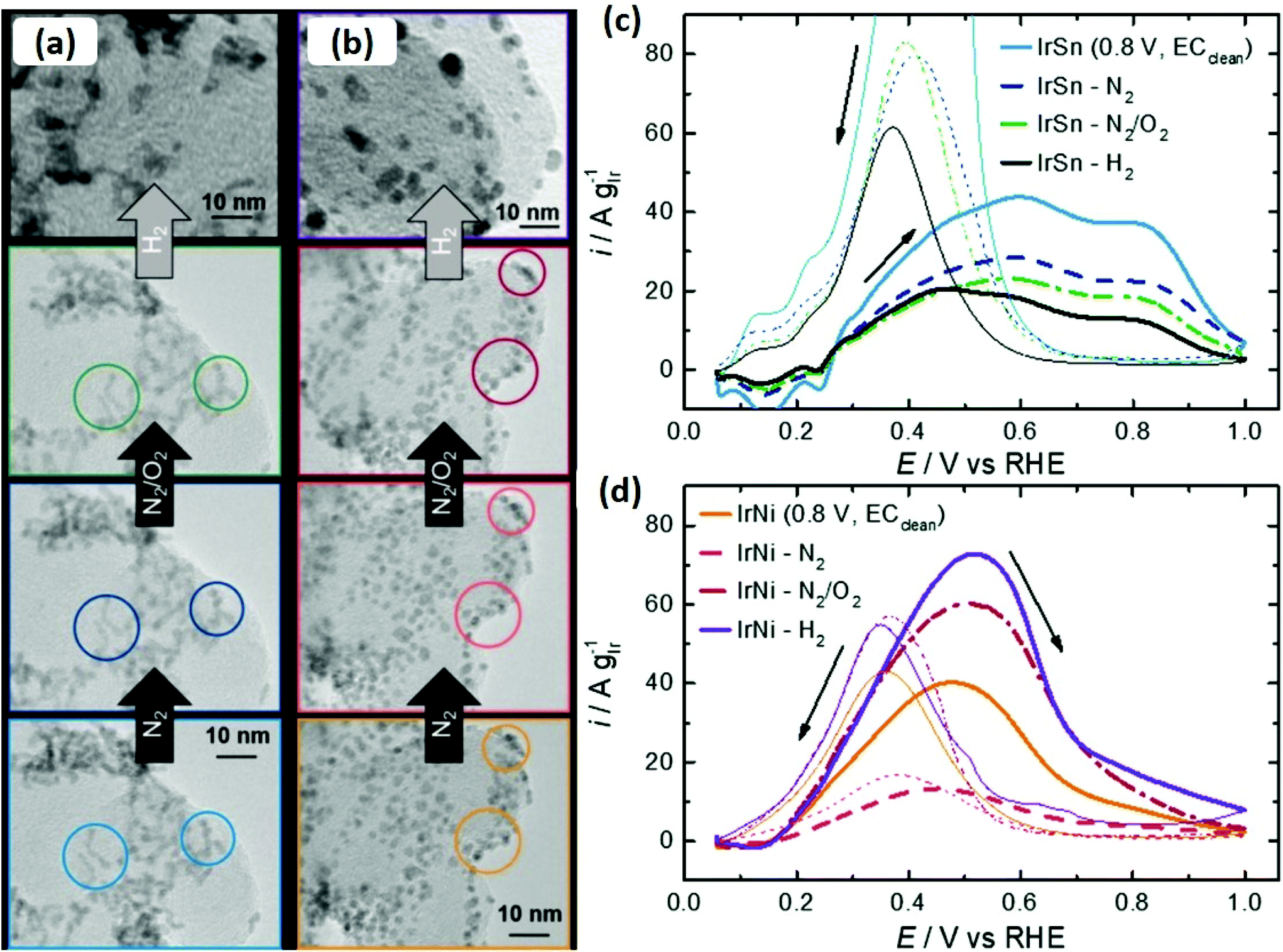 | ||
| Fig. 7 Identical location transmission electron microscopy (IL-TEM) micrographs of the initial (a) IrSn and (b) IrNi catalysts, with subsequent cleaning steps under N2 and N2/O2. The circles are indicating characteristic areas. In case of H2 treatment step, conventional TEM images are shown. CVs of ethanol oxidation reaction on (c) IrSn, IrSn-N2, IrSn-N2/O2 and IrSn-H2 and (d) IrNi, IrNi-N2, IrNi-N2/O2 and IrNi-H2 in 0.1 M KOH with 0.5 M EtOH with a scan rate of 10 mV s−1 and 84 mg cm−2 catalyst loading. Curves were corrected for CVs in N2-saturated 0.1 M KOH.72 Reproduced from ref. 70 with permission from the PCCP Owner Societies. | ||
Unlike metallic catalysts, there are very few examples of electrocatalysis at surfactant-free metal chalcogenide nanomaterials, perhaps due to the difficulties associated with the ligand removal. The surfactants cannot be removed by simple annealing in the air or by UV/ozone treatment as in the case of noble metal nanomaterials because it may result in the oxidation of the metal chalcogenide. It was demonstrated that the decomposition of the dithiocarbamate complex of molybdenum under Ar flow resulted in the formation of MoS2, whereas the decomposition of the same precursor under air, at the same temperature, yielded MoO3.73 Likewise, due to the existence of transition metals in variable oxidation states, the reducing environment (Ar/H2) may induce the formation of different sulfide phases, typically through the loss of H2S.74 Murray and co-workers suggested nitrosonium tetrafluoroborate (NOBF4), as an alternative reagent for the surface ligand removal from semiconducting nanoparticles.75 The organic ligands can be removed by simply dispersing the ligand-capped nanoparticles in hexane and mixing it with a dichloromethane solution of NOBF4. The precipitated NCs can be separated and easily dispersed in hydrophilic organic solvents such as dimethylformamide or acetonitrile. One of the advantages of this approach is that the surface of nanocrystals can be functionalized easily and any organic ligand can be introduced for functionalization. However, due to the high oxidative ability and Lewis acidity of the nitrosonium cation, this method is only suitable for materials with high chemical stability, excluding the majority of the metal chalcogenides.76 Talapin and co-workers developed a strategy based on the hard and soft acids and bases (HSAB) principle. They used inorganic ions (S2−, HS−, Se2−, HSe−, Te2−, HTe−, OH−, NH2−) to remove organic capping agents.77 It was observed that different semiconducting nanomaterials interact with the inorganic ions in agreement with the HSAB principle. For example, soft Au and Cd2+ interact with S2− and HS− stronger than the hard ligands, and the hard ligands i.e., OH− and NH2−, show stronger affinity to hard Zn2+ sites. They also investigated suitability of HBF4 and HPF6 for removal of organic ligands. However the acids can etch the nanocrystals and their general utility for ligand removal is questionable.
Helms and co-workers introduced Meerwein's and other trialkyloxonium salts as mild stripping agents for commonly used organic surfactants.78 These salts do not react with inorganic chalcogenides, and their strong alkylating nature can effectively remove the organic ligands, leaving behind nanocrystals with bare surfaces, with weakly coordinated ligands (BF4−, PF6−), through electrostatic interactions (Fig. 8(a)). This strategy was successfully used to strip different nanomaterials capped by oleic acid, oleylamine or phosphonate ligands. The use of SbCl6− was not successful due to its oxidizing nature, which indicates the sensitivity of some nanomaterials towards the oxidizing conditions. Cossairt and co-workers adopted the same methodology to improve the catalytic performance of dodecylamine-capped WSe2 nanosheets for HER (Fig. 8(b)).79 By treating WSe2 with different concentrations (0.01, 0.1 and 1.0 M) of Meerwein's reagent indicated that the 0.01 M concentration has no effect on overpotential, whereas 0.1 to 1.0 M concentration showed almost the same effect on overpotential. Furthermore, the chemically exchangeable amine from the surface was removed, whereas the intercalated amine between the sheets was not affected by the Meerwein's reagent. Although the reducing amine coverage from the surface active sites of WSe2 was enough to decrease the overpotential for HER by 180 mV. In a follow-up study, they treated TOPO capped WSe2 nanosheets with Meerwein's reagent and observed a decreased overpotential by 130 mV.80 Furthermore, it was observed that the introduction of H+, Li+, Na+ or K+ ions results in electrochemical activation of synthesized WSe2 nanosheets. The combined effect of ligand stripping and electrochemical activation resulted in a significant (400 mV) HER overpotential decrease.
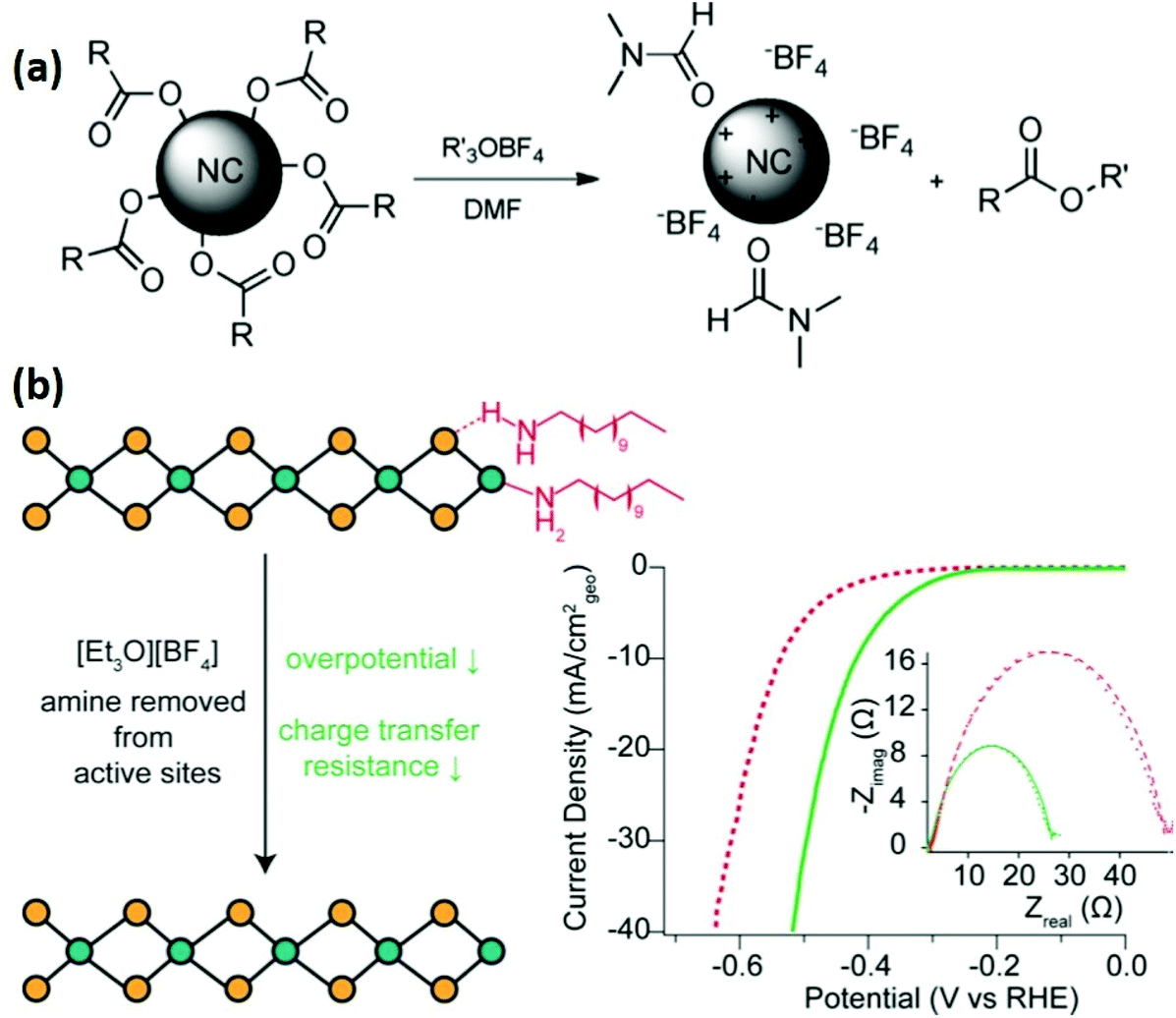 | ||
| Fig. 8 (a) Reactive ligand stripping of carboxylate-, phosphonate-, and amine-passivated nanocrystals with trialkyloxonium salts.78 (b) Illustration of ligand removal by Meerwein's reagent from WSe2 and linear sweep voltammograms (LSV) of WSe2 deposited on a carbon fiber electrode before (dashed line) and after treatment with Meerwein's reagent (solid line) in deaerated 0.5 M H2SO4; inset depicts EIS spectra recorded at 344 mV vs. RHE (reproduced with permission from ref. 77, Copyright 2017, American Chemical Society).79 | ||
Khan et al. avoided using organic ligands to prepare surface-free/uncapped metal sulfide nanomaterials via solid-state synthesis by employing metal xanthate precursors.46 The xanthate precursors were selected as they decompose via Chugaev elimination reaction, producing highly volatile by-products (alkene and carbonyl sulfide). They can be removed easily, leaving behind pure metal sulfide nanomaterials with uncapped surfaces. In a comparative study, they used nickel xanthate precursors with different alkyl chains (ethyl or octyl) and prepared NiS by solid-state decomposition.81 The electrochemical performance of the synthesized materials for water splitting and supercapacitance indicated that the bare surface NiS shows better electrochemical activity than NiS prepared by colloidal routes. It was also observed that NiS prepared from ethyl xanthate precursor showed almost similar overpotential for HER, lower overpotential for OER, and significantly better charge storage behavior than NiS prepared from octyl xanthate precursor (Fig. 9). The use of xanthate with a longer alkyl chain may result either in the deposition of some carbonaceous material or a self-capping phenomenon in which some xanthate molecules may stick to the surface of the nickel sulfide, resulting in poor charge storage behavior. It indicates that relatively, the supercapacitance is affected more than the water splitting by increasing the chain length of the starting precursor. A similar effect was visible for CoS nanoparticles prepared by decomposition of xanthate precursors with alkyl chains of different lengths (ethyl, hexyl and octyl).82 It was observed that the increasing chain length of the starting precursor reduced the size of the synthesized nanocrystals and HER/OER overpotential. However, CoS nanoparticles obtained from xanthate precursors with longer chain length showed decreased capacitance. In another study, NiS was doped with Cu, Co, and Fe by solvent-less decomposition of the respective xanthate precursors and investigated for water splitting and application in supercapacitors.83 For OER and HER, Fe-doped NiS showed the lowest overpotential, compared to other dopants, whereas Co-doped NiS showed relatively highest capacitive performance.
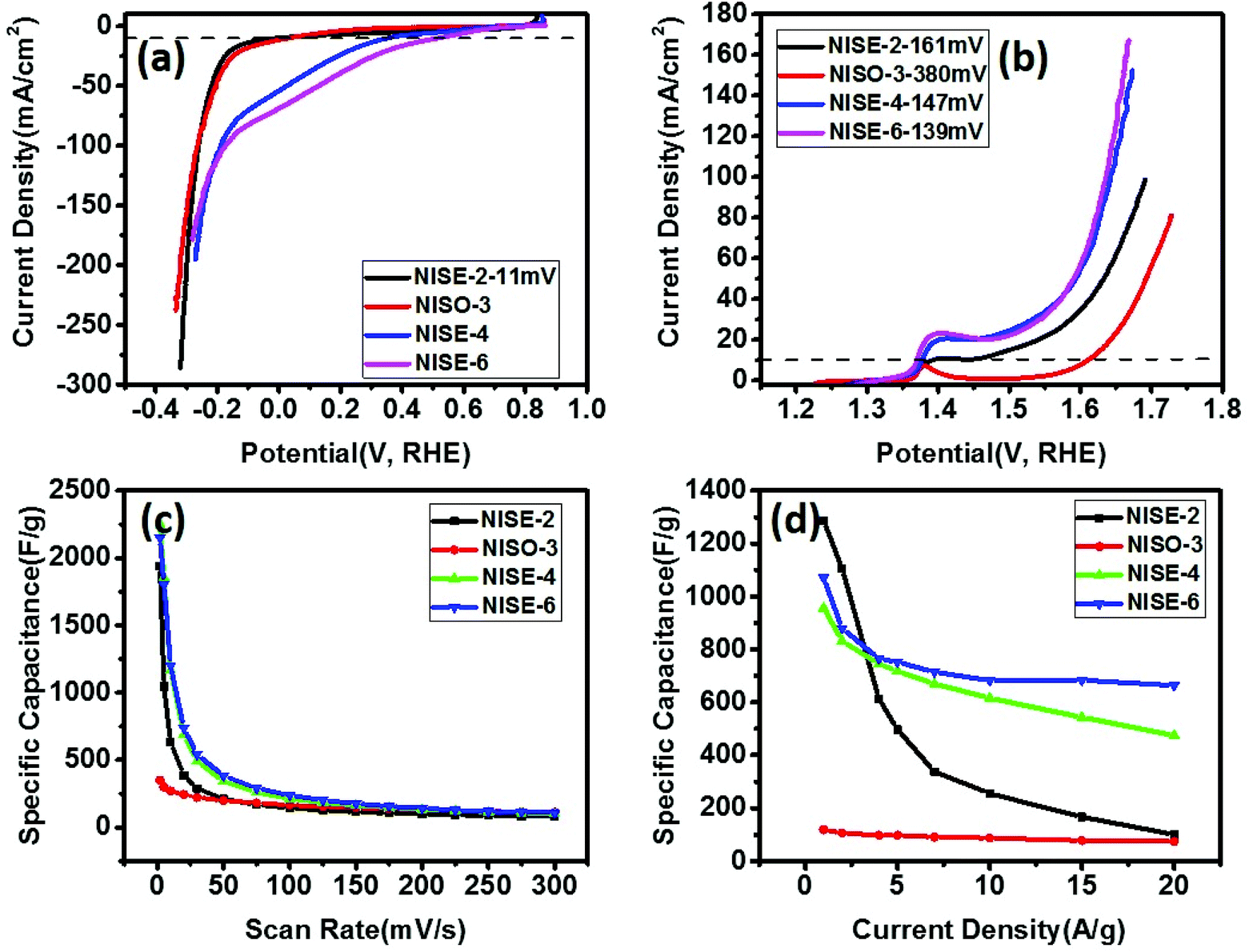 | ||
| Fig. 9 Polarization curves for (a) HER and (b) OER, in 1 M KOH, (c) variation of specific capacitance versus scan rate, (d) variation of specific capacitance versus applied current density for nickel sulfide obtained from decomposition of ethyl xanthate at 200 °C (NISE-2), at 300 °C (NISE-4), at 400 °C (NISE-6) and from octyl xanthate at 250 °C (NISO-3).81 Figure adapted with permission from ref. 46. | ||
Nonetheless, the idea of solvent-less synthesis is appealing but challenging. In the absence of surfactants, uniform size and shape control is extremely difficult. It can be tuned by designing the precursor with a suitable backbone. However, bulky groups or longer alkyl chains may cause carbon contamination. Moreover, the surface of the nanoparticles can be oxidized easily in the absence of any surface coverage.
Conclusion and future perspectives
In summary, with the development of synthetic techniques allowing judicious control of the reaction parameters, nanomaterials with desirable shape and size can be synthesized. Furthermore, the surface ligands can be modified as required and the native ligands can be stripped or re-introduced in a systematic way. Metal chalcogenides consist of a range of compounds with variable oxidation states. Therefore, in addition to size and shape, oxidation states can be tuned for enhanced electrocatalytic performance.Although the effect of ligands on the catalytic performance of metal nanoparticles has been well understood, it has to be further explored for metal chalcogenides. The first examples of bare surface electrocatalysts based on transition metal chalcogenides prepared by newly developed synthetic route were recently demonstrated. Procedures such as electrochemical surface cleaning and the stability of the metal chalcogenides in the potential range employed for this process should be adequately investigated. The use of 1-butylamine and tert-butyl amine has been reported to be efficient for obtaining a clean surface of nanoparticles but hasn't been tested yet with metal chalcogenides.84,85 Since small molecules are easy to remove by extensive washing and, their catalytic inhibition effect is relatively lower than the long-chain ligands. Likewise, various studies have shown that the adsorption of surfactants depends on the facets and nature of the surfactant. Therefore, systematic research can be carried out to study facet and surfactant selective inhibition of catalysts to identify the suitable surfactants that can control the growth and structures of catalysts without affecting their catalytic activity.
Non-oxidizing and non-corrosive reagents are required for surface modification/removal of surfactants without significantly affecting the size and shape of nanocrystals. Although some routes have been reported for specific nanocrystal systems, a generalized and efficient strategy still needs to be developed. Likewise, the electrocatalytic performance of metal chalcogenides deposited at polarized liquid–liquid interface brings new opportunities.86,87 Interfaces between two immiscible fluids are considered as flat, defect-free, and self-healing planes for immobilization and organization of nanoparticles. Besides depositing pre-synthesized nanoparticles at interfaces, metal chalcogenides can be directly synthesized at the liquid–liquid interface in the absence of surfactants.88,89 Therefore, electrocatalytic studies of metal chalcogenide nanomaterials directly synthesized at these interfaces can provide a better insight into the intrinsic performances of metal chalcogenides without the influence of surfactants and free of the effect of electrode support. For the same reason, studies of electrocatalysis in the suspension of transition metal chalcogenides have to be explored.90
The influence of ligands on electrocatalysis at nanoparticulate metal chalcogenides is still an emerging research field. Therefore the detailed studies are lacking and further research is highly recommended to understand the associated opportunities and challenges properly. Above, we have summarized different strategies for designing and developing nanocrystals, the capping ligand's role in controlling shape/size and impact on electrocatalysis, and the strategies for removing surfactants, which will assist in the synthesis of metal chalcogenide nanomaterials with better electrocatalytic properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
NR thanks the National Research Foundation (NRF, 64820) South African Research Chairs Initiative (SARChI) program. MDK and MO thank the European Union's Horizon 2020 Research and Innovation program under the Marie Skłodowska-Curie grant agreement no. 847413 for funding. Scientific work published as part of an international co-financed project founded from the program of the Minister of Science and Higher Education entitled “PMW” in the years 2020–2024; agreement no. 5005/H2020-MSCA-COFUND/2019/2.References
- Y. Xia, K. D. Gilroy, H. C. Peng and X. Xia, Angew. Chem., Int. Ed., 2017, 56, 60–95 CrossRef CAS PubMed.
- R. Samal, A. K. Samantara, S. Mahalik, J. Behera, B. Dash and K. Sanjay, New J. Chem., 2021, 45, 2795–2803 RSC.
- A. K. Ganguli, A. Ganguly and S. Vaidya, Chem. Soc. Rev., 2010, 39, 474–485 RSC.
- A. Feinle, M. S. Elsaesser and N. Huesing, Chem. Soc. Rev., 2016, 45, 3377–3399 RSC.
- F. L. Theiss, G. A. Ayoko and R. L. Frost, Appl. Surf. Sci., 2016, 383, 200–213 CrossRef CAS.
- A. Varma, A. S. Mukasyan, A. S. Rogachev and K. V. Manukyan, Chem. Rev., 2016, 116, 14493–14586 CrossRef CAS PubMed.
- J. R. Chamorro and T. M. McQueen, Acc. Chem. Res., 2018, 51, 2918–2925 CrossRef CAS PubMed.
- S. K. Gupta and Y. Mao, Prog. Mater. Sci., 2020, 100734 Search PubMed.
- E. Lewis, S. Haigh and P. O'Brien, J. Mater. Chem. A, 2014, 2, 570–580 RSC.
- S. G. Kwon and T. Hyeon, Small, 2011, 7, 2685–2702 CrossRef CAS PubMed.
- J. van Embden, A. S. Chesman and J. J. Jasieniak, Chem. Mater., 2015, 27, 2246–2285 CrossRef CAS.
- L. Polavarapu, S. Mourdikoudis, I. Pastoriza-Santos and J. Pérez-Juste, CrystEngComm, 2015, 17, 3727–3762 RSC.
- H. Duan, D. Wang and Y. Li, Chem. Soc. Rev., 2015, 44, 5778–5792 RSC.
- E. Groeneveld and C. de Mello Donegá, Nanoparticles, Springer, 2014, pp. 145–189 Search PubMed.
- S. Mourdikoudis and L. M. Liz-Marzán, Chem. Mater., 2013, 25, 1465–1476 CrossRef CAS.
- C. M. Phan and H. M. Nguyen, J. Phys. Chem. A, 2017, 121, 3213–3219 CrossRef CAS PubMed.
- T. Song, F. Gao, S. Guo, Y. Zhang, S. Li, H. You and Y. Du, Nanoscale, 2021, 13, 3895–3910 RSC.
- M. Green, J. Mater. Chem., 2010, 20, 5797–5809 RSC.
- A. Kozhakhmetov, R. Torsi, C. Y. Chen and J. A. Robinson, J. Phys.: Mater., 2020, 4, 012001 Search PubMed.
- S. R. Marri, S. Ratha, C. S. Rout and J. Behera, Chem. Commun., 2017, 53, 228–231 RSC.
- J. K. Das, A. K. Samantara, A. K. Nayak, D. Pradhan and J. Behera, Dalton Trans., 2018, 47, 13792–13799 RSC.
- S. M. Dinara, A. K. Samantara, J. K. Das, J. Behera, S. K. Nayak, D. J. Late and C. S. Rout, Dalton Trans., 2019, 48, 16873–16881 RSC.
- J. N. Behera, A. Padhy and A. K. Samantara, Sustainable Energy Fuels, 2021, 5, 3729–3736 RSC.
- R. K. Tiwari, J. Kumar and J. Behera, Chem. Commun., 2016, 52, 1282–1285 RSC.
- M. K. Sahoo, A. K. Samantara and J. Behera, Inorg. Chem., 2020, 59, 12252–12262 CrossRef CAS.
- R. K. Tripathy, A. K. Samantara and J. Behera, Dalton Trans., 2019, 48, 10557–10564 RSC.
- M. D. Khan, M. A. Malik and N. Revaprasadu, Coord. Chem. Rev., 2019, 388, 24–47 CrossRef CAS.
- M. Afzaal, D. Crouch, M. A. Malik, M. Motevalli, P. O'Brien, J. H. Park and J. D. Woollins, Eur. J. Inorg. Chem., 2004, 2004, 171–177 CrossRef.
- J. Akhtar, M. Afzaal, M. A. Vincent, N. A. Burton, J. Raftery, I. H. Hillier and P. O'Brien, J. Phys. Chem. C, 2011, 115, 16904–16909 CrossRef CAS.
- D. Crouch, P. O'Brien, M. Malik, P. Skabara and S. Wright, Chem. Commun., 2003, 1454–1455 RSC.
- T. Oyetunde, M. Afzaal, M. A. Vincent and P. O'Brien, Dalton Trans., 2016, 45, 18603–18609 RSC.
- N. Goubet, A. Jagtap, C. Livache, B. Martinez, H. Portalès, X. Z. Xu, R. P. Lobo, B. Dubertret and E. Lhuillier, J. Am. Chem. Soc., 2018, 140, 5033–5036 CrossRef CAS PubMed.
- C. Gréboval, A. Chu, N. Goubet, C. Livache, S. Ithurria and E. Lhuillier, Chem. Rev., 2021, 121, 3627–3700 CrossRef PubMed.
- S. Keuleyan, E. Lhuillier, V. Brajuskovic and P. Guyot-Sionnest, Nat. Photonics, 2011, 5, 489–493 CrossRef CAS.
- S. Keuleyan, E. Lhuillier and P. Guyot-Sionnest, J. Am. Chem. Soc., 2011, 133, 16422–16424 CrossRef CAS.
- E. Izquierdo, A. Robin, S. Keuleyan, N. Lequeux, E. Lhuillier and S. Ithurria, J. Am. Chem. Soc., 2016, 138, 10496–10501 CrossRef CAS PubMed.
- D. J. Crouch, P. M. Hatton, M. Helliwell, P. O'Brien and J. Raftery, Dalton Trans., 2003, 2761–2766 RSC.
- Y. Kojima, K. Ibi, T. Kanda, H. Ishihara, T. Murai and S. Kato, Bull. Chem. Soc. Jpn., 1993, 66, 990–992 CrossRef CAS.
- M. T. Ng and J. J. Vittal, Inorg. Chem., 2006, 45, 10147–10154 CrossRef CAS PubMed.
- S. Razzaque, M. D. Khan, M. Aamir, M. Sohail, S. Bhoyate, R. K. Gupta, M. Sher, J. Akhtar and N. Revaprasadu, Inorg. Chem., 2021, 60, 1449–1461 CrossRef CAS.
- M. D. Khan, M. Aamir, M. Sohail, M. Sher, J. Akhtar, M. A. Malik and N. Revaprasadu, Sol. Energy, 2018, 169, 526–534 CrossRef CAS.
- M. D. Khan, M. Aamir, G. Murtaza, M. A. Malik and N. Revaprasadu, Dalton Trans., 2018, 47, 10025–10034 RSC.
- M. D. Khan, M. Aamir, M. Sohail, M. Sher, N. Baig, J. Akhtar, M. A. Malik and N. Revaprasadu, Dalton Trans., 2018, 47, 5465–5473 RSC.
- J. Steggerda, J. Cras and J. Willemse, Recl. Trav. Chim. Pays-Bas, 1981, 100, 41–48 CrossRef CAS.
- G. W. Watt and B. J. McCormick, Spectrochim. Acta, 1965, 21, 753–761 CrossRef CAS.
- M. D. Khan, G. Murtaza, N. Revaprasadu and P. O'Brien, Dalton Trans., 2018, 47, 8870–8873 RSC.
- A. C. Ekennia, D. C. Onwudiwe and A. A. Osowole, J. Sulfur Chem., 2015, 36, 96–104 CrossRef CAS.
- P. D. McNaughter, S. A. Saah, M. Akhtar, K. Abdulwahab, M. A. Malik, J. Raftery, J. A. Awudza and P. O'Brien, Dalton Trans., 2016, 45, 16345–16353 RSC.
- E. A. Lewis, P. D. McNaughter, Z. Yin, Y. Chen, J. R. Brent, S. A. Saah, J. Raftery, J. A. Awudza, M. A. Malik and P. O'Brien, Chem. Mater., 2015, 27, 2127–2136 CrossRef CAS.
- N. Hollingsworth, A. Roffey, H.-U. Islam, M. Mercy, A. Roldan, W. Bras, M. Wolthers, C. R. A. Catlow, G. Sankar and G. Hogarth, Chem. Mater., 2014, 26, 6281–6292 CrossRef CAS.
- Y. K. Jung, J. I. Kim and J.-K. Lee, J. Am. Chem. Soc., 2010, 132, 178–184 CrossRef CAS PubMed.
- M. D. Khan, J. Akhtar, M. A. Malik and N. Revaprasadu, ChemistrySelect, 2016, 1, 5982–5989 CrossRef CAS.
- M. T. Ng, C. Boothroyd and J. J. Vittal, Chem. Commun., 2005, 3820–3822 RSC.
- C. Buchmaier, T. Rath, F. Pirolt, A.-C. Knall, P. Kaschnitz, O. Glatter, K. Wewerka, F. Hofer, B. Kunert and K. Krenn, RSC Adv., 2016, 6, 106120–106129 RSC.
- W. P. Lim, C. T. Wong, S. L. Ang, H. Y. Low and W. S. Chin, Chem. Mater., 2006, 18, 6170–6177 CrossRef CAS.
- C. Buchmaier, M. Glänzer, A. Torvisco, P. Poelt, K. Wewerka, B. Kunert, K. Gatterer, G. Trimmel and T. Rath, J. Mater. Sci., 2017, 52, 10898–10914 CrossRef CAS.
- W. Maneeprakorn, C. Q. Nguyen, M. A. Malik, P. O'Brien and J. Raftery, Dalton Trans., 2009, 2103–2108 RSC.
- G. E. Ayom, M. D. Khan, T. Ingsel, W. Lin, R. K. Gupta, S. J. Zamisa, W. E. van Zyl and N. Revaprasadu, Chem. – Eur. J., 2020, 26, 2693–2704 CrossRef CAS.
- W. Maneeprakorn, M. A. Malik and P. O'Brien, J. Mater. Chem., 2010, 20, 2329–2335 RSC.
- M. J. Turo and J. E. Macdonald, ACS Nano, 2014, 8, 10205–10213 CrossRef CAS.
- C. Amatore, A. Jutand and M. A. M'Barki, Organometallics, 1992, 11, 3009–3013 CrossRef CAS.
- J. S. Steckel, B. K. Yen, D. C. Oertel and M. G. Bawendi, J. Am. Chem. Soc., 2006, 128, 13032–13033 CrossRef CAS PubMed.
- S. Wang, L. Kuai, Y. Huang, X. Yu, Y. Liu, W. Li, L. Chen and B. Geng, Chem. – Eur. J., 2013, 19, 240–248 CrossRef CAS PubMed.
- Y. Lu, Y. Wang and W. Chen, J. Power Sources, 2011, 196, 3033–3038 CrossRef CAS.
- L. Lu, B. Lou, S. Zou, H. Kobayashi, J. Liu, L. Xiao and J. Fan, ACS Catal., 2018, 8, 8484–8492 CrossRef CAS.
- S. Campisi, M. Schiavoni, C. E. Chan-Thaw and A. Villa, Catalysts, 2016, 6, 185 CrossRef.
- J. Liu, J. Yin, B. Feng, T. Xu and F. Wang, Nanomaterials, 2018, 8, 955 CrossRef.
- R. M. Arán-Ais, F. J. Vidal-Iglesias, J. Solla-Gullón, E. Herrero and J. M. Feliu, Electroanalysis, 2015, 27, 945–956 CrossRef.
- D. Li, C. Wang, D. Tripkovic, S. Sun, N. M. Markovic and V. R. Stamenkovic, ACS Catal., 2012, 2, 1358–1362 CrossRef CAS.
- V. Beermann, M. Gocyla, S. Kühl, E. Padgett, H. Schmies, M. Goerlin, N. Erini, M. Shviro, M. Heggen and R. E. Dunin-Borkowski, J. Am. Chem. Soc., 2017, 139, 16536–16547 CrossRef CAS PubMed.
- Z. Liu, M. Shamsuzzoha, E. T. Ada, W. M. Reichert and D. E. Nikles, J. Power Sources, 2007, 164, 472–480 CrossRef CAS.
- C. Prössl, M. Kübler, M. A. Nowroozi, S. Paul, O. Clemens and U. I. Kramm, Phys. Chem. Chem. Phys., 2021, 23, 563–573 RSC.
- N. Zeng, D. G. Hopkinson, B. F. Spencer, S. G. McAdams, A. A. Tedstone, S. J. Haigh and D. J. Lewis, Chem. Commun., 2019, 55, 99–102 RSC.
- A. F. Hepp, M. J. Kulis, J. S. McNatt, N. V. Duffy, M. D. Hoops, E. Gorse, P. E. Fanwick, J. Masnovi, J. E. Cowen and R. N. Dominey, NASA/TM-2016-219140.
- A. Dong, X. Ye, J. Chen, Y. Kang, T. Gordon, J. M. Kikkawa and C. B. Murray, J. Am. Chem. Soc., 2011, 133, 998–1006 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- A. Nag, M. V. Kovalenko, J.-S. Lee, W. Liu, B. Spokoyny and D. V. Talapin, J. Am. Chem. Soc., 2011, 133, 10612–10620 CrossRef CAS.
- E. L. Rosen, R. Buonsanti, A. Llordes, A. M. Sawvel, D. J. Milliron and B. A. Helms, Angew. Chem., Int. Ed., 2012, 51, 684–689 CrossRef CAS PubMed.
- D. A. Henckel, O. Lenz and B. M. Cossairt, ACS Catal., 2017, 7, 2815–2820 CrossRef CAS.
- D. A. Henckel, O. M. Lenz, K. M. Krishnan and B. M. Cossairt, Nano Lett., 2018, 18, 2329–2335 CrossRef CAS.
- G. B. Shombe, M. D. Khan, C. Zequine, C. Zhao, R. K. Gupta and N. Revaprasadu, Sci. Rep., 2020, 10, 1–14 CrossRef PubMed.
- R. Akram, M. D. Khan, C. Zequine, C. Zhao, R. K. Gupta, M. Akhtar, J. Akhtar, M. A. Malik, N. Revaprasadu and M. H. Bhatti, Mater. Sci. Semicond. Process., 2020, 109, 104925 CrossRef CAS.
- G. B. Shombe, M. D. Khan, A. M. Alenad, J. Choi, T. Ingsel, R. K. Gupta and N. Revaprasadu, Sustainable Energy Fuels, 2020, 4, 5132–5143 RSC.
- J. Wu, J. Zhang, Z. Peng, S. Yang, F. T. Wagner and H. Yang, J. Am. Chem. Soc., 2010, 132, 4984–4985 CrossRef CAS.
- N. Naresh, F. Wasim, B. P. Ladewig and M. Neergat, J. Mater. Chem. A, 2013, 1, 8553–8559 RSC.
- F. Ozel, E. Aslan, A. Sarilmaz and I. Hatay Patir, ACS Appl. Mater. Interfaces, 2016, 8, 25881–25887 CrossRef CAS PubMed.
- E. Aslan, A. Sarilmaz, F. Ozel, I. Hatay Patir and H. H. Girault, ACS Appl. Nano Mater., 2019, 2, 7204–7213 CrossRef CAS.
- P. J. Thomas, G. L. Stansfield, N. Komba, D. J. Cant, K. Ramasamy, E. Albrasi, H. Al-Chaghouri, K. L. Syres, P. O'Brien and W. R. Flavell, RSC Adv., 2015, 5, 62291–62299 RSC.
- C. Rao, G. Kulkarni, V. V. Agrawal, U. K. Gautam, M. Ghosh and U. Tumkurkar, J. Colloid Interface Sci., 2005, 289, 305–318 CrossRef CAS.
- S. V. Sokolov, S. Eloul, E. Kätelhön, C. Batchelor-McAuley and R. G. Compton, Phys. Chem. Chem. Phys., 2017, 19, 28–43 RSC.
| This journal is © The Royal Society of Chemistry 2021 |