
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic, spectroscopic, structural, and electrochemical investigations of ferricenium derivatives with weakly coordinating anions: ion pairing, substituent, and solvent effects†
Firoz Shah Tuglak
Khan‡
 ,
Amy L.
Waldbusser‡
,
Maria C.
Carrasco
,
Hadi
Pourhadi
and
Shabnam
Hematian
*
,
Amy L.
Waldbusser‡
,
Maria C.
Carrasco
,
Hadi
Pourhadi
and
Shabnam
Hematian
*
Department of Chemistry and Biochemistry, University of North Carolina at Greensboro, Greensboro, NC 27402, USA. E-mail: s_hemati@uncg.edu
First published on 28th April 2021
Abstract
A facile and effective strategy for the preparation of a series of ferricenium complexes bearing either electron-donating or electron-withdrawing substituents with weakly coordinating anions such as [B(C6F5)4]− or SbF6− is reported. These systems were thoroughly investigated for their ground state electronic structures in both solution and solid states using infrared (IR) and nuclear magnetic resonance (NMR) spectroscopies as well as single crystal X-ray crystallography and electrochemical measurements. The X-ray structures of the six electron-deficient ferricenium derivatives are of particular interest as only a handful (∼5) of such derivatives have been structurally characterized to date. Comparison of the structural data for both neutral and oxidized derivatives reveals that the nature of the substituents on the cyclopentadienyl (Cp) ligands displays a more significant impact on the metal–ligand separations (Fe⋯Ct) in the oxidized species than in their neutral analogs. Our 1H-NMR measurements corroborate that in the neutral ferrocene derivatives, electron-donating ring substitutions lead to a greater shielding of the ring protons while electron-withdrawing groups via induction deshield the nearby ring protons. However, the data for the paramagnetic ferricenium derivatives reveals that this substitutional behavior is more complex and fundamentally reversed, which is further supported by our structural studies. We ascribe this reversal of behavior in the ferricenium derivatives to the δ back-donation from the iron atom into the Cp rings which can lead to the overall shielding of the ring protons. Interestingly, our NMR results for the electron-deficient ferricenium derivatives in solution also indicate a direct correlation between the solvent dielectric constant and the energy barrier for rotation around the metal–ligand bond in these systems, whereas such a correlation is absent or not significant in the case of the electron-rich ferricenium species or the corresponding neutral ferrocene analogs. In this work, we also present the electrochemical behavior of the corresponding ferricenium/ferrocene redox couples including potential values (E1/2), peak-to-peak separation (ΔE1/2), and diffusion coefficients (D) of the redox active species in order to provide a concise outline of these data in one place. Our electrochemical studies involved three different solvents and two supporting electrolytes. Notably, our findings point to the significant effect of ion-pairing in lowering the energy necessary for reduction of the ferricenium ion and E1/2 in lower-polarity media. This has significant implications in applications of the ferrocene or ferricenium derivatives as redox agents in low-polarity solvents where an accurate determination of redox potential is critical.
Introduction
Bis(η5-cyclopentadienyl) iron(II), more commonly known as ferrocene (Fc), was discovered in the mid-20th century.1 Ever since, this fascinating sandwich complex has been widely studied in terms of its remarkable structure, chemical bonding and reactivity and it is often considered to be a showpiece of modern organometallic chemistry.2 Ferrocene and its numerous derivatives have increasingly found use in catalysis, particularly those involving asymmetric and stereoselective transformations, as well as in the development of new functional materials such as optical and redox sensors, batteries, and bioconjugates for medicinal and biotechnological applications.3Ferrocene can undergo a chemically reversible, outer-sphere one-electron oxidation to generate bis(η5-cyclopentadienyl) iron(+1), also known as the ferricenium ion (Fc+).4 This metal-based outer–sphere electron transfer in the ferricenium ion/ferrocene couple is commonly employed as an internal or external reference for electrochemical measurements in organic solvents.5 It is worth noting that, recently, the first examples of the two-electron oxidized as well as the one-electron reduced form of ferrocene derivatives have also been structurally and spectroscopically characterized.6
Ferricenium salts are generally prepared through the electrochemical or chemical oxidation of the ferrocene precursors. The first isolated ferricenium salt was the dark blue crystalline ferricenium tetrachlorogallate (Fc[GaCl4]) reported by Wilkinson in 1952.7 Subsequently, a combination of theoretical and experimental efforts was focused on understanding the electronic structure and physicochemical properties of ferrocene and the ferricenium ion.8
Ferricenium salts are considered mild one-electron outer-sphere oxidants. The redox potential of the ferricenium salts can be tuned by altering the nature of the substituents on the cyclopentadienyl (Cp) rings. Thus, the ferricenium derivatives with defined redox potentials are especially useful in both redox catalysis and redox titrations or stoichiometric reactions where selective oxidation under mild conditions is desired.9 Ferricenium salts are moderately stable in acidic aqueous solutions but they rapidly decompose in many organic solvents and in air.10 The preparation of ferricenium derivatives, particularly those bearing electron-withdrawing substituents, is relatively difficult due their instability towards water, dioxygen, and nucleophilic reagents.8b,9,11 The ferricenium ions can be prepared with a variety of counter anions, such as tetra-fluoroborate (BF4−), hexafluorophosphate (PF6−), hexafluoroantimonate (SbF6−), or tetra-phenylborate (BPh4−).9 Ferricenium hexafluorophosphate (Fc[PF6]) and ferricenium tetra-fluoroborate (Fc[BF4]) are the only commercially available and the two most commonly used ferricenium salts.12 The nature of the counter anions not only alters the magnetic moment of ferricenium complexes, but also dictates their solubility in organic solvents.3e,8c,13
In more recent studies, fluoroarylborates such as tetrakis(pentafluorophenyl)borate ([B(C6F5)4]− or BArF20), or its aryl CF3-substituted analog tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (BArF24), have been used as counter anions for the preparation of ferricenium salts.14 These bulky counter anions with lower nucleophilicity exhibit remarkable chemical stability and their weaker ion-pairing interactions with the ferricenium ion result in a markedly increased solubility of the salts in lower-polarity media.15 These systems are particularly very attractive one-electron chemical redox agents for synthesis, catalysis, and kinetic and mechanistic investigations of a variety of redox systems in lower-polarity solvents, particularly low-temperature studies.9,16
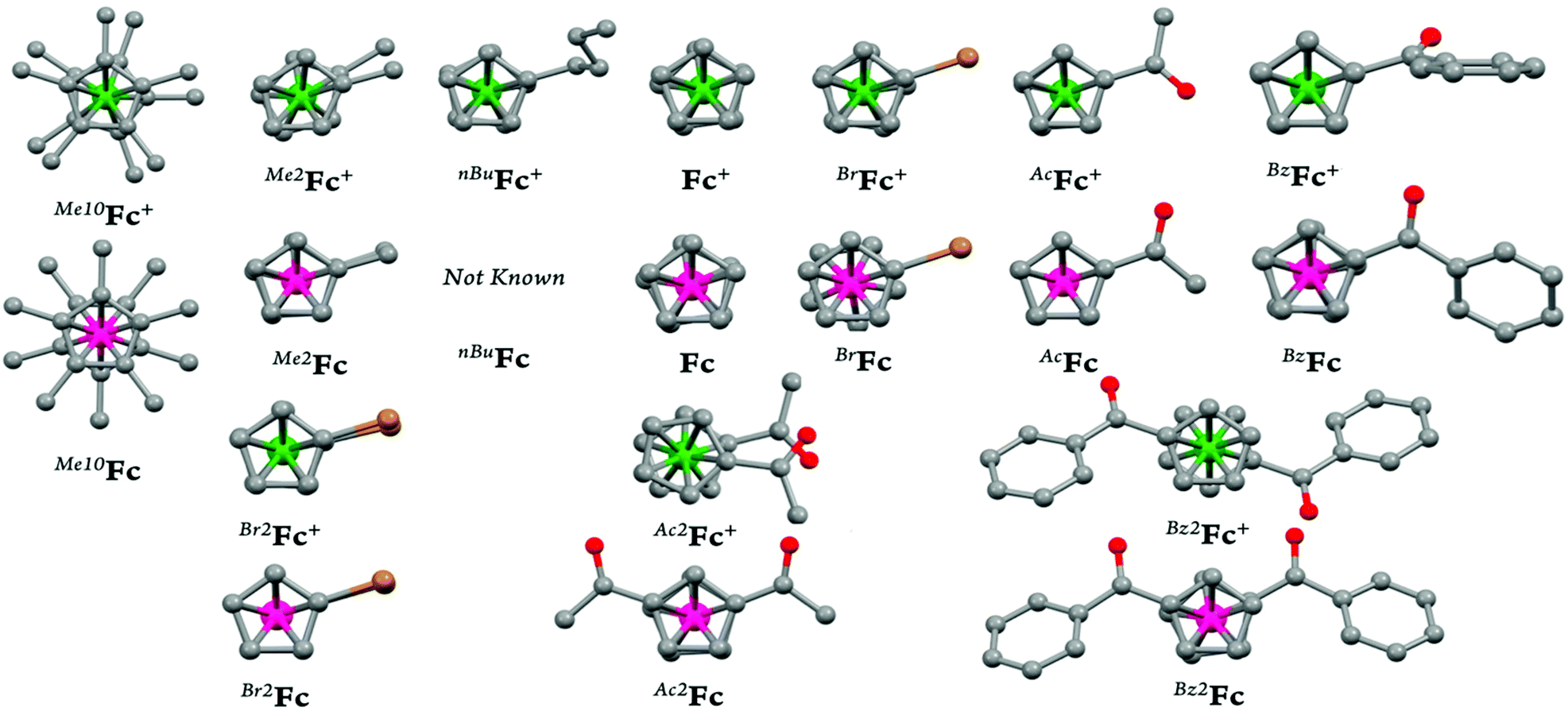
In the present study, we report a concise and straightforward strategy for preparation of a series of ferricenium complexes with electron-donating and electron-withdrawing substituents with either BArF20 or SbF6− as a counterion. Nine new ferricenium derivatives are readily isolated in high yields and all of them show exceptional solubility in lower-polarity solvents (e.g., tetrahydrofuran (THF), diethyl ether, toluene and benzene) as compared to their common PF6− or BF4− salts that are only soluble in high-polarity organic solvents such as acetonitrile, acetone, and alcohols. Scheme 1 shows the structures of ferricenium complexes described in this study.
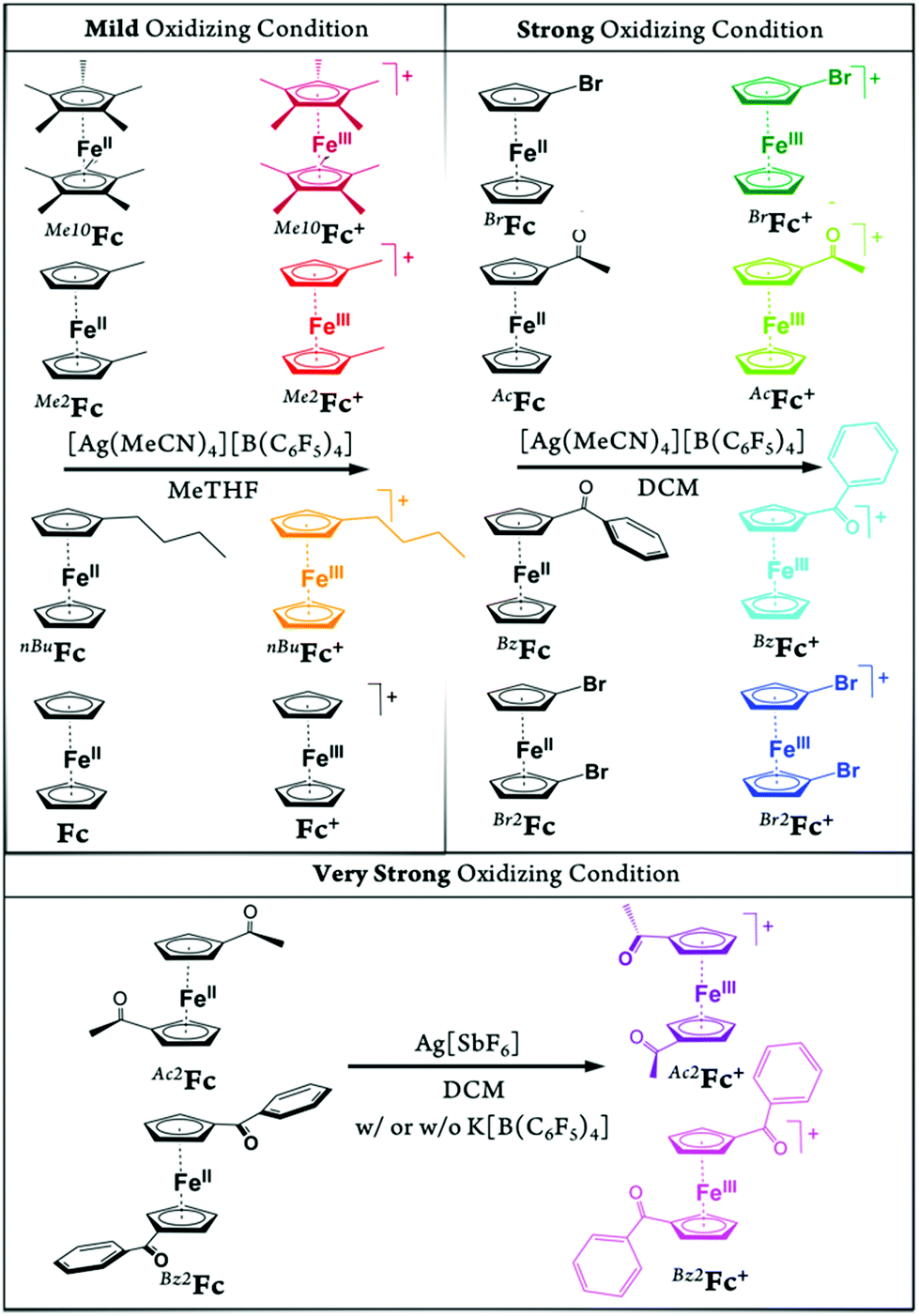 | ||
| Scheme 1 Synthetic scheme for the preparation of the ferricenium complexes described in this study. All ferricenium complexes were prepared with BArF20 as the counter anion, except for Bz2Fc[SbF6]. See the Experimental section for an alternative synthetic procedure for BzFc[B(C6F5)4] and further details. | ||
With the exception of the parent ferricenium BArF20 (that has previously been structurally characterized),17 molecular structures of all nine ferricenium derivatives used in this study were determined via single crystal X-ray crystallography. Other than the structure of 1,1′-diacetylferricenium (Ac2Fc+) that has been previously reported as the N(SO2CF3)2−, NTf2−, salt,18 the structures of five ferriceniums with electron-withdrawing substituents (i.e., BrFc+, AcFc+, BzFc+, Br2Fc+, and Bz2Fc+) are reported here for the first time.19 These systems were further studied for their ground state electronic structures using infrared (IR) and nuclear magnetic resonance (NMR) spectroscopies. The latter revealed an interesting correlation between the solvent dielectric constant and the energy barrier for rotation around the Fe-Cp axis in the electron-deficient ferricenium derivatives.
Herein, we also describe the redox behavior and potential values of the corresponding ferricenium/ferrocene (Fc+/0) redox couples in a number of organic media in order to provide a concise outline of these data in one place. Our electrochemical analyses involved three different solvents and two tetra-n-butylammonium supporting electrolytes with a more traditional anion, PF6−, as well as [B(C6F5)4]−. The results point to the significant effect of the ion-pairing in reducing the energy necessary for reduction of the ferricenium ion and the overall E1/2 potential values.
Results and discussion
Synthesis and characterization of ferricenium derivatives
We used two silver(I) salts as one-electron oxidizing agents for preparation of all of the ferricenium complexes.20 One is the BArF20 analog of silver(I) which is a mild to strong oxidant depending on the nature of the solvent. The synthesis of silver complex, [Ag(MeCN)4][B(C6F5)4], was performed using a modified procedure of Zhang et al.21 The silver(I) salt was readily prepared by metathesis of silver nitrate, AgNO3, and the commercially available potassium tetrakis(pentafluorophenyl)borate, K[B(C6F5)4], in acetonitrile (MeCN). Additionally, the molecular structure of the silver complex and coordination of four acetonitrile ligands were unambiguously confirmed by 1H-NMR and IR spectroscopies as well as X-ray crystallography, see Experimental section.The IR spectrum of [Ag(MeCN)4]+ exhibits two bands in the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching region, at 2322 and 2295 cm−1. The band at higher energy appears to arise from binary combination of the symmetric methyl deformation at 1367 cm−1 and symmetric C–C stretch at around 950 cm−1(Fig. S1†). The latter is buried under the counter anion signals and upon deuteration it shifts to 840 cm−1. These two vibration modes are both of A1 symmetry, thus forming a combination mode. In the deuterated complex, [Ag(CD3CN)4]+, the combination band is absent which further supports this supposition that only the band at 2295 cm−1 is due to the CN stretching vibration. This band appears at slightly lower frequency (2287 cm−1) in the deuterated complex (Fig. S2†). Additionally, as a result of complexation, the CN stretching frequencies are shifted to higher frequencies in [Ag(MeCN)4]+ (i.e., Δν(CN): +29 cm−1) as compared to free acetonitrile (νfree(CN): 2266 cm−1), which is well-known for nitrile adducts.22
N stretching region, at 2322 and 2295 cm−1. The band at higher energy appears to arise from binary combination of the symmetric methyl deformation at 1367 cm−1 and symmetric C–C stretch at around 950 cm−1(Fig. S1†). The latter is buried under the counter anion signals and upon deuteration it shifts to 840 cm−1. These two vibration modes are both of A1 symmetry, thus forming a combination mode. In the deuterated complex, [Ag(CD3CN)4]+, the combination band is absent which further supports this supposition that only the band at 2295 cm−1 is due to the CN stretching vibration. This band appears at slightly lower frequency (2287 cm−1) in the deuterated complex (Fig. S2†). Additionally, as a result of complexation, the CN stretching frequencies are shifted to higher frequencies in [Ag(MeCN)4]+ (i.e., Δν(CN): +29 cm−1) as compared to free acetonitrile (νfree(CN): 2266 cm−1), which is well-known for nitrile adducts.22
The other oxidizing agent is the silver(I) salt with the hexafluoroantimonate counter anion, Ag[SbF6], which is commercially available and was used as a very strong oxidizing agent. A series of ferricenium BArF20 complexes with electron-donating or electron-withdrawing groups were synthesized using three main preparative procedures with different oxidizing strengths listed here:
1. Mild oxidizing condition (<100 mV vs.Fc+/0): [Ag(MeCN)4][B(C6F5)4] in MeTHF.
2. Strong oxidizing condition (100 to 400 mV vs.Fc+/0): [Ag(MeCN)4][B(C6F5)4] in DCM.
3. Very strong oxidizing condition (>400 mV vs.Fc+/0): Ag[SbF6] in DCM.
The solvent and counter anion dependencies of the oxidation potential of silver(I) were used to provide the mild, strong, and very strong oxidizing environments in MeTHF or DCM (Scheme 1). The redox potential for the Ag+/0 couple in nonaqueous solutions is not easily measured and generally estimated values are available in the literature. For example, the formal redox potential of the Ag+/0 couple in THF (E0′ = 410 mV vs.Fc+/0) is reported to be about 0.24 V more negative than that in DCM (E0′ = 650 mV vs.Fc+/0).9,23 Our results for [Ag(MeCN)4][B(C6F5)4], already bearing the four coordinated acetonitrile in MeTHF (168 < E°′ < 244 mV vs.Fc+/0) and DCM (357 < E°′ < 533 mV vs.Fc+/0), are in agreement with the previous reports showing that, with the increase in the coordination ability of the solvent the oxidizing strength of silver(I) salts significantly decreases.
The redox potentials of the ferrocene derivatives measured in this study (vide infra) were used in the Nernst equation to predict the position of the redox equilibriums in order to assign the appropriate preparation procedures. The sub-stoichiometric oxidation of all ferrocene derivatives was accomplished by limiting the amount of the appropriate silver(I) agents to assure full consumption of the oxidants. The remaining excess ferrocene complexes were simply removed by several dry hexanes rinses.
Mild oxidation of the parent ferrocene and three of its electron-rich derivatives bearing one or more electron-donating substituents, including n-butylferrocene (nBuFc), 1,1′-dimethylferrocene (Me2Fc), and decamethylferrocene (Me10Fc), was achieved with the addition of a sub-stoichiometric amount of silver(I) BArF20 salt in MeTHF solution. In turn, the sub-stoichiometric amount of silver(I) BArF20 salt in DCM solution was employed for the oxidation of all three monosubstituted ferrocene derivatives with electron-withdrawing groups, including 1-bromoferrocene (BrFc), 1-acetylferrocene (AcFc), and 1-benzoylferrocene (BzFc), as well as one 1,1′-disubstituted derivative, 1,1′-dibromoferrocene (Br2Fc).
The corresponding ferricenium BArF20 analogs were isolated in high yields (>80%). Note: For both ketone-substituted ferrocenes, the order of addition of reagents is reversed (i.e., the solution of substituted ferrocene is gradually added to the silver(I) solution), see Experimental section. This is due to the ability of the acetyl or benzoyl groups on the Cp rings to coordinate to the silver(I) center which can hinder the electron transfer process from ferrocene to Ag(I). This is consistent with the considerably lower oxidizing ability of silver(I) salts in acetone (E°′ = 180 mV vs.Fc+/0).9 As an alternative procedure for preparation of BzFc[B(C6F5)4], a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of the 1-benzoylferrocene and K[B(C6F5)4] can be added dropwise to a solution of Ag[SbF6] in DCM.
:1 solution of the 1-benzoylferrocene and K[B(C6F5)4] can be added dropwise to a solution of Ag[SbF6] in DCM.
The other two highly electron-deficient derivatives, 1,1′-diacetylferrocene (Ac2Fc) and 1,1′-dibenzoylferrocene (Bz2Fc), were oxidized using Ag[SbF6] in DCM. The stronger oxidizing strength of this silver(I) salt in DCM was confirmed through its reactivity toward the tris (4-bromophenyl)amine (i.e., N(C6H4Br-4)3). While [Ag(MeCN)4][B(C6F5)4] does not react with the amine in DCM, the addition of Ag[SbF6] instantly oxidizes the triarylamine forming the signature blue color of the corresponding radical cation, known as Magic Blue (E°′ = 700 mV vs.Fc+/0). We also independently confirmed the redox potential of tris (4-bromophenyl)amine in DCM, in the presence of PF6− (i.e., as a model for SbF6−) and BArF20, to be about 705 and 675 mV vs.Fc+/0, respectively (Fig. S3†).
The two oxygen atoms of the ketone moieties of both Ac2Fc and Bz2Fc can also chelate to the silver(I) center forming a red complex as observed previously for similar compounds.24 Therefore, the order in which the reagents are added is critical to initiate the redox reaction. The isolated Ac2Fc[SbF6] complex was then converted to the very soluble BArF20 analog through metathesis by K[B(C6F5)4] in 1,2-difluorobenzene. The Bz2Fc[SbF6] on the other hand was used as the [SbF6]− salt due to its satisfactory solubility and stability.
Alternatively, the Ac2Fc[B(C6F5)4] complex can be readily obtained by dropwise addition of a 1:1 mixture of Ac2Fc and K[B(C6F5)4] to a solution of Ag[SbF6] in DCM. Adding the first drop of the mixture leads to the development of a light pink color indicating the transient formation of the ferrocene chelated silver(I) complex. This complex is then slowly oxidized by the excess silver(I) ions present in the solution to the green Ac2Fc+ species. After this point, each additional drop of the mixture leads to fast oxidation of the Ac2Fc and further appearance of the green color. This suggests that the initially generated ferricenium species may act as an electron transfer mediator/relay between the ferrocene-chelated silver(I) complex and the excess silver(I) pool allowing for the faster oxidation process.
Generally, removal of silver metal as the byproduct of the oxidation of the ferrocene complexes is straightforward. However, it is important to note that due to the reversibility of the ferricenium/ferrocene couple, the presence of a slight amount of silver metal impurity can result in partial re-reduction of the ferricenium sample upon dissolution in more coordinating solvents in which the silver salt is a weaker oxidant (i.e., the product distribution is governed by the Nernst equation). Since the BArF20 analogs of all these ferricenium derivatives are highly soluble in either MeTHF or DCM, the silver metal is a very finely divided precipitate and effective filtration can be achieved through the use of a filtration aid such as Celite.
It is also worth mentioning that all our synthetic procedures were performed under rigorous air-free conditions as many of the erratic results reported in literature for many ferricenium complexes including their relatively low extinction coefficients can be explained by the irreversible decomposition of ferricenium species in solutions exposed to air. It is confirmed that the yellow decomposition products reported in the earlier literature are not the starting ferrocene complexes.25 Carbon and hydrogen analyses of all nine ferricenium BArF20 complexes, as well as Bz2Fc[SbF6], indicated that the compounds were ≥99% pure, see Experimental section. Before we discuss further characterization of the ferricenium derivatives, it is helpful to point out the unique structural feature of the ferricenium ion. Due to a very small rotational energy barrier, Fc+ can adopt an eclipsed (D5h) or staggered (D5d) conformation or with the rings slightly twisted, it can even conform to an intermediate skewed (D5) geometry. We will expand on this point later in the discussion of the X-ray structures.
The signature blue or green color (λmax ∼ 621–780 nm) of the ferricenium complexes is present in all of our derivatives. This is the ligand-to-metal charge transfer (LMCT) transition which is from the e1u orbitals of Cp ligands to the hole in the essentially non–bonding e2g orbitals on the ferric center.8c,d,26 The lowest energy absorption maxima and extinction coefficient values for all ferricenium derivatives are provided in the Experimental section.
Aside from the counterion peaks (i.e., BArF20 or SbF6−), the infrared spectra of ferricenium derivatives noticeably have fewer strong bands than their neutral ferrocene counterparts (Fig. S4–S13†). Due to the one-electron oxidation, the C–H stretching frequencies of the Cp rings are shifted to higher energies by about 30–40 cm−1 in all of the ferricenium derivatives.
The carbonyl stretching modes of the ketone-substituted ferrocene species are located in the 1620–1650 cm−1 region and were assigned based on previous literature reports.24a,28 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bands of all ketone-substituted ferriceniums appear at about 35–48 cm−1 higher frequencies when compared with the neutral counterparts, indicating a significant strengthening of the carbonyl bond in the oxidized forms, see Table 1 and ESI.† This is in agreement with the shorter CO bond distances (∼1.212 Å) obtained for the oxidized complexes from our X-ray crystallography measurements as compared to those reported for the neutral ketone-substituted ferrocenes (∼1.224 Å), vide infra. The first overtone of the CO stretching band is also observed in the 3295–3380 cm−1 region in both neutral and oxidized ketone-substituted species.29
O stretching bands of all ketone-substituted ferriceniums appear at about 35–48 cm−1 higher frequencies when compared with the neutral counterparts, indicating a significant strengthening of the carbonyl bond in the oxidized forms, see Table 1 and ESI.† This is in agreement with the shorter CO bond distances (∼1.212 Å) obtained for the oxidized complexes from our X-ray crystallography measurements as compared to those reported for the neutral ketone-substituted ferrocenes (∼1.224 Å), vide infra. The first overtone of the CO stretching band is also observed in the 3295–3380 cm−1 region in both neutral and oxidized ketone-substituted species.29
O stretching frequencies and bond lengths in ketone-substituted derivatives
| Compound | ν(CO) (cm−1) |
1st overtone ν(CO) (cm−1) |
CO (Å) |
Reference for X-ray structure |
|---|---|---|---|---|
| Ac Fc | 1650 | 3297 | 1.223 | 30 |
| Ac Fc + | 1698 | 3378 | 1.209 | This work |
| Ac2 Fc | 1650 | 3296 | 1.224 | 31 |
| Ac2 Fc + | 1697 | 3376 | 1.209 | This work |
| Bz Fc | 1624 | 3242 | 1.225 | 32 |
| Bz Fc + | 1659 | 3308 | 1.215 | This work |
| Bz2 Fc | 1630 | 3252 | 1.222 | 33 |
| Bz2 Fc + | 1665 | 3315 | 1.215 | This work |
The 1H-NMR spectrum of the parent diamagnetic Fc displays one sharp resonance at 4.2 ppm for the Cp rings (Fig. S14†). The position and multiplicity of the signal for Cp protons are sensitive to ring substitution. In the neutral ferrocene derivatives, electron-donating ring substitutions give rise to a greater shielding of the ring protons (δ = 3.9–4.1 ppm) while electron-withdrawing groups via induction deshield the nearby Cp protons (δ = 4.3–4.9 ppm). This substitutional behavior is more complex and reversed in ferricenium derivatives (Fig. 1). We propose that in the oxidized complexes, the δ back-donation from the iron dx2−y2 and dxy orbitals to the Cp ring (i.e., the increase in bonding character of the e2g molecular orbitals) is responsible for this reversal of behavior. In the substituted ferricenium, the ring protons of the Cp ligand with the more stabilized orbitals experience more significant shielding due to a better energy match (and overlap) with the iron and stronger δ back-donation.
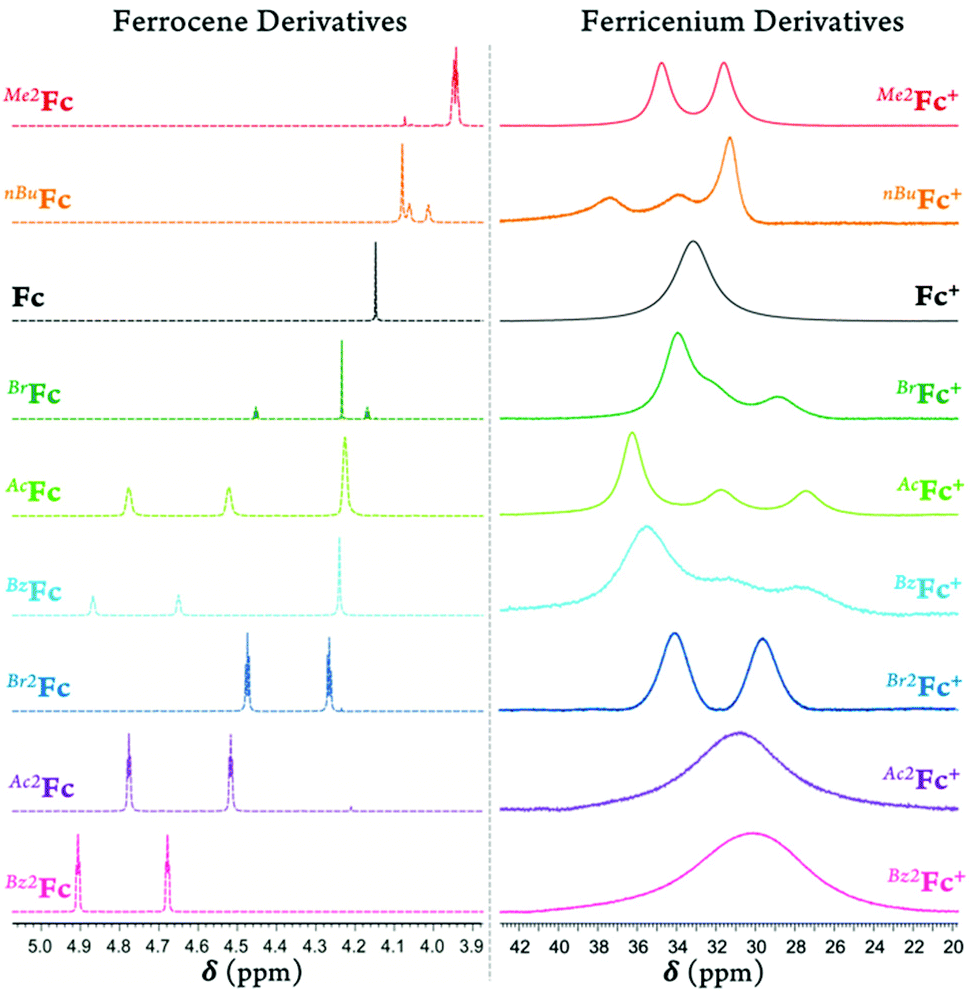 | ||
| Fig. 1 Part of the 1H-NMR spectra of (left) the ferrocene derivatives and (right) the ferricenium analogs in acetone-d6 at room temperature. Due to reaction with acetone, the spectra of Ac2Fc+ and Bz2Fc+ were recorded in CD2Cl2. | ||
The 1H-NMR signal of the parent paramagnetic Fc+ complex (S = 1/2) appears as a single broad peak at 33.2 ppm, which is shifted ∼29 ppm downfield relative to that of Fc (Fig. S15†). Interestingly, the introduction of electron-donating group(s) in the ferricenium species have a net deshielding effect on the ring protons, whereas electron-withdrawing substituents tend to shield the substituted ring protons. The 1H-NMR spectrum of the electron-rich Me10Fc, with no Cp ring protons, only displays a singlet at 1.66 ppm for the protons of the methyl groups. In the paramagnetic Me10Fc+ counterpart, the singlet methyl proton signal shifts to a lower frequency (δ = –37.6 ppm), see Fig. S16 and S17.†
Mono- or 1,1′-di-substituted ferrocenes typically exhibit NMR signals which can be readily assigned except for those from protons in the 2,5- and 3,4-positions. The assignment of these ring protons for some substituted ferrocenes have previously been achieved through specific deuteration, heteronuclear differential nuclear Overhauser effect difference (NOE) spectroscopy, or 1J(13C–13C) coupling measurements along with selective proton decoupling.35 By analogy, we can assign the 2,5- and 3,4-protons of all mono- and 1,1′-di-substituted ferrocene species described in this study (Table 2). In deuterated acetone at room temperature, the resonances representing the protons in 2,5- and 3,4-positions in nearly all of our substituted ferrocenes appear as a pair of apparent triplets with the coupling constant, 3J(1H–1H), value of about 2 Hz. Table S4† lists the coupling constants of the Cp protons for all the substituted ferrocene species. Table 2 summarizes the 1H-NMR chemical shifts for the cyclopentadienyl protons of both ferrocene derivatives and their ferricenium counterparts.
a
| δ (ppm) | |||||||
|---|---|---|---|---|---|---|---|
| Compound | HCp | H2,5 | H3,4 | Compound | HCp | H2,5 | H3,4 |
Numbering scheme:  bOnly one single broad resonance. bOnly one single broad resonance. |
|||||||
| Me2 Fc | — | 3.96 | 3.94 | Me2 Fc + | — | 34.8 | 31.6 |
| nBu Fc | 4.08 | 4.06 | 4.01 | nBu Fc + | 31.3 | 37.4 | 33.9 |
| Fc | 4.20 | — | — | Fc + | 33.2 | — | — |
| Br Fc | 4.23 | 4.45 | 4.17 | Br Fc + | 34.0 | 28.8 | 32.0 |
| Ac Fc | 4.23 | 4.78 | 4.52 | Ac Fc + | 36.3 | 27.5 | 31.8 |
| Bz Fc | 4.24 | 4.87 | 4.65 | Bz Fc + | 35.4 | 27.6 | 31.3 |
| Br2 Fc | — | 4.47 | 4.27 | Br2 Fc + | — | 29.6 | 34.2 |
| Ac2 Fc | — | 4.81 | 4.58 |
Ac2
Fc
+
a |
— | 30.5b | |
| Bz2 Fc | — | 4.98 | 4.68 |
Bz2
Fc
+a |
— | 30.2b | |
For Me2Fc, the protons of two methyl substituents resonate at 1.95 ppm and the lower symmetry of the substituted ring system predictably leads to a pair of triplets for the Cp protons (δ = 3.94 and 3.96 ppm; Fig. S18 and S19†). In nBuFc, the 2,5-protons and, to a greater extent, the 3,4-protons experience the shielding associated with the electron-donating substituent,34a resulting in an upfield shift of these substituted ring protons relative to that of the unsubstituted Cp ring. The elongated n-butyl chain gives rise to three separate (–CH2–) resonances at 2.34 (t, 2H, a-H), 1.48 (m, 2H, b-H), and 1.34 (m, 2H, c-H) ppm plus a triplet centered around 0.9 ppm for the three protons of the CH3 group (Fig. S20 and S21†).
The presence of electron-withdrawing substituents such as bromo, acetyl, or benzoyl groups on the cyclopentadienyl rings of the neutral complexes leads to more pronounced changes in chemical shifts between the 2,5- and the 3,4-protons of the substituted Cp rings (Fig. 1 and S22–S29†). In these electron-deficient systems, both resonances are shifted to a lower field compared to the Cp resonance of the parent ferrocene. In the case of BrFc, the 2,5-proton signal appears at a lower field and the 3,4-protons resonate at a slightly higher field compared to the resonance for unsubstituted Cp ring.
For the oxidized species, in most cases, the different Cp protons can still be distinguished in spite of the broadened resonances. For example, both 1,1′-disubstituted Me2Fc+ and Br2Fc+ complexes show two broad downfield Cp proton resonances. This is in agreement with our X-ray crystallography data that confirmed their “locked” eclipsed structures in the solid state (vide infra) and supporting that this conformation is retained in both solution and solid state.
In deuterated acetone, the 2,5- and 3,4-proton peaks for Me2Fc+ appear at 31.6 and 34.8 ppm and for Br2Fc+ at 29.6 and 34.2 ppm, respectively. Going from deuterated acetone to DCM, the peak separation for Br2Fc+ diminishes from 4.6 to 1.8 ppm (i.e., for Br2Fc+δ = 32.7 and 34.5 ppm in CD2Cl2; ΔΔδ = –2.8 ppm) while the peak separation for Me2Fc+ remains essentially the same (i.e., for Me2Fc+δ = 32.5 and 35.8 ppm in CD2Cl2; ΔΔδ = 0.1 ppm), see Fig. 2 and Fig. S30–S33.† Here, the lower dielectric constant of the media results in lowering the rotational barrier of the substituted rings in Br2Fc+ to some degree and not in Me2Fc+. This may be due to the different nature of the bonding and overall spin density delocalization in these ferricenium complexes. The methyl protons of Me2Fc+ resonate in the upfield region as a rather sharp singlet, i.e., δ = –10.5 ppm in acetone-d6 and δ = –9.0 ppm in CD2Cl2.
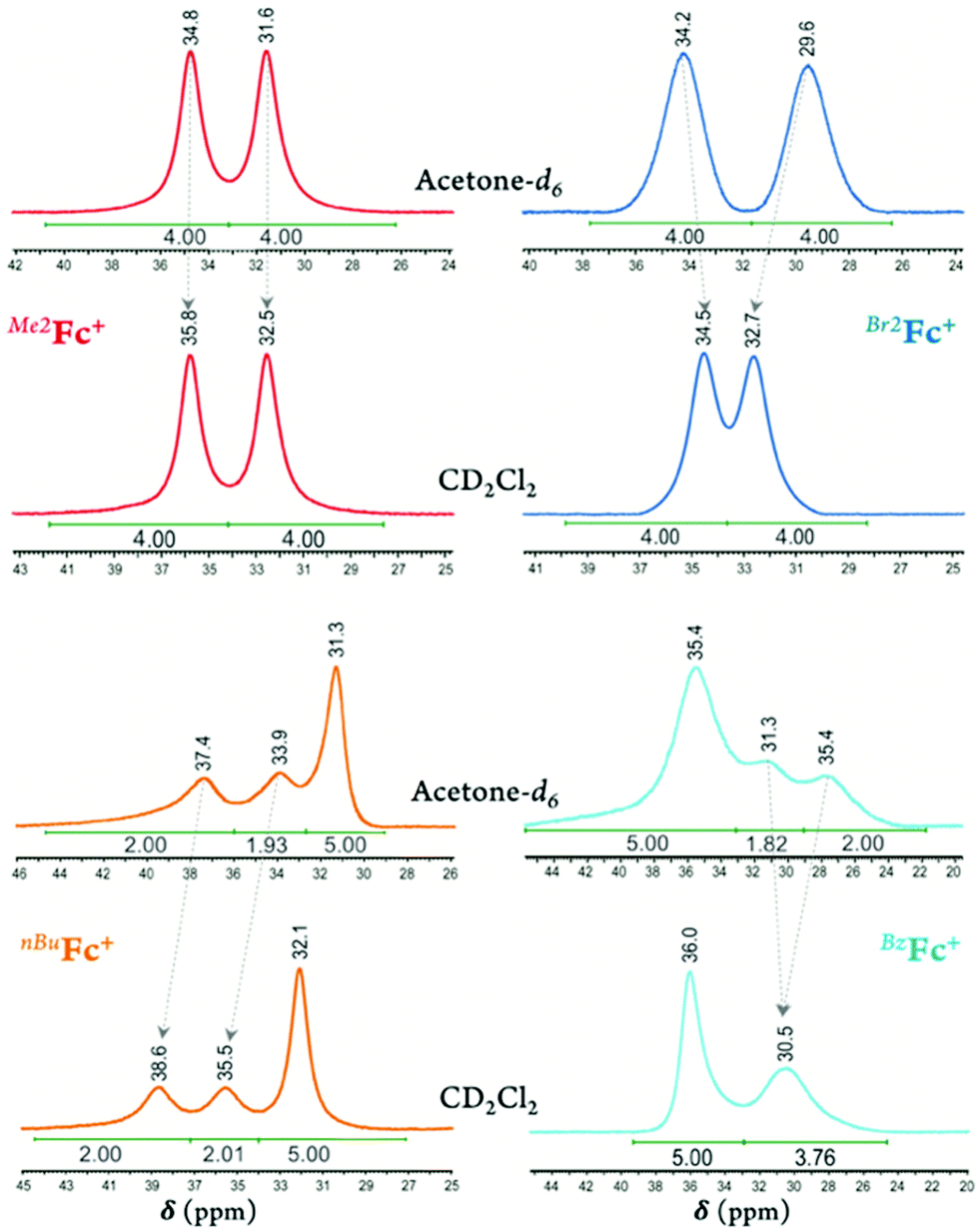 | ||
| Fig. 2 Part of the 1H-NMR spectra of two electron-rich ferricenium derivatives (left; i.e., Me2Fc+ and nBuFc+) and two electron-deficient analogs (right; i.e., Br2Fc+ and BzFc+) recorded in deuterated acetone (ε ≈ 21) vs. dichloromethane (ε ≈ 9) at room temperature. The behaviors of the 2,5- and 3,4-protons of the substituted Cp ligand are compared. | ||
On the other hand, in the case of the 1,1′-diketone-substituted ferricenium species such as Ac2Fc+ and Bz2Fc+, only one broad Cp proton resonance is observed in CD2Cl2 (Fig. 1, S34 and S35†). Apparently, the ketone-substituted Cp rings in these complexes have lower rotational barriers and rotation rates are sufficiently high, exceeding the NMR time scale and preventing the observation of separate resonances by the 2,5- and 3,4-protons. Our X-ray crystallography data for the oxidized forms of these complexes also support a less restricted rotation around the Fe-Cp axis in these systems, vide infra.
In all four monosubstituted ferricenium species the 2,5- and 3,4-protons of the substituted ring along with the unsubstituted Cp protons resolve into three broad peaks. Here, the Cp ring carrying the substituent faces a larger rotational barrier in acetone-d6 and the reduced rotation rates lie within the timescale of NMR measurements. For the electron-rich nBuFc+, the unsubstituted Cp ring resonance moves upfield up to 1.9 ppm from that of the parent ferrocene Cp protons. In turn, the 2,5-protons of the n-butyl-substituted ring and, to a lesser extent, the 3,4-protons are deshielded relative to that of the parent Fc+ protons (Table 2, Fig. S36 and S37†). The protons of the n-butyl substituent resonate in the upfield region as four separate signals at −6.8 (2H, a-H), −18.3 (2H, b-H), 1.2 (2H, c-H), and −1.0 (3H, d-H) ppm.
On passing from the electron-rich monosubstituted ferricenium ion to electron-deficient monosubstituted ferricenium species bearing a bromo, acetyl, or benzoyl group, the unsubstituted Cp ring signal shifts to a lower field (Δδ = 0.8–3.1 ppm) in reference to the Cp ring signal of the parent ferricenium complex (Fig. S38–S43†). Additionally, the protons of the 2,5-positions and to a lesser degree, the 3,4-positions of the substituted Cp ring become increasingly shielded with the increasing electronegativity of the substituent and resonate at a higher field relative to that of the parent Fc+ protons. Here again, in CD2Cl2, the rotational barrier of the substituted rings decreases more significantly in the electron-deficient ferricenium species (i.e., BrFc+, AcFc+, and BzFc+) than in the electron-rich system such as nBuFc+, and the faster ring rotation results in much more severe broadening and overlap of the 2,5- and 3,4-protons of the substituted Cp ligand, Fig. 2.
It is also worth noting that the peak separations for the 2,5- and the 3,4-protons of the substituted Cp rings in both higher and lower polarity organic solvents (e.g., acetone-d6vs. CDCl3) stay the same across the series of neutral ferrocene derivatives discussed in this study. This points to the significant difference in bonding and electronic structures of the one electron oxidized and neutral species.
The 19F-NMR spectra of the paramagnetic ferricenium salts were also recorded. The BArF20 anion of all ferricenium species in deuterated acetone gives rise to three 19F-NMR resonances at −133.0, −164.4, and −168.4 ppm for the ortho-, para-, and meta-fluorines in a ratio of around 8:4:8, respectively. In a lower polarity solvent such as CD2Cl2, these 19F-NMR signals are more shielded, and the largest shift is observed for the ortho-fluorines by no more than −2.4 ppm, see Fig. S44–S58.† This confirms that although the ferricenium derivatives and BArF20 anion are unpaired in acetone, they are likely ion paired in CD2Cl2. As previously described, the BArF20 anion lacks specificity in where to ion pair to the cation.15b
The 19F-NMR spectrum of Bz2Fc+ displays one broad resonance at 133.2 ppm for the SbF6− counterion with a sextet pattern (1J(19F–121Sb) ∼2 kHz). Given that the two most abundant isotopes of antimony are both quadrupolar, 121Sb I = 5/2 and 123Sb I = 7/2, broadening of the 19F-NMR signal in SbF6− due to the quadrupolar relaxation is expected. In the presence of Ag[SbF6], the splitting pattern of 19F resonance disappears giving a single broadened signal that is shifted to −134.7 ppm which points to a fast mode of exchange in this system (Fig. S59†).
X-Ray crystallography
A couple of decades ago, Geiger and coworkers first introduced tetra-n-butylammonium BArF20, [(nBu)4N][B(C6F5)4], as an ideal non-coordinating supporting electrolyte for electrochemical studies in low-polarity solvents.36 Here, we describe an alternative procedure for preparation of this electrolyte. Diffraction quality crystals of [(nBu)4N][B(C6F5)4] were obtained by slow liquid diffusion of hexanes into the DCM solution of the electrolyte and its structure was determined via single crystal X-ray crystallography at 100 K (Table S5†). The electrolyte, which was previously reported by Bolte and coworkers at 173 K,37 crystallizes in the monoclinic space group Cc, with one tetra-n-butylammonium moiety paired with one BArF20 anion per asymmetric unit (Fig. S60†).The crystals of [Ag(MeCN)4][B(C6F5)4] were grown by keeping a saturated solution of the complex in acetonitrile at −35 °C. Although the X-ray structure of this complex was recently reported,38 we were able to obtain higher quality data (Table S5†). The X-ray structure clearly shows the ligation of four acetonitrile molecules to the silver(I) center in a pseudo-tetrahedral fashion (i.e., ∠N–Ag–N is in the range of 91 to 136°) with one slightly bent acetonitrile ligand while BArF20 remains in the crystal lattice as the counter anion, residing near the largest N–Ag–N angle in the silver complex (Fig. S61†).
As briefly discussed earlier, both ferrocene and ferricenium derivatives possess a high degree of molecular flexibility, adopting a variety of conformations. This conformation flexibility is typically in response to different electronic, steric, or crystal packing forces in different structures. In addition to their highly flexible geometries, the ferrocene or ferricenium derivatives, even symmetrically substituted ones, can become chiral to some extent or exhibit conformational chirality.39 In the following, we analyze the overall conformational arrangements of all ferricenium derivatives reported here according to the four geometrical parameters shown in Fig. 3. We also compare these values with those of the corresponding ferrocene counterparts (Tables S9 and S10†).
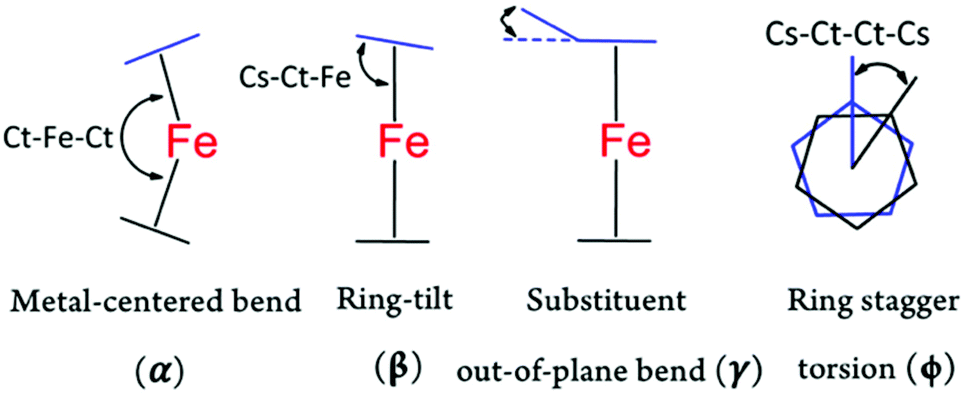 | ||
| Fig. 3 Schematic representation of the geometrical parameters used to describe the conformations of ferrocene and ferricenium derivatives. Ct refers to the Cp ring centroid while Cs indicates the substituted C-atom on the ring. In the case of monosubstituted structures, the second Cs is the closest C-atom on the unsubstituted ring. | ||
Fig. 4 depicts the molecular structure of the ferricenium derivatives bearing electron-donating groups in the solid state. The most electron-rich ferricenium complex described in this work, Me10FcBArF20, crystallizes as dark green single crystals in a P21/c space group and the crystallographic asymmetric unit contains two ferricenium entities and two BArF20 counterions. The two entities exist in slightly different relative conformations. The Cp rings in one of the ferricenium centers are almost perfectly staggered with a torsional angle of 35.9° between the two opposing methyl groups while the second ferricenium entity has the rings slightly skewed by an angle of 16.9°. Interestingly, in the case of the neutral Me10Fc, the Cp rings only adopt absolute staggered orientation (ϕ = 36°).31 The Fe–Cavg bond distance in Me10FcBArF20 was found to be 2.101 Å which is about 0.05 Å larger than that of Me10Fc (Table S9†). The distance between the iron center and Cp ring centroid (i.e., Fe⋯Ctavg) is also about 0.06 Å longer in the ferricenium complex which highlights the small elongation of the Fe–C bonds upon oxidation. A very similar trend is observed for the neutral and oxidized states of the other ferrocene derivatives (Fig. S71†). This constancy of Fe–C bond length in going from the neutral ferrocene state to the ferricenium state in all derivatives has been ascribed to the very weakly bonding nature of the e2g orbitals.8c,d,26a The methyl substituents in Me10FcBArF20 are slightly out of the Cp planes and away from the iron center (γ = 1.4°) which is also observed in the neutral analog, Me10Fc, (γ = 1.8°) pointing to the steric constraints imposed by the substitution of all Cp protons by methyl groups.
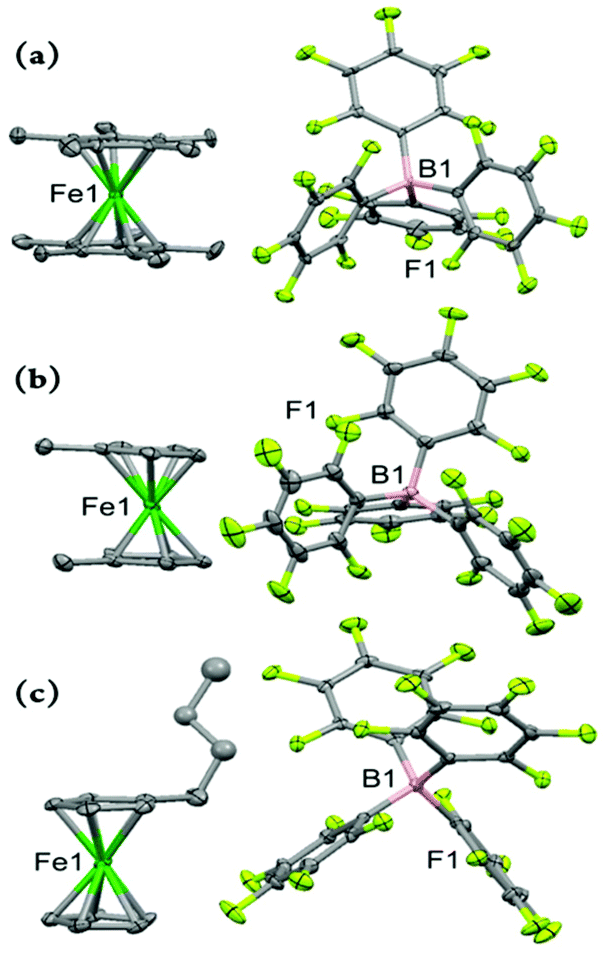 | ||
| Fig. 4 Perspective views of the electron-rich ferricenium derivatives at 100 K: (a) Me10FcBArF20, (b) Me2FcBArF20, and (c) nBuFcBArF20 showing 50% thermal contours for all non-hydrogen atoms. Hydrogen atoms have been omitted for clarity. | ||
The dark blue single crystals of Me2FcBArF20 were obtained in an orthorhombic system with the Pbcn space group. The asymmetric unit contains three molecules and one out of the three Me2Fc+ entities is disordered over two different orientations and the occupancy factor of the major component refines to 0.618(3). The methyl groups are closer to the eclipsed conformation (ϕ = 13.6°, −14.9°, or −19.2) in the oxidized complex and the torsion angle becomes even smaller on going to the neutral analog, Me2Fc, (ϕ = –3.6°).31 Here, an increase of 0.06 Å in Fe–Cavg bond length is observed for Me2FcBArF20 relative to Me2Fc which is the largest elongation of the Fe–C bonds upon oxidation observed in all the derivatives discussed in this study. The separation of the Cp rings also increases by 0.17 Å, in going from Me2Fc to Me2FcBArF20. Unsurprisingly, in the oxidized complex, Me2FcBArF20, with the greater Cp ring separation (i.e., 3.463 Å), both methyl groups tend to come within the Cp plane with an average out-of-plane displacement of 0.01 Å as compared to 0.06 Å in Me2Fc.
The peacock blue nBuFcBArF20 complex crystallizes in a triclinic crystal system with P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The asymmetric unit contains two molecules and the n-butyl moieties of both ferricenium entities are disordered over two different orientations. The occupancy factor of the major component in disorder is 0.552(9). The nBuFc complex is a brownish orange liquid at room temperature and no crystal structure is available for this neutral counterpart for comparison. Instead, we used the structural data reported for the two closely related ferrocene derivatives, n-tetradecylferrocene (nC14Fc) and 1,8-bis(ferrocenyl)octane (Fc–(CH2)8–Fc).40 In the nBuFcBArF20 complex, the substituted and unsubstituted Cp rings are nearly eclipsed (ϕ = 7.9° or −1.2°) and the iron center is about 1.702 and 1.703 Å away from the centroids of the substituted and unsubstituted Cp ligands, respectively. In the neutral analogs, the rings maintain the eclipsed structure (ϕ = –0.2° in nC14Fc and −7.8° in Fc–(CH2)8–Fc) and both metal–ligand separations are reduced by about 0.5 Å (Fe⋯Ct sub. = 1.648 Å and Fe⋯Ct unsub. = 1.650 Å). See Tables S9 and S10† for further structural details.
space group. The asymmetric unit contains two molecules and the n-butyl moieties of both ferricenium entities are disordered over two different orientations. The occupancy factor of the major component in disorder is 0.552(9). The nBuFc complex is a brownish orange liquid at room temperature and no crystal structure is available for this neutral counterpart for comparison. Instead, we used the structural data reported for the two closely related ferrocene derivatives, n-tetradecylferrocene (nC14Fc) and 1,8-bis(ferrocenyl)octane (Fc–(CH2)8–Fc).40 In the nBuFcBArF20 complex, the substituted and unsubstituted Cp rings are nearly eclipsed (ϕ = 7.9° or −1.2°) and the iron center is about 1.702 and 1.703 Å away from the centroids of the substituted and unsubstituted Cp ligands, respectively. In the neutral analogs, the rings maintain the eclipsed structure (ϕ = –0.2° in nC14Fc and −7.8° in Fc–(CH2)8–Fc) and both metal–ligand separations are reduced by about 0.5 Å (Fe⋯Ct sub. = 1.648 Å and Fe⋯Ct unsub. = 1.650 Å). See Tables S9 and S10† for further structural details.
In moving on to the electron-deficient ferricenium derivatives, we obtained molecular structures of three monosubstituted systems bearing a bromo, acetyl, or benzoyl substituent on one of the Cp rings (Fig. 5). These are the first examples of X-ray crystal structures of the ferricenium derivatives with these electron-withdrawing substituents. To date, only a very limited number of structural data for electron-deficient ferricenium species (∼5) are deposited in the Cambridge Structural Database (CSD), which is most likely a reflection of challenges in their preparation due to the necessity of meticulous exclusion of potential reactive nucleophiles and reducing reagents (i.e., air, moisture, coordinating and redox-active solvents and counterions).
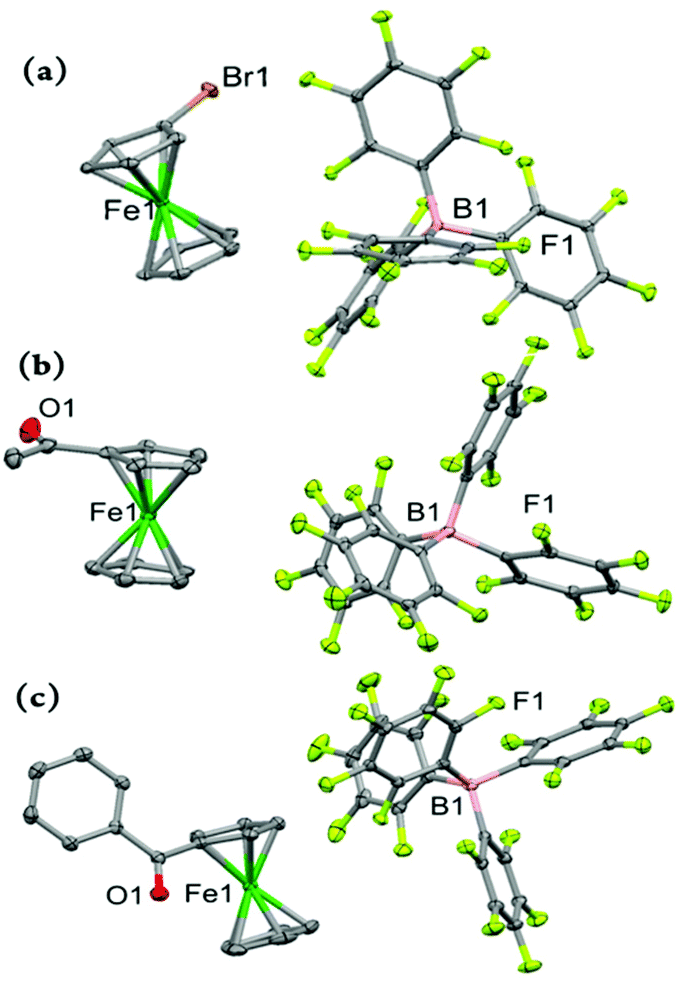 | ||
| Fig. 5 Perspective views of the electron-deficient monosubstituted ferricenium derivatives at 100 K: (a) BrFcBArF20, (b) AcFcBArF20, and (c) BzFcBArF20 showing 50% thermal contours for all non-hydrogen atoms. Hydrogen atoms have been omitted for clarity. | ||
Our first monosubstituted ferricenium derivative of this class is BrFcBArF20 (Fig. 5a) which was obtained from DCM/hexanes. This complex crystallizes in a monoclinic crystal system with P21/n space group and the asymmetric unit contains one molecule. The Cp rings in BrFc+ adopt an approximately eclipsed conformation with a torsion angle of 9.1° which is in the range of those found for the neutral complex, BrFc (ϕ = –2.6° or 28.6°).41 In BrFc+, the Fe–Cavg and Fe⋯Ct distances are 2.089 and 1.706 Å for the bromosubstituted Cp ring while 2.082 and 1.701 Å for the unsubstituted ring, respectively. As expected, the neutral complex, BrFc, possesses smaller Fe–Cavg and Fe⋯Ct distances. Here, the oxidation results in a larger degree of elongation of the Fe⋯Ct distance for the substituted ring as compared to unsubstituted ring, while in the electron-rich nBuFc+ complex both substituted and unsubstituted rings experienced similar degrees of displacement upon oxidation (i.e., Δ(Fe⋯Ct) ≈ 0.05 Å), vide supra.
Both electron-deficient mono-ketone-substituted ferricenium complexes, AcFcBArF20 and BzFcBArF20, crystallize in the triclinic crystal system with the P space group. Similarly, one electron oxidation imposes larger separation (i.e., by ∼0.1 Å) of the iron center from the substituted Cp ring as compared to the unsubstituted ligand in these complexes. The Cp rings in both AcFc+ and AcFc are nearly eclipsed with the torsion angles of about −3.4° and 0.6°, respectively.30 The acetyl group shows a rotation of 180° around the C–C bond of the Cp ring and acetyl group in going from the neutral to oxidized complex while retaining a similar degree of out-of-plane displacement in both forms.
Interestingly, the one-electron oxidation of BzFc also triggers a significant rearrangement of the benzoyl substituent. In the oxidized form, the carbonyl group of the benzoyl moiety bows toward the iron center (i.e., an out-of-plane bend of 1.8 Å) with the phenyl group having an interplanar angle of 81.8°, as compared to the significantly smaller interplanar angle of 37.7° (i.e., the benzyl moiety has an out-of-plane bend of 0.33 Å away from the iron center) in the neutral BzFc counterpart.32 Alternatively, this substantial difference in the orientation of benzoyl groups may be due to changes in the molecular packing of the neutral and oxidized forms.
The molecular structures of the three 1,1′-disubstituted ferricenium derivatives bearing electron-withdrawing groups are shown in Fig. 6. All these complexes crystallize in centrosymmetric space groups and contain no more than one independent molecule of the compound in the asymmetric unit. The Br2FcBArF20 complex crystallizes in a monoclinic crystal system with the P21/n space group.
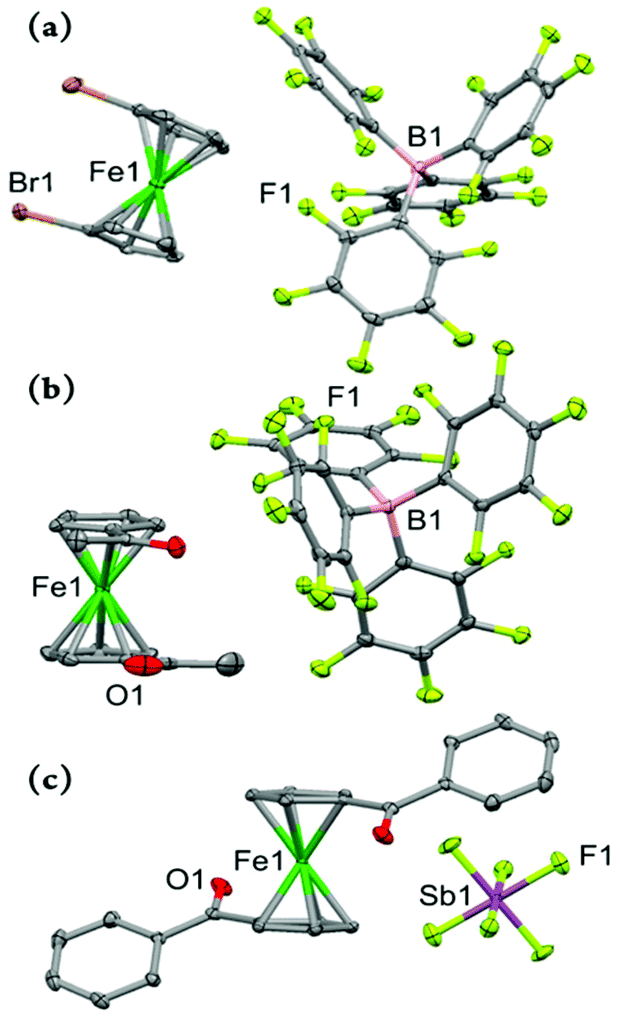 | ||
| Fig. 6 Perspective views of the electron-deficient disubstituted ferricenium derivatives at 100 K: (a) Br2FcBArF20, (b) Ac2FcBArF20, and (c) Bz2Fc[SbF6] showing 50% thermal contours for all non-hydrogen atoms. Hydrogen atoms have been omitted for clarity. | ||
The Cp rings in Br2Fc+ are approximately eclipsed with a torsion angle of about 3.5° between the two bromo substituents which is larger than that of Br2Fc (ϕ = 0.6°).42 Due to the smaller torsion angle and shorter Ct⋯Ct distance (∼3.298 Å) in the neutral analog, the two Br-atoms are forced out of the Cp planes with out-of-plane displacement of 0.14 Å and 0.08 Å (i.e., γ = 2.7 and 4° away from the ferrous center) and a Br⋯Br non-bonding separation of 3.617 Å. By comparison, the smaller torsion angle restraints and greater Ct⋯Ct distance (∼3.405 Å) in Br2Fc+ lead to a decrease of the out-of-plane displacement of both Br-atoms to less than 0.02 Å (i.e., γ = 1.1° and 1.3° toward the ferric center) and the Br⋯Br non-bonding separation is about 3.773 Å.
Among all the derivatives discussed in this study, the dibromo substituted system, in either oxidized or neutral form, exhibits the most significant metal-centered bending (α = 177.0° in Br2Fc+ and 177.7° in Br2Fc+). This is consistent with the overall trend observed in ferrocene derivatives highlighting that the structures with eclipsed conformations reach considerably larger bending angles.39,43
The difference between structures of Ac2FcBArF20 and its neutral analog, Ac2Fc is even more pronounced. The Ac2Fc+ complex crystallizes in the monoclinic space group P21/c, with the Cp rings in staggered arrangements (ϕ = –26.4°) in contrast to the nearly eclipsed conformation observed for Ac2Fc (ϕ = 139.6°).31 Here, upon one–electron oxidation, the two acetyl groups on the rings drastically move toward and pass each other (Δϕ = 166°). The average out–of–plane displacement of the acetyl moieties in Ac2Fc is about 0.098 Å and it decreases to 0.042 Å in Ac2FcBArF20, which in turn slightly affects the relative position of the Cp rings in a way that the interplanar angle between the Cp rings changes from 1.4° in Ac2Fc to 2.6° in Ac2FcBArF20.
As mentioned earlier, only a handful of ferricenium derivatives with electron-withdrawing groups have been structurally characterized to date. Among the electron-deficient derivatives presented in this work, only the structure of Ac2Fc+ as the N(SO2CF3)2−, NTf2−, salt has been previously reported.18 The Fe–Cavg and Ct⋯Ct distances in this Ac2FcNTf2 complex are 2.093 and 3.416 Å, respectively, which are very similar to those of Ac2FcBArF20 reported here. The major structural discrepancy for these two complexes is found in the relative orientation of the acetyl substituents on the Cp rings. The torsion angle of 180.0° in Ac2FcNTf2 clearly indicates that acetyl groups lie in perfectly opposite positions from each other, while the acetyl groups in Ac2FcBArF20 are only 26.4° apart. Another interesting observation is the rotation of the C–C bond between one of the Cp rings and the attached acetyl group in Ac2FcNTf2 and both in the case of Ac2FcBArF20 as compared to the structure of neutral complex (Fig. 7).
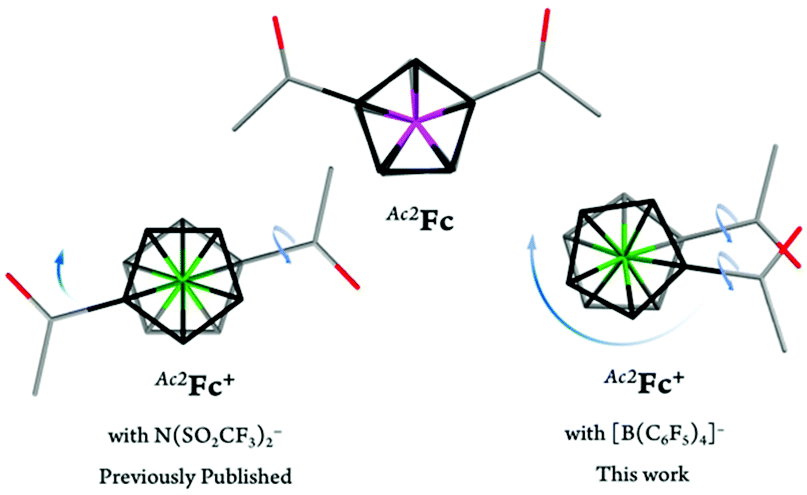 | ||
| Fig. 7 Top view of the capped stick representations of the molecular structures of Ac2Fc,31Ac2FcNTf2,18 and Ac2FcBArF20. The top and bottom Cp rings are shown in black and grey, respectively. Counterions and hydrogen atoms are omitted for clarity. Arrows represent the conformational rearrangements required for the oxidized complex to adopt a similar conformation as the neutral complex. | ||
The complex Bz2Fc[SbF6] crystallizes in a triclinic crystal system with the P space group and the asymmetric unit contains one half of the molecule where the Fe and Sb atoms are located on the crystallographic inversion center. The torsion angle between the two substituted Cp rings in Bz2Fc[SbF6] is 180.0° which is significantly larger than that of the neutral counterpart, Bz2Fc (ϕ = 130.4°).33 From the top view of the two complexes shown in Fig. 8, it is clear that the Cp rings in Bz2Fc[SbF6] are almost perfectly staggered whereas in the neutral counterpart they adopt a close to eclipsed conformation. The Fe–Cavg and Ct⋯Ct distances are about 2.042 and 3.296 Å in Bz2Fc while for the oxidized species, Bz2Fc[SbF6], they increase to 2.089 and 3.410 Å, respectively. Since in the neutral and oxidized forms, the substituents are far apart from each other, the Cp rings are highly coplanar in both cases with interplanar angles of 0.4° and 0.0°, respectively. We will return to this point later in the electrochemistry discussion.
 | ||
| Fig. 8 Ball and stick representation of the top view of the molecular structures of ferrocene and corresponding ferricenium derivatives depicting the changes in the ring stagger torsion angle that accompany the oxidation process. The ferric and ferrous centers are shown in green and pink, respectively. Hydrogen atoms are omitted for clarity. | ||
A comparison of the iron-ring centroid distances for both ferrocene and ferricenium derivatives is shown in Fig. 9. As discussed earlier, the removal of one electron from the metal e2g orbitals gives rise to a larger separation between the iron and Cp ligands. This is consistent with the very weakly bonding character of the e2g orbitals. Additionally, the largest Fe⋯Ct elongation (by ∼0.085 Å) is observed for the oxidation of the dimethyl substituted system.
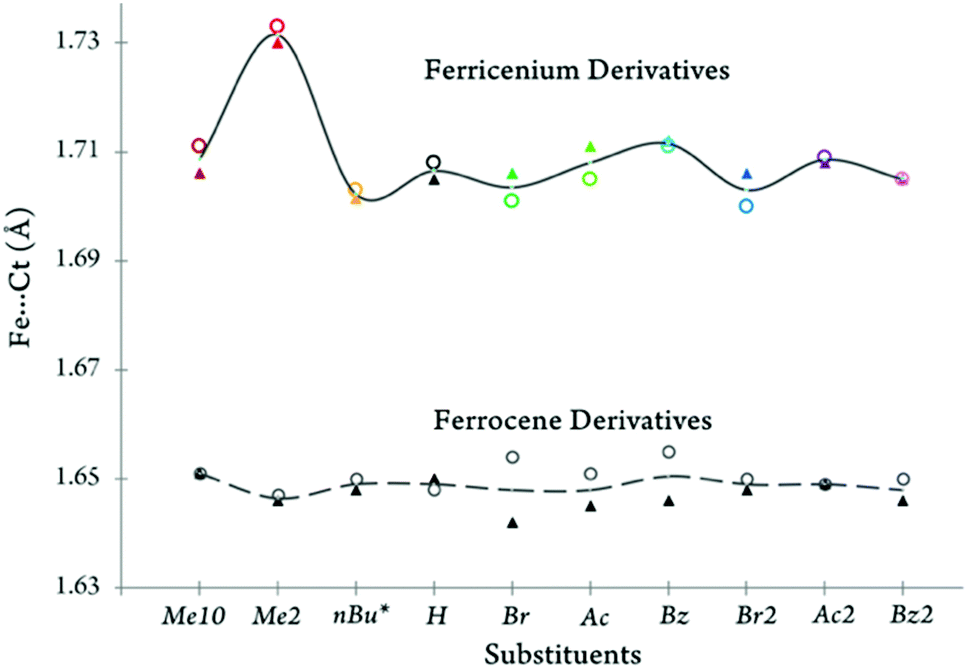 | ||
| Fig. 9 The separation of the iron and centroid (Fe⋯Ct) of the top (▲) or bottom (○) cyclopentadienyl ring is plotted for all of ferrocene and ferricenium derivatives discussed in this study. *For the neutral form average of the Fe⋯Ct distances for two closely related derivatives, nC14Fc and Fc–(CH2)8–Fc, was used. | ||
The oxidation of ferrocene leads to more significant shortening of the C–C bond lengths in the cyclopentadienyl ligands in the parent ferricenium complex (i.e., ∼0.033 Å) relative to those of their substituted analogs (i.e.,<0.015 Å). For example, the C–C bond length stays nearly intact throughout the oxidation of 1-benzoylferrocene, see Table S9† for details on the average change in C–C bond length across all ferrocene and ferricenium derivatives.
A closer look at this metal–ligand separation also reveals that the nature of the substituents has a more significant effect on the Fe⋯Ct distances in the oxidized species (i.e., Δ(Fe⋯Ct) ≈ 0.03 Å) than in their neutral counterparts (i.e., Δ(Fe⋯Ct) ≈ 0.01 Å). Another interesting finding is that, in the monosubstituted ferrocene derivatives bearing an electron-withdrawing group (e.g., BrFc, AcFc, and BzFc), the distance between the ferrous center and the unsubstituted ring is slightly larger than that of the electron-deficient substituted ring. Although smaller in magnitude, a reversal of behavior is observed in the oxidized counterparts of these electron-deficient monosubstituted systems. This reversal of behavior is in excellent agreement with our NMR results (Fig. 1).
Electrochemical analyses
In order to further understand the redox behavior of the ferrocene and ferricenium derivatives, we conducted cyclic voltammetry measurements under 5 different conditions of solvent and supporting electrolyte. All electrochemical experiments were performed in dry and oxygen-free MeCN, DCM, or MeTHF containing 0.1 M of one of two chosen supporting electrolytes, i.e., tetra-n-butylammonium tetrakis(pentafluorophenyl)-borate, [(nBu)4N][B(C6F5)4], or a more traditional electrolyte, tetra-n-butylammonium hexafluorophosphate, [(nBu)4N][PF6] (Fig. 10 and S72–S75†). The latter is not soluble in MeTHF.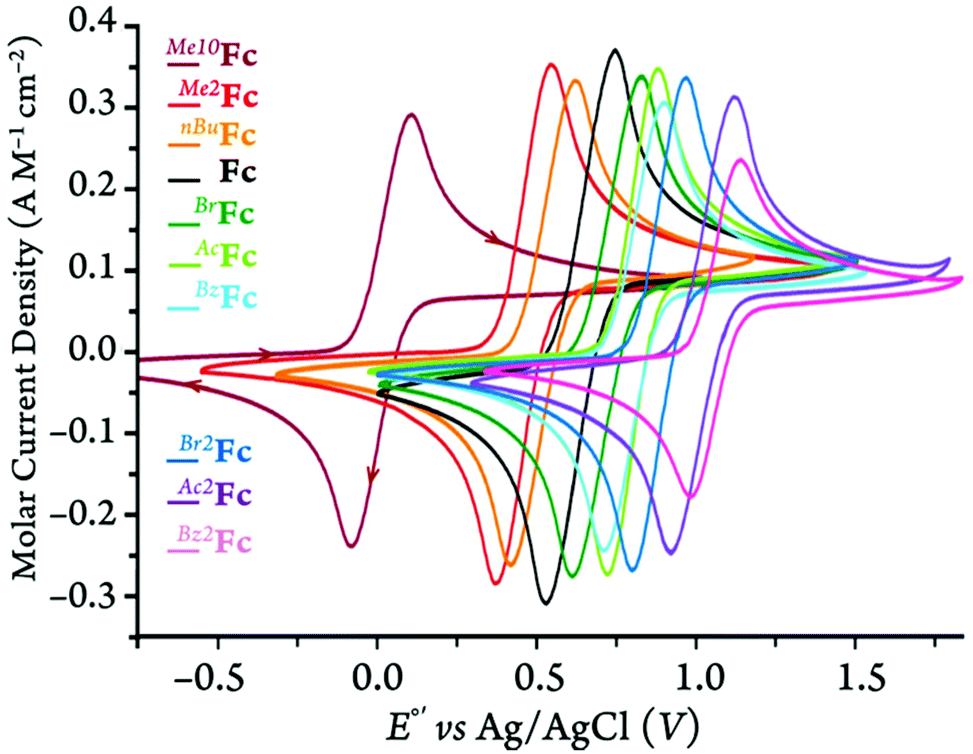 | ||
| Fig. 10 Normalized cyclic voltammograms of ferrocene and its derivatives in DCM with 100 mM of [(nBu)4N][PF6] at 100 mV·s−1 scan rate. | ||
Prior to each cyclic voltammetry experiment, the uncompensated solution resistance (Ru) of each solvent/electrolyte combination was measured using potentiostatic electrochemical impedance spectroscopy (PEIS) at frequencies ranging from 1 MHz to 100 mHz at open circuit potential. Our results for five different media followed the trends reported in the literature,44 see ESI† for further details. In DCM, using [(nBu)4N][B(C6F5)4] as the supporting electrolyte results in lower solution resistance compared to [(nBu)4N][PF6], and an opposite trend was observed in MeCN. Additionally, for a 0.1 M solution of tetra-n-butylammonium perchlorate at room temperature, the specific resistance in MeCN (ρ = 132 Ω cm) is shown to be significantly smaller than in DCM (ρ = 725 Ω cm).45 Such data has not been reported for MeTHF, however, the fact that the specific resistance value obtained in THF (ρ = 2670 Ω cm) is considerably larger than in DCM, infers that MeTHF follows a similar trend. To avoid instabilities in the potentiostat, the iR drop was corrected for only 85% of the uncompensated solution resistance during the cyclic voltammetry measurements through positive feedback using the Bio-Logic EC-Lab software. The half-wave potential, E1/2, (V vs. Ag/AgCl) and peak-to-peak separation, ΔE1/2, of each ferricenium/ferrocene couple in various media are listed in Table 3.
| MeCN | DCM | MeTHF | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| [(nBu)4N][PF6] | [(nBu)4N][B(C6F5)4] | [(nBu)4N][PF6] | [(nBu)4N][B(C6F5)4] | [(nBu)4N][B(C6F5)4] | ||||||
| E 1/2 | ΔE1/2 | E 1/2 | ΔE1/2 | E 1/2 | ΔE1/2 | E 1/2 | ΔE1/2 | E 1/2 | ΔE1/2 | |
| a The values were obtained at 100 mV s−1 scan rate. | ||||||||||
| Me10 Fc | −0.060 | 0.066 | −0.068 | 0.076 | 0.014 | 0.186 | 0.028 | 0.156 | 0.047 | 0.134 |
| Me2 Fc | 0.347 | 0.080 | 0.341 | 0.093 | 0.462 | 0.174 | 0.513 | 0.164 | 0.460 | 0.170 |
| nBu Fc | 0.396 | 0.076 | 0.386 | 0.100 | 0.523 | 0.203 | 0.538 | 0.142 | 0.515 | 0.160 |
| Fc | 0.450 | 0.076 | 0.451 | 0.089 | 0.550 | 0.217 | 0.577 | 0.142 | 0.589 | 0.183 |
| Br Fc | 0.628 | 0.081 | 0.630 | 0.092 | 0.725 | 0.215 | 0.747 | 0.163 | 0.757 | 0.162 |
| Ac Fc | 0.700 | 0.094 | 0.689 | 0.086 | 0.803 | 0.158 | 0.861 | 0.175 | 0.833 | 0.132 |
| Bz Fc | 0.705 | 0.077 | 0.697 | 0.094 | 0.810 | 0.185 | 0.878 | 0.130 | 0.806 | 0.150 |
| Br2 Fc | 0.763 | 0.087 | 0.751 | 0.122 | 0.887 | 0.166 | 0.934 | 0.133 | 0.900 | 0.165 |
| Ac2 Fc | 0.925 | 0.093 | 0.930 | 0.129 | 1.020 | 0.203 | 1.110 | 0.150 | 1.037 | 0.125 |
| Bz2 Fc | 0.927 | 0.102 | 0.903 | 0.100 | 1.070 | 0.157 | 1.230 | 0.236 | 1.003 | 0.124 |
As expected, the incorporation of various electron-donating or -withdrawing substituents on the Cp rings altered the redox potential cathodically or anodically. Electron-rich systems with one or more alkyl substituent(s) such as Me10Fc, Me2Fc, and nBuFc all possess redox potentials lower than that of Fc, while having electron-withdrawing groups on the rings creates an electron-deficient system such as BrFc, AcFc, BzFc, Br2Fc, Ac2Fc, and Bz2Fc, hence increasing the E°′ values.
The cyclic voltammograms of all the derivatives in DCM with the [(nBu)4N][PF6] supporting electrolyte are shown in Fig. 10. For the cyclic voltammograms collected in the other solvent/electrolyte combinations, see section 3 of the ESI.† The neutral ferrocene derivatives are generally very soluble in the three solvents chosen for this study, i.e., DCM, MeTHF, and MeCN. Only Me10Fc has a limited (ca. 10−3 M) solubility in acetonitrile. For that reason, the cyclic voltammograms of all ferrocene derivatives are plotted using the molar current density (A M−1 cm−2) rather than the current alone. This was done in order to compare the cyclic voltammetry measurements independent of the ferrocene concentration and surface area of the working electrode.
The correlation between the redox potentials of the substituted ferrocenes and the sum of Hammett substituent constants in the MeTHF solution is shown in Fig. 11. Typically, the E1/2 data of substituted ferrocenes correlate linearly with the sum of the Hammett values, Σσp,m, which is a combination of para- and meta-substituents (i.e., σp and σm).
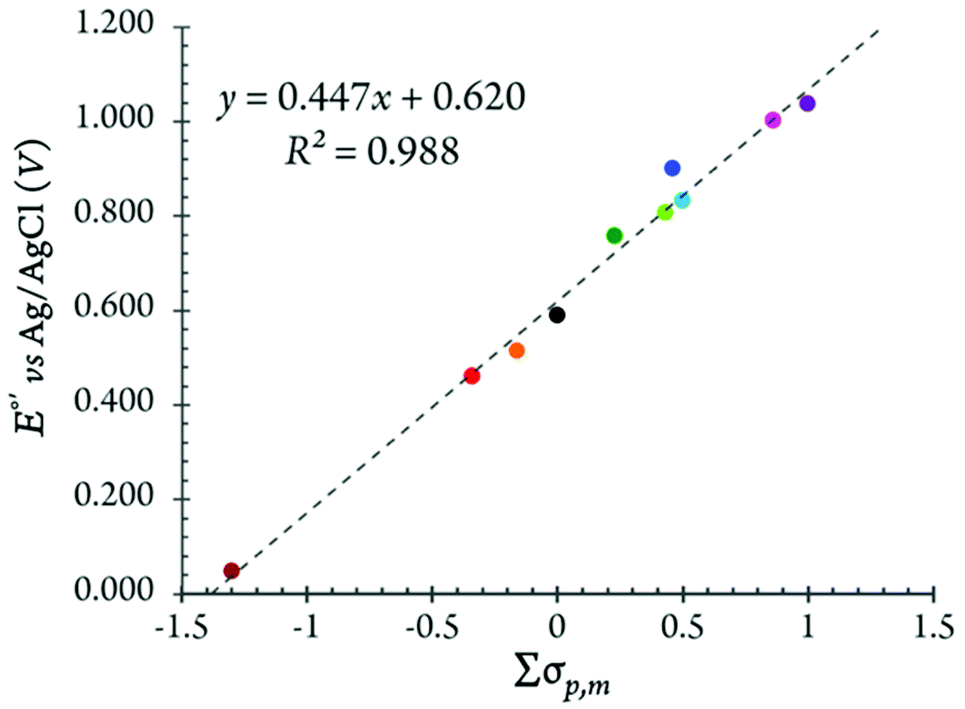 | ||
| Fig. 11 The half-wave potential, E1/2, of all ferrocene derivatives discussed in this study in MeTHF with 100 mM of [(nBu)4N][B(C6F5)4] plotted vs. sum of the Hammett values, Σσp,m. | ||
For all of our mono- and 1,1′-di-substituted ferrocenes, only σp was taken into account. The σp values for methyl, n-butyl, bromo, benzoyl and acetyl groups are −0.17, −0.16, +0.23, +0.43, +0.50, respectively.46 The impact of the substitution in 3- or 4- (and 3′- or 4′) positions of a Cp ring is included using σm.35b,47 For example, in Me10Fc, the Σσp,m value contains contributions of both para and meta methyl substituents (i.e., σm for a methyl group is −0.07 and Σσp,m = [6 × (−0.17) + 4 × (−0.07)] = –1.3). One apparent discrepancy that merits special attention at this point is that, based on the σp values, the benzoyl substituted ferrocenes are expected to be less electron-deficient than the acetyl substituted analogs. This trend holds true in MeTHF but not in DCM, see Table 3.
The one-electron transfer redox processes showed quasi-reversible behavior with peak-to-peak separation values greater than 57 mV (i.e., ΔE ranging from 66 to 236 mV; see Table 3) and anodic/cathodic peak current ratios between 0.96 and 1.09 (Table S14†), except for the two 1,1′-diketone-substituted ferrocenes in MeCN (ipa/ipc = 1.04–1.24), vide infra. Our Randles–Sevcik analysis of the peak current vs. the square root of the scan rate confirmed that in all cases the species involved in the redox reactions were freely diffusing through the electrochemical cell (Fig. 12 and S76–S125†), rather than adsorbed on the surface of the working electrode.
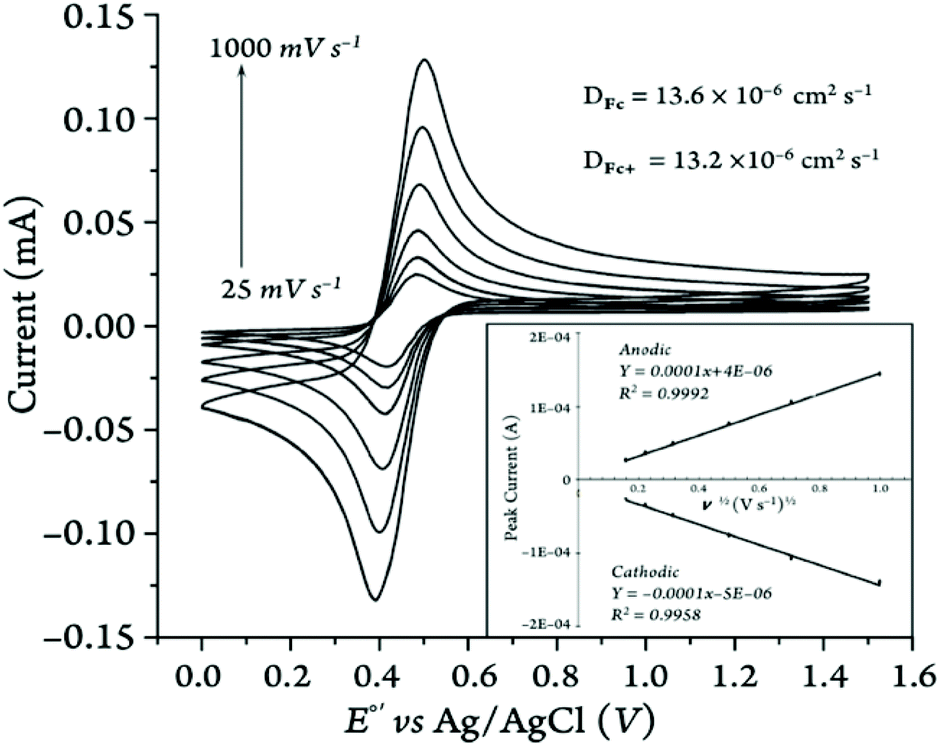 | ||
| Fig. 12 Representative cyclic voltammograms of the parent Fc+/Fc couple (2 mM) at various scan rates in MeCN with [(nBu)4N][PF6] as the supporting electrolyte (100 mM). The inset shows the Randles–Sevcik plot of the CV data. | ||
The effect of the medium on the redox potential, ΔE1/2, and diffusion of the redox active species is a complicated function of the interactions between solvent and solute and their overall ion-pairing.44,48 Some of the most important properties that govern these interactions are dielectric constant (ε), dipole moment (μ), donor number (DN), acceptor number (AN), and absolute viscosity (η). The relevant parameters for the solvents chosen for this study are listed in Table 4. Acetonitrile (MeCN), which has by far the largest dielectric constant (ε = 36.6), is historically favored for most electrochemical measurements in organic systems. Here, in addition to MeCN, we employed two lower polarity solvents with dielectric constants of less than 10 (i.e., DCM and MeTHF). For the latter, using [(nBu)4N] [B(C6F5)4] as the supporting electrolyte, rather than the more traditional analogs such as [(nBu)4N][PF6], can remarkedly enhance the conductivity and decrease the overall ohmic drop.44
| MeCN | DCM | MeTHF | |
|---|---|---|---|
| a From ref. 48a. b From ref. 49. c The AN is not reported for MeTHF, although it can be inferred to be close to values reported for tetrahydrofuran (8.0) and diethyl ether (3.9).50 d From ref. 51. e From ref. 52. | |||
| Dielectric constant, ε | 36.6a | 8.93a | 6.97b |
| Dipole moment, μ (D) | 3.92a | 1.60a | 1.36b |
| Donor number DN | 14a | (0)a | 18b |
| Acceptor number AN | 19a | 20a | 3.9–8.0c |
| Absolute viscosity, η (mPa s) | 0.375d | 0.426d | 0.492e |
As shown in Fig. 13 and Table 3, the redox potentials of the ferrocene derivatives vary with the nature of the solvent. Considering solely the solvent polarity for a given electrolyte, the redox potentials are predicted to be the lowest in MeCN compared to those in DCM and MeTHF. All of our data follow the expected trend in acetonitrile. When comparing the two lower polarity solvents, DCM and MeTHF, the donor and acceptor numbers of the solvents are taken into consideration in predicting the redox behavior of the ferrocene derivatives in solution.
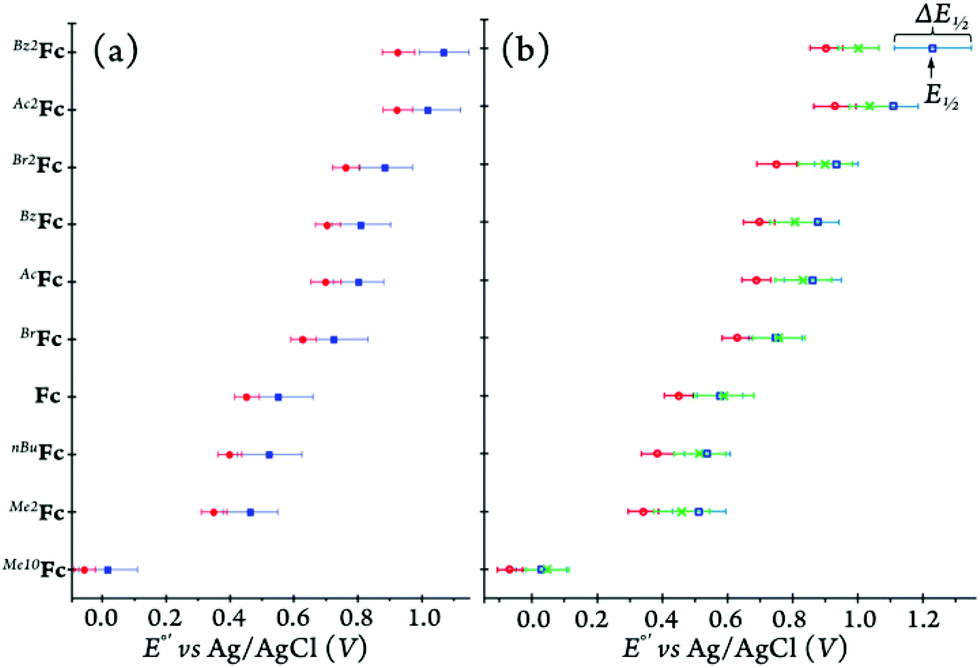 | ||
| Fig. 13 The half-wave potential, E1/2, and peak-to-peak separation, ΔE1/2, of all ferricenium/ferrocene couples discussed in this study in different media: (a) with [(nBu)4N][PF6] as the supporting electrolyte in MeCN (red) and DCM (blue) and (b) with [(nBu)4N][B(C6F5)4] as the supporting electrolyte in MeCN (red), MeTHF (green), and DCM (blue). | ||
The donor and acceptor numbers of DCM are reported to be around 0 and 20, respectively, while MeTHF has a donor number of 18 and an estimated acceptor number in the range of 3.9 to 8.0.50 The differences in the donor and acceptor properties of these two solvent influence electrolyte dissociation. Additionally, higher donor number indicates that MeTHF can act more as a Lewis base stabilizing the oxidized species as well as improving the thermodynamics of the ion paring between ferricenium and the anion of the supporting electrolyte, in turn lowering the redox potential. On the other hand, the higher acceptor number of DCM suggests a higher degree of Lewis acidity, stabilizing the neutral form and in turn increasing the potential needed to oxidize the ferrocene derivatives. The pattern of higher redox potentials in DCM compared to MeTHF was observed for most ferrocene derivatives except for Me10Fc, Fc, and BrFc. Lay and coworkers previously ascribed the considerably weaker solvent effects on the redox potential of Me10Fc to the shell of the methyl substituents that can protect the iron center against close interactions with solvents and electrolytes.53 However, it is not clear as to why Fc and BrFc also do not follow the pattern and further understanding of solvent and solute interactions other than electrostatic effects is necessary to explain their behavior.
The role of the two electrolyte anions, PF6− and [B(C6F5)4]−, in altering redox potentials and peak-to-peak separations in both MeCN and DCM was investigated. As described in the Introduction, the high degree of charge delocalization in a large weakly coordinating anion such as [B(C6F5)4]− makes it a weak nucleophile and generally well soluble in lower-polarity solvents. This enhanced solubility can minimize adsorption problems with cationic electrode products (e.g., the ferricenium derivatives). The [B(C6F5)4]− anion is also considered a weakly ion-pairing anion.48a
In MeCN, the nature of the electrolyte anion has limited effect on the E1/2 and ΔE1/2 values, as the differences observed for all the ferrocene derivatives with the two electrolyte anions, PF6− and [B(C6F5)4]−, on average are about 7 mV and 15 mV, respectively (Table 3). This is likely due to the high polarity of MeCN minimizing the ion-pairing effects of the different electrolytes.44 As for DCM, there is a more observable trend wherein Me10Fc, nBuFc, Fc, and BrFc experience the smallest electrolyte-induced change in E1/2 (i.e., 14–27 mV) while the di-substituted ferrocenes Me2Fc and Br2Fc show differences of 47 and 51 mV, respectively.
The largest anodic shifts in E1/2 (i.e., 58–160 mV) are seen for mono- and 1,1′-di-ketone-substituted ferrocene derivatives going from [(nBu)4N][PF6] to [(nBu)4N][B(C6F5)4] in DCM. Quite curiously, the electrolyte-induced changes of ΔE1/2 follow a very different trend (Table 3). For example, the largest difference of about 77 mV in ΔE1/2 is observed for the parent Fc and its most electron-deficient derivative, Bz2Fc. However, in the presence of [B(C6F5)4]− as compared to PF6−, the peak-to-peak separation becomes markedly smaller for the parent compound while it significantly increases for the Bz2Fc derivative, see Table 3.
As shown in Fig. 14, the anodic peak of the parent ferrocene is barely affected (i.e., only by 8 mV) by the nature of electrolyte anion in DCM while the cathodic peak shifts by about 67 mV. This again emphasizes the fact that in low-polarity solvents the ferricenium species can be further stabilized when the electrolyte anion is changed from weakly (i.e., [B(C6F5)4]−) to relatively strongly (PF6−) ion-pairing, highlighting the often overlooked effects of counter anions in governing the redox potentials. Also, the scan rate has the least influence on the ΔE1/2 values of different ferrocene derivatives in acetonitrile (Fig. S126–S135†) due to negligible incomplete iR compensation.54
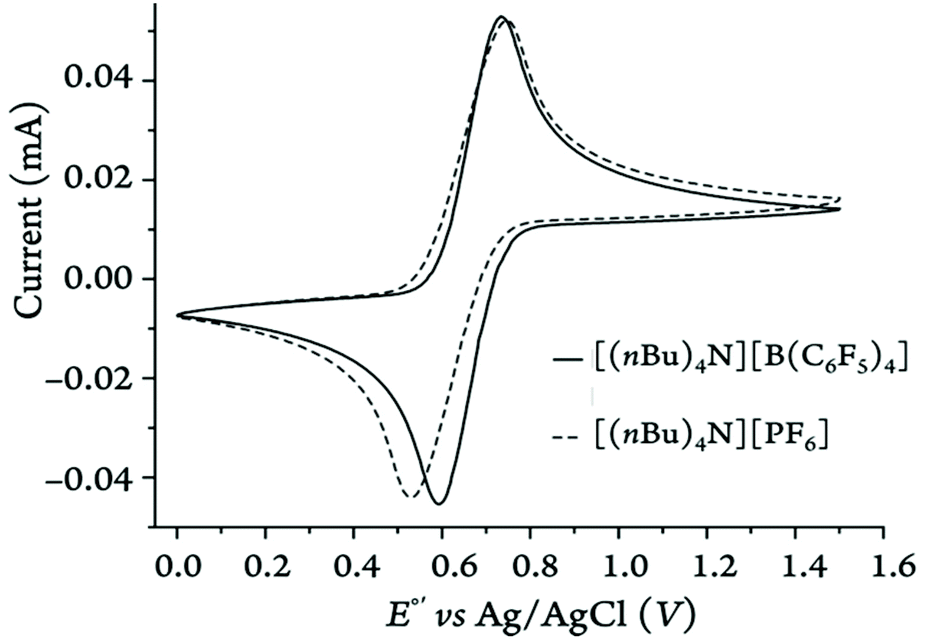 | ||
| Fig. 14 Representative cyclic voltammograms recorded for Fc (2 mM) in DCM on a glassy carbon disk electrode at 100 mV·s−1 in two different electrolytes. | ||
The cyclic voltammetry measurements of Bz2Fc in MeCN with either electrolytes revealed a possible ErCi process which means that Bz2Fc+ reacts in a homogenous chemical reaction upon oxidation, thus, being chemically irreversible. While scanning at 100 mV s−1, the peak current moved closer to the baseline (Fig. 15). The voltammograms become more reversible at scan rates above 100 mV s−1. It was found that 1500 mV s−1 was the optimal scan rate, as that is where the cathodic/anodic peak current ratio reached the highest value at 0.81. This is a notable feature for ErCi reactions, at higher scan rates, the chemical reaction following the initial electron transfer step is too slow to compete, leaving more oxidized species to become subsequently reduced in the reverse scan.55
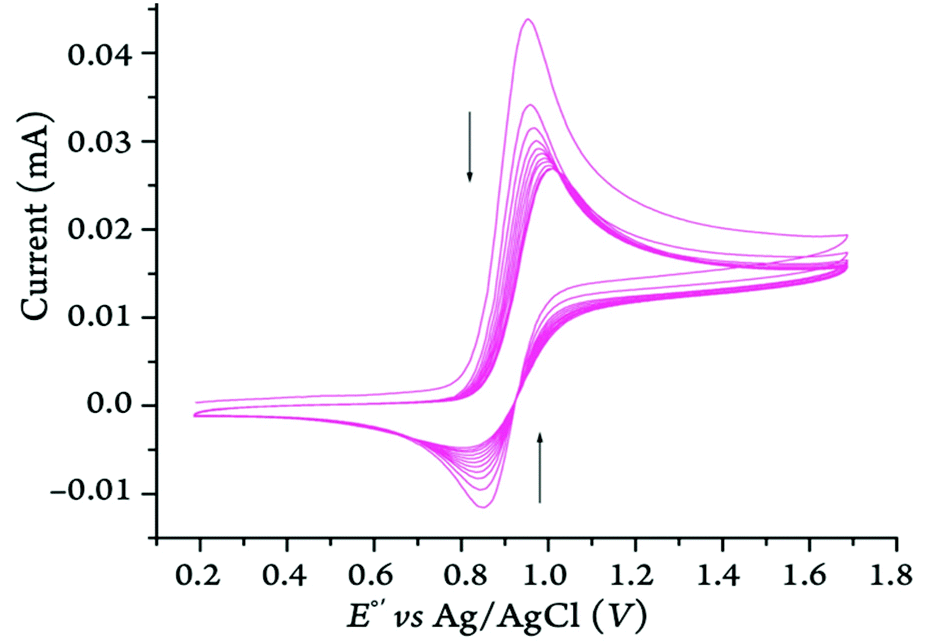 | ||
| Fig. 15 Cyclic voltammograms of Bz2Fc (2 mM) in MeCN at 100 mV·s−1 with 100 mM of [(nBu)4N][B(C6F5)4] as the supporting electrolyte. Arrows indicate the decay of the current towards the baseline as the scans progressed (i.e., an ErCi process). | ||
As Ac2Fc demonstrated a similar behavior, a series of faster scan rates was also used in MeCN to outrun the chemical reaction that accompany the electron transfer process for this derivative. Therefore, for the studies of Bz2Fc and Ac2Fc in MeCN with both electrolytes, the scan rates were varied at 1500, 1250, 1000, 500, 250, and 100 mV s−1 and the working electrode was cleaned between each scan rate variation. These results are consistent with our direct observation of the reactivity of the chemically synthesized ketone-substituted ferricenium derivatives toward acetonitrile. The exact mechanism of this reactivity is outside the scope of this report and will be discussed elsewhere.
As mentioned earlier, the electron transfer processes were diffusion-controlled for all the ferrocene and ferricenium derivatives used in this study in five different media. The diffusion coefficients (D) are presented in Table 5.
| 106 × D [(cm2 s−1)] in MeCN | 106 × D [(cm2 s−1)] in DCM | 106 × D [(cm2 s−1)] in MeTHF | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| [(nBu)4N][PF6] | [(nBu)4N][B(C6F5)4] | [(nBu)4N][PF6] | [(nBu)4N][B(C6F5)4] | [(nBu)4N][B(C6F5)4] | ||||||
| Anodic | Cathodic | Anodic | Cathodic | Anodic | Cathodic | Anodic | Cathodic | Anodic | Cathodic | |
| Me10 Fc | 12.96 ± 0.00 | 13.86 ± 0.00 | 12.96 ± 0.00 | 12.66 ± 0.00 | 5.62 ± 0.01 | 6.22 ± 0.01 | 3.69 ± 0.01 | 3.36 ± 0.01 | 4.78 ± 0.01 | 3.91 ± 0.01 |
| Me2 Fc | 17.83 ± 0.00 | 18.79 ± 0.00 | 14.18 ± 0.01 | 15.59 ± 0.01 | 9.40 ± 0.04 | 9.05 ± 0.01 | 5.03 ± 0.02 | 2.66 ± 0.02 | 5.51 ± 0.01 | 5.92 ± 0.01 |
| nBu Fc | 18.34 ± 0.00 | 18.60 ± 0.00 | 14.12 ± 0.01 | 14.84 ± 0.01 | 7.92 ± 0.02 | 7.64 ± 0.01 | 8.07 ± 0.01 | 6.93 ± 0.01 | 4.54 ± 0.01 | 4.52 ± 0.01 |
| Fc | 13.67 ± 0.00 | 13.28 ± 0.01 | 8.80 ± 0.01 | 13.19 ± 0.00 | 9.27 ± 0.02 | 9.37 ± 0.02 | 12.3 ± 0.01 | 12.2 ± 0.01 | 4.66 ± 0.01 | 4.59 ± 0.01 |
| Br Fc | 13.28 ± 0.00 | 12.74 ± 0.00 | 12.31 ± 0.01 | 13.04 ± 0.00 | 7.81 ± 0.01 | 7.73 ± 0.01 | 6.70 ± 0.01 | 4.55 ± 0.01 | 5.82 ± 0.00 | 5.17 ± 0.01 |
| Ac Fc | 15.30 ± 0.01 | 12.35 ± 0.01 | 13.60 ± 0.00 | 14.55 ± 0.01 | 9.81 ± 0.01 | 9.32 ± 0.01 | 5.77 ± 0.01 | 3.96 ± 0.01 | 7.20 ± 0.01 | 6.74 ± 0.01 |
| Bz Fc | 13.29 ± 0.01 | 11.00 ± 0.01 | 13.38 ± 0.01 | 13.72 ± 0.01 | 6.61 ± 0.01 | 6.48 ± 0.01 | 6.11 ± 0.01 | 5.69 ± 0.01 | 5.98 ± 0.01 | 5.16 ± 0.01 |
| Br2 Fc | 18.08 ± 0.00 | 14.98 ± 0.01 | 11.18 ± 0.01 | 9.94 ± 0.01 | 9.15 ± 0.01 | 8.76 ± 0.01 | 10.7 ± 0.00 | 9.71 ± 0.01 | 4.60 ± 0.01 | 2.66 ± 0.01 |
| Ac2 Fc | 12.51 ± 0.01 | 10.49 ± 0.02 | 12.85 ± 0.00 | 9.78 ± 0.02 | 5.60 ± 0.01 | 5.68 ± 0.01 | 2.75 ± 0.00 | 1.30 ± 0.01 | 6.14 ± 0.00 | 3.99 ± 0.01 |
| Bz2 Fc | 9.37 ± 0.01 | 6.72 ± 0.01 | 7.30 ± 0.00 | 5.67 ± 0.02 | 2.95 ± 0.01 | 2.29 ± 0.02 | 1.33 ± 0.00 | 0.21 ± 0.01 | 4.65 ± 0.00 | 3.04 ± 0.01 |
The diffusion coefficients of the neutral ferrocene derivatives and their ferricenium counterparts are typically not very different.56 As expected, the species moved the fastest in MeCN compared to DCM or MeTHF. This can be attributed to the high polarity of the solvent, which minimizes the analyte-electrolyte ion-pairing and subsequently decreases the resistance of the solution.48a Another factor that can facilitate the movement of analytes in the solution is the lower absolute viscosity of acetonitrile compared to DCM and MeTHF (Table 4).49,51 Among the ferrocene derivatives described here, both oxidized and neutral forms of Me2Fc, nBuFc, and Br2Fc diffuse faster than the parent ferrocene and ferricenium by about 35% through the acetonitrile media.
The diffusion coefficients obtained in this study for both oxidized and neutral parent ferrocene species in MeCN with [(nBu)4N][PF6] are lower (i.e., DFc = 1.367 × 10−5 cm2 s−1) than most previously reported values (i.e., DFc ≈ 2 × 10−5–2.7 × 10−5 cm2 s−1),55,57 which we contribute to our rigorous efforts to minimize the amount of water present in the solutions during our electrochemical measurements,58 see ESI† for more details.
Some of the complexes (i.e., nBuFc, Fc, BrFc, BzFc, and Br2Fc) are found to diffuse slowest in the MeTHF solution which is in line with the higher viscosity and lower dielectric constant of the solvent compared to DCM. However, the remaining derivatives such as Me10Fc, Me2Fc, AcFc, Ac2Fc, and Bz2Fc follow a reverse trend and have larger diffusion constants in MeTHF than DCM. Perhaps this stems from the fact that the substituents in these derivatives may disrupt the “normal” charge density distribution throughout the complex, interfering with ion-pairing interaction in MeTHF, therefore, disrupting the expected trend of the solvent/analyte interactions prevalent in solvents of low-polarity.53
Furthermore, the diffusion coefficient of a given solute is inversely proportional to the size (i.e., hydrodynamic radius) of the solute and, hence, to its molecular weight assuming all species are hard spheres and have the same density.59 Gonzalez and coworkers described a linear correlation between the diffusion coefficient and molecular weight.57a As the molecular weight increases, the diffusion coefficient decreases. This was generally observed in our experiments, although not perfectly, with the highest correlation obtained in the DCM solution with [(nBu)4N][PF6] as the supporting electrolyte. Slight outliers are labeled in Fig. S136.†
As mentioned earlier, all the ferrocene derivatives described here show a quasi-reversibility of the redox behavior which is in agreement with our crystallographic data confirming that the difference in Fe–C bond length between their oxidized and neutral form is not larger than 0.05 Å, except for Me2Fc which experiences about 0.06 Å Fe–C bond elongation upon oxidation (Table S9†). Additionally, the relatively lower current density observed for Bz2Fc and to a lesser extent for Ac2Fc, particularly in DCM with [B(C6F5)4]− as the counter anion (Fig. S72†), can be the consequence of the much slower diffusion (Table 5) and significant rearrangements of the substituents that accompany the electron transfer process. This is in agreement with our crystallographic data and the one broad Cp proton resonance observed for their oxidized forms, Ac2Fc+ and Bz2Fc+, in our 1H-NMR studies.
Conclusion
Despite the enormous number of publications in the field of ferrocenes and their redox counterparts, ferriceniums, information on the synthesis and characterization of these complexes is relatively sparse and in most cases the reports lack the relevant experimental details. Moreover, most efforts have been focused on the electron-rich ferricenium species and very limited data are available on the electron-deficient systems. This represents a surprising knowledge gap in the literature. Herein, we attempted to address some of that gap through a systematic and thorough evaluation of a library of highly organic soluble ferricenium derivatives.Our 1H-NMR measurements revealed that the substitutional behavior in the paramagnetic ferricenium derivatives is more complex and fundamentally reversed as compared to the neutral ferrocene counterparts. We proposed that the δ back-donation from the iron atom into the substituted Cp rings leads to the overall shielding of the ring protons in the ferricenium derivatives. This shielding through δ back-donation is more pronounced in the electron-deficient rings with lower energy molecular orbitals. Our data for the electron-deficient ferricenium derivatives in solution also drew a direct correlation between the solvent dielectric constant and the rotation of the cyclopentadienyl ligands around the Fe–Cp bond in these systems.
Here, nine new X-ray structures are also added to the library of ferricenium derivatives, five of which presented the first examples of molecular structures of such derivatives. Structural comparison of the neutral ferrocene derivatives and their oxidized counterparts revealed that the oxidation of ferrocene results in more substantial shortening of the C–C bond lengths in the Cp rings of the parent ferricenium complex relative to those of their substituted counterparts. Interestingly, our results also show that the nature of the substituents has a more significant effect on the metal–ligand separations in the oxidized species than in their neutral analogs. For the ketone-substituted ferricenium derivatives, the increase in the oxidation state was reflected in a significant strengthening of the carbonyl bond(s) by about 35–48 cm−1 when compared with the neutral ferrocene counterparts.
Additionally, the redox behavior of the corresponding ferricenium/ferrocene (Fc+/0) redox couples such as potential values (E1/2), peak-to-peak separation (ΔE1/2), and diffusion coefficients (D) of the redox active species in three different solvents and two supporting electrolytes are reported in this work. The results point to the significant effect of the ion-pairing in lowering the energy necessary for reduction of the ferricenium species and the overall half-wave potential.
In order to explain some of the observed spectroscopic and structural features of the ferricenium systems, particularly those bearing electron withdrawing substituents, further investigations are required; as is often the case in bonding and electronic structures, “the devil is in the details”. Additional experimental and computational efforts are currently underway in our laboratory.
Experimental section
General methods
All chemicals and solvents were of commercially available grade, unless otherwise noted. Acetonitrile (MeCN), 2-methyltetrahydrofuran (MeTHF) (inhibitor free, 673277), dichloromethane (DCM) and hexanes were purchased from Sigma-Aldrich. 1,2-Difluorobenzene was purchased from Alfa Aesar. All solvents were further purified by passing through a 60 or 18 cm-long column of activated alumina under argon using an Innovative Technologies or Inert PureSolv Micro solvent purification system. The solvents were further deoxygenated by either repeated freeze/pump/thaw cycles or bubbling with argon for 45–60 min followed by storage over 3 or 5 Å molecular sieves for at least 72 hours prior to use. Air- and moisture-sensitive compounds were synthesized and handled under a dry oxygen-free argon atmosphere using standard Schlenk techniques or in a Vacuum Atmospheres OMNI-Lab inert atmosphere (<0.5 ppm of O2 and H2O) glovebox filled with nitrogen.Bench-top UV-vis spectra were recorded with a Cary-60 spectrophotometer using a 2 or 10 mm modified Schlenk cuvette. Infrared (IR) spectra were obtained using a Thermo Scientific Nicolet iS5 Fourier Transform IR (FT-IR) spectrometer equipped with an iD7 attenuated total reflection (ATR) accessory. 1H-NMR spectra were recorded on a JEOL 400 or 500 MHz instrument. The chemical shifts were referenced against tetramethyl-silane (TMS, δ = 0.00 ppm). The NMR solvent residual peaks were used as a secondary reference. Elemental analysis was accomplished at Midwest Microlab (Indianapolis, IN). Electrochemical data was collected using a Bio-Logic SP-200 potentiostat. Single-crystal X-ray data were collected using a Gemini R (Agilent Technologies) diffractometer at the X-ray diffraction facility of the Joint School of Nanoscience and Nanoengineering (JSNN). The temperature of the data collection was controlled using the system Cryojet (manufactured by Oxford Instruments).
The compounds, 1-acetylferrocene (97%), and decamethylferrocene (99%) were purchased from Alfa Aesar; 1-benzoylferrocene (>98%), 1,1′-dibromoferrocene (>98%), and 1,1′-dibenzoylferrocene (98%) from Tokyo Chemical Industry (TCI); ferrocene (98%), 1-bromoferrocene (98.8%), 1,1′-diacetylferrocene (97%), and ferrocenecarboxylic acid (99%) from Sigma-Aldrich; 1,1′-dimethylferrocene (98%), and n-butylferrocene (99%) from Stream Chemicals. Potassium tetrakis(pentafluorophenyl)borate, K[B(C6F5)4], (99.9%) was purchased from Boulder Scientific Company; AgNO3 (99.99%) from Alfa Aesar; Ag[SbF6] (98%) and tris (4-bromophenyl)amine from Sigma-Aldrich; tetra-n-butylammonium hexafluorophosphate (98%) from Oakwood Chemicals. tetra-n-Butylammonium chloride hydrate (99.84%) and potassium chloride (99%) were purchased from Alfa Aesar.
Deuterated solvents including acetone-d6 (D, 99.9%) and methylene chloride-d2 (D, 99.8%) were purchased from Cambridge Isotope Laboratories. Distilled water was further purified by a PURELAB flex 1 Analytical Ultrapure Water System (ELGA) to obtain the specific resistance of 18.2 MΩ cm at 25 °C.
Synthesis and characterization
N) = 2295 (Fig. S1†). The deuterated complex, [Ag(CD3CN)4][B(C6F5)4], was prepared by the same procedure described above using deuterated acetonitrile, CD3CN. FT-IR (solid; cm−1): ν(CN) = 2287 (Fig. S2†). 1H-NMR (acetone-d6, 500 MHz; δ, ppm): 2.04 (s, 12H) (Fig. S137†). 19F-NMR (acetone-d6, 470 MHz; δ, ppm): −133.0, −164.3, −168.2 (Fig. S138†).
The BArF20 salts of the parent ferricenium complex and a series of ferricenium derivatives bearing electron-donating substituents were prepared following the general procedure described here. The complexes include Me10FcBArF20, Me2FcBArF20, nBuFcBArF20, and FcBArF20. Details are given for decamethylferricenium BArF20, as a representative case.
The BArF20 salts of a series of ferricenium derivatives bearing one electron-withdrawing substituents as well as the 1,1′-dibromo substituted ferricenium were prepared following the general procedure described here. Those complexes include BrFcBArF20, AcFcBArF20, BzFcBArF20, and Br2FcBArF20. Details are given for 1-bromoferricenium BArF20, as a representative case. Note: For both mono-ketone-substituted ferrocenes, the order of addition of reagents is reversed (i.e., the solution of substituted ferrocene is gradually added to the silver(I) solution). An alternative procedure for the preparation of BzFc[B(C6F5)4] is also included.
O):Ac = 1698 (Fig. S9†). 1H-NMR (acetone-d6, 500 MHz; δ, ppm): 36.3 (s, vbr, 5H), 31.8 (s, vbr, 2H), 27.5 (s, vbr, 2H), −15.9 (s, br, 3H) (Fig. S40†). 19F-NMR (acetone-d6, 470 MHz; δ, ppm): −133.0, −164.4, −168.4 (Fig. S52†). 1H-NMR (CD2Cl2, 500 MHz; δ, ppm): 37.2 (s, vbr, 5H), 32.4 (s, vbr, 2H), 30.4 (s, vbr, 2H), −13.0 (s, br, 3H) (Fig. S41†). 19F-NMR (CD2Cl2, 470 MHz; δ, ppm): −135.2, −164.1, −169.0 (Fig. S53†).
O):Bz = 1658 (Fig. S10†). 1H-NMR (acetone-d6, 500 MHz; δ, ppm): 35.4 (vbr, 5H), 31.3 (vbr, 2H), 27.6 (vbr, 2H), 7.5 (s, br, 2H), 6.3 (s, 2H), 6.2 (s, H) (Fig. S42†). 19F-NMR (acetone-d6, 470 MHz; δ, ppm): −133.0, −164.3, −168.3 (Fig. S54†). 1H-NMR (CD2Cl2, 500 MHz; δ, ppm): 36.0 (vbr, 5H), 30.5 (vbr, 4H), 12.4 (s, br, 2H), 7.3 (s, 2H), 6.7 (s, H) (Fig. S43†). 19F-NMR (CD2Cl2, 470 MHz; δ, ppm): −134.9, −164.4, −168.5 (Fig. S55†).
O):Ac = 1697 (Fig. S12†). 1H-NMR (CD2Cl2, 500 MHz; δ, ppm): 30.69 (vbr, 8H), −9.43 (s, br, 6H) (Fig. S34†). 19F-NMR (CD2Cl2, 470 MHz; δ, ppm): −134.7, −163.8, −168.5 (Fig. S58†).
O):Bz = 1665; ν(SbF6) = 651 (Fig. S13†). 1H-NMR (CD2Cl2, 500 MHz; δ, ppm): 30.2 (s, vbr, 8H), 10.3 (s, br, 4H), δ 7.0 (s, 4H), δ 6.7 (s, 2H) (Fig. S35†). 19F-NMR (CD2Cl2, 470 MHz; δ, ppm): −133.2 (1J(19F–121Sb) ≈ 2 kHz) (Fig. S59†).
Crystallographic studies
Suitable X-ray quality single crystals were grown in the glovebox by layering hexanes onto the solution of the ferricenium derivatives in either MeTHF or DCM in 5 mm glass tubes at RT. The complexes Me10FcBArF20, Me2FcBArF20, nBuFcBArF20 and Br2FcBArF20 were crystallized from MeTHF/hexanes while all other ferricenium derivatives were crystallized using DCM/hexanes. All reflection intensities were measured at 100(2) K using a Gemini R diffractometer (equipped with Atlas detector) with Mo Kα radiation (λ = 0.71073 Å) under the program CrysAlisPro (Version CrysAlisPro 1.171.38.43f, Rigaku OD, 2015). The same program (but a different version viz. CrysAlisPro 1.171.40.53, Rigaku OD, 2019) was used to refine the cell dimensions and for data reduction. The structures were solved with the program SHELXT-2018/2 and were refined on F2 by full-matrix least-squares technique using the SHELXL-2018/3 program package.60 Numerical absorption correction based on Gaussian integration was applied using a multifaceted crystal model by CrysAlisPro. Non-hydrogen atoms were refined anisotropically. In the refinement, hydrogen was treated as riding atoms using SHELXL default parameters.Electrochemical measurements
A three-electrode setup was used for all voltammetry experiments with a 3.0 mm glassy carbon disk working electrode, a carbon rod counter electrode, and a leak-free Ag/AgCl reference electrode inside an inert atmosphere box. Three separate 2.0 mm diameter reference electrodes were filled with 3.4 M KCl aqueous solutions (i.e., LF2) from Innovative Instruments, Inc. The electrodes were stored in either a 0.05 M H2SO4 aqueous solution or a saturated KCl aqueous solution between experiments. All potentials were referenced to the leak-free Ag/AgCl electrode by measuring the ferrocene/ferricenium couple under identical conditions. The electrodes were cleaned with acetone and ultrapure water. The solvents used were MeCN, DCM, and MeTHF. The electrolytes, [(nBu)4N][B(C6F5)4] and [(nBu)4N][PF6], were further purified by recrystallization from MeCN and ethanol, respectively. The solutions (1 or 2 mM analyte, 100 mM electrolyte) were scanned anodically then cathodically within a 1.5–2 V potential range at varying scan rates. (For further details, see ESI†).Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are most thankful to the University of North Carolina at Greensboro for the financial support provided in the form of startup funds and the New Faculty Research Award. The Joint School of Nanoscience and Nanoengineering is gratefully acknowledged for providing access to the X-ray diffraction facility. We are also indebted to Professor Harry B. Gray and Dr. Maxime A. Siegler for helpful discussions.References
- (a) T. J. Kealy and P. L. Pauson, A New Type of Organo-Iron Compound, Nature, 1951, 168(4285), 1039–1040 CrossRef CAS; (b) G. Wilkinson, M. Rosenblum, M. C. Whiting and R. B. Woodward, The Structure of Iron Bis-Cyclopentadienyl, J. Am. Chem. Soc., 1952, 74(8), 2125–2126 CrossRef CAS.
- (a) A. Haaland, Molecular Structure and Bonding in the 3d Metallocenes, Acc. Chem. Res., 1979, 12(11), 415–422 CrossRef CAS; (b) S. Cantrill, An Iron-Clad Structure, Nature, 2014, 511(7509), 11–11 CrossRef; (c) J. I. Seeman and S. Cantrill, Wrong But Seminal, Nat. Chem., 2016, 8(3), 193–200 CrossRef CAS PubMed.
- (a) S. Fery-Forgues and B. Delavaux-Nicot, Ferrocene and Ferrocenyl Derivatives in Luminescent Systems, J. Photochem. Photobiol., A, 2000, 132(3), 137–159 CrossRef CAS; (b) D. R. van Staveren and N. Metzler-Nolte, Bioorganometallic Chemistry of Ferrocene, Chem. Rev., 2004, 104(12), 5931–5986 CrossRef CAS; (c) A. Togni and T. Hayashi, Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science, Wiley, 2008 Search PubMed; (d) H. Hagen, P. Marzenell, E. Jentzsch, F. Wenz, M. R. Veldwijk and A. Mokhir, Aminoferrocene-Based Prodrugs Activated by Reactive Oxygen Species, J. Med. Chem., 2012, 55(2), 924–934 CrossRef CAS PubMed; (e) D. Astruc, Why is Ferrocene so Exceptional?, Eur. J. Inorg. Chem., 2017, 2017(1), 6–29 CrossRef CAS; (f) M. Patra and G. Gasser, The Medicinal Chemistry of Ferrocene and Its Derivatives, Nat. Rev. Chem., 2017, 1(9), 0066 CrossRef CAS; (g) K. P. Fitzpatrick, C. B. Schwamb, C. T. Check, K.-P. Jang, D. N. Barsoum and K. A. Scheidt, Development of Ferrocene-Based Planar Chiral Imidazopyridinium Salts for Catalysis, Organometallics, 2020, 39(14), 2705–2712 CrossRef CAS.
- Soon after the discovery of the bis(η5-cyclopentadienyl) iron(II) complex in 1951, the term “ferrocene” was coined by Woodward and Whiting. They established the aromatic character of the cyclopentadienyl rings of ferrocene, coining its now ingrained name having the suffix “-ene” in analogy to organic aromatic compounds such as benzene, naphthalene, etc. Additionally, the prefix “ferro-” was used to specify the iron(II) oxidation state in ferrocene while the prefix “ferri-” was assigned to the one-electron oxidized counterpart with the iron(III) center and thus named the ferricenium ion. A common alternative spelling for the latter is ferricinium. Our review of the past seven decades of literature revealed that with a dramatic increase in applications of ferrocene and ferricenium ion in a variety of fields including materials, medicine and energy, the original name of the oxidized complex gradually fell out of common usage and was superseded by the name ferrocenium. The ratio of the usage of the names ferricenium/ferricinium and ferrocenium has reversed over the past several decades. Today, the oxidized ferrocene is commercially marketed as “ferrocenium” and either names are found in the current literature. In 2005, IUPAC strongly recommended the name bis(η5-cyclopentadienyl) iron(+1) to avoid any ambiguity.
- (a) R. R. Gagne, C. A. Koval and G. C. Lisensky, Ferrocene As an Internal Standard for Electrochemical Measurements, Inorg. Chem., 1980, 19(9), 2854–2855 CrossRef CAS; (b) G. Gritzner and J. Kuta, Recommendations on Reporting Electrode Potentials in Nonaqueous Solvents (Recommendations 1983), Pure Appl. Chem., 1984, 56(4), 461–466 Search PubMed; (c) J. C. Kotz, The Electrochemistry of Transition Metal Organometallic Compounds, in Topics in Organic Electrochemistry, ed. A. J. Fry and W. E. Britton, Springer, Boston, MA, 1986, pp. 81–176 Search PubMed.
- (a) M. Malischewski, M. Adelhardt, J. Sutter, K. Meyer and K. Seppelt, Isolation and Structural and Electronic Characterization of Salts of the Decamethylferrocene Dication, Science, 2016, 353(6300), 678–682 CrossRef CAS PubMed; (b) C. A. P. Goodwin, M. J. Giansiracusa, S. M. Greer, H. M. Nicholas, P. Evans, M. Vonci, S. Hill, N. F. Chilton and D. P. Mills, Isolation and Electronic Structures of Derivatized Manganocene, Ferrocene and Cobaltocene Anions, Nat. Chem., 2021, 13(3), 243–248 CrossRef CAS PubMed.
- R. B. Woodward, M. Rosenblum and M. C. Whiting, A New Aromatic System, J. Am. Chem. Soc., 1952, 74(13), 3458–3459 CrossRef CAS.
- (a) R. Prins and F. J. Reinders, Electron Spin Resonance of the Cation of Ferrocene, J. Am. Chem. Soc., 1969, 91(17), 4929–4931 CrossRef CAS; (b) R. Prins, Electronic Structure of the Ferricenium Cation, Mol. Phys., 1970, 19(5), 603–620 CrossRef CAS; (c) Y. S. Sohn, D. N. Hendrickson and H. B. Gray, Electronic Structure of Ferricenium Ion, J. Am. Chem. Soc., 1970, 92(10), 3233–3234 CrossRef CAS; (d) M. D. Rowe and A. J. McCaffery, Electronic Structure of Ferricenium Ion from Absorption, MCD, and ESR Studies, J. Chem. Phys., 1973, 59(7), 3786–3794 CrossRef CAS; (e) P. S. Bagus, U. I. Walgren and J. Almlof, A Theoretical Study of the Electronic Structure of Ferrocene and Ferricinium: Application to Mössbauer Isomer Shifts, Ionization Potentials, and Conformation, J. Chem. Phys., 1976, 64(6), 2324–2334 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chemical Redox Agents for Organometallic Chemistry, Chem. Rev., 1996, 96(2), 877–910 CrossRef CAS PubMed.
- (a) M. Sato, T. Yamada and A. Nishimura, Electrolytic Oxidation of Ferrocene In the Presence of Trace Amounts of Dissolved Oxygen, Chem. Lett., 1980, 9(8), 925–926 CrossRef; (b) R. Prins, A. R. Korswagen and A. G. T. G. Kortbeek, Decomposition of the Ferricenium Cation by Nucleophilic Reagents, J. Organomet. Chem., 1972, 39(2), 335–344 CrossRef CAS; (c) G. Zotti, G. Schiavon, S. Zecchin and D. Favretto, Dioxygen-Decomposition of Ferrocenium Molecules in Acetonitrile: The Nature of the Electrode-Fouling Films During Ferrocene Electrochemistry, J. Electroanal. Chem., 1998, 456(1), 217–221 CrossRef CAS; (d) A. Singh, D. R. Chowdhury and A. Paul, A Kinetic Study of Ferrocenium Cation Decomposition Utilizing an Integrated Electrochemical Methodology Composed of Cyclic Voltammetry and Amperometry, Analyst, 2014, 139(22), 5747–5754 RSC.
- H. Choi, J. W. Hershberger, A. R. Pinhas and D. M. Ho, Isomerization of a Vinylcyclobutene to a Cyclohexadiene: A Nickel(I) Promoted Rearrangement, Organometallics, 1991, 10(8), 2930–2936 CrossRef CAS.
- Today, ferrocene is readily available from more than 45 different commercial sources and it is as cheap as $0.11 g−1, so very few people choose to synthesize it in research laboratories. Ferricenium ion, however, is only available as the PF6− or BF4− salts through less than half the number of commercial suppliers and costs about 100 times more than the parent ferrocene complex.
- H. B. Gray, D. N. Hendrickson and Y. S. Sohn, Magnetic Susceptibility Study of Various Ferricenium and Iron(III) Dicarbollide Compounds, Inorg. Chem., 1971, 10(8), 1559–1563 CrossRef CAS.
- Notably, a quick survey of the literature on the salts of ferricenium and its derivatives reveals that despite their wide application, especially those with weakly coordinating anions such as hexafluoroantimonate or fluoroarylborates analogs in recent reports, to date, information on the synthesis and characterization of these complexes is sparse and the reports lack the relevant experimental details.
- (a) I. Krossing and I. Raabe, Noncoordinating Anions—Fact or Fiction? A Survey of Likely Candidates, Angew. Chem., Int. Ed., 2004, 43(16), 2066–2090 CrossRef CAS PubMed; (b) K. E. Aldrich, B. S. Billow, D. Holmes, R. D. Bemowski and A. L. Odom, Weakly Coordinating yet Ion Paired: Anion Effects on an Internal Rearrangement, Organometallics, 2017, 36(7), 1227–1237 CrossRef CAS.
- (a) K. Mase, K. Ohkubo and S. Fukuzumi, Efficient Two-Electron Reduction of Dioxygen to Hydrogen Peroxide with One-Electron Reductants with a Small Overpotential Catalyzed by a Cobalt Chlorin Complex, J. Am. Chem. Soc., 2013, 135(7), 2800–2808 CrossRef CAS PubMed; (b) M. A. Ehudin, L. B. Gee, S. Sabuncu, A. Braun, P. Moënne-Loccoz, B. Hedman, K. O. Hodgson, E. I. Solomon and K. D. Karlin, Tuning the Geometric and Electronic Structure of Synthetic High-Valent Heme Iron(IV)-Oxo Models in the Presence of a Lewis Acid and Various Axial Ligands, J. Am. Chem. Soc., 2019, 141(14), 5942–5960 CrossRef CAS PubMed; (c) T. J. Zerk, C. T. Saouma, J. M. Mayer and W. B. Tolman, Low Reorganization Energy for Electron Self-Exchange by a Formally Copper(III,II) Redox Couple, Inorg. Chem., 2019, 58(20), 14151–14158 CrossRef CAS PubMed; (d) V. M. Krishnan, D. Y. Shopov, C. J. Bouchey, W. D. Bailey, R. Parveen, B. Vlaisavljevich and W. B. Tolman, Structural Characterization of the [CuOR]2+Core, J. Am. Chem. Soc., 2021, 143(9), 3295–3299 CrossRef CAS PubMed.
- (a) A. R. O'Connor, C. Nataro, J. A. Golen and A. L. Rheingold, Synthesis and Reactivity of [N(C6H4Br)3 B(C6F5)4]: the X-ray Crystal Structure of [Fe(C5H5)2] [B(C6F5)4], J. Organomet. Chem., 2004, 689(14), 2411–2414 CrossRef; (b) A. L. Rheingold and C. Nataro, CSD Private Communication (CCDC 1441545), 2015.
- N. M. Mews, A. Berkefeld, G. Hörner and H. Schubert, Controlling Near-Infrared Chromophore Electronic Properties through Metal–Ligand Orbital Alignment, J. Am. Chem. Soc., 2017, 139(7), 2808–2815 CrossRef CAS PubMed.
- It is also worth mentioning that although ferricenium salts have been the subject of a variety of spectroscopic, electrochemical, and structural investigations over the past seven decades, these efforts have been mainly focused on the electron-rich ferricenium species and the very limited data available on the electron-deficient systems are sometimes ambiguous. This represents a surprising knowledge gap in the literature and the current study attempts to address some of that gap through a systematic and thorough evaluation of a series of highly organic soluble ferricenium derivatives.
- Use of other very strong oxidizing agents such as nitrosonium, [NO]+, salts or triarylaminium radical cations such as Magic Blue resulted in unwanted side reactions and will be discussed elsewhere.
- Y. Zhang, A. M. Santos, E. Herdtweck, J. Mink and F. E. Kühn, Organonitrile Ligated Silver Complexes with Perfluorinated Weakly Coordinating Anions and Their Catalytic Application for Coupling Reactions, New J. Chem., 2005, 29(2), 366–370 RSC.
- (a) J. C. Evans and G. Y. S. Lo, Raman and Infrared Studies of Acetonitrile Complexed with Zinc Chloride, Spectrochim. Acta, 1965, 21(6), 1033–1038 CrossRef CAS; (b) K. F. Purcell and R. S. Drago, Studies of the Bonding in Acetonitrile Adducts1, J. Am. Chem. Soc., 1966, 88(5), 919–924 CrossRef CAS.
- L. Song and W. C. Trogler, [(CO)3(PPh3)2OsAg(O2CCF3)]: A Model for an Intermediate on the Reaction Coordinate in Electron Transfer, Angew. Chem., Int. Ed., 1992, 31(6), 770–772 CrossRef.
- (a) P. Carty and M. F. A. Dove, The Reaction of Some Ferrocenyl Ketones with Anhydrous Silver Tetrafluoroborate, a New Route to Substituted Ferricenium salts, J. Organomet. Chem., 1971, 28(1), 125–132 CrossRef CAS; (b) C. Bittner, D. Bockfeld and M. Tamm, Formation of Alkyne-Bridged Ferrocenophanes Using Ring-Closing Alkyne Metathesis on 1,1′-Diacetylenic Ferrocenes, Beilstein J. Org. Chem., 2019, 15, 2534–2543 CrossRef CAS PubMed.
- E. S. Yang, M.-S. Chan and A. C. Wahl, Rate of Electron Exchange Between Ferrocene and Ferricenium Ion from Nuclear Magnetic Resonance Studies, J. Phys. Chem., 1975, 79(19), 2049–2052 CrossRef CAS.
- (a) H. B. Gray, Y. S. Sohn and N. Hendrickson, Electronic Structure of Metallocenes, J. Am. Chem. Soc., 1971, 93(15), 3603–3612 CrossRef; (b) S. E. Anderson and R. Rai, Mechanism of Spin Delocalization and Nature of the Ground state in Ferricenium Cations, Chem. Phys., 1973, 2(2), 216–225 CrossRef CAS; (c) D. M. Duggan and D. N. Hendrickson, Electronic Structure of Various Ferricenium Systems as Inferred from Raman, Infrared, Low-Temperature Electronic Absorption, and Electron Paramagnetic Resonance Measurements, Inorg. Chem., 1975, 14(5), 955–970 CrossRef CAS.
- (a) A. F. Reid, D. E. Scaife and P. C. Wailes, The Characterisation of Solid Cyclopentadienyl Metal Compounds by Their Near Infrared Reflection Spectra, Spectrochim. Acta, 1964, 20(8), 1257–1268 CrossRef CAS; (b) F. A. Cotton and T. J. Marks, Infrared Study of the Structures of Cyclopentadienyl Compounds of Copper(I) and Mercury(II), J. Am. Chem. Soc., 1969, 91(26), 7281–7285 CrossRef CAS.
- W. Ding, C. T. Sanderson, R. C. Conover, M. K. Johnson, I. J. Amster and C. Kutal, Characterization of the Low-Energy Electronic Excited States of Benzoyl-Substituted Ferrocenes, Inorg. Chem., 2003, 42(5), 1532–1537 CrossRef CAS PubMed.
- (a) A. S. Wexler, Integrated Intensities of Absorption Bands in Infrared Spectroscopy, Appl. Spectrosc. Rev., 1967, 1(1), 29–98 CrossRef CAS; (b) G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, John Wiley & Sons, Ltd., 3rd ed., 2001 Search PubMed.
- M. Zeller, M. W. Lufaso, L. Curtin and A. D. Hunter, CSD Private Communication (CCDC 252665). 2004, CSD Private Communication (CCDC 252665).
- A. M. Makal, D. Plażuk, J. Zakrzewski, B. Misterkiewicz and K. Woźniak, Experimental Charge Density Analysis of Symmetrically Substituted Ferrocene Derivatives, Inorg. Chem., 2010, 49(9), 4046–4059 CrossRef CAS PubMed.
- I. R. Butler, W. R. Cullen, S. J. Rettig and J. Trotter, Structure of Benzoylferrocene, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44(9), 1666–1667 CrossRef.
- J. F. Gallagher, G. Ferguson, S. Z. Ahmed, C. Glidewell and A. Lewis, Accurate Redeterminations of 1,1′-Dibenzoylferrocene and (4-Nitrophenyl)ferrocene, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53(12), 1772–1775 CrossRef.
- (a) J. Bluemel, N. Hebendanz, P. Hudeczek, F. H. Koehler and W. Strauss, Synthesis and NMR Spectroscopy of Metallocenium Ions. Support for a New Ferromagnetic Coupling Mechanism in Decamethylmetallocenium Tetracyanoethenides, J. Am. Chem. Soc., 1992, 114(11), 4223–4230 CrossRef CAS; (b) T. Helgaker, M. Jaszuński and K. Ruud, Ab Initio Methods for the Calculation of NMR Shielding and Indirect Spin−Spin Coupling Constants, Chem. Rev., 1999, 99(1), 293–352 CrossRef CAS PubMed.
- (a) D. W. Slocum, P. S. Shenkin, T. R. Engelmann and C. R. Ernst, Fundamental Studies of Substituted Ferrocene Systems. I. Electronic Effects of Electron Donating Groups on the Ferrocene System, Tetrahedron Lett., 1971, 12(46), 4429–4432 CrossRef; (b) D. W. Slocum and C. R. Ernst, Electronic Effects in Metallocenes and Certain Related Systems, in Adv. Organomet. Chem, ed. F. G. A. Stone and R. West, Academic Press, 1972, vol. 10, pp. 79–114 Search PubMed; (c) T. E. Pickett and C. J. Richards, Assignment of 1H-NMR Chemical Shifts in 1,2- and 1,1′-disubstituted ferrocenes, Tetrahedron Lett., 1999, 40(28), 5251–5254 CrossRef CAS.
- R. J. LeSuer and W. E. Geiger, Improved Electrochemistry in Low-Polarity Media Using Tetrakis(pentafluorophenyl)borate Salts as Supporting Electrolytes, Angew. Chem., Int. Ed., 2000, 39(1), 248–250 CrossRef CAS PubMed.
- H.-W. Lerner, F. Schödel, M. Bru Roig and M. Bolte, Tetra-n-butyl-ammonium Tetra-kis(penta-fluoro-phen-yl)borate, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62(1), o372–o373 CrossRef.
- E. H. Kim, H. M. Lee, M. S. Jeong, J. Y. Ryu, J. Lee and B. Y. Lee, Methylaluminoxane-Free Chromium Catalytic System for Ethylene Tetramerization, ACS Omega, 2017, 2(3), 765–773 CrossRef CAS PubMed.
- A. W. Kaspi-Kaneti and I. Tuvi-Arad, Twisted and Bent Out of Shape: Symmetry and Chirality Analysis of Substituted Ferrocenes, Organometallics, 2018, 37(19), 3314–3321 CrossRef CAS.
- (a) K. Wedeking, Z. Mu, G. Kehr, R. Fröhlich, G. Erker, L. Chi and H. Fuchs, Tetradecylferrocene: Ordered Molecular Array of an Organometallic Amphiphile in the Crystal and in a Two-dimensional Assembled Structure on a Surface, Langmuir, 2006, 22(7), 3161–3165 CrossRef CAS PubMed; (b) K. Wedeking, Z. Mu, G. Kehr, J. Cano Sierra, C. Mück Lichtenfeld, S. Grimme, G. Erker, R. Fröhlich, L. Chi, W. Wang, D. Zhong and H. Fuchs, Oligoethylene Chains Terminated by Ferrocenyl End Groups: Synthesis, Structural Properties, and Two-Dimensional Self-Assembly on Surfaces, Chem. – Eur. J., 2006, 12(6), 1618–1628 CrossRef CAS PubMed.
- A. S. Romanov, J. M. Mulroy, V. N. Khrustalev, M. Y. Antipin and T. V. Timofeeva, Monohalogenated Ferrocenes C5H5FeC5H4X (X=Cl, Br and I) and a Second Polymorph of C5H5FeC5H4I, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65(11), m426–m430 CrossRef CAS PubMed.
- C. A. Hnetinka, A. D. Hunter, M. Zeller and M. J. G. Lesley, 1,1′-Di-bromo-ferrocene, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60(12), m1806–m1807 CrossRef CAS.
- C. A. Morrison, S. F. Bone, D. W. H. Rankin, H. E. Robertson, S. Parsons, R. A. Coxall, S. Fraser, J. A. S. Howell, P. C. Yates and N. Fey, Conformational Properties of Substituted Ferrocenes: Experimental and Theoretical Studies of the Molecular Structures of 1,1′-Di-tert-butylferrocene and Isopropylferrocene, Organometallics, 2001, 20(11), 2309–2320 CrossRef CAS.
- R. J. LeSuer, C. Buttolph and W. E. Geiger, Comparison of the Conductivity Properties of the Tetrabutylammonium Salt of Tetrakis(pentafluorophenyl)borate Anion with Those of Traditional Supporting Electrolyte Anions in Nonaqueous Solvents, Anal. Chem., 2004, 76(21), 6395–6401 CrossRef CAS PubMed.
- K. M. Kadish, J. Q. Ding and T. Malinski, Resistance of Nonaqueous Solvent Systems Containing Tetraalkylammonium Salts. Evaluation of Heterogeneous Electron Transfer Rate Constants for The Ferrocene/Ferrocenium Couple, Anal. Chem., 1984, 56(9), 1741–1744 CrossRef CAS PubMed.
- (a) H. H. Jaffé, A Reëxamination of the Hammett Equation, Chem. Rev., 1953, 53(2), 191–261 CrossRef; (b) C. Hansch, A. Leo and R. W. Taft, A Survey of Hammett Substituent Constants and Resonance and Field Parameters, Chem. Rev., 1991, 91(2), 165–195 CrossRef CAS.
- S. D. Waniek, J. Klett, C. Förster and K. Heinze, Polysubstituted Ferrocenes as Tunable Redox Mediators, Beilstein J. Org. Chem., 2018, 14, 1004–1015 CrossRef CAS PubMed.
- (a) F. Barrière and W. E. Geiger, Use of Weakly Coordinating Anions to Develop an Integrated Approach to the Tuning of ΔE1/2 Values by Medium Effects, J. Am. Chem. Soc., 2006, 128(12), 3980–3989 CrossRef PubMed; (b) W. E. Geiger and F. Barrière, Organometallic Electrochemistry Based on Electrolytes Containing Weakly-Coordinating Fluoroarylborate Anions, Acc. Chem. Res., 2010, 43(7), 1030–1039 CrossRef CAS PubMed.
- D. F. Aycock, Solvent Applications of 2-Methyltetrahydrofuran in Organometallic and Biphasic Reactions, Org. Process Res. Dev., 2007, 11(1), 156–159 CrossRef CAS.
- V. Gutmann, Solvent Effects on the Reactivities of Organometallic Compounds, Coord. Chem. Rev., 1976, 18(2), 225–255 CrossRef CAS.
- O. Ivanciuc, T. Ivanciuc, P. A. Filip and D. Cabrol-Bass, Estimation of the Liquid Viscosity of Organic Compounds with a Quantitative Structure−Property Model, J. Chem. Inf. Comput. Sci., 1999, 39(3), 515–524 CrossRef CAS.
- F. Comelli, R. Francesconi, A. Bigi and K. Rubini, Molar Heat Capacities, Densities, Viscosities, and Refractive Indices of Dimethyl Sulfoxide+Tetrahydropyran and+2-Methyltetrahydrofuran at (293.15, 303.15, and 313.15) K, J. Chem. Eng. Data, 2007, 52(2), 639–644 CrossRef CAS.
- I. Noviandri, K. N. Brown, D. S. Fleming, P. T. Gulyas, P. A. Lay, A. F. Masters and L. Phillips, The Decamethylferrocenium/Decamethylferrocene Redox Couple: A Superior Redox Standard to the Ferrocenium/Ferrocene Redox Couple for Studying Solvent Effects on the Thermodynamics of Electron Transfer, J. Phys. Chem. B, 1999, 103(32), 6713–6722 CrossRef CAS.
- D. O. Wipf, E. W. Kristensen, M. R. Deakin and R. M. Wightman, Fast-Scan Cyclic Voltammetry as a Method to Measure Rapid Heterogeneous Electron-Transfer Kinetics, Anal. Chem., 1988, 60(4), 306–310 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, A Practical Beginner's Guide to Cyclic Voltammetry, J. Chem. Educ., 2018, 95(2), 197–206 CrossRef CAS.
- Y. Wang, E. I. Rogers and R. G. Compton, The Measurement of the Diffusion Coefficients of Ferrocene and Ferrocenium and their Temperature Dependence in Acetonitrile Using Double Potential Step Microdisk Electrode Chronoamperometry, J. Electroanal. Chem., 2010, 648(1), 15–19 CrossRef CAS.
- (a) D. P. Valencia and F. J. González, Estimation of Diffusion Coefficients by Using a Linear Correlation Between the Diffusion Coefficient and Molecular Weight, J. Electroanal. Chem., 2012, 681, 121–126 CrossRef CAS; (b) A. Paul, R. Borrelli, H. Bouyanfif, S. Gottis and F. Sauvage, Tunable Redox Potential, Optical Properties, and Enhanced Stability of Modified Ferrocene-Based Complexes, ACS Omega, 2019, 4(12), 14780–14789 CrossRef CAS.
- Y. Hui and R. D. Webster, Absorption of Water into Organic Solvents Used for Electrochemistry under Conventional Operating Conditions, Anal. Chem., 2011, 83(3), 976–981 CrossRef CAS PubMed.
- (a) H. L. Friedman and R. Mills, Hydrodynamic Approximation for Distinct Diffusion Coefficients, J. Solution Chem., 1986, 15(1), 69–80 CrossRef CAS; (b) L. Costigliola, D. M. Heyes, T. B. Schrøder and J. C. Dyre, Revisiting the Stokes-Einstein Relation without a Hydrodynamic Diameter, J. Chem. Phys., 2019, 150(2), 021101 CrossRef PubMed; (c) R. Evans, Z. Deng, A. K. Rogerson, A. S. McLachlan, J. J. Richards, M. Nilsson and G. A. Morris, Quantitative Interpretation of Diffusion-Ordered NMR Spectra: Can We Rationalize Small Molecule Diffusion Coefficients?, Angew. Chem., Int. Ed., 2013, 52(11), 3199–3202 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXL-2018: Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 2018 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details concerning spectroscopy including IR and NMR as well as electrochemical data and crystallographic details. CCDC 2045745–2045755. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt01192h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |