
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Platinum(IV)-azido monocarboxylato complexes are photocytotoxic under irradiation with visible light†
Evyenia
Shaili
a,
Marίa J.
Romero
ab,
Luca
Salassa
 acde,
Julie A.
Woods‡
f,
Jennifer S.
Butler
a,
Isolda
Romero-Canelón
ag,
Guy
Clarkson
a,
Abraha
Habtemariam
a,
Peter J.
Sadler
*a and
Nicola J.
Farrer
*ah
acde,
Julie A.
Woods‡
f,
Jennifer S.
Butler
a,
Isolda
Romero-Canelón
ag,
Guy
Clarkson
a,
Abraha
Habtemariam
a,
Peter J.
Sadler
*a and
Nicola J.
Farrer
*ah
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: p.j.sadler@warwick.ac.uk
bDepartamento de Didácticas Aplicadas, Facultade de Formación do Profesorado, Universidade de Santiago de Compostela, 27002, Lugo, Spain
cDonostia International Physics Center, Paseo Manuel de Lardizabal 4, Donostia, 20018, Spain
dPolimero eta Material Aurreratuak: Fisika, Kimika eta Teknologia, Kimika Fakultatea, Euskal Herriko Unibertsitatea UPV/EHU, Paseo Manuel de Lardizabal 3, Donostia, 20018, Spain
eIkerbasque, Basque Foundation for Science, Bilbao, 48011, Spain
fPhotobiology Unit, Department of Dermatology and Photobiology, Ninewells Hospital, Dundee, DD1 9SY, UK
gSchool of Pharmacy, Institute of Clinical Sciences, College of Medical and Dental Sciences, Sir Robert Aitken Institute for Medical Research, University of Birmingham, Birmingham, B15 2TT, UK
hChemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: Nicola.Farrer@chem.ox.ac.uk; Tel: +44 (0)1865 285131
First published on 12th July 2021
Abstract
Complexes trans,trans,trans-[Pt(N3)2(OH)(OCOR)(py)2] where py = pyridine and where OCOR = succinate (1); 4-oxo-4-propoxybutanoate (2) and N-methylisatoate (3) have been synthesized by derivation of trans,trans,trans-[Pt(OH)2(N3)2(py)2] (4) and characterised by NMR and EPR spectroscopy, ESI-MS and X-ray crystallography. Irradiation of 1–3 with green (517 nm) light initiated photoreduction to Pt(II) and release of the axial ligands at a 3-fold faster rate than for 4. TD-DFT calculations showed dissociative transitions at longer wavelengths for 1 compared to 4. Complexes 1 and 2 showed greater photocytotoxicity than 4 when irradiated with 420 nm light (A2780 cell line IC50 values: 2.7 and 3.7 μM) and complex 2 was particularly active towards the cisplatin-resistant cell line A2780cis (IC50 3.7 μM). Unlike 4, complexes 1–3 were phototoxic under green light irradiation (517 nm), with minimal toxicity in the dark. A pKa(H2O) of 5.13 for the free carboxylate group was determined for 1, corresponding to an overall negative charge during biological experiments, which crucially, did not appear to impede cellular accumulation and photocytotoxicity.
Introduction
Approximately 40% of the cancer patients treated with chemotherapy receive a platinum(II)-based medicine such as cisplatin, carboplatin or oxaliplatin.1 Despite the wide-spread use of platinum-based drugs2 – particularly in combination therapies – disadvantages exist, including the development of resistance and serious side-effects of treatment.3 Octahedral, Pt(IV) prodrugs with a d6 electronic configuration typically demonstrate greater kinetic inertness than Pt(II) complexes, as well as offering additional ligand sites for derivation.4Pt(IV) prodrugs are classically regarded to exert their cytotoxic effect following reduction to Pt(II) species in vivo, although Pt(IV) complexes may also form adducts with biomolecules.5 Photoactivated chemotherapy (PACT) provides both spatial and temporal control over Pt(IV) prodrug reduction. Photoreduction of Pt(IV) prodrugs of carboplatin,6 oxaliplatin7 and cisplatin8 – either by direct irradiation of the Pt(IV) complex, or by employing separate photosensitisers – have been reported. The ligands coordinated to the Pt(IV) prodrug influence the solubility, lipophilicity and ease of reduction of the complex, whilst the ligands themselves have the potential to be biologically active, through mechanisms which are distinct from the resultant platinum species, following dissociation. The reduction of Pt(IV) complexes is greatly affected by the coordinated ligands; for a series of complexes with chlorido, hydroxido and carboxylato anionic axial ligands [Pt(en)Cl2(X)2] (where X = axial ligand), cathodic reduction potentials varied by more than 650 mV, with the rates of reduction following the order X = Cl− > OC(O)CH3− > OH−.5 It is notable that for several Pt(IV) complexes – including Satraplatin (ctc-[Pt(NH3)(CHA)(OAc)2Cl2] where CHA = cyclohexylamine) – equatorial anionic ligand hydrolysis can occur more readily than axial anionic ligand hydrolysis.9 The most important biological reductants, the mechanism(s) and the rate of Pt(IV) reduction in a biological environment for a given complex cannot readily be predicted from the reduction potential.10 A range of Pt(IV) complexes undergo photoreduction, typically requiring short wavelengths of light i.e. UVA.11 The coordination of azido (N3) ligands to Pt(IV) results in highly photosensitive complexes, which can be kinetically inert in the dark and – depending on judicious ligand choice – can be photoactivated with longer (visible) wavelengths of light.12 Penetration of light into tissue is wavelength-dependent, and extending the wavelength of light which can initiate photoreaction of Pt(IV) complexes – whilst retaining stability in the dark – is therefore a key goal. Replacing the axial OH ligands of Pt(IV) diazido complexes with acetato (OAc) ligands has been demonstrated by TD-DFT to lower the energy of the LUMO and LUMO+1, reducing the energy gap between the HOMO and LUMO.13 This has the effect of extending the strongest absorption band to longer wavelengths. Since this modification also has the potential to lead to more facile spontaneous reduction in the dark, evaluation of stability of the complex in the absence of irradiation is important. Modification of the axial ligands of photoactivatable Pt(IV) diazido complexes to incorporate biotin,14 guanidinoneomycin,15 coumarins16 and naphthalimides,17 as well as an RGD peptide to target integrin18 have been reported. However, the synthesis and extensive characterisation of 1 itself has not been reported previously, and we report it here for the first time along with two other complexes. Hydroxido groups are common Pt(IV) axial ligands because oxidation of Pt(II) to Pt(IV) is readily accomplished through the use of H2O2 and also due to the favourable solubility and stability properties imparted by inclusion of the hydroxido ligand. Derivatisation of one hydroxido ligand of a dihydroxido Pt(IV) complex to a carboxylate can be achieved through a number of methods19 including reaction with anhydrides,20–23 acid chlorides,24 and reaction with carboxylic acids in acetic acid solvent.25 Terminal carboxylic acids which have been produced through reaction with anhydrides can be used to introduce targeting peptides.26 Other functional groups, such as carbamates can also be axially introduced.27,28
The aim of the present work was to explore the effect of axial ligand modification on the photochemistry and photobiology of platinum(IV) azido complexes.12
We report the synthesis, characterisation, photochemical and photobiological studies of the mono-carboxylato compounds: trans,trans,trans-[Pt(N3)2(OH)(succ)(py)2] (1) where succ = succinate and py = pyridine; trans,trans,trans-[Pt(N3)2(OH)(succ-Pr)(py)2] where succ-Pr = 4-oxo-4-propoxybutanoate (2) and trans,trans,trans-[Pt(N3)2(OH)(N-MI)(py)2] where N-MI = N-N-methylisatoate (3), in comparison with trans,trans,trans-[Pt(N3)2(OH)2(py)2] (4), which we have reported previously29 (Fig. 1). The N-MI ligand was chosen since it introduces an additional absorption band (ca. 350 nm) and increased absorption towards longer wavelengths. We therefore anticipated that complex (3) might display increased photosensitivity on irradiation with visible light.
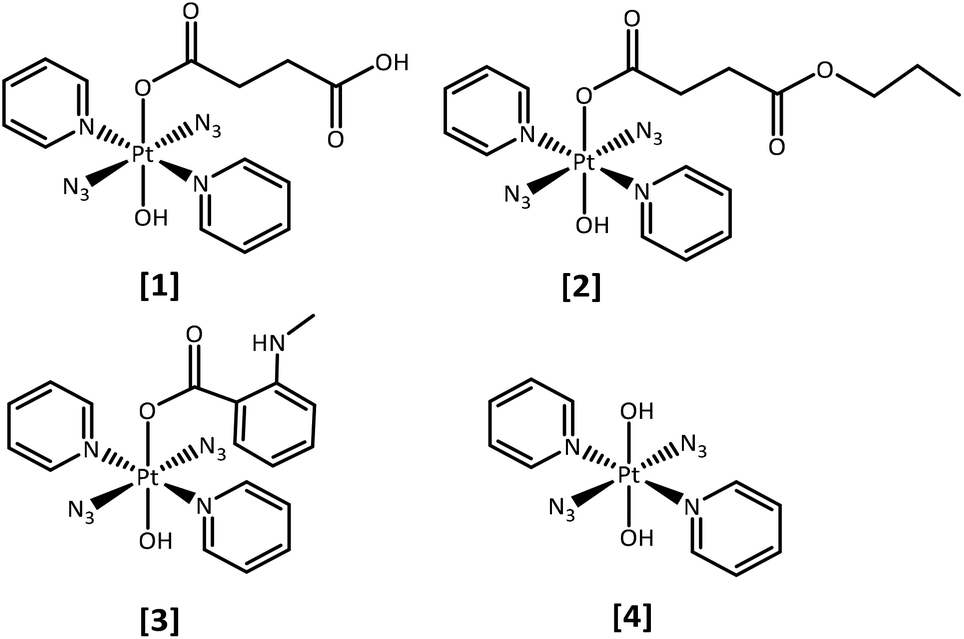 | ||
| Fig. 1 Platinum(IV) azido complexes reported here (axial group): 1 (succinate), 2 (4-oxo-4-propoxybutanoate), 3 (N-methylisatoate), and 4 (hydroxide, previously reported).29 | ||
Results
Synthesis and characterisation
Precursor compounds N-methylisatoic acid (N-MIA) (5), trans-[PtCl2(py)2] (6), trans-[Pt(N3)2(py)2] (7) and complex 4 were synthesized as reported previously; characterisation was consistent with previous reports.29 Complexes 1–3 were synthesised from 4 and characterised by 1H, 13C, 195Pt-NMR spectroscopy, ESI-MS and UV-vis spectroscopy (ESI) and single crystal X-ray diffraction.X-ray diffraction
Single crystals suitable for X-ray diffraction were obtained for complexes trans,trans,trans-[Pt(N3)2(OH)(succ)(py)2] 1, trans,trans,trans-[Pt(N3)2(OH)(succ-Pr)(py)2] 2 and trans,trans,trans-[Pt(N3)2(OH)(N-MI)(py)2] 3 as described in the Experimental section. ORTEP diagrams for the molecular structures are shown in Fig. 2 and the crystallographic data, including selected bond distances and angles are summarized in Table S2.†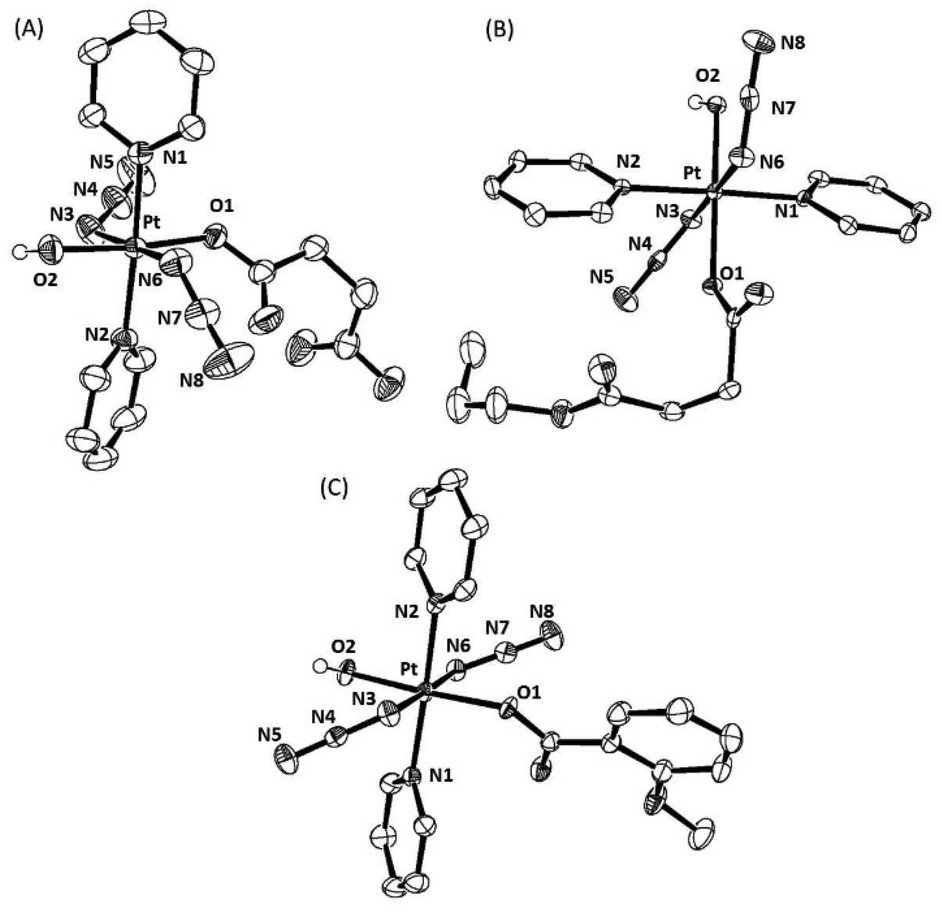 | ||
| Fig. 2 ORTEP diagrams of the complexes: (A) trans,trans,trans-[Pt(N3)2(OH)(succ)(py)2] 1; (B) trans,trans,trans-[Pt(N3)2(OH)(succ-Pr)(py)2] 2; and (C) trans,trans,trans-[Pt(N3)2(OH)(N-MI)(py)2] 3. The ellipsoids are set to 50% probability level. Atom labels do not represent the numbering in the cif files (CCDC1998067(1); CCDC1998069 (2) and CCDC1998068 (3)†) but are used to facilitate comparison between the complexes. All hydrogen atoms have been omitted for clarity, except for the axial hydroxido hydrogen atoms. | ||
Compounds 1, 2 and 3 show a distorted octahedral geometry around the Pt(IV) ion and differ only in the nature of one of the oxygen donor ligands coordinated in an axial position. The equatorial plane is defined by four nitrogen atoms from two pyridine molecules trans to each other (N1 and N2) and two azido anions trans (N3 and N6), with an oxygen atom (O2) from a hydroxido ligand located in one axial position. The sixth position is occupied by an oxygen atom (O1) from a monoanionic ligand: a succinate anion in 1, a 4-oxo-4-propoxybutanoate anion in 2 and a N-methylisatoate anion in the case of 3. The distortion from octahedral geometry is demonstrated by the O2–Pt–O1 bond angle being 174.21(7) Å in 1, 175.52(9) Å in 2 and 176.50(11) Å in 3, which are all considerably less linear than the corresponding angle in complex 4 at 179.58(11) Å. The Pt–O1 (carboxylato) bond lengths for the monocarboxylato, monohydroxido complexes 1–3 are all elongated in comparison to 4 and similar to values for previously reported dicarboxylato complexes trans,trans,trans-[Pt(N3)2(OAc)2(py)2] (2.007(5) Å)30 and trans,trans,trans-[Pt(N3)2(CF3COO)2(py)2] (2.0093(15) Å).15 In complexes 1–3 the Pt–O1 (carboxyl) bond is longer than the Pt–O2 (hydroxido) bond. However, a difference between both axial Pt–OH bonds was also observed in the crystallographic structure of 4 (Pt–O1 1.990(3) Å and Pt–O2 2.027(3) Å). In 4, one of the hydroxido ligands is involved in two hydrogen bonds (as a donor and acceptor) with hydroxido and azido groups of a neighbouring molecule, which correlates with the lengthened Pt–OH bond, whereas the second hydroxido ligand acts only as a donor. The intermolecular interactions in complexes 1, 2 and 3 are shown in the ESI (Fig. S2–S4†).
pKa of complex 1
The of complex 1 (1.1 mM in D2O) was determined by plotting the change in the 1H NMR chemical shift of the pyridine protons against pH*, with dioxane as an internal reference. The pH* was measured at ambient temperature, directly in the NMR tube. The pH* was adjusted to the desired value using DClO4. 1H NMR spectra were recorded at 25 °C. The data were fitted to the Henderson–Hasselbalch equation (r2 = 0.99), and the
of complex 1 (1.1 mM in D2O) was determined by plotting the change in the 1H NMR chemical shift of the pyridine protons against pH*, with dioxane as an internal reference. The pH* was measured at ambient temperature, directly in the NMR tube. The pH* was adjusted to the desired value using DClO4. 1H NMR spectra were recorded at 25 °C. The data were fitted to the Henderson–Hasselbalch equation (r2 = 0.99), and the  of the free carboxyl group was determined to be 5.07 (Fig. S5†) in D2O, which was converted to a final value of pKa(H2O) 5.13.32
of the free carboxyl group was determined to be 5.07 (Fig. S5†) in D2O, which was converted to a final value of pKa(H2O) 5.13.32
UV-vis spectroscopy
UV-vis spectra of complexes 1–3 (in H2O for 1 and 2; in 5% DMSO/95% H2O for 3 due to solubility constraints) were measured, with extinction coefficients determined by ICP-MS analysis of platinum concentration (Fig. S6†).DFT and TD-DFT calculations
DFT and TD-DFT calculations were performed on complex 1 to characterise the electronic transitions giving rise to the UV-vis absorption spectrum and to rationalize the photoactivity of 1, particularly in comparison to complex 4.29 Ground-state geometry and lowest-lying triplet geometry optimizations were performed in the deprotonated state, since the pKa determined (above) for 1 (5.13, H2O) suggests it would be largely deprotonated at physiological pH. The singlet excited states, as well as the orbital contributions (%) in each case are summarised in Table S3,† along with molecular orbital representations and Electron Difference Density Maps (EDDMS) of singlet excited-state transitions in H2O (Table S4†) and molecular orbitals for the ground state (Table S5†). The UV-vis spectra were simulated by calculation of 32 singlet states using water as a solvent (cpcm)33,34 and the TD-DFT method (Table 1) and are overlaid with the experimental data in Fig. 3.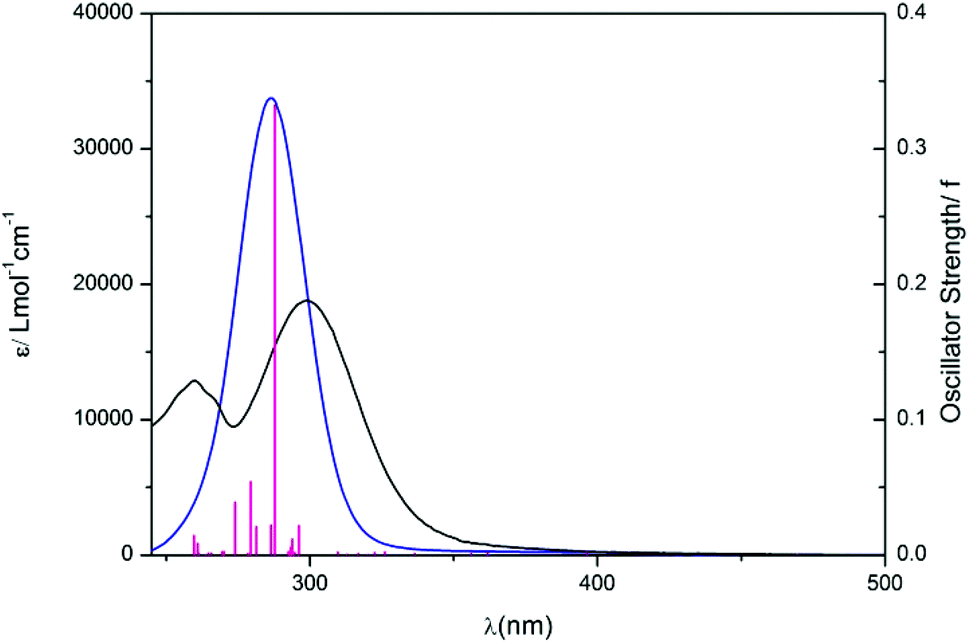 | ||
| Fig. 3 Experimental (black, H2O) and theoretical (blue) UV-vis spectra of complex 1 in the deprotonated state. Vertical (red) bars represent the oscillator strengths of the excited states. | ||
| Bond | Bond length | ||
|---|---|---|---|
| X-ray structure (Å) | Ground-state (deprotonated) | Lowest lying triplet | |
| Pt–N3(azide) | 2.049(2) | 2.088 | 2.359 |
| Pt–N6(azide) | 2.045(2) | 2.069 | 2.508 |
| Pt–O2 (OH) | 1.9852(15) | 2.015 | 2.015 |
| Pt–O1(CO) | 2.0196(16) | 2.048 | 2.065 |
| Pt–N1(py) | 2.040(2) | 2.068 | 2.053 |
| Pt–N2(py) | 2.032(2) | 2.027 | 2.040 |
| N6–N7 | 1.206(3) | 1.213 | 1.200 |
| N3–N4 | 1.202(3) | 1.211 | 1.189 |
| N7–N8 | 1.137(3) | 1.149 | 1.164 |
| N4–N5 | 1.126(4) | 1.148 | 1.170 |
The longest wavelength singlet–singlet transitions in this complex extend up to 469 nm, whereas in 4 they reach to 414 nm.29 The transitions S1, S2 and S3 of 1 show that there is a transfer of electron density from the succinate to the platinum centre (1LMCT) and also contain 1IL (inter-ligand transitions) character. Such transitions (S1, S2, S3) as well as some of the higher energy involve the LUMO, which is antibonding in character with regard to the four equatorial ligands but mainly the azides. Interestingly the LUMO+1, to which electron density is promoted in the higher wavelength transitions (e.g. S8, S14) is orthogonal with respect to the LUMO, making it strongly antibonding mostly towards the pyridines and also the hydroxido and the succinate ligand.
This suggests that occupation of the LUMO or LUMO+1 could potentially lead to dissociation of two out of the six ligands. These two ligands would be in a trans position to each other. Singlet–singlet transitions in the UV region (e.g. S14, S19) occur from lower energy HOMOs and can be assigned as mixed 1LMCT/1IL. In general, all transitions for complex 1 show some d–d character, as metal orbitals always contribute to the frontier molecular orbitals.
As is commonly observed in DFT calculations,44–46 we found that the bond lengths of the ground state of complex 1 are overestimated compared to X-ray, the largest discrepancy being the two Pt–azido bonds, with differences of 0.035 and 0.028 Å (Table 1). In the lowest lying excited-state geometry, Pt–azido bonds are significantly elongated with respect of the ground state (up to 0.44 Å), while variations much less pronounced for the other ligands (Table 1).
Solution reactivity
Complexes 1–3 showed no signs of decomposition in aqueous solution in the absence of light for at least 1 h at ambient temperature.Irradiation studies
 | ||
| Fig. 4 Plot of the decrease in intensity of the N3 → Pt LMCT band of compounds 1–4 over time, when irradiated with green light (λirr 517 nm). | ||
Complex 1 was also irradiated with longer wavelength green light (550 nm, filtered below 530 nm with GG530 filter, 4 nm slits, 2.5 mW cm−2) to investigate the wavelength limit of photoactivation of the complex (Fig. S8E†). No significant change in the UV-vis spectrum of 1 was observed under these conditions.
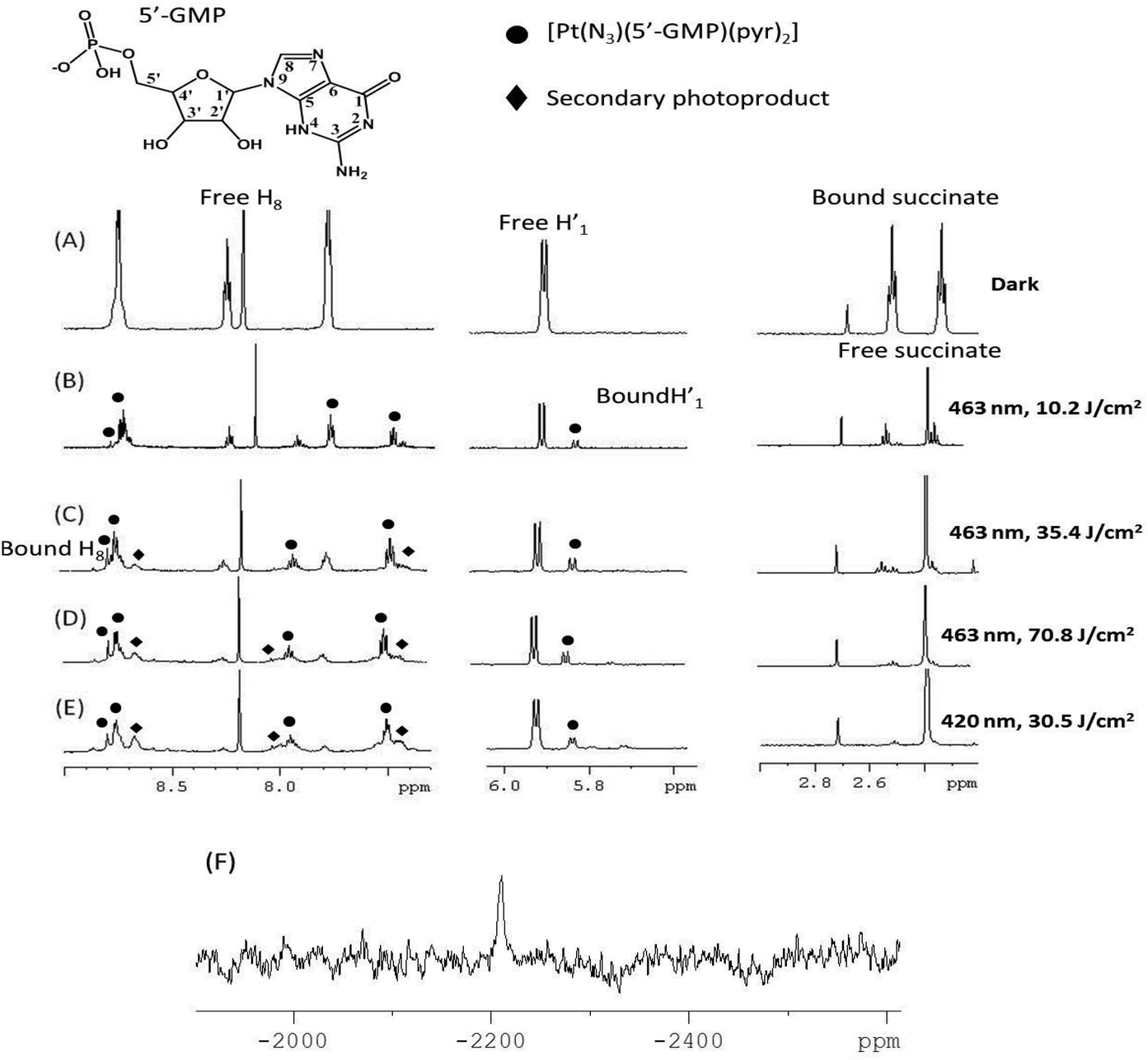 | ||
| Fig. 5 1H NMR spectra (500 MHz, PBS in D2O, pH 7.4, dioxane) of complex 1 and 5′-GMP (2 mol eq.) under the following conditions: (A) dark; then following irradiation for increasing (total) doses of 463 nm light: (B) 10.2 J cm−2; (C) 35.4 J cm−2; (D) 70.8 mJ cm−2; followed by an additional dose of shorter wavelength (420 nm) light (E) 30.5 J cm−2. ● and ◀ denote the two main photoproducts. The sharp singlet at 2.40 ppm corresponds to free succinate. (F) 195Pt NMR spectrum (129 MHz) following irradiation, showing resonance at −2210 ppm, assignable to the previously reported trans-[Pt(N3)(5′-GMP)(py)2].29 | ||
The photoactivity of complex 1 was also investigated through monitoring the release of the succinate ligand in the presence and absence of 5′-GMP. Photorelease of the succinate ligand from 1 occurred ca. 3× more slowly in the presence of 5′-GMP than in its absence (Fig. S10†). A precipitate was also observed in the absence – but not in the presence – of 5′-GMP, consistent with previous reports.29
Due to the limited aqueous solubility of 3, irradiation studies in the presence of 5′-GMP were carried out in 75% MeOH-d4/25% D2O solution. In contrast to complex 1, very little 5′-GMP binding was observed for 3 following irradiation, and pyridine release was clearly observed (Fig. S11,† top).
To establish whether the release of pyridine was solvent-dependent, complex 4 was irradiated in 75% d4-MeOH/25% D2O in the presence of 5′-GMP, with either 420 nm or UVA light. Pyridine release was also observed from 4 under these conditions (Fig. S11,† bottom), which was not observed when 4 was irradiated in D2O or H2O solutions with no MeOH-d4 present.29 The release of the axial N-methylisatoate (N-MIA) ligand from complex 3 in aqueous solution was also monitored by fluorescence emission spectroscopy (Fig. S12†), upon excitation of complex 3 at 320 nm, which demonstrated an increase in fluorescence consistent with ligand release.
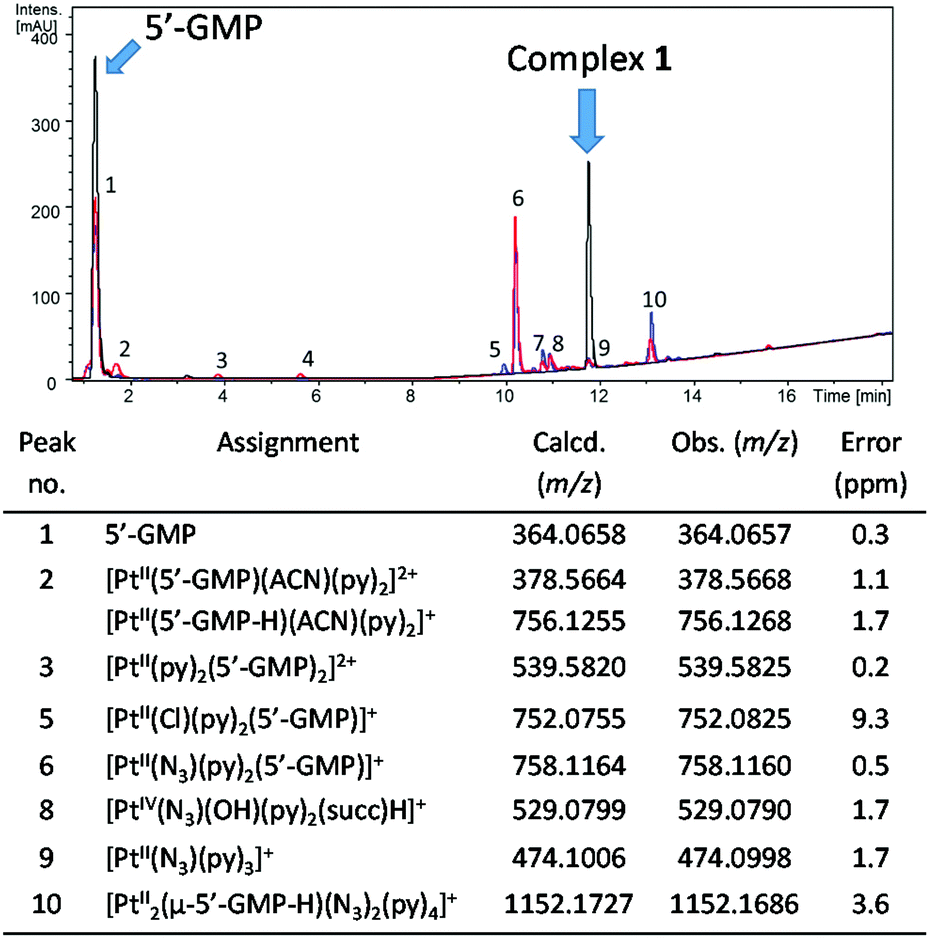 | ||
| Fig. 6 Chromatogram (λdetection 240 nm, ACN/H2O, 0.1% formic acid) for complex 1 in the dark (black trace) or after irradiation in the presence of 2 mol. eq of 5′-GMP in H2O (red trace, 1 (0.5 mM), λirr 420 nm, 30.5 J cm−2) and in PBS (blue trace, 1 (1.5 mM) λirr 463 nm, 153.6 J cm−2) with identifiable peaks assigned. Corresponding mass spectral data for numbered peaks are given in Table S6.† | ||
The photoproducts from irradiation (λirr 420 nm, 45 min) of complex 3 (1.5 mM) plus 5′-GMP (2 mol. eq) in MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (75%:25%, v/v) were also analysed by LCMS (Fig. 7). In addition to the platinum photoproducts, photoproducts corresponding to loss of pyridine were observed, and free N-MI ligand was also detected (peak 7) at 152.0715 m/z.
:H2O (75%:25%, v/v) were also analysed by LCMS (Fig. 7). In addition to the platinum photoproducts, photoproducts corresponding to loss of pyridine were observed, and free N-MI ligand was also detected (peak 7) at 152.0715 m/z.
 | ||
| Fig. 7 Chromatogram (λdetection 240 nm, ACN/H2O, 0.1% formic acid) for complex 3 in the dark (black line) and after irradiation with 420 nm light in the presence of 2 mol. eq of 5′-GMP in H2O (red line) with identifiable peaks assigned. Corresponding mass spectral data for numbered peaks are given in Table S7.† | ||
EPR spectroscopy
Compounds 1, 2 and 4 (1 mM, in PBS) were irradiated with green light (517 nm, 33 mW cm−2) in the presence of the spin-trap 5,5-dimethyl-pyrroline N-oxide (DMPO) (2 mM). The yield of trapped azidyl radicals (DMPO-N3) was determined by the double integration of the EPR signals, quantified by comparison to Tempol (Fig. 8A). EPR spectra of the solutions were also recorded in the dark, during which time no azido radical trapping was observed. Analysis of the DMPO-N3 production at regular time-points (0, 7, 21, 28, 35 min) during the irradiation demonstrated that the two compounds with carboxylate ligands (1 and 2) produced azido radicals when irradiated with 517 nm light, whereas the dihydroxido compound 4 did not produce an appreciable concentration of azidyl radicals. Complexes 1–4 were then investigated for their ability to generate azidyl radicals in DMF/H2O (75%/25% v/v), since it was a suitable solvent system for allowing direct comparison of all complexes. | ||
| Fig. 8 Time dependent trapping of azidyl radicals with DMPO (2 eq.) upon continuous irradiation of complexes with green light (517 nm, 33 mW cm−2); (A) complexes 1, 2 and 4 PBS (1 mM) and (B) complexes 1–4 in DMF/H2O (75%/25% v/v, 2 mM). | ||
The yield of azido radicals was determined at regular time-points during irradiation (0, 7, 14, 21, 28, 35 min), following a similar trend to the PBS solution, with compounds 1 and 2 giving rise to more intense EPR signals than compound 4, which, with the inclusion of DMF, did produce azidyl radicals under green light irradiation.
Compound 3 produced only a very low yield of azidyl radicals (Fig. 8B). To investigate the effect of the N-MI ligand on the yield of azidyl radicals, a solution of complex 4 (2 mM, DMF/H2O (75%/25% v/v)) was irradiated with shorter wavelength light (blue, 463 nm, 64 mW cm−2) in the presence of free N-MI (2 mol eq., 4 mM) and DMPO (2 eq., 4 mM) and the azido radical yield was measured at regular time-points. This demonstrated a marked decrease in azido radical trapping in presence of the free ligand compared to in its absence (Fig. 9).
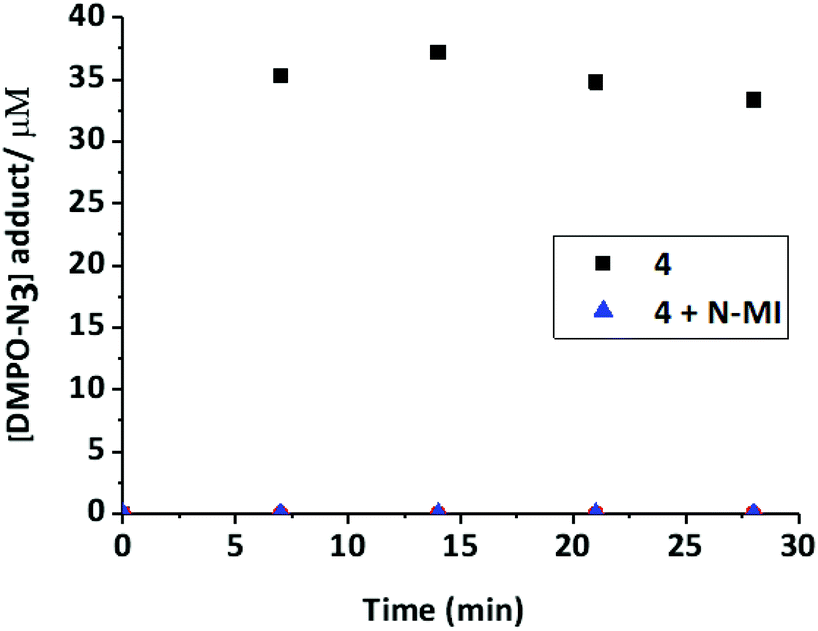 | ||
| Fig. 9 [DMPO-N3] signal upon continuous irradiation (463 nm) of complex 4 (2 mM) alone, or in the presence of 2 mol eq. of the N-MI ligand in DMF/H2O (75%/25%) at ambient temperature. | ||
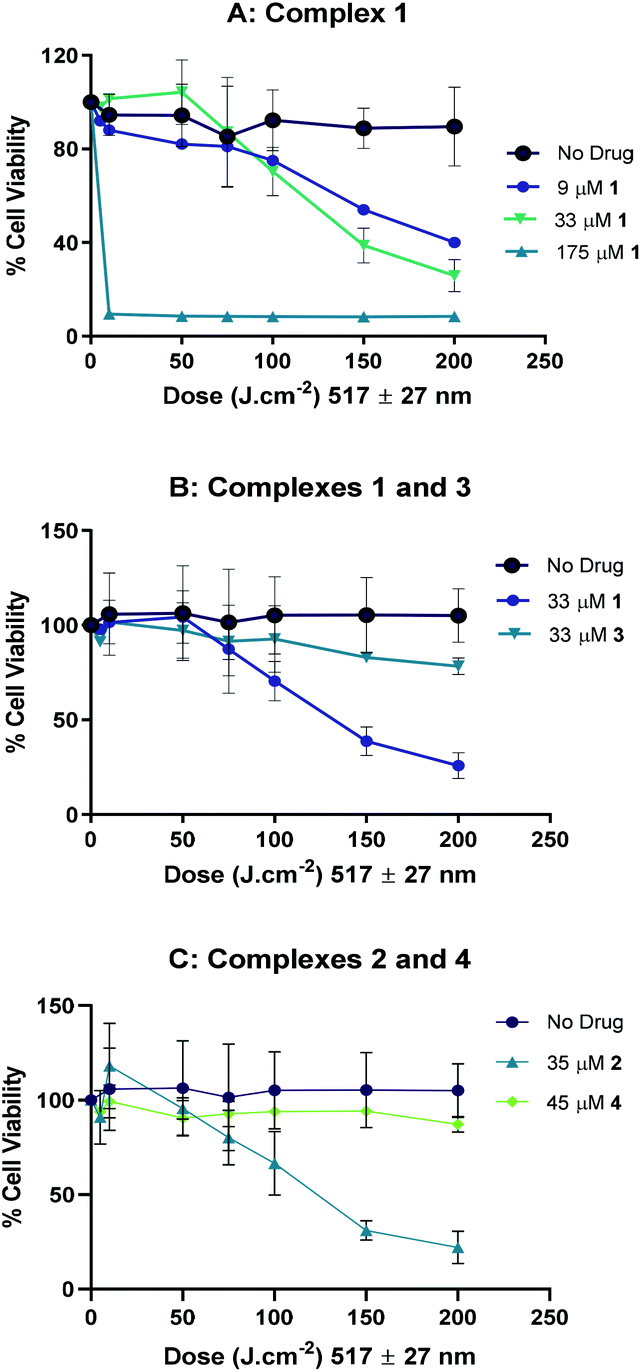 | ||
| Fig. 10 Anti-proliferative activity of complexes 1–4 after photoactivation with green light (517 ± 27 nm) in A2780 cells: (A) in the presence of increasing concentrations of complex 1; (B) in the presence of complexes 1 or 3 and (C) in the presence of complexes 2 or 4. Data points represent the mean ± SD of two independent experiments performed in triplicate. | ||
| Complex | Cell linea | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A2780 | A2780cis | OE19 | ||||||||||
| IC50/μM | 95% CI | Viability ± SE at MAD (%) | PI | IC50 /μM | 95% CI | Viability ± SE at MAD (%) | PI | IC50/μM | 95% CI | Viability ± SE at MAD (%) | PI | |
| a IC50: concentration of drug that inhibited dye uptake by 50%. 95% CI: 95% confidence interval of the IC50 value. MAD: maximum administered dose. In the absence of a cytotoxic IC50 value, the reported PI values are ‘at least’ the value stated (denoted by ‘>’). Results are the mean of 2–5 independent experiments performed in triplicate. | ||||||||||||
| 1 | 2.7 | 2.2–3.4 | 65.5 ± 13.8 | >49 | 5.4 | 4.3–6.7 | 75.4 ± 2.3 | >37 | 8.2 | 5.3–12.7 | 89.3 ± 16.4 | >21 |
| 2 | 3.7 | 3.0–4.6 | 62.2 ± 5.1 | >44 | 4.0 | 2.9–5.4 | 59.9 ± 6.8 | >41 | 10.3 | Wide | 74.4 ± 3.5 | >15 |
| 3 | 13.4 | 6.0–30.3 | 79.5 ± 5.6 | >2 | 13.0 | 6.8–24.6 | 77.5 ± 1.6 | >2 | 17.5 | 10.3–29.8 | 85.5 ± 2.1 | >1.9 |
| 4 | 5.1 | 3.7–7.0 | 98.7 ± 8.4 | >41 | 8.0 | 5.2–12.3 | 81.4 ± 5.3 | >26 | 8.4 | 6.5–10.8 | 98.6 ± 5.1 | >25 |
| Complex | Without tryptophana | With 1 mM tryptophana | Protection factor | ||
|---|---|---|---|---|---|
| IC50 (μM) | 95% CI | IC50 (μM) | 95% CI | ||
| a IC50: concentration of drug that inhibited dye uptake by 50%. 95% CI: 95% confidence interval of the IC50 value. Results are the mean of 2 independent experiments performed in triplicate. | |||||
| 1 | 2.4 | 2.1–2.7 | 7.3 | 2.3–23.3 | 3.0 |
| 4 | 5.3 | 3.1–9.2 | 22.5 | 7.6–67.0 | 4.2 |
Complexes 3 (33 μM) and 4 (45 μM) demonstrated negligible photoactivity under these conditions (Fig. 10B and C respectively). No reduction in cell viability by complex 1 was observed under irradiation with even longer-wavelength light (570 ± 27 nm, 175 μM complex 1: Fig. S14†). These experiments also demonstrate the lack of dark toxicity of the complexes compared to the control cells treated with light only.
The phototoxicity of complex 1 correlated with the results of a comet assay (which was not performed with complex 2 as well, due to time constraints), which showed increased DNA migration when A2780 cells were irradiated (100 J cm−2 420 nm light) in the presence of the drug (Fig. 11A). This increased migration might be due to reactive species produced on photoactivation, but is more likely due to cell death, as this combination of drug and light is cytotoxic. DNA migration was not increased when the same concentration of drug was activated with longer-wavelength (100 J cm−2 517 nm) light (Fig. 11B) which could be due to reduced toxicity at this wavelength (ca. 50% cell death) or the nature of the reactive species produced.
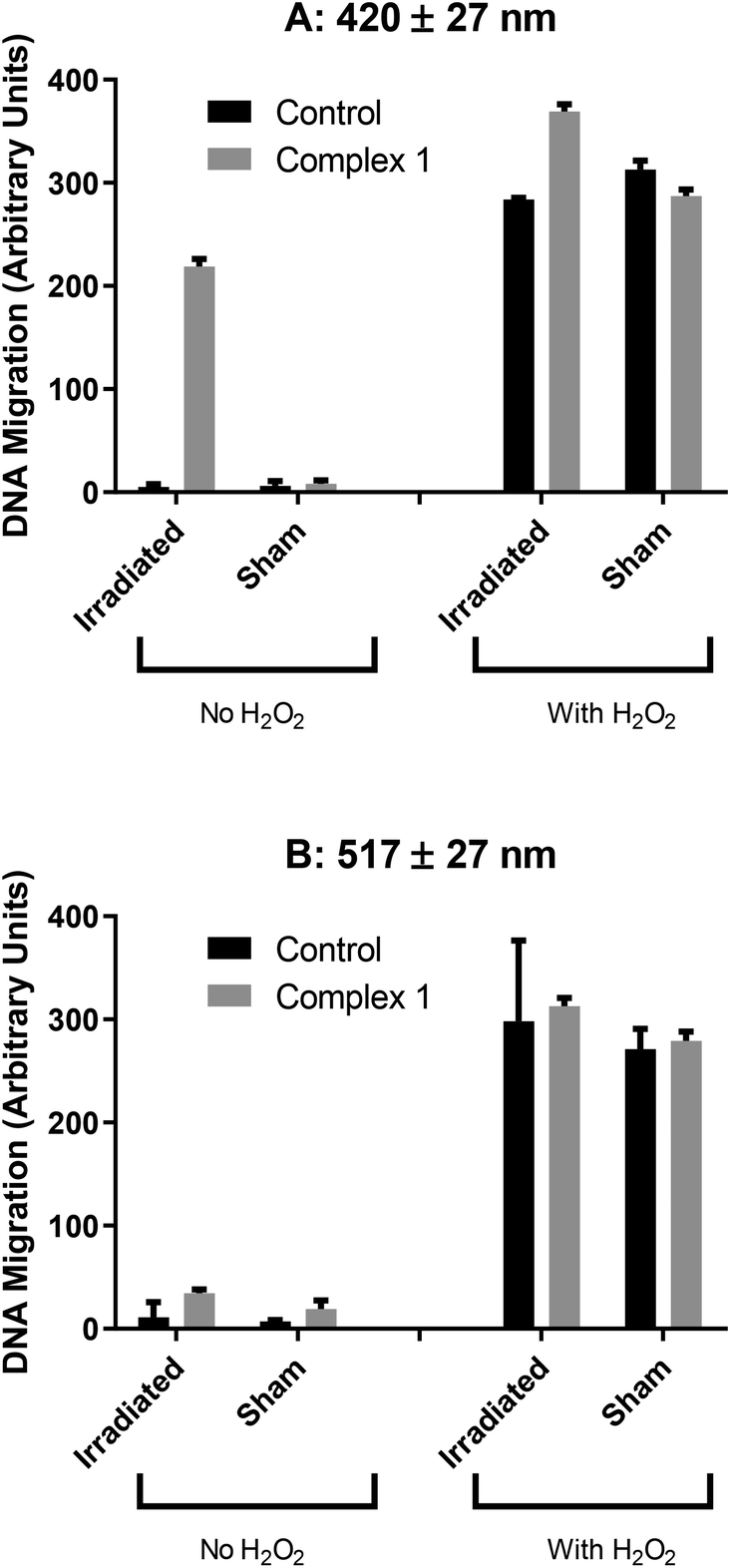 | ||
| Fig. 11 DNA migration in A2780 cells following photoactivation of complex 1 (35 μM) with 10 J cm−2 of either (A) blue 420 nm or (B) green 517 nm light. | ||
Cellular uptake studies
Cellular uptake studies were carried out for complexes 1–4 with the established Pt(II) anti-cancer complex cisplatin as a control, in the A2780 cell line. The concentration used for the uptake of all of the complexes was 20 μM, since no dark toxicity was observed at this concentration for any of the compounds. Uptake of Pt from the compounds at 37 °C followed the order (from highest to lowest): 3 (300 ng Pt/106 cells) ≫ 4 (32 ng Pt/106 cells) > cisplatin (29 ng Pt/106 cells) > 2 (25 ng Pt/106 cells) > 1 (15 ng Pt/106 cells) (Fig. 12, top). The temperature-dependence of cellular uptake was studied by measuring uptake at 4 °C, 18 °C and 37 °C (Fig. 12, bottom) for compounds 3 and 4. Both compounds followed the same trend, with uptake increasing with increasing temperature.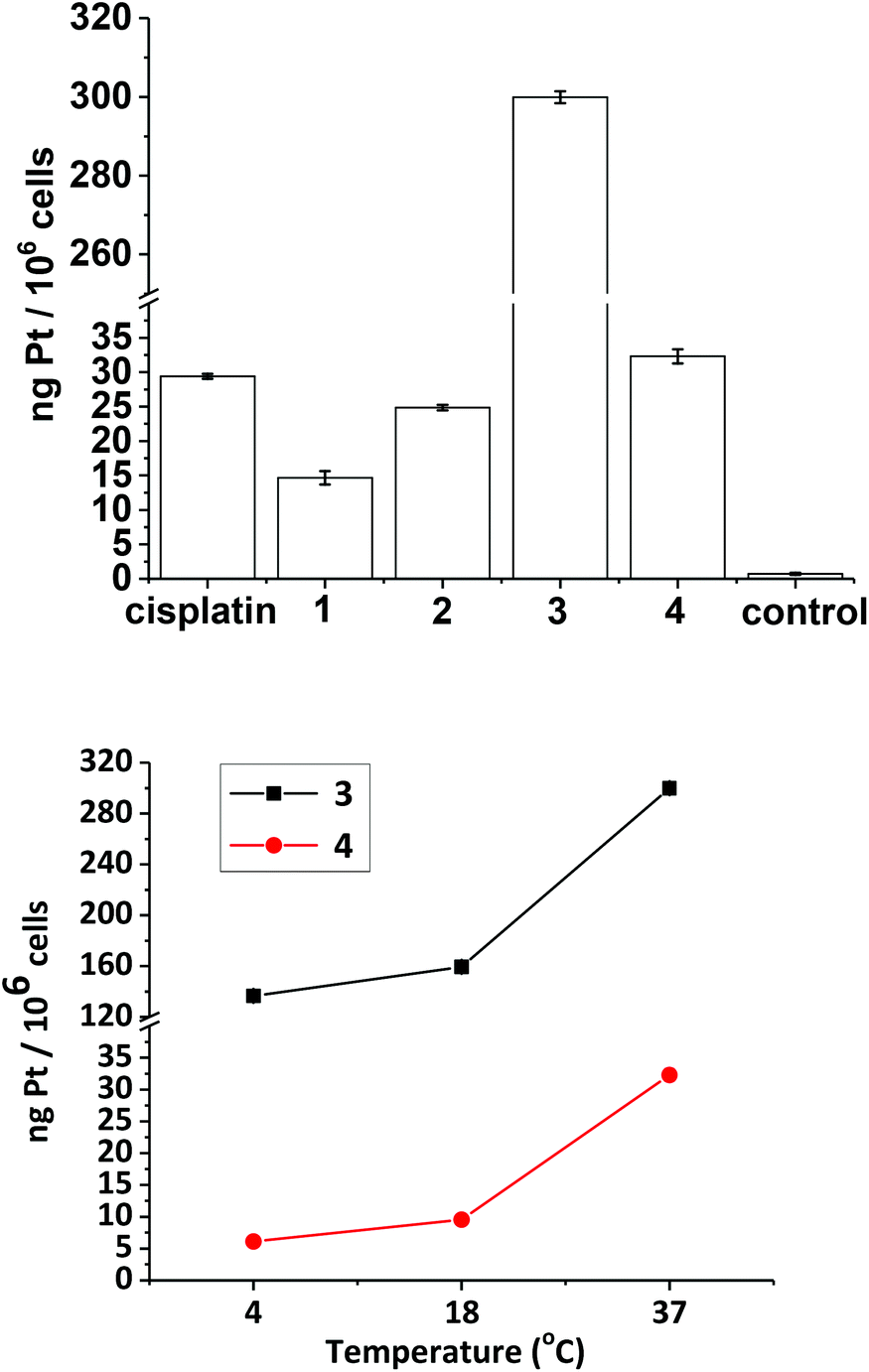 | ||
| Fig. 12 Top: cellular uptake studies of compounds 1–4 (ng Pt/106 cells) at 37 °C with cisplatin for comparison. Bottom: temperature-dependence on the cellular uptake of complexes 3 and 4, after 1 h drug incubation in the dark. Each experimental condition was carried out in triplicate in the A2780 human ovarian cancer cell line. | ||
Discussion
Synthesis and characterisation
trans-Dihydroxido Pt(IV) complexes can be axially functionalised through use of anhydrides, acid chlorides, pyrocarbonates, and isocyanates to form carboxylates, carbonates and carbamates, respectively.27,37–39 Three novel complexes 1–3 have been synthesised, axial ligation was achieved through reaction of 4 with the corresponding anhydride (either succinic anhydride or N-methylisatoic anhydride) through a ring-opening reaction. Complex 2 was then obtained from 1via esterification of the succinate ligand with sodium propanolate, resulting in formation of a 4-oxo-4-propoxybutanoate group. Incorporation of the N-methylisatoate ligand in complex 3 was chosen as it was anticipated that it could impart greater photosensitivity to the complex.NMR spectroscopy and X-ray crystallography
The higher 195Pt NMR chemical shifts (D2O) for complexes 1 (1059 ppm) and 2 (1065 ppm) in comparison to 4 (942 ppm) indicate that the 195Pt nucleus is more deshielded following carboxylation; complex 3 was measured in DMSO-d6 due to solubility constraints and therefore a direct comparison for 3 is less helpful. Lengthening of the Pt–P bond in the case of platinum complexes containing phosphine ligands has been reported to increase the 195Pt NMR chemical shift.40 Here, the Pt–O bonds to carboxylate ligands are systematically marginally longer than the Pt–OH bond for 1–3 (Table S2†), which may account for the increase in the chemical shift.Activation of prodrugs with longer-wavelength light is desirable to achieve greater penetration into tissues. Therefore, the longer wavelength absorption bands in the UV-vis spectra are of particular interest for photoactivatable metallodrugs. Green light has greater clinical relevance, penetrating ca. 50% more deeply into tissues than shorter wavelength blue light. Complexes 1–4 show two absorption bands at ca. 260 nm and 300 nm (Fig. S6 and S8†), composed mainly of transitions with 1LMCT character. The absorption bands in compounds 1–3 are red-shifted in comparison with those in the precursor 4, indicating that modification of the axial group has the potential to enhance the photolability of the Pt(IV)-diazido prodrugs when irradiated at longer wavelengths. The nature of the axial ligand also affects the pharmacological properties of the drugs, such as lipophilicity, water solubility and stability. Biological reducing agents in the cell and in the blood plasma include ascorbate, glutathione as well as cysteine-rich metallothioneins.41 Glutathione levels vary widely, but in healthy adults, mean GSH concentrations are reported as 849 ± 63 μM in whole blood and 3.39 ± 1.04 μM in blood plasma.42 Intracellular levels are typically higher, around 2–8 mM in the cytosol.43 Compared to disease-free tissue, glutathione levels tend to be elevated in ovarian tumours.44 It is known that complexes with axial acetate ligands have the potential to be more readily reduced than those with hydroxido ligands: the monocarboxylato Pt(IV) analogue of oxoplatin (cis,trans,cis-[PtCl2(OH)2(NH3)2]) is reduced faster in the presence of glutathione than oxoplatin.41 We determined that 1 showed good stability in the presence of 2 mol eq. of GSH, which is important for its potential development as a photoactivatable Pt(IV) prodrug, since these need to be relatively stable in a biological environments following administration before being activated by irradiation.
DFT and TD-DFT
The theoretical UV-vis maximum appears at 286 nm (33748 mol−1 cm−1) compared to the experimental maximum at 299 nm (ε = 18796 mol−1 cm−1). TD-DFT calculations therefore slightly underestimate the absorption maximum by ca. 10 nm and overestimate the value of the extinction coefficient. The method used for these TD-DFT calculations is the same as that used for 4, for which a closer match between the theoretical and the experimental extinction coefficient was found.30 The larger difference between calculation and experiment for 1 is therefore ascribed to the inclusion of the succinate ligand. Although functional and basis set benchmarking testing might improve the agreement between calculated and experimental transitions, the results obtained are still satisfactory to characterise the nature of the UV-Vis absorption bands. Overall, our results show that light excitation would results in the formation of low-energy singlet states of mixed nature, in which the strong 1LMCT component and the nature of the LUMOs involved is consistent with a variability in the photoproducts.
Besides, transition metal complexes such as d6-Pt(IV) derivatives usually undergo intersystem crossing (IC) to triplet states upon light-excitation. Therefore, the nature of the lowest-lying triplet state is crucial to understand their photochemical behavior, since such a state is significantly populated before the molecule relaxes to the ground state. Elongation of bonds in the triplet state indicates that they could break upon photoexcitation, and therefore suggests which ligands are most likely to dissociate. The difference between the lowest-lying excited-state geometry and the ground state demonstrates that for complex 1, both Pt-azido bonds show the biggest elongation, the Pt–OH bond does not elongate, the Pt-succinate bond is elongated, and one of the Pt-pyridine bonds is elongated whereas the other is shortened (Table S2†). This is consistent with the experimental observations which include release of the succinate ligand (Fig. 5) and azido radicals (Fig. 8).
Solution reactivity
Photoreductions of 1–4 were monitored by UV-vis spectroscopy (spectral output of the light sources are given in Fig. S7†), showing a decrease of the N3 → Pt ligand–to–metal charge-transfer (LMCT) band (at ca. 300 nm) due to the loss of an azido ligand(s) (Fig. S8†), following an exponential decay profile (Fig. 4, 4). The photoreduction occurred faster for the monocarboxylato complexes 1–3 than for the hydroxido complex 4, indicating that the carboxylate functionality enhances the photoreactivity of these complexes. Furthermore, complex 2, showing the fastest photodegradation kinetics, shows the longest Pt–O (carboxylate) bond, which photodissociates upon irradiation. The increased rate of photodegradation for the monocarboxylate complexes correlates with the extinction coefficient, which is higher at longer wavelengths (Fig. S6†).EPR spectroscopy
The formation of azidyl radicals when Pt(IV) diazido complexes are irradiated is thought to play a key role in the mechanism of action of these prodrugs.35 Therefore the photoproduction of azidyl radicals from 1–4 was studied by EPR spectroscopy. Under green (517 nm) light irradiation, PBS solutions of complex 4 did not generate trapped radicals, whereas 1 and 2 produced radicals at concentrations up to 68 μM and 55 μM, respectively. The identity of the trapped radical was confirmed as DMPO-N3 by matching the hyperfine splitting constants (HFSC) to the theoretical data reported previously.47,48 Hydroxyl or carboxylate radicals, which could potentially be released from the Pt(IV) complex, would give a 1:2:2:1 pattern when trapped by DMPO; this signal was not detected in the spectrum. Initial production of radicals (after 7 min of irradiation) was lower for 1 than for 2, although the azidyl radical yield after 14 min of irradiation was slightly higher for 1 than for 2. Both complexes 1 and 2 gave rise to significantly greater azido radical production under green light irradiation than the parent dihydroxido complex 4. A similar experiment was carried out in DMF/H2O (75%/25% v/v) to enable comparison between all four complexes and to investigate the effect of solvent. Compounds 1, 2 and 4 in DMF/H2O (75%/25% v/v) gave broadly similar results as seen in PBS, following prolonged irradiation, with 1 giving the highest and 4 the lowest azidyl radical yield, although at the first measurement (7 min) in DMF/H2O 1 showed greater azide radical production than 2, whereas 2 had showed greater initial azide radical production than 1 in PBS. Irradiation of compounds 3 and 4 resulted in a similar, although modest, concentration of azide radicals being trapped (which for 4 was higher than the radical production observed in PBS), despite the rate of photodegradation of 3 being ∼3-fold higher than that of 1 (Fig. 4). The N-methylisatoate ligand (axially bound in 3) may act as a radical scavenger, which could explain the low levels of azido radical trapping for 3. In order to confirm this hypothesis, complex 4 was irradiated (λ = 463 nm, 64 mW cm−2) in the presence of N-methylisatoic acid (2 mol eq.). The production of azidyl radicals was entirely quenched by the presence of N-methylisatoate acid, suggesting that upon release, this ligand may readily react with N3˙ Fig. 9. Methyl anthranilate, which is structurally related to the N-methylisatoate ligand, has been shown to quench singlet oxygen due to the presence of the anilinic moiety.49
Biological data
The phototoxicity of complexes 1, 2 and 4 towards A2780 cells after irradiation with green 517 nm light did not correlate with levels of Pt uptake by the cells (Fig. 12), but did correlate with azidyl radical production in PBS (Fig. 8), implicating the azide radicals in the phototoxic mechanism of action of the complexes. If the azide radicals are scavenged by the N-methylsatoate ligand this could explain the lower relative phototoxicity of complex 3, despite cellular uptake studies demonstrating a Pt content in A2780 cells for 3 at least 10-fold that detected for 1, 2, and 4. Furthermore, co-incubating A2780 cells with 1 mM tryptophan protected against blue-light induced phototoxicity in cells which had been treated with either 1 or 4, with slightly more protection afforded to the cells treated with 4. Tryptophan is an efficient scavenger of singlet oxygen50 and can also quench azidyl radicals.36 It should be noted that blue light itself will photosensitise endogenous cellular chromophores, a process which could be affected by tryptophan, but the sensitivity of the cells themselves to blue light was insignificant (survival 103 ± 5.6%) and this was not significantly altered by the presence of tryptophan.The irradiation of both 1 and 4 in the presence of 5′-GMP (2 mol eq.) in PBS produced the mono-5′-GMP Pt(II) adduct trans-[Pt(py)2(5′-GMP)(N3)] (Fig. S9†). However, 23% of 5′-GMP was bound to platinum in the case of 1, whereas only 9% was bound in the case of compound 4, as judged by analysis of the starting/product 5′-GMP resonance S1′/P1′. This result, along with the UV-Vis and EPR spectroscopy studies demonstrates the enhanced photoreactivity of the carboxylate complex 1versus the dihydroxido complex 4. TD-DFT calculations gave an insight into the enhanced photoactivity, showing that 1 has dissociative transitions which can extend up to 469 nm. The photoproducts from the irradiation of 1 with blue light (463 nm) were characterised by 1H NMR and 195Pt-NMR spectroscopy as well as LCMS. The release of succinate as well as azidyl radicals was observed (Fig. S10†). The reduction of Pt(IV) to Pt(II) requires the gain of two electrons, however, the only radical detected by EPR spectroscopy was the azidyl radical. Since the major product generated after irradiation is a mono-azido Pt(II) complex, the loss of both azides as azidyl radicals cannot be the main photochemical pathway to products in PBS. The second electron could originate from either the hydroxido or the carboxylato ligands, which could react rapidly with another radical or solvent molecule and thus be quenched.
The assignment of trans-[Pt(py)2(5′-GMP)(N3)] was readily confirmed as the 1H and 195Pt-NMR spectral data match those chemical shifts assigned previously for trans-[Pt(py)2(5′-GMP)(N3)].29 Irradiation of 1 with slightly shorter wavelength light (420 nm vs. 463 nm) did not produce in any significant change in the photoproducts, the change in irradiation wavelength appeared to only affect the rate of formation of products. Similar to the results obtained upon excitation at 463 nm, apart from the major product trans-[Pt(py)2(5′-GMP)(N3)], only very low intensity signals corresponding to other 5′-GMP adducts were visible in the 1H-NMR spectra.
LC-MS analysis was carried out after the irradiation of compound 1 in the presence of 2 mol eq. of 5′-GMP, with the majority of the species successfully assigned (Tables S1 and S6†). In H2O, in addition to trans-[Pt(py)2(5′-GMP)(N3)], other 5′-GMP adducts were observed including trans-[Pt(ACN)(GMP)(py)2]2+trans-[Pt(ACN)(GMP-H)(py)2]+, [Pt(N3)(py)3]+ and the di-platinum species [Pt2(μ-5′-GMP-H)(N3)2(py)4]+. The ACN adducts form due to use of acetonitrile as HPLC solvent, but in biological media, it is anticipated this coordination site would be readily occupied by another ligand. Irradiation in PBS (139 mM chloride) resulted in a chloride readily binding to give [PtII(Cl)(py)2(5′-GMP)]+. This is consistent with a report that the amount of platinated DNA was significantly reduced when 4 was irradiated in the presence of 100 mM chloride.51 The [M − N3]+ species observed for 1 in the mass spectrum before irradiation (529.0750 m/z, Fig. S1,† top) was also observed post-irradiation, but only at low intensity (529.0790 m/z, peak 8, Fig. 6 and Table S6†). We suggest that in-source fragmentation and loss of azide may occur in the mass spectrometer in the absence of irradiation, but this does not seem to be significantly enhanced by irradiation. The corresponding [M − N3]+ species was also detected for 3, before (562.1210 m/z, Fig. S1,† bottom) and following irradiation (562.1171 m/z, peak 8, Fig. 7 and Table S7†). This could be a Pt(III) complex or a metallacyclic species in which the N-methylanthranilate ligand is doubly deprotonated and coordinates to Pt(IV) in a bidentate chelating mode. Pt(III) intermediates are not unusual in mass spectrometry, but the elution of the species from the HPLC column suggests that it is stable, and therefore may be more likely to correspond to a metallacyclic Pt(IV) complex. Very little 5′-GMP binding was observed for 3 in 75% MeOH-d4/25% D2O (v/v) following irradiation, and photoproducts of 3 detected by LC-MS included the axial ligand (N-MI) (Fig. 7 and Table S7†), whereas for 1, no assignable photoproducts retained the succinate ligand. In general, the photoproducts detected by LCMS were consistent with those previously seen following photoplatination of the synthetic oligodeoxynucleotide 5′dCATGGCT (containing two consecutive guanine nucleobases) by 1.15
The attachment of fluorescent tags to platinum compounds is useful to study their temporal and spatial distribution in living cells. Frequently, the attachment of fluorescent tags to Pt(IV) results in fluorescence quenching, when reduced to Pt(II), within the cell, fluorescence enhancement occurs.52,53 The fluorescence emission spectrum of 3 – which incorporates a fluorescent ligand (N-MI) in the axial position – showed a single broad band with maximum at 420 nm, similar to the maximum emission wavelength of the free ligand N-MIA, and repeated excitation at 320 nm caused an enhancement in the fluorescence emission at 420 nm, which is consistent with the detection of free N-MIA ligand by LCMS (Fig. S12†).
Pyridine release was not observed when complex 4 was irradiated with UVA light in aqueous solution.29 However, irradiation of either 3 or 4 in 75% MeOH-d4/25% D2O (v/v), released of pyridine, particularly upon UVA irradiation (Fig. S11†). This highlights the importance of the solvent and the environment on the photodecomposition pathways of these complexes; if these drugs accumulate in a hydrophobic, non-aqueous environment within the cell (e.g. membranes) different photoproducts may be produced. The release of pyridine is consistent with TD-DFT calculations performed on compounds 1 and 429 which showed that the LUMO and LUMO+1 are strongly antibonding towards all ligands.
Complexes 1 and 2 showed promising photocytotoxicity towards ovarian cancer cells (A2780) with low-dose blue light (420 nm, 5 J cm−2). The phototoxicity of 1 and 2 are 2-fold higher compared to compound 4. Complex 3, was the least phototoxic with an IC50 value of 13.4 μM. This is despite complex 3 showing ca. 10-fold greater uptake in comparison to the rest of the compounds (Fig. 12, top). The comparatively modest IC50 for 3 may be related to the low yield of azidyl radicals or other factors, such as sub-cellular distribution. Cell uptake of 2 is ca. 2-fold higher than that of 1 in A2780 cells (from 15 to 25 ng/106), attributable to the increased hydrophobicity arising from the propyl ester functionality in 2. However, only a slight improvement in the cytotoxicity was observed (IC50 values of 2.7 μM for 1 and 3.7 μM for 2). Complex 4 showed greater accumulation than both 1 and 2, but lower photocytotoxicity, underlining the importance of triggered reduction and production of azido radicals in the biological effects.
The transport of molecules across the membrane of a cell can be either active (energy-dependent) or passive (energy-independent).54 Platinum(II) complexes are taken up by both passive and active transport. For cisplatin, copper transporters (e.g. Ctr1) play a major role.55–57 Organic cation transporters constitute another class of important transporters, responsible for the uptake of picoplatin in mice.58 Cisplatin shows minimal cellular uptake at 4 °C (0.005 ng of Pt per 106 cells) consistent with predominantly energy-dependent transport.59 In contrast, complexes 3 and 4 accumulated to a greater extent at this temperature, with accumulation increasing with temperature (Fig. 12, bottom). The uptake of these complexes is therefore likely to be through a combination of both passive and active transport, the passive transport appears to be enhanced greatly by the incorporation of an aromatic ligand in the axial position, as shown by complex 3.
Complexes 1 and 2 were also more effective against the Pt-resistant A2780cis cell line compared to complex 4. These three complexes all showed activity against OE19 oesophageal carcinoma cells with blue light (420 nm). Blue light is already in widespread clinical use for treating actinic keratoses – precancerous lesions on the skin, and has been shown to be more appropriate for treating superficial oesophageal cancerous lesions in animal models than green light, since it causes less damage to the underlying healthy tissue.60 For other applications however, longer-wavelength light activation is more desirable, and therefore the response of 1 and 2 to irradiation with green (517 nm) light and photocytotoxic activity towards both in ovarian cancer (and cisplatin-resistant ovarian cancer) cell lines is encouraging. The photocytotoxicities (λmax 420 nm) of 1–3 are broadly consistent with guanidinoneomycin-derivatised succinate complexes in SK-MEL-28 cancer cell lines; in the SK-MEL-28 line the succinate complex 1 was less active than complex 4,15 whereas complex 1 was slightly more active than 4 in the A2780, A2780cis and OE19 cell lines reported here.
Conclusions
Complexes trans,trans,trans-[Pt(N3)2(OH)(OCOR)(py)2] where OCOR = succinate, 4-oxo-4-propoxybutanoate and N-methylisatoate (1, 2 and 3, respectively), were synthesized and characterised by NMR spectroscopy, MS, UV-vis spectrometry and X-ray crystallography. Photoactivation studies comparing complexes 1–3 to their dihydroxido synthetic precursor trans,trans,trans-[Pt(N3)2(OH)2(py)2] 4 indicate a 3-fold faster rate of photodegradation for 1–3 through monitoring loss of the LMCT band in the UV-vis absorbance spectrum when irradiated with green light (517 nm). This is consistent with TD-DFT transitions indicating greater absorption at longer wavelengths for 1 in comparison to 4. Consistent with this, complex 1 also showed significantly greater (2.6-fold) 5′-GMP complexation than 4 when irradiated with the same dose of blue (463 nm) light. EPR spectroscopic studies of 1, 2 and 4 demonstrated that considerably higher quantities of azidyl radicals were generated under green light (517 nm) irradiation of 1 and 2, with correspondingly lower (DMF/H2O) or minimal (PBS) radical production for 4. Complex 3 incorporates the fluorescent ligand N-methylisatoate (N-MIA). Irradiation of 3 also resulted in an increase in fluorescence, consistent with platinum(IV) reduction and N-MIA release. LC-MS studies upon blue light irradiation (420 nm) resulted in the release of the axial ligands (succinate for 1 and N-MIA for 3) and formation of a variety of photoproducts. Irradiation of 3 and 4 in non-aqueous solvent (75% MeOH-d4/25% D2O) in the presence of 5′-GMP also resulted in release of pyridyl ligand(s) from the complexes, which is not observed for 4 in aqueous or buffered solutions. The pKa of the pendant carboxylate of complex 1 (5.13), suggests it may be negatively charged under biological conditions, which could explain the lower cellular uptake of 1 in comparison to 2 and 4. Although complex 3 showed high levels of cellular accumulation, it showed poor aqueous solubility in comparison to 1 and 2, and also lower azido radical trapping upon illumination with green light, possibly as a result of quenching by the N-MIA ligand. Cellular uptake studies indicated that complexes 3 and 4 may enter cells through a combination of passive diffusion and active transport.Phototoxicity studies with 420 nm (blue) irradiation on A2780 cells showed that complexes 1 and 2 (IC50 = 2.7 and 3.7 μM) were ∼2-fold more photocytotoxic than 4 (IC50 = 5.1 μM). These complexes were also more effective towards the platinum-resistant A2780cis cell line in comparison to 4, particularly complex 2. The propyl derivative 2 was synthesised to produce a neutral compound, potentially also with greater lipophilicity than 1, and although this demonstrated greater cellular accumulation in A2780 cells, this did not translate to greater photocytotoxicity than 1 in this cell line. Crucially, both complexes 1 and 2 also efficiently killed A2780 cells when activated by green (517 nm) light. Of the novel complexes, 3 was the least photocytotoxic (13.4 μM). Although the photoproducts formed by complexes 1, 2 and 4 are similar, the improved photoactivity that the carboxylate ligand confers to 1 and 2 results in rapid generation of Pt(II) photoproducts and higher concentrations of azide radicals for a given irradiation dose.
The derivatisation of one axial position of platinum(IV) azido complexes with carboxylate ligands enables them to be activated with green (517 nm) light, whilst remaining stable in the presence of biological reductants (GSH). Complexes 1 and 2 have the potential to be further modified or developed in their own right as phototherapeutics, particularly for superficial cancers; complex 2 in particular showed promising activity towards the cisplatin-resistant A2780cis cell line. The production of azido radicals by the complexes is clearly of crucial importance to their photocytotoxic mechanism of action.
Experimental
Synthesis and characterisation of synthetic precursors 5, 6 and 7 and 4 are given in the ESI.† The NMR spectroscopic assignment of specific hydrogens and carbons for complexes 1–3 is illustrated in Fig. S13.†Caution! No problems were encountered during this work, however heavy metal azides are known to be shock-sensitive detonators, therefore it is essential that platinum azides are handled with care. The Pt-diazido complexes were synthesised and handled under dim lighting conditions.
trans,trans,trans-[Pt(N3)2(OH)(succ)(py)2](1)
Carboxylation of one of the axial ligands of 4 was achieved by modifying a published method.61trans,trans,trans-[Pt(N3)2(py)2(OH)2] 3 (0.150 g, 0.318 mmol) was dissolved in anhydrous DMSO (1.8 mL) under N2, in the presence of activated molecular sieves. Succinic anhydride (1.1 mol eq., 0.035 g) was added and the reaction stirred in the dark at 40 °C for 48 h. The suspension was filtered and water (∼2 mL) was added. The yellow solution was allowed to stand at room temperature until a crystalline solid precipitated (3–4 h). The remaining solution was decanted and the crystalline precipitate was washed with diethyl ether and then filtered (yield = 0.096 g, 52%). Crystals suitable for single-crystal X-ray diffraction were obtained by slow diffusion of water into the DMSO reaction mixture at room temperature. Purity (HPLC, UV-vis absorbance): 94%.1H-NMR (D2O, 600 MHz, pH 5.18, ppm): δ = 8.77 (dd, 3J1H1H = 5.78 Hz, 3J195Pt1H = 11 Hz, 4H, Ho), δ = 8.27 (t, 3J1H1H = 7.64 Hz, 2H, Hp), δ = 7.80 (t, 3J1H1H = 7.04 Hz, 4H, Hm,), δ = 2.58 (t, 3J1H1H = 6.90 Hz, 2H, H1), δ = 2.48 (t, 3J1H1H = 6.90 Hz, 2H, H2). 13C-NMR (d4-MeOD, 150 MHz, ppm): δ = 178.7, (Ca), δ = 177.57 (Cd), δ = 150.89, (Ce), δ = 143.15 (Cg), δ = 127.37 (Cf). 195Pt-NMR (D2O, 129 MHz): δ = 1059 ppm.
ESI-MS: [M + Na]+ (m/z) calc. (C14H16N8NaO5Pt), 594.0741; found, 594.0783.
trans,trans,trans-[Pt(N3)2(OH)(succ-Pr)(py)2](2)
trans,trans,trans-[Pt(N3)2(OH)(Succ-Pr)(py)2] 4 (0.100 g, 0.176 mmol) was pre-dried using P2O5 and then dissolved in dry DMF (1 mL) under argon. A stock solution of CDI (1,1′-carbonyldiimidazole) in DMF (0.194 M) was prepared under dry conditions and 1.2 mL was added (1.2 mol eq.) to the solution of the platinum complex and allowed to stir at 50 °C for 20 min, under continuous purging with argon. Then, sodium propanolate (2 mL), formed by the dissolution of sodium metal (0.02 g) in absolute propanol (20 mL), was added and the reaction carried out at room temperature for 48 h. DMF was removed under vacuum and the crude product was purified by column chromatography (eluent: EtOAc:MeOH, 11:1). After solvent removal from the combined fractions, the solid was recrystallised from DCM/diethyl ether (yield = 0.060 g, 56%). Crystals suitable for X-ray crystallography were grown by slow evaporation from H2O at ambient temperature. Purity (HPLC, UV-Vis absorbance): 96%.
1H-NMR (D2O, 600 MHz, dioxane, ppm): δ = 8.76 (dd, 3J1H1H = 5.64 Hz, 3J195Pt1H = 25.40 Hz, 4H, H6), δ = 8.28 (t, 3J1H1H = 7.73 Hz, 2H, H7), δ = 7.81 (t, 3J1H1H = 7.14 Hz, 4H, H8), δ = 3.95 (t, 3J1H1H = 6.72, 1H, H3), δ = 2.64, 2.63 (m, 2H, H1,2), δ = 1.54 (s, 3J1H1H = 7.28 Hz, 1H, H4), δ = 0.84 (t, 3J1H1H = 7.50 Hz, 1H, H5). 13C-NMR (D2O, 150 MHz, dioxane, ppm): δ = 179.29 (Cd), δ = 176.22 (Ca), δ = 149.84 (Ch), δ = 143.31 (Cj), δ = 127.64 (Ci), δ = 67.90 (Ce), δ = 31.78 (Cb), δ = 30.88 (Cc), δ = 21.91 (Cf), δ = 10.17 (Cg). 195Pt-NMR (D2O, 107 MHz, dioxane): 1065 ppm. ESI-MS: [M + Na]+ (m/z) calc. (C17H22N8NaO5Pt). 636.1210; found, 636.2.
trans,trans,trans-[Pt(N3)2(OH)(N-MI)(py)2](3)
trans,trans,trans-[Pt(N3)2(py)2(OH)2] 3 (0.086 g, 18.25 mmol) was dissolved in anhydrous DMSO (2 mL) and N-methylisatoic anhydride (0.162 g, 91.23 mmol, 5 mol eq.) was added. The reaction was stirred for 72 h at 40 °C under nitrogen with an outlet to allow the release of CO2(g). The isolation of the product was achieved by the addition of water (10 mL) at which point the compound precipitated. For the removal of excess ligand, repeated washing with diethyl ether was necessary and carried out via centrifugation (yield = 0.020 g, 18%). Crystals suitable for X-ray diffraction were grown by slow evaporation of a solution in DCM/MeOH at ambient temperature. Purity (HPLC, UV-vis absorbance): 94%.1H-NMR (DMSO-d6, 500 MHz, ppm): δ = 8.86 (dd, 3J1H1H = 5.54 Hz, 3J195Pt1H = 27.00 Hz, 4H, H7), δ = 8.30 (t, 3J1H1H = 7.56 Hz, 2H, H9), δ = 7.88 (m, 5H, 4H8, H4), δ = 7.62 (q, 3J1H1H = 5.07 Hz, 1H, H5), δ = 7.26 (t, 3J1H1H = 7.78 Hz, 1H, H3), δ = 6.53 (d, 3J1H1H = 7.53 Hz, 1H, H1), δ = 6.51 (t, 3J1H1H = 7.53 Hz, 1H, H2), δ = 3.88 (d, 2J195Pt1H = 14.8 Hz, H10), δ = 2.70 (d, 3J1H1H = 5.00 Hz, 3H, H6). 13C-NMR (DMSO-d6, 125 MHz, ppm): δ = 171.26 Ca, δ = 150.82 Cc, δ = 149.73 Cb, δ = 149.38 Ci, δ = 142.18 Ck, δ = 132.86 Cg, δ = 132.09 Cf, δ = 126.42 Cj, δ = 113.74 Cd, 110.00 Ce, δ = 29.07 Ch. 195Pt-NMR (DMSO-d6, 107 MHz,): δ = 990 ppm. ESI-MS: [M + H]+ (m/z) calc. (C18H20N9O3Pt) 605.1288; found, 605.1338.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
NF thanks the Wellcome Trust (201406/Z/16/Z); Cancer Research UK (CR-UK) grant number C5255/A18085 through the Cancer Research UK Oxford Centre and the John Fell Fund for funding. NF thanks Profs. Stephen Faulkner for support. PJS and NF thank the EPSRC (for grant EP/P030572/1 and studentship for ES), PJS also thanks the ERC (grant 247450). L. S. performed this work under the Severo Ochoa Centres of Excellence Programme run by the Spanish State Research Agency, grant no. CEX2018-000867-S (DIPC). L. S. also thanks the Spanish Multi-MetDrugs network (RED2018-102471-T) for fruitful discussion.References
- M. Galanski, Recent Pat. Anticancer Drug Discov., 2006, 1, 285 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573 CrossRef CAS.
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645 RSC.
- M. D. Hall, C. T. Dillon, M. Zhang, P. Beale, Z. Cai, B. Lai, A. P. J. Stampfl and T. W. Hambley, J. Biol. Inorg. Chem., 2003, 8, 726 CrossRef CAS PubMed.
- L. T. Ellis, H. M. Er and T. W. Hambley, Aust. J. Chem., 1994, 48, 793 CrossRef.
- S. Alonso-de Castro, A. Terenzi, S. Hager, B. Englinger, A. Faraone, J. C. Martínez, M. Galanski, B. K. Keppler, W. Berger and L. Salassa, Sci. Rep., 2018, 8, 1 CAS.
- Z. Deng, N. Wang, Y. Liu, Z. Xu, Z. Wang, T.-C. Lau and G. Zhu, J. Am. Chem. Soc., 2020, 142, 7803 CrossRef CAS PubMed.
- V. E. Y. Lee, C. F. Chin and W. H. Ang, Dalton Trans., 2019, 48, 7388 RSC.
- A. Kastner, I. Poetsch, J. Mayr, J. V. Burda, A. Roller, P. Heffeter, B. K. Keppler and C. R. Kowol, Angew. Chem., Int. Ed., 2019, 58, 7464 CrossRef CAS PubMed.
- J. Z. Zhang, E. Wexselblatt, T. W. Hambley and D. Gibson, Chem. Commun., 2012, 48, 847 RSC.
- W. P. Fehlhammer and W. Beck, Z. Anorg. Allg. Chem., 2013, 639, 1053–1082 CrossRef CAS.
- H. Shi, C. Imberti and P. J. Sadler, Inorg. Chem. Front., 2019, 6, 1623 RSC.
- H.-C. Tai, Y. Zhao, N. J. Farrer, A. E. Anastasi, G. Clarkson, P. J. Sadler and R. J. Deeth, Chem. – Eur. J., 2012, 18, 10630 CrossRef CAS PubMed.
- H. Shi, C. Imberti, H. Huang, I. Hands-Portman and P. J. Sadler, Chem. Commun., 2020, 56, 2320 RSC.
- E. Shaili, M. Fernández-Giménez, S. Rodríguez-Astor, A. Gandioso, L. Sandín, C. García-Vélez, A. Massaguer, G. J. Clarkson, J. A. Woods, P. J. Sadler and V. Marchán, Chem. – Eur. J., 2015, 21, 18474 CrossRef CAS.
- S. Huayun, C. Imberti, G. J. Clarkson and P. J. Sadler, Inorg. Chem. Front., 2020, 7, 3533 RSC.
- H. Shi, J. Kasparkova, C. Soulie, G. J. Clarkson, C. Imberti, N. Olga, M. J. Paterson, V. Brabec and P. J. Sadler, Chem. – Eur. J., 2021 DOI:10.1002/chem.202101168.
- A. Gandioso, E. Shaili, A. Massaguer, G. Artigas, A. González-Cantó, J. A. Woods, P. J. Sadler and V. Marchán, Chem. Commun., 2015, 51, 9169 RSC.
- J. J. Wilson and S. J. Lippard, Chem. Rev., 2014, 114, 4470–4495 CrossRef CAS PubMed.
- Q. Zhang, G. Kuang, S. He, H. Lu, Y. Cheng, D. Zhou and Y. Huang, Nano Lett., 2020, 20(5), 3039 CrossRef CAS.
- M. Ravera, E. Gabano, I. Zanellato, I. Bonarrigo, E. Escribano, V. Moreno, M. Font-Bardia, T. Calvet and D. Osella, Dalton Trans., 2012, 41, 3313 RSC.
- A. Alvarez-Valdes, J. M. Perez, I. Lopez-Solera, R. Lannegrand, J. M. Continente, P. Amo-Ochoa, M. J. Camazon, X. Solans, M. Font-Bardia and C. Navarro-Ranninger, J. Med. Chem., 2002, 45, 1835 CrossRef CAS PubMed.
- K. R. Barnes, A. Kutikov and S. J. Lippard, Chem. Biol., 2004, 11, 557 CrossRef CAS PubMed.
- C. F. Chin, Q. Tian, M. I. Setyawati, W. Fang, E. S. Q. Tan, D. T. Leong and W. H. Ang, J. Med. Chem., 2012, 55, 7571 CrossRef CAS PubMed.
- M. X. Tan, Z. F. Wang, Q. P. Qin, B. Q. Zou and H. Liang, Dalton Trans., 2020, 49, 1613 RSC.
- S. Mukhopadhyay, C. M. Barnés, A. Haskel, S. M. Short, K. R. Barnes and S. J. Lippard, Bioconjugate Chem., 2008, 19, 39 CrossRef CAS PubMed.
- C. M. Giandomenico, M. J. Abrams, B. A. Murrer, J. F. Vollano, M. I. Rheinheimer, S. B. Wyer, G. E. Bossard and J. D. Higgins, Inorg. Chem., 1995, 34, 1015 CrossRef CAS PubMed.
- T. Babu, A. Sarkar, S. Karmakar, C. Schmidt and D. Gibson, Inorg. Chem., 2020, 59, 5182 CrossRef CAS PubMed.
- N. J. Farrer, J. A. Woods, L. Salassa, Y. Zhao, K. S. Robinson, G. Clarkson, F. S. Mackay and P. J. Sadler, Angew. Chem., Int. Ed., 2010, 49, 8905 CrossRef CAS PubMed.
- F. S. Mackay, N. J. Farrer, L. Salassa, H.-C. Tai, R. J. Deeth, S. A. Moggach, P. A. Wood, S. Parsons and P. J. Sadler, Dalton Trans., 2009, 2315 RSC.
- E. Shaili, L. Salassa, J. A. Woods, G. Clarkson, P. J. Sadler and N. J. Farrer, Chem. Sci., 2019, 10, 8610 RSC.
- A. Krezel and W. Bal, J. Inorg. Biochem., 2004, 98, 161 CrossRef CAS PubMed.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS PubMed.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324 CrossRef CAS PubMed.
- J. S. Butler, J. A. Woods, N. J. Farrer, M. E. Newton and P. J. Sadler, J. Am. Chem. Soc., 2012, 134, 16508 CrossRef CAS PubMed.
- C. Vallotto, E. Shaili, H. Shi, J. S. Butler, C. J. Wedge, M. E. Newton and P. J. Sadler, Chem. Commun., 2018, 54, 13845 RSC.
- M. Galanski and B. K. Keppler, Inorg. Chem., 1996, 35, 1709 CrossRef CAS PubMed.
- W. H. Ang, S. Pilet, R. Scopelliti, F. Bussy, L. Juillerat-Jeanneret and P. J. Dyson, J. Med. Chem., 2005, 48, 8060 CrossRef CAS PubMed.
- J. J. Wilson and S. J. Lippard, Inorg. Chem., 2011, 50, 3103 CrossRef CAS PubMed.
- B. M. Still, P. G. A. Kumar, J. R. Aldrich-Wright and W. S. Price, Chem. Soc. Rev., 2007, 36, 665 RSC.
- Y. Shi, S.-A. Liu, D. J. Kerwood, J. Goodisman and J. C. Dabrowiak, J. Inorg. Biochem., 2012, 107, 6 CrossRef CAS PubMed.
- F. Michelet, R. Gueguen, P. Leroy, M. Wellman, A. Nicolas and G. Siest, Clin. Chem., 1995, 41, 1509 CrossRef CAS.
- L. Qin, H. Bartlett, H. R. Griffiths, F. Eperjesi, R. A. Armstrong and D. Gherghel, Invest. Ophthalmol. Visual Sci., 2011, 52, 5029 CrossRef CAS PubMed.
- M. P. Gamcsik, M. S. Kasibhatla, S. D. Teeter and O. M. Colvin, Biomarkers, 2012, 17, 671 Search PubMed.
- L. Salassa, T. Ruiu, C. Garino, A. M. Pizarro, F. Bardelli, D. Gianolio, A. Westendorf, P. J. Bednarski, C. Lamberti, R. Gobetto and P. J. Sadler, Organometallics, 2010, 29, 6703 CrossRef CAS.
- A. Vlček and S. Záliš, Coord. Chem. Rev., 2007, 251, 258 CrossRef.
- M. Branch, Can. J. Chem., 1980, 58, 1592 CrossRef.
- R. S. Partridge, S. M. Monroe, J. K. Parks, K. Johnson, J. Parker, W. Davis, G. R. Eaton and S. S. Eaton, Arch. Biochem. Biophys., 1994, 310, 210 CrossRef CAS PubMed.
- C. Gambetta, J. Natera, W. A. Massad and N. A. García, J. Photochem. Photobiol., A, 2013, 269, 27 CrossRef CAS.
- M. J. Davies, Photochem. Photobiol. Sci., 2004, 3, 17 CrossRef CAS PubMed.
- A. F. Westendorf, A. Bodtke and P. J. Bednarski, Dalton Trans., 2011, 40, 5342 RSC.
- J. J. Wilson and S. J. Lippard, Inorg. Chim. Acta, 2012, 389, 77 CrossRef CAS PubMed.
- E. J. New, R. Duan, J. Z. Zhang and T. W. Hambley, Dalton Trans., 2009, 3092 RSC.
- S. P. Oldfield, M. D. Hall and J. A. Platts, J. Med. Chem., 2007, 50, 5227 CrossRef CAS PubMed.
- M. D. Hall, M. Okabe, D.-W. Shen, X.-J. Liang and M. M. Gottesman, Annu. Rev. Pharmacol. Toxicol., 2008, 48, 495 CrossRef CAS PubMed.
- D. Sinani, D. J. Adle, H. Kim and J. Lee, J. Biol. Chem., 2007, 282, 26775 CrossRef CAS PubMed.
- S. Ishida, J. Lee, D. J. Thiele and I. Herskowitz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14298 CrossRef CAS PubMed.
- S. S. More, S. Li, S. W. Yee, L. Chen, Z. Xu, D. M. Jablons and K. M. Giacomini, Mol. Cancer Ther., 2010, 9, 1058 CrossRef CAS PubMed.
- I. Romero-Canelón, A. M. Pizarro, A. Habtemariam and P. J. Sadler, Metallomics, 2012, 4, 1271 CrossRef PubMed.
- A. Radu, R. Conde, C. Fontolliet, G. Wagnieres, H. Van den Bergh and P. Monnier, Gastrointest. Endosc., 2003, 57, 897 CrossRef PubMed.
- S. Dhar, W. L. Daniel, D. A. Giljohann, C. A. Mirkin and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 14652 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, synthesis and characterisation data, X-ray crystallographic tables, biological data. CCDC 1998067, 1998068 and 1998069. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt01730f |
| ‡ This paper describes experimental work performed by Dr Julie A Woods but she is unaware that it has been submitted for publication. Dr Julie A Woods therefore, does not take any responsibility for the submission. |
| This journal is © The Royal Society of Chemistry 2021 |