
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Immobilising molecular Ru complexes on a protective ultrathin oxide layer of p-Si electrodes towards photoelectrochemical CO2 reduction†
Maxime
Laurans
 ,
Jordann A. L.
Wells
and
Sascha
Ott
*
,
Jordann A. L.
Wells
and
Sascha
Ott
*
Department of Chemistry – Ångström Laboratory, Uppsala University, Box 523, 75120 Uppsala, Sweden. E-mail: sascha.ott@kemi.uu.se
First published on 16th June 2021
Abstract
Photoelectrochemical CO2 reduction is a promising approach for renewable fuel generation and to reduce greenhouse gas emissions. Owing to their synthetic tunability, molecular catalysts for the CO2 reduction reaction can give rise to high product selectivity. In this context, a RuII complex [Ru(HO-tpy)(6-mbpy)(NCCH3)]2+ (HO-tpy = 4′-hydroxy-2,2′:6′,2′′-terpyridine; 6-mbpy = 6-methyl-2,2′-bipyridine) was immobilised on a thin SiOx layer of a p-Si electrode that was decorated with a bromide-terminated molecular layer. Following the characterisation of the assembled photocathodes by X-ray photoelectron spectroscopy and ellipsometry, PEC experiments demonstrate electron transfer from the p-Si to the Ru complex through the native oxide layer under illumination and a cathodic bias. A state-of-the-art photovoltage of 570 mV was determined by comparison with an analogous n-type Si assembly. While the photovoltage of the modified photocathode is promising for future photoelectrochemical CO2 reduction and the p-Si/SiOx junction seems to be unchanged during the PEC experiments, a fast desorption of the molecular Ru complex was observed. An in-depth investigation of the cathode degradation by comparison with reference materials highlights the role of the hydroxyl functionality of the Ru complex to ensure its grafting on the substrate. In contrast, no essential role for the bromide function on the Si substrate designed to engage with the hydroxyl group of the Ru complex in an SN2-type reaction could be established.
Introduction
Limiting anthropogenic CO2 emissions from fossil fuel combustion would contain climate change and contribute to stabilising the global mean temperature. Consequently, converting CO2 emissions into a renewable alternative energy source with sunlight as the energy input is a promising alternative to diminish atmospheric CO2 concentration and lower its impact.1,2The development of molecular catalysts for the CO2-reduction reaction (CO2RR) and the conversion of CO2 into fuels has been widely studied3–12 to circumvent the formation of the highly energetic radical anion CO2*−, the product of one-electron reduction of CO2.3–5 Molecular catalysts lower the activation energy barriers and facilitate the (photo)electrochemical formation of various fuels. The fine tuning of molecular catalysts by synthetic modifications permits the selective reduction of CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 and has the potential to outcompete H2 evolution which occurs at similar electrochemical potentials.
13 and has the potential to outcompete H2 evolution which occurs at similar electrochemical potentials.
Although molecular catalysts can present higher selectivity in the CO2RR than solid-state catalysts, their degradation in solution is frequently problematic. Thus, their immobilisation onto various solid supports to afford heterogeneous electro/photocatalytic devices has moved into the centre of interest.14–23 Heterogenisation of molecular catalysts on photoelectrodes would combine the advantages of homogeneous catalysts with high CO2RR selectivity with those of heterogeneous systems that simplify product isolation and catalyst recycling.4,9,12,14,16 This approach can also render water-insoluble molecular catalysts useable in aqueous devices.
Silicon is a low-cost narrow band gap (1.1 eV) semiconductor that can harvest light from a large part of the solar spectrum. Since the first use of a heavily-doped p-Si photocathode for H2 evolution by Candea et al. in 1976, p-Si has been an attractive candidate for the assembly of photoelectrodes.24–28 Recently, examples of silicon-based photocathodes modified by molecular catalysts have been developed for proton reduction29–34 and the CO2RR.4,16,19,35 Some of these studies have been carried out by grafting the molecular catalyst directly onto freshly etched Si.30,36,37 However, silicon surface functionalisation remains sensitive to oxidation and requires the strict exclusion of air and moisture during assembly and when performing catalysis.36–38
Without specific etching procedures, silicon surfaces present a native silicon oxide layer that is suggested to bring advantages to the photocathode. Indeed, the presence of a thin insulative layer would improve the open-circuit potential compared to a silicon–coating interface by avoiding current leakage of the majority carrier flow (holes on p-Si) that goes in the opposite direction to the minority carrier flow (electrons for p-Si), towards the anode. Moreover, native silicon oxide layers thinner than 3 nm are permeable to minority carriers by tunnelling (electrons for p-Si).39,40 Additionally, silicon oxide is thermodynamically stable at a wide pH range and would provide a stable supportive substrate.41 Therefore, the immobilisation of molecular complexes onto native silicon oxide could be defined as an MIS (Metal–Insulator–Semiconductor) inspired junction in which the complex layer plays a similar role to the metal in that it acts as a charge extraction layer combined with potential catalytic features. We envisaged that the presence of the insulative oxide layer would still allow a significant photovoltage for the CO2RR and potentially limit the back-electron transfer after catalyst reduction at the surface.
Recently, we and others have developed a family of Ru-based CO2RR catalysts, [Ru(tpy)(bpy)(NCCH3)] (tpy = 2,2′:6′, 2′′-terpyridine and bpy = 2,2′-bipyridine), that present a high CO2-to-CO selectivity.42–45 The presence of a methyl group at the bipyridine facilitates the dissociation of the acetonitrile ligand upon one-electron reduction and promotes the electrochemical CO2RR via a low-overpotential ECE mechanism. The methyl group was also included in the present study, and we report a new member of this catalyst family, [Ru(HO-tpy)(6-mbpy)(NCCH3)]2+ (1) (HO-tpy = 4′-hydroxy-2,2′:6′,2′′-terpyridine and 6-mbpy = 6-methyl-2,2′-bipyridine), that contains a hydroxyl group for grafting on native silicon oxide coated p-Si flat electrodes. The silicon based molecular junctions were prepared in a bottom-up approach. First, a bromine-terminated reactive layer was grafted on flat p-Si/SiOx through a siloxane linkage. Subsequently, this decorated surface was reacted with (1) bearing the complementary hydroxyl function for an ether linkage. The structural characterisation of the assembly was performed by X-ray photoelectron spectroscopy (XPS) and ellipsometry. The electrochemical properties of the assembly were investigated by cyclic voltammetry while the stability of the adducts under the photoelectrochemistry (PEC) conditions was explored by XPS.
Synthesis and characterisation of (1)
The complex [Ru(HO-tpy)(6-mbpy)(NCCH3)]2+ (1) bears a hydroxyl group in the 4′ position of the terpyridyl moiety that is envisaged to engage in ether bond formation with the bromide-terminated supportive layer described below. Complex (1) was synthesised by adapting literature procedures (Fig. 1).43,46,47 As a key step, the ruthenium dimer (3) that was formed from the reduction of (4) in the presence of triethylamine could be easily separated from unidentified impurities by simple filtration.46,48 Complex (3) is a dark purple-black solid which upon reaction with 6-mbpy gives rise to (2). The indirect pathway via (3) allows easier purification of (2) by chromatography on alumina than the direct synthesis of (2) from (4).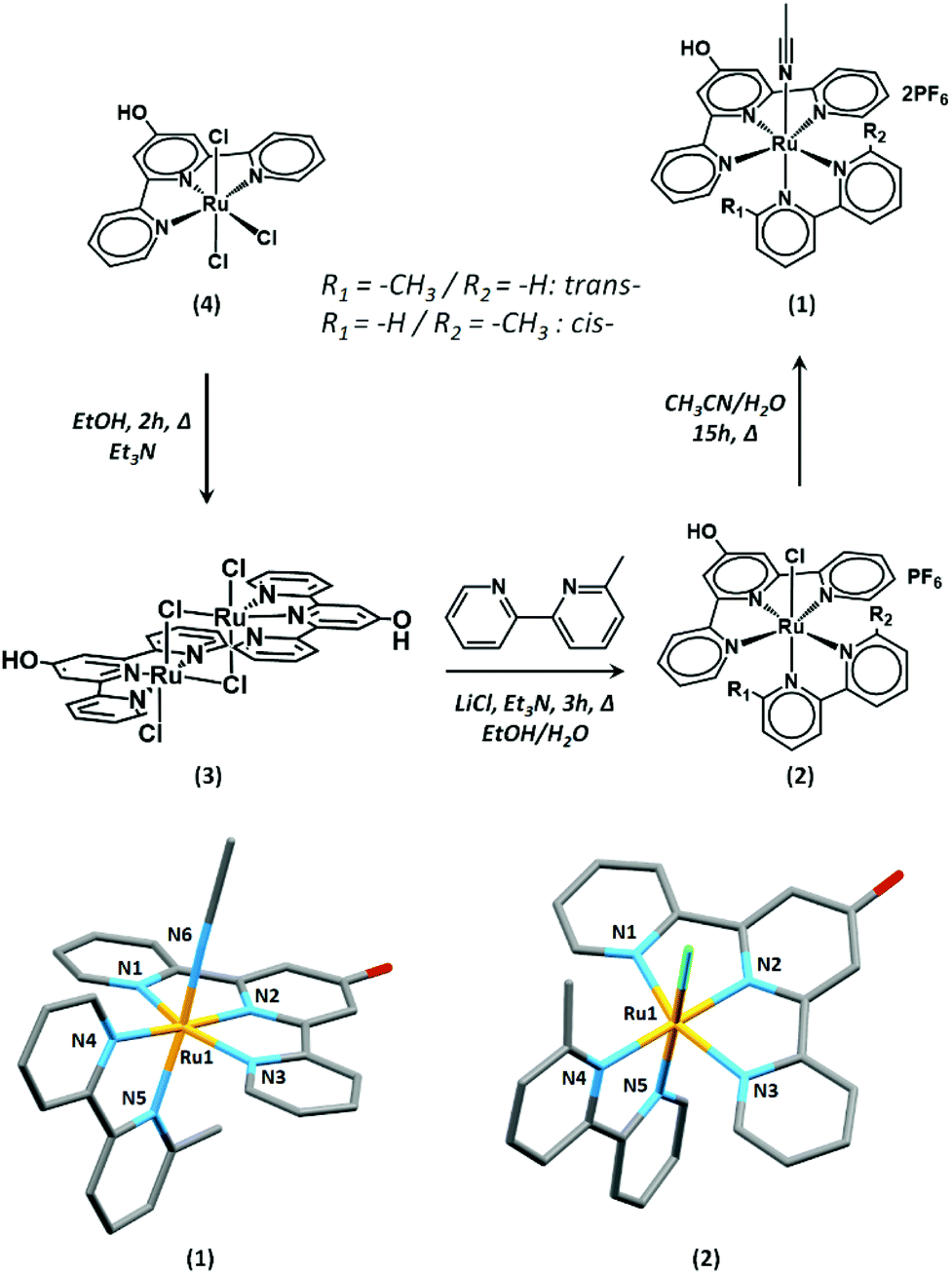 | ||
| Fig. 1 Synthetic scheme for the preparation of (1) and single-crystal solid-state structures of trans-(1) (bottom left) and cis-(2) (bottom right). All atoms are displayed as capped sticks, and H atoms and non-coordinating Cl− and PF6− counterions have been omitted for clarity. | ||
As reported by Johnson et al., the asymmetric 6-methyl-2,2′-bipyridine ligand leads to the formation of cis- and trans-isomers.43 Both isomers of (1) could be observed by 1H NMR spectroscopy, with the trans-isomer being the major component (Fig. S1†). The trans-isomers of this family of complexes have been shown to promote the CO2RR at a lower overpotential than the cis-isomers. Due to the difficulties in their separation and potential interconversions, the subsequent study was conducted on a mixture of cis- and trans-isomers of (1).
High-resolution mass spectrometry in the positive mode attests the formation of complex (1). The main signal at m/z: 260.5384 (calc. 260.5389) corresponds to the dication (1) with the loss of the acetonitrile ligand. The assignment of all other peaks is presented in the Experimental section.
Single crystals of (1) and (2) suitable for X-ray diffraction analysis were obtained by vapor diffusion of diethyl ether into a concentrated acetonitrile solution of (1) and by layering a CD3Cl solution of (2) with aqueous conc. HCl, respectively. The asymmetric unit of (2) contains a single complex, while that of (1) contains two non-equivalent complexes. In both (1) and (2), the Ru centers exhibit a six-coordinate, distorted octahedral geometry (Fig. 1). The HO-tpy ligand occupies a meridional plane and the 6-mbpy and chloride or CH3CN co-ligands satisfy the metal coordination sphere. The 6-mbpy ligands exist both in the cis- and trans- conformations with respect to the chloride and CH3CN ligands in the solid-state, resulting in a positional disorder in the solid-state structures. The bond lengths in (1) are comparable to those of the related complexes. The average Ru–Nterpy distance of 2.040(3) Å is within the range of the previously reported examples (1.958–2.083 Å), while the average Ru–Nbipy length of 2.086(8) Å are slightly elongated (2.019–2.085 Å).49–51 Unfortunately, very low intensity data were obtained from single crystals of (2), which did not allow for the structure to be discussed beyond connectivity information. Further details of both structures are provided in the ESI.† The average C–O bond length of 1.333(6)–1.351(7) Å in (1) is consistent with that of a protonated oxygen atom as previously reported for related complexes.47
Cyclic voltammetry in solution
In order to verify the redox properties of (1) prior to surface anchorage, the complex was studied by cyclic voltammetry (CV) in the homogeneous phase (0.1 M TBAPF6 in CH3CN). As visible in Fig. 2, several cathodic features can be observed between −1.78 and −2.26 V (all potentials are given vs. Fc+/0). The first reductive process is an irreversible wave at Epc = −1.78 V. Its attenuation on the second consecutive scan at ν = 200 mV s−1 and its disappearance in the presence of 1.1 equivalents of (iPr)2EtN (DIPEA, Fig. 2 left) suggest that this process is a ligand-based reduction with concomitant deprotonation. Indeed, Fujita and co-workers recently reported the absence of the most anodic reduction wave on the second scan or after the chemical deprotonation of the hydroxyl-modified polypyridyl-Ru complexes similar to (1).52 They assigned this phenomenon to a ligand-based reduction that is accompanied by the loss of a proton. Tanaka and co-workers also reported a comparable behaviour in the presence of Et3N.53 | ||
| Fig. 2 Cyclic voltammograms of (1) (1 mM). Left: in the absence and presence of 1.1 equivalent of (iPr)2EtN under Ar. Right: under CO2 (dashed line) compared to the response under Ar (solid line). All experiments were done in 0.1 M TBAPF6 in CH3CN, ν = 200 mV s−1. | ||
Upon the addition of (iPr)2EtN, a new wave appears at Epc = −1.86 V (Fig. 2, left) that is attributed to the reduction of an ammonium cation formed by the deprotonation of (1). This assignment is based on the work by Tanaka and co-workers, who reported the reduction of Et3N-H+ formed by the deprotonation of a complex similar to that of (1) at E = −1.75 V vs. Ag+/0.53
Irrespective of the presence of (iPr)2EtN, a second irreversible reduction can be observed at Epc = −1.95 V which can be attributed to the reduction of the deprotonated complex (1) (Fig. 2, left). As a result of the negative charge, this reduction is cathodically shifted compared to that of the mother complex (5) (Chart 1) that lacks the hydroxyl group (Fig. S3†).53,54 The introduction of the hydroxyl function on (1) is compatible with the CO2RR as current enhancement can be observed at Epc = −1.95 V when the CV is conducted under CO2 (Fig. 2, right).
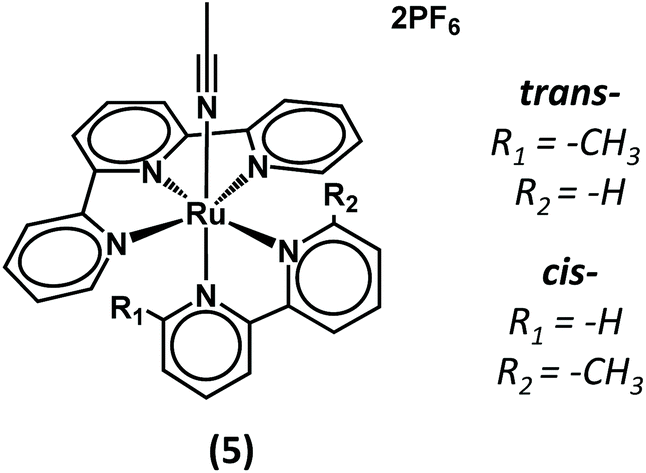 | ||
| Chart 1 Representation of complex (5). | ||
Having proven that the catalytic activity of the complex is not greatly altered by the presence of the hydroxyl group, the grafting of (1) was pursued.
Immobilisation on silicon substrate
The immobilisation of (1) onto silicon wafers followed a two-step process. For the work presented herein, we decided to take advantage of the native oxide layer that covers the silicon wafers. This thin oxide layer is envisaged to protect the underlying semiconductor, while acting as a tunnelling barrier for charge extraction by (1).In the first step, a supportive molecular layer of 3-bromopropyltrimethoxysilane (Br-PTMS) was installed onto the native silicon oxide (p-Si/SiOx/Br, Fig. 3). The second step included the anchorage of complex (1) on the modified surface to afford p-Si/SiOx/[Ru]. The bromide groups are complementary to the hydroxyl group of (1) for ether bond formation via an SN2 reaction in the presence of (iPr)2EtN.
 | ||
| Fig. 3 Representation of the grafting steps to achieve p-Si/SiOx/Br and p-Si/SiOx/[Ru] (Key: Ru (orange), N (blue), O (red), and C (grey)). | ||
Halide terminated monolayers have previously been shown to engage in SN2 substitution reactions with strong nucleophiles such as azide, thiocyanate, or alkoxide anions.55–58 Fryxell et al. have shown that such SN2 reactivity is decreased when the halide is positioned at the terminus of highly ordered monolayers that are obtained with long carbon chains as the rigidity of such monolayers blocks the attack of the incoming nucleophile at the halide-carbon.59,60 To guarantee the flexibility of the monolayer and allow for a successful SN2 coupling, we chose to immobilise the short carbon chain Br-PTMS onto p-Si/SiOx substrates. Moreover, we conducted the grafting under an ambient atmosphere and high concentration to lead to a potential disorganised and flexible molecular supportive layer.61,62
The SN2 coupling of an alcohol decorated–Ru complex with a flexible Cl-terminated trialkoxysilane layer has previously been performed on SiOx solid supports by thermal activation.57,58 It is well known that the formation of robust monodentate alcohol monolayers on SiOx requires a thermal activation.61,63 Therefore, to favour the SN2 coupling and limit a potential direct interaction of (1) with SiOx, we chose to perform the coupling at room temperature in the presence of an activating base. The poorly nucleophilic and sterically hindered base DIPEA was chosen for the SN2 reaction to avoid the potential formation of an ammonium layer on the Br-terminated p-Si/SiOx/Br layer.64,65 DIPEA (pKa = 10.75) is sufficiently basic to deprotonate (1), the hydroxyl group of which is expected to have a similar pKa as [Ru(tpy)(HO-tpy)]2+ (pKa = 5.85).47,66
The thickness of the native SiOx layer was determined by ellipsometry before grafting with Br-PTMS and was found to range between 12 and 17 Å from Si wafer to wafer. The p-Si/SiOx/Br surfaces were obtained by grafting Br-PTMS using 0.1 M solutions in dry toluene overnight on a clean native silicon oxide surface p-Si/SiOx (Fig. 3). The ellipsometry measurements showed a slight increase in thickness of 8–9 Å, consistent with the deposition of a molecular layer with a theoretical thickness close to 6 Å for an ideally organised monolayer (Fig. 3).
The XP spectra of the substrate did not feature any signals that could be assigned to the bromide from the molecular layer (Fig. S4†). However, this behaviour is not surprising and has been observed before by Sato et al., who demonstrated the degradation of 3-chloro- and 3-bromomethoxysilane layers on silicon oxide during XPS measurements.67 In analogy, we assigned the absence of a Br3d (5/2) contribution in the XP spectrum to C–Br bond degradation during the measurement. Nevertheless, the thickness increase as determined by ellipsometry indicates the successful formation p-Si/SiOx/Br which was thus submitted to grafting with (1).
The p-Si/SiOx/Br coated surfaces were placed in a solution of (1) in the presence of 2.5 equivalents of DIPEA in dry acetonitrile for 4 days at room temperature under ambient conditions.68 After a thorough rinsing step with acetonitrile, the p-Si/SiOx/[Ru] surfaces were characterised by ellipsometry and XPS.
The thickness of (1)-modified surfaces was expected to be 30–35 Å (Fig. 3). In agreement with these theoretical values, the experimental thicknesses varied from 28 to 39 Å, thus indicating the successful formation of p-Si/SiOx/[Ru].
XPS measurements were performed to further confirm the composition of the p-Si/SiOx/[Ru] electrodes. All constitutive elements of (1) were detected by XPS (Table 1) and the high-resolution spectra of the C1s, Ru3d, and N1s regions are displayed in Fig. 4. A unique contribution is observed in the N1s region at 399.9 eV. The C1s and Ru3d regions feature a contribution at 280.9 eV attributed to Ru3d (5/2). The photopeak at higher binding energy encompasses the C1s and Ru3d (3/2) contributions. The latter was set at 285.1 eV in agreement with the Ru3d (5/2) contribution (see the ESI†). Two C1s contributions correspond to the C–H and C–C bonds from the alkyl chain and the inherent C1s contamination at 284.6 eV and the C–N, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–O bonds from the complex at 285.5 eV. The atomic percentage of N/Ru shows a ratio of 5.9 N atoms for 1 Ru atom, consistent with the presence of intact (1) at the surface (Table 1). No significant nitrogen contribution is present in the spectrum of a control experiment of p-Si/SiOx/Br after 4 days in a 2.5 mM DIPEA solution in acetonitrile and in the absence of (1) (Fig. S5†), thus proving the absence of any potential DIPEA-based quaternary ammonium ions.64,65
C and C–O bonds from the complex at 285.5 eV. The atomic percentage of N/Ru shows a ratio of 5.9 N atoms for 1 Ru atom, consistent with the presence of intact (1) at the surface (Table 1). No significant nitrogen contribution is present in the spectrum of a control experiment of p-Si/SiOx/Br after 4 days in a 2.5 mM DIPEA solution in acetonitrile and in the absence of (1) (Fig. S5†), thus proving the absence of any potential DIPEA-based quaternary ammonium ions.64,65
 | ||
| Fig. 4 High-resolution XP spectra of the C1s, Ru3d, and N1s regions of a p-Si/SiOx/[Ru] electrode (calibrated against Si2p at 99.4 eV). | ||
| Additional post-coupling proceduresa | Atomic ratio N/Ru | Binding energies/eV (ref. Si 99.4 eV) | |||
|---|---|---|---|---|---|
| C | N | Brb | |||
| a All procedures were preceded and followed by copious rinsing with acetonitrile. b Values determined from the survey spectrum. | |||||
| 1 | None | 5.9 | 280.9 | 399.9 | 68.4 |
| 2 | Sonication in CH3CN (5 min) | 5.9 | 281.1 | 400.0 | 68.8 |
| 3 | Sonication in TBAPF6 0.1 M in CH3CN (5 min) | 6.1 | 281.2 | 400.3 | 68.1 |
| 4 | TBAPF6 1 M in CH3CN (20 min) | 5.6 | 281.0 | 399.0 | 68.6 |
Interestingly, the survey spectrum of p-Si/SiOx/[Ru] also shows a Br3d contribution between 68.1 and 68.8 eV that is different from the typical range for C–Br bonds at 70.6 and 71.6 eV, and that is assigned to a bromide anion (Table 1, entries 1–4).60 Bromide is the leaving group in the SN2 reaction and may be trapped as a counter ion to the Ru complex. Alternatively, bromide may also result from the decomposition of unreacted C–Br bonds during the XPS scan as described above but may be trapped under the layer of (1) on p-Si/SiOx/[Ru].
To assess the robustness of the modified photocathodes prior to the PEC experiments, the p-Si/SiOx/[Ru] electrodes were exposed to several treatment procedures (Table 1), using the Ru3d:N1s ratio as determined by XPS as stability markers. The electrodes are stable to sonication in acetonitrile (5 min), even in the presence of TBAPF6 (0.1 M), and the Ru3d (5/2) and N1s photopeaks retain the 1:6 ratio as expected for (1) (Table 1, entries 2 & 3; Fig. S6 and S7†). The p-Si/SiOx/[Ru] electrodes are also stable under the conditions of the PEC experiments (1 M TBAPF6 in acetonitrile), and no substantial desorption of (1) is observed (Table 1, entry 4; Fig. S6†).
Photoelectrochemistry study
The CVs of p-Si/SiOx/[Ru] were recorded under one sun illumination (Fig. 5). Compared to the p-Si/SiOx and p-Si/SiOx/Br reference electrodes, a significant photocurrent with a cathodic peak potential at Epc = −1.58 V is observed for p-Si/SiOx/[Ru] (Fig. 5a). This effect can be explained by the presence of (1) at the native oxide and an effect of potential DIPEA traces is dismissed (Fig. S8†). The p-Si/SiOx/[Ru] electrode can be described as an MIS junction where complex (1) extracts the electrons photogenerated in the bulk of the p-Si semiconductor.41 The thin silicon oxide layer does not block the electron transfer but acts merely as a tunnelling barrier for charge extraction.41,69 The reduction of (1) at p-Si/SiOx can be explained by the Fermi level pinning enhanced by the presence of a high density of interfacial traps at p-Si/SiOx70,71 or by the unpinning of the band edges of p-Si with the formation of an inversion layer making the p-Si electrode behave as a metal electrode under an applied bias and illumination.41,72 To determine the effect of doping in p-Si/SiOx/[Ru] upon illumination, an analogous conductive n+-Si/SiOx/[Ru] electrode was prepared in a procedure analogous to that described for p-Si/SiOx/[Ru]. Comparing the CVs of n+-Si/SiOx/[Ru] and p-Si/SiOx/[Ru] shows a photovoltage of ca. +570 mV at J = 100 μA cm−2 (Fig. 5b). This photovoltage is similar to that observed for the reduction of [Re((tBu)2bpy)(CO)3Cl] (530 mV) and [Fe2(μ-bdt)(CO)6] (500 mV; bdt = benzene-1,2-dithiolate) in a homogeneous solution at an illuminated p-Si electrode.73,74 In terms of molecular acceptors that are anchored on p-Si, Rose and co-workers observed photovoltages varying from +200 to +235 mV for a nickel catalysed proton reduction scheme.36 A table with reported photovoltages obtained for molecular species at illuminated p-Si is included in the ESI (Table S4†). | ||
| Fig. 5 Cyclic voltammograms of (a) p-Si/SiOx, p-Si/SiOx/Br, and p-Si/SiOx/[Ru]; (b) p-Si/SiOx/[Ru] (solid line), and n+-Si/SiOx/[Ru] (dashed line); and (c) p-Si/SiOx/[Ru] in the dark (0) and under one sun illumination (1–3) (TBAPF6 1 M in CH3CN, ν = 1 V s−1) under an Ar atmosphere. | ||
Consecutive cathodic scans of p-Si/SiOx/[Ru] result in the rapid disappearance of the photocurrent (Fig. 5c). The XPS measurements of p-Si/SiOx/[Ru] after the PEC experiment revealed the disappearance of all N1s and Ru3d (5/2) contributions, while they were very prominent prior to the PEC experiment (Fig. 6). The absence of these contributions indicates a loss of the entire complex during the PEC study. In contrast, the Si2p region of the XP spectrum remained unchanged, highlighting the stability of the SiOx layer during the PEC experiments (Fig. 7).41 The propensity of Si/SiOx/[Ru] to drive the photoelectrochemical reduction of CO2 was probed by conducting the CV in a CO2-saturated electrolyte solution under one-sun illumination. Unfortunately, no significant photocurrent increase was observed relative to that of p-Si/SiOx/[Ru] under argon (Fig. S9†). Such a current enhancement would be expected for CO2RR activity at p-Si/SiOx/[Ru] during PEC measurements in analogy to the observations with the homogeneous complex (1) under CO2-staturated conditions. Its absence is most likely due to the rapid detachment of (1) from the photoelectrode under an applied bias. Johnson et al. have demonstrated that CO2 fixation on the analogous complex (5) (Chart 1) is preceded at least by one reduction step. Therefore, it is very likely that p-Si/SiOx/[Ru] is not stable after the first reduction even under CO2-saturated conditions and that CO2 fixation by the immobilised complex is not ensured at the interface.
 | ||
| Fig. 6 High-resolution XPS of C1s/Ru3d and the N1s regions of p-Si/SiOx/[Ru] before (a and b), and after (c and d) the PEC experiments. | ||
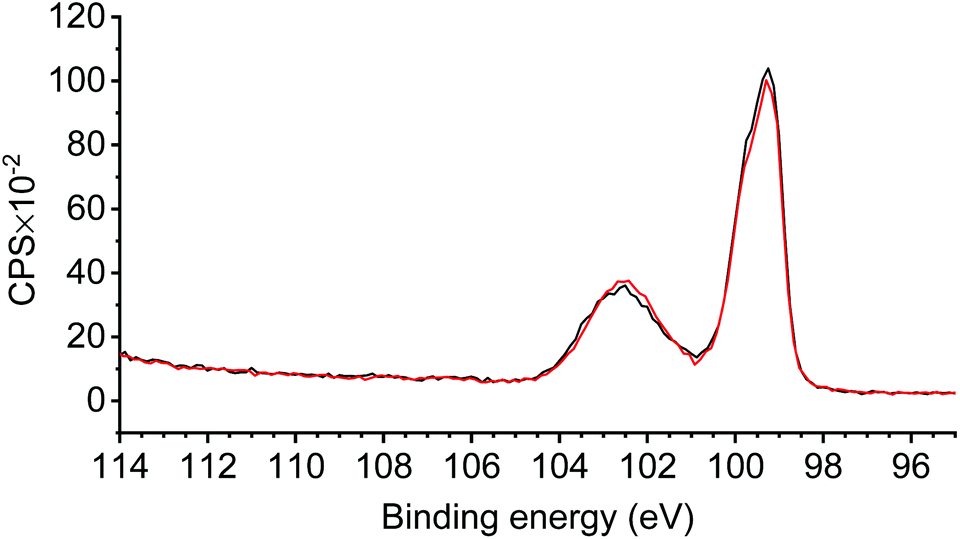 | ||
| Fig. 7 High-resolution XPS of the Si2p region of p-Si/SiOx/[Ru] before (black) and after (red) the PEC experiments. | ||
Discussion
From the PEC experiments, it is clear that the immobilised molecular layers are sensitive to the application of a bias. Consequently, it is legitimate to question the efficiency of the SN2 coupling reaction, even though all the XPS data prior to PEC confirm the presence of (1). To shine more light on the nature of the bond between (1) and the underlying substrate, different control experiments were performed, in particular, to delineate the role of the bromide and the hydroxyl functional groups.If the immobilisation of (1) proceeds through ether linkage formation as envisaged, removing either of the functional groups should decrease the grafting yield dramatically. Therefore, an n-propyltrimethoxysilane (n-PTMS) modified surface p-Si/SiOx/CH3 was prepared in analogy to the procedure described for p-Si/SiOx/Br and exposed to a 1 mM solution of (1) in the presence of DIPEA (2.5 mM). Although the modified supporting substrate was lacking bromide, significant Ru and N contributions were observed on the XP spectra of the modified substrate (Table 2, entry 2; and Fig. S10†). These results indicate the immobilisation of (1) even in the absence of a terminal bromide. The presence of (1) on p-Si/SiOx/CH3 might be due to the interaction between the hydroxyl function of (1) and the residual Si–O–R (R = Me or H, if hydrolysed) bonds that did not react with the oxide surface.61,63 In addition, the decreased N:Ru ratio of 5:1 may suggest different anchorage modes of (1) on p-Si/SiOx/CH3 than on p-Si/SiOx/Br, and in some cases, a ligand may be replaced by Si–O–R of the supportive layer. The control experiment shows that the bromide function does not play an essential role in the immobilisation of (1).
| Supportive layer | Complex | Ratios N/Ru | Binding energies/eV | ||
|---|---|---|---|---|---|
| C | N | ||||
| All samples were subjected to sonication (5 min) in 0.1 M TBAPF6 in CH3CN, and preceded and followed by copious rinsing with CH3CN after coupling the substrate in 1 mM of complex and 2.5 mM of DIPEA in CH3CN at RT for 4 days. PTMS: propyltrimethoxysilane. Binding energy values are referenced to Si 2p at 99.4 eV. | |||||
| 1 | Br-PTMS | 4 | 6.1 | 281.2 | 400.3 |
| 2 | n-PTMS | 4 | 5.2 | 281.0 | 399.9 |
| 3 | SiOx | 4 | 5.9 | 281.3 | 400.2 |
| 4 | Br-PTMS | 5 | — | — | — |
Next, we investigated the possible interaction of (1) with a non-modified p-Si/SiOx. Thus, a bare p-Si/SiOx substrate was placed in a 1 mM solution of (1) in the presence of DIPEA. Surprisingly, Ru3d and N1s contributions were also observed in this case, even after sonication in an electrolytic solution (Table 2, entry 3; Fig. S11†). Their presence demonstrates that a strong interaction occurs even between the bare surface and (1).
In summary, these control experiments exclude the necessity to have a bromide group present on the substrate for immobilisation of (1). In order to elucidate the role of the hydroxyl group on (1) for immobilisation, p-Si/SiOx/Br was sensitised with complex (5) that is lacking the hydroxyl group (Table 2).43 The high-resolution XP spectra of the resulting material did not show any contribution of the Ru3d and N1s regions (Table 2, entry 4; Fig. S12†). The absence of Ru and N signals indicates that without the hydroxyl moiety, no interaction occurs between the terminal bromide or the potential pendant Si–O bonds. This experiment clearly highlights the crucial role of the hydroxyl group of (1) to ensure immobilisation onto p-Si/SiOx/Br.
Conclusions
In summary, we presented the immobilisation of the complex [Ru(HO-tpy)(6-mbpy)(NCCH3)]2+ (1) on a p-Si based electrode through a molecular bottom-up approach. The presence of a thin insulative native oxide layer, which is intended to minimise back electron transfer, allows efficient charge extraction giving rise to a state-of-the-art photovoltage of +570 mV. The overall architecture can be compared to an MIS junction in which complex (1) takes the role of a metal layer. The study illustrates the possibility of using p-Si to diminish the overpotential that is needed for CO2 reduction by (1). Furthermore, the underlying supportive photocathode p-Si/SiO2 architecture is not altered by the applied potential. Despite these encouraging photoelectrochemistry results, severe desorption of complex (1) from the photocathode was observed upon applying the potential, thereby preventing further in-depth photoelectrochemical CO2 reduction studies. In other words, the detachment of (1) is faster than any catalytic CO2RR, which could thus not be demonstrated for p-Si/SiOx/[Ru]. Although the exact interactions that are responsible for the anchorage of (1) onto p-Si/SiOx/Br remained unclear, the necessity of (1) to bear a hydroxyl group for anchorage was clearly demonstrated. The challenge remains to produce more robust photocathodes to ensure efficient catalytic devices.Experimental
Instrumentation
NMR spectra were recorded using a JEOL (400YH Magnet) 400 MHz spectrometer at 293 K in CD3CN. Chemical shifts (δ) are quoted as parts per million (ppm) and the coupling constants J are reported in Hz. All 1H NMR spectra are referenced to the solvent residual signal (δ = 1.94 ppm). HR-NSI-MS analyses were performed at the Organisch Chemisches Institut WWU Münster.Crystallography measurements were performed using graphite-monochromatised Mo Kα radiation at 170 K using a Bruker D8 APEX-II equipped with a CCD camera. The structure was solved by direct methods (SHELXT-2014),75 and refined by full-matrix least-squares techniques against F2 (SHELXL-2018)76 in Olex277 The non-hydrogen atoms were refined with anisotropic displacement parameters. The H atoms of the CH2/CH groups were refined with common isotropic displacement parameters for the H atoms of the same group and idealised geometry. The H atoms of the methyl groups were refined with common isotropic displacement parameters for the H atoms of the same group and idealised staggered geometry. CCDC 2059979 and 2059980† contain the supplementary crystallographic data for this paper.
Ellipsometry measurements were recorded using a J.A. Woollam Co. RC2-XI spectroscopic ellipsometer equipped with a 75 W Xenon Arc lamp. The measurement angles were 60, 65 and 70°. Data fitting was assured by using the Complete EASE software using a Cauchy model (A = 1.450 and B = 0.010) on the silicon surface. The molecular layer thickness was determined by subtraction of the oxide thickness measured before the chemical modification.
XPS measurements were performed using a PHI Quantera SXH spectrometer with a monochromatic Al Kα source (hν = 1486.6 eV) operated with a 25 W electron beam power and a beam diameter of 100 μm. The spectra were collected with a 112 eV pass energy at a take-off angle of 45°. The binding energies were referred to the Si2p binding energy calibrated at 99.4 eV. The spectra were analysed by using the Casa XPS v 2.3.22PR1.0 software (Casa Software Ltd, UK) and all the elemental peak intensities were corrected by Scofield factors (Table S3†).
Homogeneous electrochemical studies were performed on an Autolab PGSTAT100 controlled by the GPES 4.9 software using a standard three-electrode set-up with glassy carbon as the working electrode. Photoelectrochemical studies were performed on an Autolab PGSTAT204 controlled by the NOVA software using a three-electrode set-up with a custom Teflon cell (Pine Instrument) equipped with a quartz window. The modified silicon surfaces were used as the working electrodes. The electrical contact of the silicon surfaces consisted of a drop of gallium-indium eutectic covered by a conductive copper tape (Electron Microscopy Science) in contact with a stainless-steel back-contact. All electrochemical set-up encompassed an Ag/Ag+ (0.01 M AgNO3 in CH3CN, calibrated against Fc+/Fc potential) nonaqueous reference electrode and a glassy carbon rod auxiliary electrode.
The modified silicon surfaces were illuminated by a solar simulator (SS-F5-3A Enlitech) equipped with a 300 W Xe light source and an AM 1.5G spectral correction filter and calibrated towards an NREL-traceable silicon solar cell (Ref SRC-2020-SRC-00124 Enlitech).
Synthesis
All chemicals were purchased from commercial sources and used as received. The boron p-doped silicon (111) wafers of thickness 265 μM and a resistivity of 0.3–0.5 Ω cm were ordered from Łukasiewicz ITME. Acetonitrile was dried by distillation over calcium hydride before electrochemical studies and surface preparation. [(4′-Hydroxy-2,2′:6′,2′′-terpyridyl)(trichloro)ruthenium(III)] and [(2,2′:6′,2′′-terpyridyl)(6-methyl-2,2′-bipyridyl)acetonitrile ruthenium(II)] hexafluorophosphate have been synthesised according to the literature procedure.43,47 [Bis-((4′-hydroxy-2,2′:6′,2′′-terpyridyl)(chloro)ruthenium) dichloride] synthesis was adapted from the literature and led to a dark purple powder that was used without further characterisation.46,47[(4′-Hydroxy-2,2′:6′,2′′-terpyridyl)(6-methyl-2,2′-bipyridyl)chlororuthenium(II)] hexafluorophosphate (39.0 mg, 0.056 mmol) was placed in a acetonitrile/water (3/1) (40/10 mL) mixture at 90 °C overnight. The solution was concentrated under reduced pressure and cooled down. A red-orange precipitate appeared after the addition of an excess of saturated ammonium hexafluorophosphate aqueous solution. The solid was filtered off, thoroughly washed with cold water and copiously rinsed with diethyl ether to obtain 41 mg of (1) (86%).
1H NMR (400 MHz, CD3CN): δ = 8.40 (d, J = 8.02 Hz, 1H), 8.28 (m, 3H), 8.15 (t, J = 7.82 Hz, 1H), 7.95 (m, 4H), 7.87 (d, J = 7.82 Hz,1H), 7.76 (m, 3H), 7.45 (d, J = 5.57 Hz, 1H), 7.31(m, 2H), 7.10 (t, J = 6.35 Hz, 1H), 3.08 (s, 3H), 2.00 (s, 3H).
NSI-MS(+): m/z: 260.5384 ([M − 2PF6 − ACN]2+; calc. 260.5389); 281.0514 ([M − 2PF6]2+; calc. 281.0522); 281.0514 ([M − 2PF6]2+; calc. 281.0522); 520.0696 ([M − 2PF6 − ACN − H+]+; calc. 520.0706); 540.0757 ([M − 2PF6 − ACN + H2O + H+]+; calc. 540.0968); 561.0959 ([M − 2PF6 − H+]+; calc. 561.0971); 707.0680 ([M − PF6]+; calc. 707.0691).
Surface preparation
Pieces of ca. 1 × 1 cm were cut from a p-Si wafer. Prior to coating, the surfaces were washed with water and then with isopropanol which was followed by sonication (5 min) in isopropanol. After another iso-propanol rinsing, a 5 min sonication in dichloromethane was performed which was followed by a final rinsing step using dichloromethane. Final cleaning was performed in piranha solution for 15 min to eliminate traces of organic residues. Caution: piranha solution is a very corrosive mixture and thorough precaution must be considered when handled. This was followed by a copious washing step with water and 5 min sonication in water, followed by water rinsing and drying under an Ar flow. All the grafting processes were performed in the dark.p-Si/SiOx/Br preparation
The freshly cleaned surfaces were placed overnight at room temperature in a 0.1 M solution of 3-bromopropyltrimethoxysilane in dry toluene in air. The coated surfaces were thoroughly rinsed with toluene and dried under an Ar flow.p-Si/SiOx/[Ru] preparation
The freshly prepared p-Si/SiOx/Br were placed for 4 days at room temperature in 0.001 M [Ru]-(3) solution in acetonitrile in the presence of 2.5 eq. of DIPEA. The surfaces were then copiously rinsed with acetonitrile (10 mL for each substrate) and dried with an Ar flow. If needed, they were kept under an Ar atmosphere before further characterisation.Author contributions
Maxime Laurans: original draft writing, synthesis and characterisation, surface modification, PEC study, and XPS study. Jordann A. L. Wells: crystal structures and writing (review and editing). Sascha Ott: supervision, conceptualisation, funding, and writing (review and editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the Swedish Energy Agency (grant no. 11674-8) and the NordForsk foundation (No. 85378) for the Nordic University hub NordCO2. The authors acknowledge Dr Hemlata Agarwala for providing the material precursor of complex (5) and for fruitful scientific discussions.Notes and references
- S. J. Davis, K. Caldeira and H. D. Matthews, Science, 2010, 329, 1330–1333 CrossRef CAS PubMed.
- Y. Xu, V. Ramanathan and D. G. Victor, Nature, 2018, 564, 30–32 CrossRef CAS PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70–88 CrossRef CAS.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS PubMed.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- D. Ghosh, T. Kajiwara, S. Kitagawa and K. Tanaka, Eur. J. Inorg. Chem., 2020, 2020, 1814–1818 CrossRef CAS.
- T.-T. Li, B. Shan, W. Xu and T. J. Meyer, ChemSusChem, 2019, 12, 2402–2408 CrossRef CAS PubMed.
- K. Maeda, Adv. Mater., 2019, 31, 1808205 CrossRef PubMed.
- J. J. Leung, J. Warnan, K. H. Ly, N. Heidary, D. H. Nam, M. F. Kuehnel and E. Reisner, Nat. Catal., 2019, 2, 354–365 CrossRef CAS.
- T.-T. Li, B. Shan and T. J. Meyer, ACS Energy Lett., 2019, 4, 629–636 CrossRef CAS.
- Y. Wang, S. L. Marquard, D. Wang, C. Dares and T. J. Meyer, ACS Energy Lett., 2017, 2, 1395–1399 CrossRef CAS.
- E. Torralba-Peñalver, Y. Luo, J.-D. Compain, S. Chardon-Noblat and B. Fabre, ACS Catal., 2015, 5, 6138–6147 CrossRef.
- P. Ding, T. Jiang, N. Han and Y. Li, Mater. Today Nano, 2020, 10, 100077 CrossRef.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef PubMed.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- A. M. Beiler, B. D. McCarthy, B. A. Johnson and S. Ott, Nat. Commun., 2020, 11, 5819 CrossRef CAS.
- D. Zhang, J. Shi, W. Zi, P. Wang and S. (Frank) Liu, ChemSusChem, 2017, 10, 4324–4341 CrossRef CAS PubMed.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- W. Peng, S. M. Rupich, N. Shafiq, Y. N. Gartstein, A. V. Malko and Y. J. Chabal, Chem. Rev., 2015, 115, 12764–12796 CrossRef CAS PubMed.
- R. M. Candea, M. Kastner, R. Goodman and N. Hickok, J. Appl. Phys., 1976, 47, 2724–2726 CrossRef CAS.
- J. J. Leung, J. Warnan, D. H. Nam, J. Z. Zhang, J. Willkomm and E. Reisner, Chem. Sci., 2017, 8, 5172–5180 RSC.
- C. M. Hanna, R. T. Pekarek, E. M. Miller, J. Y. Yang and N. R. Neale, ACS Energy Lett., 2020, 5, 1848–1855 CrossRef CAS.
- C. Nie, C. Liu, L. Gong and M. Wang, J. Mater. Chem. A, 2021, 9, 234–238 RSC.
- B. Shan, M. K. Brennaman, L. Troian-Gautier, Y. Liu, A. Nayak, C. M. Klug, T.-T. Li, R. M. Bullock and T. J. Meyer, J. Am. Chem. Soc., 2019, 141, 10390–10398 CrossRef CAS PubMed.
- N. Queyriaux, N. Kaeffer, A. Morozan, M. Chavarot-Kerlidou and V. Artero, J. Photochem. Photobiol., C, 2015, 25, 90–105 CrossRef CAS.
- M. Wang, Y. Yang, J. Shen, J. Jiang and L. Sun, Sustainable Energy Fuels, 2017, 1, 1641–1663 RSC.
- B. Shan, S. Vanka, T.-T. Li, L. Troian-Gautier, M. K. Brennaman, Z. Mi and T. J. Meyer, Nat. Energy, 2019, 4, 290–299 CrossRef CAS.
- J. M. Gurrentz and M. J. Rose, J. Am. Chem. Soc., 2020, 142, 5657–5667 CrossRef CAS PubMed.
- J. R. C. Lattimer, J. D. Blakemore, W. Sattler, S. Gul, R. Chatterjee, V. K. Yachandra, J. Yano, B. S. Brunschwig, N. S. Lewis and H. B. Gray, Dalton Trans., 2014, 43, 15004–15012 RSC.
- J. Seo, R. T. Pekarek and M. J. Rose, Chem. Commun., 2015, 51, 13264–13267 RSC.
- K. Sun, S. Shen, Y. Liang, P. E. Burrows, S. S. Mao and D. Wang, Chem. Rev., 2014, 114, 8662–8719 CrossRef CAS PubMed.
- J. Shewchun, R. Singh and M. A. Green, J. Appl. Phys., 1977, 48, 765–770 CrossRef CAS.
- D. V. Esposito, I. Levin, T. P. Moffat and A. A. Talin, Nat. Mater., 2013, 12, 562–568 CrossRef CAS PubMed.
- B. A. Johnson, S. Maji, H. Agarwala, T. A. White, E. Mijangos and S. Ott, Angew. Chem., Int. Ed., 2016, 55, 1825–1829 CrossRef CAS PubMed.
- B. A. Johnson, H. Agarwala, T. A. White, E. Mijangos, S. Maji and S. Ott, Chem. – Eur. J., 2016, 22, 14870–14880 CrossRef CAS PubMed.
- Z. Chen, C. Chen, D. R. Weinberg, P. Kang, J. J. Concepcion, D. P. Harrison, M. S. Brookhart and T. J. Meyer, Chem. Commun., 2011, 47, 12607–12609 RSC.
- Z. Chen, P. Kang, M.-T. Zhang and T. J. Meyer, Chem. Commun., 2013, 50, 335–337 RSC.
- D. C. Marelius, S. Bhagan, D. J. Charboneau, K. M. Schroeder, J. M. Kamdar, A. R. McGettigan, B. J. Freeman, C. E. Moore, A. L. Rheingold, A. L. Cooksy, D. K. Smith, J. J. Paul, E. T. Papish and D. B. Grotjahn, Eur. J. Inorg. Chem., 2014, 2014, 676–689 CrossRef CAS.
- K. A. Maghacut, A. B. Wood, W. J. Boyko, T. J. Dudley and J. J. Paul, Polyhedron, 2014, 67, 329–337 CrossRef CAS.
- J. J. Concepcion, J. W. Jurss, M. R. Norris, Z. Chen, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2010, 49, 1277–1279 CrossRef CAS PubMed.
- S. Watabe, Y. Tanahashi, M. Hirahara, H. Yamazaki, K. Takahashi, E. A. Mohamed, Y. Tsubonouchi, Z. N. Zahran, K. Saito, T. Yui and M. Yagi, Inorg. Chem., 2019, 58, 12716–12723 CrossRef CAS PubMed.
- R. Tatikonda, M. Cametti, E. Kalenius, A. Famulari, K. Rissanen and M. Haukka, Eur. J. Inorg. Chem., 2019, 2019, 4463–4470 CrossRef CAS.
- M. Yagi, S. Tajima, M. Komi and H. Yamazaki, Dalton Trans., 2011, 40, 3802–3804 RSC.
- L. Duan, G. F. Manbeck, M. Kowalczyk, D. J. Szalda, J. T. Muckerman, Y. Himeda and E. Fujita, Inorg. Chem., 2016, 55, 4582–4594 CrossRef CAS PubMed.
- T. Tomon, T. Koizumi and K. Tanaka, Eur. J. Inorg. Chem., 2005, 2005, 285–293 CrossRef.
- T. A. White, S. Maji and S. Ott, Dalton Trans., 2014, 43, 15028–15037 RSC.
- S. Onclin, B. J. Ravoo and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2005, 44, 6282–6304 CrossRef CAS PubMed.
- A. Gulino, S. Bazzano, G. G. Condorelli, S. Giuffrida, P. Mineo, C. Satriano, E. Scamporrino, G. Ventimiglia, D. Vitalini and I. Fragalà, Chem. Mater., 2005, 17, 1079–1084 CrossRef CAS.
- F. Lupo, M. E. Fragalà, T. Gupta, A. Mamo, A. Aureliano, M. Bettinelli, A. Speghini and A. Gulino, J. Phys. Chem. C, 2010, 114, 13459–13464 CrossRef CAS.
- B. W.-K. Chu and V. W.-W. Yam, Langmuir, 2006, 22, 7437–7443 CrossRef CAS PubMed.
- G. E. Fryxell, P. C. Rieke, L. L. Wood, M. H. Engelhard, R. E. Williford, G. L. Graff, A. A. Campbell, R. J. Wiacek, L. Lee and A. Halverson, Langmuir, 1996, 12, 5064–5075 CrossRef CAS.
- J. Böhmler, A. Ponche, K. Anselme and L. Ploux, ACS Appl. Mater. Interfaces, 2013, 5, 10478–10488 CrossRef PubMed.
- S. P. Pujari, L. Scheres, A. T. M. Marcelis and H. Zuilhof, Angew. Chem., Int. Ed., 2014, 53, 6322–6356 CrossRef CAS PubMed.
- S. Desbief, L. Patrone, D. Goguenheim, D. Guérin and D. Vuillaume, Phys. Chem. Chem. Phys., 2011, 13, 2870–2879 RSC.
- A. W. H. Lee and B. D. Gates, Langmuir, 2017, 33, 8707–8715 CrossRef CAS.
- T. Masuda, K. Shimazu and K. Uosaki, J. Phys. Chem. C, 2008, 112, 10923–10930 CrossRef CAS.
- S. Li and M. Mrksich, Langmuir, 2018, 34, 6713–6718 CrossRef CAS PubMed.
- D. Gimenez, A. Dose, N. L. Robson, G. Sandford, S. L. Cobb and C. R. Coxon, Org. Biomol. Chem., 2017, 15, 4081–4085 RSC.
- M. Sato, T. Furusawa, T. Hotta, H. Watanabe and N. Suzuki, Surf. Interface Anal., 2006, 38, 838–841 CrossRef CAS.
- S. Yin, H. Sun, Y. Yan, H. Zhang, W. Li and L. Wu, J. Colloid Interface Sci., 2011, 361, 548–555 CrossRef CAS.
- Q. Li, S. Surthi, G. Mathur, S. Gowda, V. Misra, T. A. Sorenson, R. C. Tenent, W. G. Kuhr, S. Tamaru, J. S. Lindsey, Z. Liu and D. F. Bocian, Appl. Phys. Lett., 2003, 83, 198–200 CrossRef CAS.
- A. G. Scheuermann, J. P. Lawrence, K. W. Kemp, T. Ito, A. Walsh, C. E. D. Chidsey, P. K. Hurley and P. C. McIntyre, Nat. Mater., 2016, 15, 99–105 CrossRef CAS PubMed.
- A. J. Bard, A. B. Bocarsly, F. R. F. Fan, E. G. Walton and M. S. Wrighton, J. Am. Chem. Soc., 1980, 102, 3671–3677 CrossRef CAS.
- J. A. Turner, J. Manassen and A. J. Nozik, Appl. Phys. Lett., 1980, 37, 488–491 CrossRef CAS.
- B. Kumar, J. M. Smieja and C. P. Kubiak, J. Phys. Chem. C, 2010, 114, 14220–14223 CrossRef CAS.
- B. Kumar, M. Beyler, C. P. Kubiak and S. Ott, Chem. – Eur. J., 2012, 18, 1295–1298 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. a. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- D. J. Wasylenko, C. Ganesamoorthy, B. D. Koivisto, M. A. Henderson and C. P. Berlinguette, Inorg. Chem., 2010, 49, 2202–2209 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2059979 and 2059980. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt01331a |
| This journal is © The Royal Society of Chemistry 2021 |