
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel primary phosphinecarboxamides derived from diamines†
Erica N.
Faria
,
Andrew R.
Jupp
 and
Jose M.
Goicoechea
*
and
Jose M.
Goicoechea
*
Department of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: jose.goicoechea@chem.ox.ac.uk
First published on 28th April 2021
Abstract
We describe the synthesis of N-functionalised phosphinecarboxamides obtained by reaction of the 2-phosphaethynolate anion (PCO−) with diamines, specifically hydrazine, methylenediamine and ethylenediamine, in the presence of acid. The resulting neutral compounds can be deprotonated to generate phosphide anions that, when further reacted with electrophiles, form secondary phosphines.
1. Introduction
Primary phosphines (H2PR) are interesting chemical feedstocks that have found applications in asymmetric catalysis,1 medicinal chemistry,2 polymer science,3 and the synthesis of extended solids.4 However, despite their enormous versatility, the reactivity of this class of compounds remains underexplored compared to primary amines (H2NR). This is largely due to the noxious character, propensity towards oxidation, and the pyrophoric nature of the vast majority of primary phosphines. In recent years, a significant amount of research has been conducted exploring the synthesis and reactivity of air-stable primary species in an effort to circumvent many of the pitfalls associated with their manipulation.5–7 For example, the use of sterically encumbering substituents has been shown to kinetically stabilise such compounds, allowing for a more thorough exploration of their highly reactive P–H bonds.8We recently reported on a unique class of primary phosphines, so-called phosphinecarboxamides or carbamoylphosphines, (RHNC(O)PH2), that can be accessed by the reaction of the 2-phosphaethynolate anion (PCO−) with amines in the presence of an acid.9–12 This was made possible due to the availability of a phosphorus-containing congener of the cyanate anion, PCO−, which while first reported in 1992,13 has only come to the fore as a chemical reagent this last decade.14 As with other primary phosphines, H2NC(O)PH2 may also be employed as a precursor for metal phosphides, for example in the synthesis of Zn3P2 thin films.15 The main advantage of this family of phosphines is that the carbamoyl group imparts a high degree of stability to such species, making their manipulation in air straightforward. Further research into the synthesis of phosphinecarboxamides has included mechanochemical methods,16 metal-catalysed reactions,17 direct reaction of secondary phosphines with cyanates,18 and phosphinoboration of isocyanates.19 The reactivity of such species has also been demonstrated by coordination to metal centres,20 functionalization of P–H bonds to afford secondary phosphines,11,21 and their use as precursors to generate phosphonates.22
These findings prompted us to investigate the accessibility of di-substituted phosphinecarboxamides by employing diamines as starting materials. For this purpose, we studied the reactivity of [Na(dioxane)x][PCO] (x = 1.97–2.08) towards hydrazine, methylenediamine and ethylenediamine (NH2(CH2)nNH2, where n = 0, 1, 2, respectively). Herein we report the synthesis of new N-functionalised phosphinecarboxamides, their corresponding phosphides (accessed by deprotonation) as well as their secondary phosphine derivatives.23
2. Results and discussion
2.1. Synthesis of phosphinecarboxamides
In the first instance we explored the reactivity of hydrazine hydrochloride, NH2NH2·HCl, towards [Na(dioxane)x][PCO] in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Scheme 1). This reaction gives rise to a product with a triplet resonance at −139.9 ppm (1JP–H = 206 Hz) in the 31P NMR spectrum, accompanied by a side product observed at −128.2 ppm as a triplet of doublets (1JP–H = 218 Hz; 3JP–H = 5 Hz). Both triplets collapse to singlets on proton decoupling, suggesting the formation of the trans- and cis-isomers of H2NHNC(O)PH2 (1).‡
:1 ratio (Scheme 1). This reaction gives rise to a product with a triplet resonance at −139.9 ppm (1JP–H = 206 Hz) in the 31P NMR spectrum, accompanied by a side product observed at −128.2 ppm as a triplet of doublets (1JP–H = 218 Hz; 3JP–H = 5 Hz). Both triplets collapse to singlets on proton decoupling, suggesting the formation of the trans- and cis-isomers of H2NHNC(O)PH2 (1).‡
 | ||
| Scheme 1 Reaction of NH2NH2·HCl and [Na(dioxane)x][PCO] to afford 1 as its trans (1a) and cis (1b) isomers. | ||
The peak at −139.9 ppm can be assigned to the trans isomer of the monosubstituted hydrazine phosphinecarboxamide (or phosphinecarbohydrazide; 1a) and the peak at −128.2 ppm corresponds to the cis isomer (1b). In the case of the latter species, the PH2 group and the carboxamide proton are in a vicinal trans arrangement to one another relative to the C–N bond, allowing for the resolution of the 3JP–H coupling. This data is in agreement with that of the parent species (H2PC(O)NH2), where only the coupling to the trans proton of the amide could be observed.9
Accordingly, the 1H NMR spectrum of this mixture reveals two doublets centred at 3.72 ppm (1JH–P = 206 Hz) and 4.02 ppm (1JH–P = 218 Hz) corresponding to the PH2 protons in 1a and 1b, respectively. These resonances collapse into singlets upon selective phosphorus decoupling. The 13C{1H} NMR spectrum is also consistent with the proposed structures, with the most diagnostic resonances being in the downfield region of the spectrum, where a doublet at 172.56 ppm (1JC–P = 9 Hz) and a singlet at 181.51 ppm corresponding to the carbonyl carbon in 1a and 1b were observed. The coupling between carbon and phosphorus could not be resolved for 1b, therefore 1H–13C HMBC 2D NMR experiments were used to unambiguously assign the peak.
Single crystals of 1a were obtained from a toluene solution (Fig. 1). X-ray diffraction studies revealed that the proton attached to the α-nitrogen is cis to the phosphorus atom relative to the C–N bond. The phosphorus presents a pyramidal geometry, while the (NH2)NH–CO fragment is planar. There are two molecules in the asymmetric unit and, consequently, all the bond metric data are discussed as average values. Bond metric data show a C–P bond length of 1.864(av) Å, which is consistent with a single bond between these atoms, and the C–N bond length is 1.334(av) Å, suggesting multiple bond character between carbon and nitrogen.24,25
 | ||
| Fig. 1 Molecular structure of the trans isomer of hydrazine mono-phosphinecarboxamide 1a. Thermal ellipsoids pictured at the 50% probability level. Selected bond distances (Å) and angles (deg): P1–C1: 1.864(av); C1–O1: 1.224(av); C1–N1: 1.334(av); N1–N2: 1.431(av); O1–C1–P1: 119.2(av); N1–C1–P1: 116.5(av); O1–C1–N1: 124.2(av); C1–N1–N2: 121.4(av). | ||
Single crystals of cis-isomer, 1b, could also be obtained by co-crystallisation of the mixture with 18-crown-6, which forms hydrogen bonds with the product (Fig. 2). Bond metric data for this species were similar to those recorded for 1a.
 | ||
| Fig. 2 Molecular structure of the cis isomer of the hydrazine mono-phosphinecarboxamide in 1b·(18-crown-6)0.5. Thermal ellipsoids pictured at the 50% probability level. Note that 1b crystallises with 0.5 equivalents of 18-crown-6 in the asymmetric unit, for clarity, the full 18-crown-6 molecule is pictured. Selected bond distances (Å) and angles (deg): P1–C1: 1.869(av); C1–O1: 1.222(av); C1–N1: 1.339(av); N1–N2: 1.407(av); O1–C1–P1: 122.2(av); N1–C1–P1: 114.3(av); O1–C1–N1: 123.4(av); C1–N1–N2: 118.7(av). | ||
The barrier to rotation around the C–N bond was calculated using density functional theory (DFT) at the PBE0/6-31G(d,p) level of theory (Fig. S35†). The transition state was found to be 92 kJ mol−1 higher in energy than the more stable isomer. The trans isomer 1a is more stable than the cis1b by 11 kJ mol−1. This value is in good agreement with the spectroscopic data obtained, which suggest that 1a is the major product of the reaction. The relatively high barrier precludes interconversion between isomers due to inherent thermal instability of phosphinecarboxamides.
In an attempt to synthesise a bifunctionalised phosphinecarboxamide derived from hydrazine, i.e. PH2C(O)NHNHC(O)PH2, the reaction of NH2NH2·2HCl and two equivalents of [Na(dioxane)x][PCO] was attempted. 31P{1H} NMR spectroscopy revealed that the product of this reaction was a mixture of 1a and 1b. Attempts to force the reaction to completion by heating resulted in decomposition. It is likely that the desired compound could not be obtained due to decreased nucleophilicity of the β-nitrogen in 1a/1b caused by the presence of one phosphinecarboxamide moiety. Consequently, nucleophilic attack of the β-nitrogen at a second HPCO molecule is slow. HPCO is unstable and short-lived in solution and rapidly decomposes.26
We next aimed to extend this research to bridged diamines, by exploring the reactivity of methylenediamine towards [Na(dioxane)x][PCO]. Interestingly, the product obtained from the reaction of NH2CH2NH2·2HCl and one equivalent of [Na(dioxane)x][PCO] was shown to be the parent phosphinecarboxamide species, H2PC(O)NH2,9 and hexamethylenetetramine, also known as urotropine.27 We propose that the desired phosphinecarboxamide product is initially formed but methylene imine is quickly eliminated to form the parent species, and that the imine, in turn, oligomerises to yield the urotropine and ammonia (Scheme 2).
 | ||
| Scheme 2 Proposed decomposition pathway of the methylenediamine mono-phosphinecarboxamide derivative. | ||
The relative energies of the two proposed stages of this decomposition reaction have been calculated (Fig. S36/Table S11†). The final products, C, are 8 kJ mol−1 more stable than if the reaction were to stop at the targeted phosphinecarboxamide, A, and 53 kJ mol−1 more stable than intermediates B, which explains why urotropine is observed in the 1H NMR spectrum. This pathway is also supported by the presence of a small peak at 0.05 ppm in the 1H NMR spectrum that corresponds to the ammonia formed.28
Finally, when NH2CH2CH2NH2·2HCl and two equivalents of [Na(dioxane)x][PCO] were reacted, the di-substituted phosphinecarboxamide 2 was obtained (Scheme 3). The 31P NMR spectrum exhibits a triplet at −132.9 ppm (1JP–H = 208 Hz) that collapses to a singlet on proton decoupling. The 1H NMR spectrum shows a doublet centred at 3.72 ppm (1JH–P = 208 Hz) that also collapses to a singlet upon selective phosphorus decoupling. The 13C{1H} NMR spectrum shows a doublet at 173.52 ppm (1JC–P = 7 Hz) that corresponds to the carbonyl carbons of the two phosphinecarboxamide moieties coupling to the phosphorus atoms.
 | ||
| Scheme 3 Reaction of NH2CH2CH2NH2·2HCl and [Na(dioxane)x][PCO] to afford 2. | ||
Single crystals obtained by slow diffusion of hexane into a pyridine solution of the product confirm the incorporation of two phosphinecarboxamide units to the ethylenediamine (Fig. 3). Bond metric data are consistent with 1a and 1b.
 | ||
| Fig. 3 Molecular structure of 2. Thermal ellipsoids pictured at the 50% probability level. Symmetry operation ‘: −x, −y, 1 − z. Selected bond distances (Å) and angles (deg): P1–C1: 1.8633(12); C1–O1: 1.2275(15); C1–N1: 1.3256(15); N1–C2: 1.4549(15); C2–C2′: 1.519(2); O1–C1–P1: 118.38(9); N1–C1–P1: 118.47(8); O1–C1–N1: 123.06(11). | ||
2.2. Deprotonation and alkylation studies
We sought to investigate the relative acidity of the P–H bonds in these novel phosphinecarboxamides by exploring their reactivity towards strong non nucleophilic bases such as benzyl potassium (BnK) or potassium bis(trimethylsilyl)amide (KHMDS).Addition of one equivalent of BnK and 18-crown-6 to 1 in d5-pyridine gave rise to a broad doublet at −109.4 ppm (1JP–H = 150 Hz) and a triplet at −128.6 ppm (1JP–H = 190 Hz) in the 31P NMR spectrum (Fig. 4). In d8-THF these resonances were observed at −114.0 and −132.5 ppm, respectively. These resonances collapse to singlets upon proton decoupling and have an integration ratio of 0.3:1, respectively, and they are consistent with deprotonation at the phosphorus and nitrogen sites of the molecule to yield two different anionic species, [K(18-crown-6)][3] (Scheme 4).
 | ||
| Fig. 4 31P{1H} (top) and 31P (bottom) NMR spectra (in d8-THF) of the reaction between 1 and BnK in the presence of 18-crown-6. | ||
 | ||
| Scheme 4 Reaction of 1, one equivalent of BnK and 18-crown-6 to yield [K(18-crown-6)][3]. | ||
In the 1H NMR spectrum, two well defined doublets were observed centred at 2.95 ppm (1JP–H = 150 Hz), corresponding to the phosphide product, and another at 4.08 ppm (1JP–H = 190 Hz) which corresponds to the amide. These resonances collapse to singlets upon phosphorus decoupling.
Compound 1 possesses three possible deprotonation sites that can yield eight isomers of 3 either in their cis or trans forms relative to the C–N and C–P bonds. The energy of all isomers of 3a–3c (Fig. 5) were calculated using DFT. Calculations revealed that the phosphide trans–cis-3a is the most stable isomer. Based on these results, the broad doublet in the 31P NMR spectrum was assigned as trans–cis-3a, as this is the most stable phosphide isomer, and the triplet has been assigned as trans-3c, resulting from deprotonation of the NHNH2 site, which is 58 kJ mol−1 higher in energy (structures highlighted with boxes in Fig. 5).
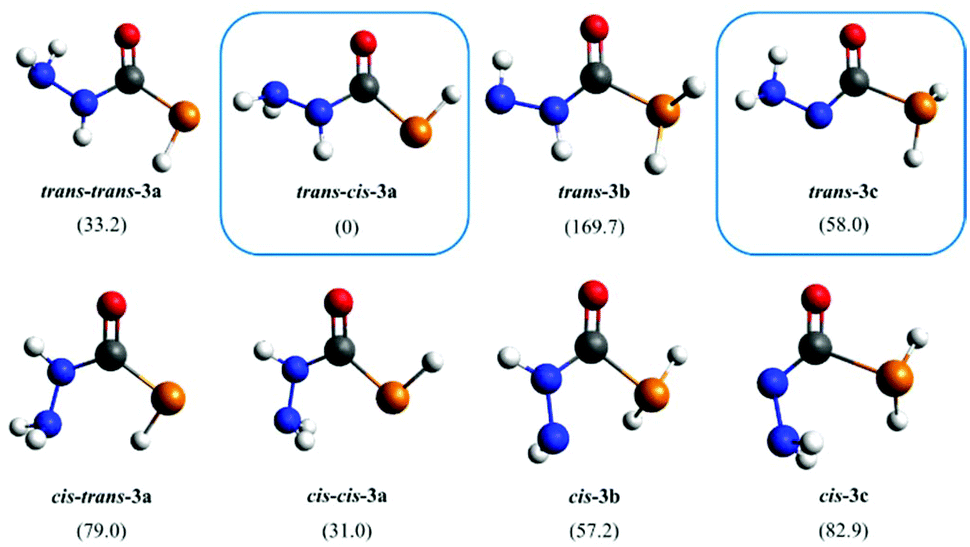 | ||
| Fig. 5 Possible deprotonation isomers of 3 relative to the C–N and C–P bonds. The calculated relative ΔG values are given in brackets in kJ mol−1. The isomers obtained in solution assigned according to NMR data are highlighted with boxes. | ||
A variable temperature 31P NMR experiment was performed on a mixture of 1, KHMDS and 18-crown-6 in d8-THF over the range of −80 to +60 °C (Fig. S16†). At −80 °C, it is possible to observe a broad doublet at −97.0 ppm (1JP–H = 157 Hz), a triplet at −130.1 ppm (1JP–H = 190 Hz), a triplet of doublets at −132.9 ppm (1JP–H = 217 Hz, 3JP–H = 8 Hz), and another triplet at −135.6 ppm (1JP–H = 197 Hz). The broad doublet shifts from −97.0 ppm to −114.9 ppm as the temperature increases and two of the triplets disappear over time leaving only one broad triplet at +60 °C. Interestingly, even though the broad doublet shifted 18 ppm upfield, no coalescence between the doublet and the triplet could be observed as the temperature was increased from −80 °C to room temperature. This observation suggests that at room temperature, a slow exchange between these two species might be occurring.
The inability to observe coalescence in this system indicates that if exchange is taking place, it is slow on the NMR timescale. To further investigate this behaviour, a 31P{1H} 1D EXSY exchange spectroscopy NMR experiment was conducted at room temperature (Fig. S17†) where the phosphide peak was inverted while the signal of the phosphine is monitored as a function of time as the inverted signal relaxes back to equilibrium. As the first peak relaxes, the intensity of the non-inverted signal is observed to decrease and, with time, the phosphide resonance regains its initial intensity while the phosphine increases its intensity, which suggests exchange between the two species at room temperature.
Single crystals suitable for X-ray diffraction were obtained from slow diffusion of hexane into a pyridine solution of [K(18-crown-6)][3]. Interestingly, only [K(18-crown-6)][cis–cis-3a] crystallized from the solution (Fig. 6).
 | ||
| Fig. 6 Molecular structure of [K(18-crown-6)][cis–cis-3a]. Thermal ellipsoids pictured at the 50% probability level. Selected bond distances (Å) and angles (deg): P1–C1: 1.792(2); C1–O1: 1.248(3); C1–N1: 1.383(3); N1–N2: 1.393(3); O1–K1: 2.679(2); O1–C1–P1: 128.0(2); N1–C1–P1: 115.26(18); O1–C1–N1: 116.7(2); C1–N1–N2: 122.8(2). | ||
Bond metric data indicate that the P–C, C–N and C–O bond lengths are 1.792(2), 1.383(3) and 1.248(3) Å, respectively. Upon deprotonation at the phosphorus, the degree of multiple bond character between phosphorus and carbon increases (1.793(3) vs. 1.869(av) Å in 1b), while there is a decrease in the multiple bond character between the carbon and nitrogen atoms (1.384(3) vs. 1.339(av) Å in 1b). These data are consistent with the bond metrics of previously reported carbamoylphosphides.11
Even though cis–cis-3a is 31 kJ mol−1 less stable than trans–cis-3a, the fact that it crystalizes preferentially can be explained by the presence of hydrogen bonds with the neighbouring molecules that favours crystal packing. It should also be mentioned that our gas phase calculations do not take into consideration the presence of the cation or the sequestering agent.
Addition of two equivalents of KHMDS to 2 in pyridine in the presence of two equivalents of 18-crown-6 promotes exclusive deprotonation at the phosphorus atoms, affording compound [K(18-crown-6)]2[4] (Scheme 5).
 | ||
| Scheme 5 Reaction of 2, two equivalents of KHMDS and 18-crown-6 to yield [K(18-crown-6)]2[4]. | ||
This selectivity is evident in the 31P NMR spectrum, which exhibits a broad doublet centred at −98.8 ppm (1JP–H = 150 Hz). With longer reaction times, we observe the presence of a broad triplet at −256.5 ppm (1JP–H = 134 Hz) that corresponds to KPH2, a decomposition product. These resonances collapse to singlets upon proton decoupling.
The 1H NMR spectrum shows a doublet centred at 2.78 ppm (1JP–H = 150 Hz) that corresponds to the phosphide moiety. The 13C{1H} NMR spectrum reveals a doublet at 203.79 ppm (1JC–P = 59 Hz). The 1JC–P coupling constant found for this anion is significantly larger than that of the neutral species 2 (1JC–P = 7 Hz) presumably due to contraction of the P–C bond with an increase of multiple bond character between these two atoms upon deprotonation.
The deprotonated species can be used as precursors to form secondary phosphines. In order to probe this possibility, iodomethane (MeI) was chosen as a simple electrophile to generate P-functionalised phosphinecarboxamides.
Iodomethane was added to a [K(18-crown-6)][3] solution to yield the corresponding trans (5a) and cis (5b) isomers of the methylated products in solution (Scheme 6).
 | ||
| Scheme 6 Reaction of [K(18-crown-6)][3] and MeI to yield 5a and 5b. | ||
The 31P NMR spectrum revealed one major doublet of quartets at −85.4 ppm (1JP–H = 207 Hz, 2JP–H = 3 Hz) and a minor multiplet at −86.8 ppm (1JP–H = 228 Hz, 2JP–H = 3 Hz) that are approximately 23 ppm downfield-shifted relative to trans–cis-3a and have been assigned to 5a and 5b, respectively. These resonances are in a 1:0.4 ratio and collapse to singlets upon phosphorus decoupling suggesting that methylation has taken place at the phosphorus centre upon addition of base. Despite [K(18-crown-6)][3] being deprotonated at both nitrogen and phosphorus atoms, there is no evidence of methylation at the nitrogen.
In the 1H NMR spectrum it is possible to observe two clear sets of doublets of quartets centred at 4.04 ppm (1JP–H = 207 Hz, 3JH–H = 8 Hz) and 4.60 ppm (1JP–H = 228 Hz, 3JH–H = 7 Hz). These correspond to 5a and 5b. As expected, these resonances collapse to quartets upon phosphorus decoupling. The presence of two doublets of doublets at 1.37 ppm (2JH–P = 3 Hz, 3JH–H = 7 Hz) and 1.36 ppm (2JH–P = 4 Hz, 3JH–H = 7 Hz) corresponding to the methyl protons was also noted and, with the help of 1H–31P HMBC experiment, they have been assigned as belonging to 5a and 5b, respectively. The 13C{1H} NMR spectrum reveals doublets from the methyl carbons at 1.53 ppm (1JC–P = 9 Hz) and 1.83 ppm (1JC–P = 6 Hz) that, with the help of 1H–13C HMBC, have been attributed to 5a and 5b, respectively. The carbonyl carbons of 5a and 5b could also be observed as doublets at 177.68 ppm (1JC–P = 13 Hz) and 185.08 ppm (1JC–P = 6 Hz).
The 31P NMR spectrum also displays 1% of the product of double methylation of the phosphorus atom which is apparent as a broad peak at −44.2 ppm. This happens when there is an excess of KHMDS and MeI in solution. Excess base promotes a second deprotonation at the phosphorus that is then attacked by a second equivalent of electrophile.
Two equivalents of KHMDS were added to a mixture of MeI and 2 to yield the corresponding methylated product (6). The 31P NMR spectrum exhibits three overlapping doublet of quartets, two of equal integration centred at −81.08 and −81.11 ppm (1JP–H = 207 Hz, 3JH–H = 3 Hz), and a smaller one at −81.08 ppm (1JP–H = 207 Hz, 3JH–H = 3 Hz) that is only apparent in the 31P{1H} NMR spectrum. These resonances collapse to singlets upon proton decoupling and are 51.5 ppm downfield-shifted relative to 2 and 17.6 ppm downfield-shifted relative to 4. We propose that the two main overlapping resonances observed correspond to the trans–cis isomers of the product of single methylation at the phosphorus atoms (6a) and that the minor product is the trans–trans isomer (6b) (Fig. 7).
 | ||
| Fig. 7 Trans–cis (6a) and trans–trans (6b) isomers of 6. | ||
Other than the desired products obtained, it is also possible to observe the presence of two overlapping septets at −41.0 and −41.1 ppm accompanied by a triplet at −132.7 ppm in the 31P NMR spectrum. These resonances are complementary and correspond to the product of double methylation of one of the phosphorus atoms in a molecule while leaving the other phosphorus untouched.
In the 1H NMR spectrum, the doublets originating from the PH2 moiety in 2 are replaced by two overlapping doublets of quartets centred at 4.02 ppm (1JH–P = 207 Hz, 3JH–H = 3 Hz; and 1JH–P = 207 Hz, 3JH–H = 3 Hz) that correspond to the P–H protons in the trans–cis isomer (6a). These resonances also overlap with the methylene units from each isomer, but they become very clear in the 1H{31P} NMR spectrum, where they collapse to quartets upon phosphorus decoupling. The presence of a doublet of doublets at 1.34 ppm (2JH–P = 3 Hz, 3JH–H = 7 Hz) corresponding to the methyl protons was also noted. The 13C{1H} NMR spectrum reveals doublets from the methyl carbons at 1.74 ppm (1JC–P = 8 Hz) and these appear to have the same chemical shift for the two isomers (6a). The carbonyl carbons of 6a were also observed as two overlapping doublets at 178.82 and 178.80 ppm (1JC–P = 11 Hz).
3. Conclusions
We have shown that the synthesis and characterization of two novel phosphinecarboxamides and their subsequent functionalization is possible by reacting diamines with sodium phosphaethynolate salts. It has been shown that, in general, despite having two possible deprotonation sites, the phosphorus-bound protons are more acidic than the amide protons and that phosphinecarboxamides are not able to tolerate basic media. The phosphides obtained can be further reacted with simple electrophiles rendering P-functionalised phosphinecarboxamides. The generation of molecules featuring multiple phosphinecarboxamide moieties, such as 2, highlights the potential of this method for the synthesis of bespoke chelating ligands from abundant poly(amine) precursors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) of the Brazilian government (studentship ENF; Process number 233778/2014-4) and the EPSRC (DTA studentship ARJ) for funding.Notes and references
- (a) X. Li, K. D. Robinson and P. P. Gaspar, J. Org. Chem., 1996, 61, 7702–7710 CrossRef CAS PubMed; (b) A. Marinetti and J.-P. Genêt, C. R. Chim., 2003, 6, 507–514 CrossRef CAS; (c) T. Clark and C. Landis, Tetrahedron: Asymmetry, 2004, 15, 2123–2137 CrossRef CAS; (d) G. Hoge and B. Samas, Tetrahedron: Asymmetry, 2004, 15, 2155–2157 CrossRef CAS; (e) D. J. Brauer, K. W. Kottsieper, S. Roßenbach and O. Stelzer, Eur. J. Inorg. Chem., 2003, 2003, 1748–1755 CrossRef.
- K. V. Katti, H. Gali, C. J. Smith and D. E. Berning, Acc. Chem. Res., 1998, 32, 9–17 CrossRef.
- (a) T. N. Hooper, M. A. Huertos, T. Jurca, S. D. Pike, A. S. Weller and I. Manners, Inorg. Chem., 2014, 53, 3716–3729 CrossRef CAS PubMed; (b) H. Dorn, R. A. Singh, J. A. Massey, A. J. Lough and I. Manners, Angew. Chem., Int. Ed., 1999, 38, 3321–3323 CrossRef CAS.
- J. T. Scheper, K. C. Jayaratne, L. M. Liable-Sands, G. P. A. Yap, A. L. Rheingold and C. H. Winter, Inorg. Chem., 1999, 38, 4354–4360 CrossRef CAS.
- M. Brynda, Coord. Chem. Rev., 2005, 249, 2013–2034 CrossRef CAS.
- B. Stewart, A. Harriman and L. J. Higham, Organometallics, 2011, 30, 5338–5343 CrossRef CAS.
- J. T. Fleming and L. J. Higham, Coord. Chem. Rev., 2015, 297–298, 127–145 CrossRef CAS.
- Selected examples of the use of sterically encumbered substituents to stabilize primary phosphines: (a) M. Yoshifuji, K. Shibayama and N. Inamoto, J. Am. Chem. Soc., 1983, 105, 2495–2497 CrossRef CAS; (b) K. Tsuji, S. Sasaki and M. Yoshifuji, Heteroat. Chem., 1998, 9, 607–613 CrossRef CAS; (c) B. Twamley, C.-S. Hwang, N. J. Hardman and P. Power, J. Organomet. Chem., 2000, 609, 152–160 CrossRef CAS; (d) For more examples, see ref. 5 and the references therein.
- A. R. Jupp and J. M. Goicoechea, J. Am. Chem. Soc., 2013, 135, 19131–19134 CrossRef CAS PubMed.
- T. P. Robinson and J. M. Goicoechea, Chem. – Eur. J., 2015, 21, 5727–5731 CrossRef CAS PubMed.
- A. R. Jupp, G. Trott, É. Payen De La Garanderie, J. D. G. Holl, D. Carmichael and J. M. Goicoechea, Chem. – Eur. J., 2015, 21, 8015–8018 CrossRef CAS PubMed.
- E. N. Faria, A. R. Jupp and J. M. Goicoechea, Chem. Commun., 2017, 53, 7092–7095 RSC.
- G. Becker, W. Schwarz, N. Seidler and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 612, 72–82 CrossRef CAS.
- For a recent review of the chemistry of the 2-phosphaethynolate ion see: J. M. Goicoechea and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 CrossRef CAS PubMed.
- S. V. F. Beddoe, S. D. Cosham, A. N. Kulak, A. R. Jupp, J. M. Goicoechea and G. Hyett, Dalton Trans., 2018, 47, 9221–9225 RSC.
- (a) Y.-H. Wu, Z.-F. Li, W.-P. Wang, X.-C. Wang and Z.-J. Quan, Eur. J. Org. Chem., 2017, 5546–5553 CrossRef CAS; (b) Z.-F. Li, W.-P. Wang, X.-C. Wang and Z.-J. Quan, Chin. J. Org. Chem., 2020, 40, 1563–1570 CrossRef.
- (a) H. R. Sharpe, A. M. Geer, W. Lewis, A. J. Blake and D. L. Kays, Angew. Chem., Int. Ed., 2017, 56, 4845–4848 CrossRef CAS PubMed; (b) A. C. Behrle and J. A. R. Schmidt, Organometallics, 2013, 32, 1141–1149 CrossRef CAS.
- (a) G. P. Papp and S. A. Buckler, J. Org. Chem., 1966, 31, 588–589 CrossRef CAS; (b) L. G. Vaughan and R. V. Lindsey Jr., J. Org. Chem., 1968, 33, 3088–3089 CrossRef CAS; (c) M. Itazaki, T. Matsutani, T. Nochida, T. Moriuchi and H. Nakazawa, Chem. Commun., 2020, 56, 443–445 RSC.
- S. J. Geier, J. H. W. LaFortune, D. Zhu, S. C. Kosnik, C. L. B. Macdonald, D. W. Stephan and S. A. Westcott, Dalton Trans., 2017, 46, 10876–10885 RSC.
- (a) M. B. Geeson, A. R. Jupp, J. E. McGrady and J. M. Goicoechea, Chem. Commun., 2014, 50, 12281–12284 RSC; (b) M. Navrátil, E. N. Faria, G. Panahy, I. Císařová, J. M. Goicoechea and P. Štěpnička, Dalton Trans., 2020, 49, 8645–8651 RSC.
- X.-G. Chen, Q.-L. Wu, F. Hou, X.-C. Wang and Z.-J. Quan, Synlett, 2019, 73–76 Search PubMed.
- (a) Y.-H. Wu, Q.-L. Wu, W.-P. Wang, X.-C. Wang and Z.-J. Quan, Adv. Synth. Catal., 2018, 360, 2382–2388 CrossRef CAS; (b) F. Hou, X.-P. Du, A. I. Alduma, Z.-F. Li, C.-D. Huo, X.-C. Wang, X.-F. Wu and Z.-J. Quan, Adv. Synth. Catal., 2020, 362, 4755–4760 CrossRef CAS.
- For review articles on phosphinecarboxamides and related species see: (a) O. Kühl, Coord. Chem. Rev., 2006, 250, 2867–2915 CrossRef; (b) P. Štěpnička, Chem. Soc. Rev., 2012, 41, 4273–4305 RSC.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- R. Hinz, C. Labbow, C. Rennick, A. Schulz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2017, 56, 3911–3915 CrossRef PubMed.
- K. Eller, E. Henkes, R. Rossbacher and H. Höke, “Amines, Aliphatic” in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH, 2020 Search PubMed.
- F. Haase and J. Sauer, J. Phys. Chem., 1994, 98, 3083 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, analytical data, spectra and computational methods. CCDC 2076831–2076834. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt01198g |
| ‡ See ESI† for full experimental details. |
| This journal is © The Royal Society of Chemistry 2021 |