
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMixed H2O/H2 plasma-induced redox reactions of thin uranium oxide films under UHV conditions†
Ghada
El Jamal
 *a,
Thomas
Gouder
b,
Rachel
Eloirdi
b,
Evgenia
Tereshina-Chitrova
cd,
Lukáš
Horákd
d and
Mats
Jonsson
a
*a,
Thomas
Gouder
b,
Rachel
Eloirdi
b,
Evgenia
Tereshina-Chitrova
cd,
Lukáš
Horákd
d and
Mats
Jonsson
a
aSchool of Engineering Sciences in Chemistry, Biotechnology and Health (CBH), Department of Chemistry, Applied Physical Chemistry, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: ghadaej@kth.se
bEuropean Commission, Joint Research Centre, Postfach 2340, DE-76215 Karlsruhe, Germany
cInstitute of Physics, ASCR, Prague, Czech Republic
dFaculty of Mathematics and Physics, Charles University, 12116 Prague, Czech Republic
First published on 1st July 2021
Abstract
X-ray photoelectron spectroscopy (XPS) has been used to study the effect of mixed H2O/H2 gas plasma on the surfaces of UO2, U2O5 and UO3 thin films at 400 °C. The experiments were performed in situ under ultra-high vacuum conditions. Deconvolution of the U4f7/2 peaks into U(IV), U(V) and U(VI) components revealed the surface composition of the films after 10 min plasma exposure as a function of H2 concentration in the feed gas of the plasma. Some selected films (unexposed and exposed) were also analysed using grazing-incidence X-ray diffraction (GIXRD). The XPS results show that U(V) is formed as a major product upon 10 minutes exposure of UO3 by a mixed H2O/H2 plasma in a fairly wide H2 concentration range. When starting with U(V) (U2O5), rather high H2 concentrations are needed to reduce U(V) to U(IV) in 10 minutes. In the plasma induced oxidation of UO2, U(V) is never observed as a major product after 10 minutes and it would seem that once U(V) is formed in the oxidation of UO2 it is rapidly oxidized further to U(VI). The grazing incidence X-ray diffraction analysis shows that there is a considerable impact of the plasma and heating conditions on the crystal structure of the films in line with the change of the oxidation state. This structural difference is proposed to be the main kinetic barrier for plasma induced transfer between U(IV) and U(V) in both directions.
Introduction
Many countries using nuclear power are considering deep geological repositories as the solution for the storage of highly radioactive used nuclear fuel. Most geological repository concepts are based on a mixture of natural and engineered barriers1,2 to prevent intrusion of groundwater into the canisters containing the nuclear fuel and migration of radionuclides out of the canisters.3 The used fuel is mainly UO2 (95%) and 5% of heavier actinides and fission products.4 Noble metals like Mo, Ru, Pd, Tc, and Rh are among the fission products which form metallic alloy particles commonly known as ε-particles.5 Although groundwaters at many potential repository sites are reducing and UO2 has very low solubility under reducing conditions, the inherent radioactivity of the used fuel can also drive its dissolution. Ionizing radiation emitted by the fuel and absorbed by the adjacent groundwater (in the event of canister failure) will induce water radiolysis producing oxidizing (H2O2, ˙OH, HO2˙) and reducing species (H2, H˙, eaq−).6,7 Since the fuel matrix is mainly in its reduced form (U(IV)), the radiolytic oxidants will initially dominate the chemistry at the fuel surface and drive the oxidative dissolution process where the fission products and actinides will subsequently be released.8 In the process of anaerobic corrosion of iron in the canister, large quantities of H2 will be produced.9,10 H2 has been shown to efficiently inhibit the radiation driven oxidative dissolution of used nuclear fuel,11–14 an effect that can mainly be attributed to ε-particle catalysed reduction of oxidized uranium in the fuel matrix.15,16 The reduction competes with the dissolution of oxidized uranium but becomes dominating at fairly low H2 concentrations. The other inhibiting effect of H215,17–26 is the impact it has on water radiolysis. H2 reacts with hydroxyl radicals producing H2O and hydrogen atoms. This reduces the yield of oxidants and increases the yield of reductants and thereby reduces the rate of oxidative dissolution.As dissolution and precipitation of secondary phases occur in parallel with the redox processes, it is difficult to draw reliable mechanistic conclusions for the individual processes from experiments performed in solution. To circumvent this problem, we have recently developed an approach to simulate the radiolysis by a vacuum ECR (electron cyclotron resonance) water plasma (the products are to a large extent the same as the aqueous radiolysis products) to which we expose uranium oxide model films. In situ X-ray photoelectron spectroscopy (XPS) analysis was used to monitor the evolution of the freshly reacted surfaces, without interference of laboratory atmosphere, dissolution or precipitation reactions. Our previous work using this methodology has shown that a pure water plasma oxidizes UO2 and U2O5 (U(V)) to UO3 (U(VI)). In a series of exposures of UO2 films to the water plasma for different times we could evaluate the dynamics of the process. The results showed that UO2 is initially oxidized to U(V) and U(VI) and the latter becomes the major product after 10–15 minutes exposure. For longer exposures, U(VI) is slowly reduced back to U(V). These findings were discussed in detail and a simple kinetic model was developed to describe the dynamics of the process.27 While we have previously only used this methodology for exposures to pure water plasma,28 we have extended in the present study the experimental conditions by using a mixed H2O/H2 plasma.
The use of a mixed H2O/H2 plasma does not simply mimic the impact of H2 on water radiolysis since, in water radiolysis, the radiation energy is completely absorbed by water and H2 only reacts with the aqueous radiolysis products. In the mixed plasma, the plasma is generated both from H2O and H2 and the products may react with each other inside the plasma generator, while outside the plasma generator the pressure is too low to allow bimolecular processes before the plasma products reach the oxide surface. The use of a mixed H2O/H2 plasma enables fine-tuning of the redox properties of the plasma, which opens up the possibility for more detailed studies of the redox chemistry of uranium oxide films under UHV conditions.
In this work, we have exposed UO2, U2O5 and UO3 films to mixed H2O/H2 plasmas and analysed the films after 10 min plasma exposure using XPS complemented by grazing-incidence x-ray diffraction (GIXRD). The results are discussed in terms of intrinsic kinetic barriers for the transfer between different oxidation states.
Experimental
Sample preparation
Uranium oxide UO2.0 films were prepared in situ using optimized conditions of direct current (DC) sputtering from a uranium metal target in a gas mixture of Ar (6N) and O2 (5N). The Ar pressure used was 5 × 10−3 mbar and the O2 pressure ranged between 10−6 mbar and 2 × 10−6 mbar. The uranium target voltage was fixed at −700 V. The films of 20 nm thickness were deposited at 400 °C on polycrystalline Au substrates, cleaned by annealing to 200 °C for 10 min. The deposition time was 15 min. The plasma in the diode source was maintained by injecting electrons of 25–50 eV energy (triode setup), allowing working at a low Ar pressure in the absence of stabilizing magnetic fields. UO3 films were prepared by exposing the UO2 films to the oxygen plasma produced by the electron cyclotron resonance (ECR) source.ECR plasma source
In the ECR source, the excitation of electrons is based on stochastic heating by microwave radiation in magnetic fields produced by permanent magnets. The electrons gain sufficient energy to ionize the gas and sustain the plasma when the resonance condition between the electrons and the microwave electric field is fulfilled. A reactive plasma environment is formed of excited species, free radicals, and ions. The temperature of samples during exposure was maintained at 400 °C using a pyrolytic boron nitride (PBN) heater installed below the sample holder. The water vapour used as a feed gas was in some cases mixed with H2 gas. Gas pressures were kept constant by a flowmeter setup to ensure stable plasma conditions.More details about the ECR source and different gas plasma characterization methods with a RGA-Mass spectrometer are reported in a previous study.29
Surface characterisation
High-resolution X-ray photoelectron spectroscopy (XPS) was used to analyse the chemical surface composition of the films before and after each treatment. The spectra were obtained using a Specs Phoibos 150 hemispherical analyser, using Al-Kα (1486.6 eV) radiation, produced by an XRC-1000 μ-focus source, equipped with a monochromator and operating at 120 W. The background pressure in the analysis chamber was 2 × 10−10 mbar. The Au4f7/2 line of the Au metal (83.9 eV BE) and the Cu-2p3/2 line of the Cu metal (932.7 eV BE) were used for calibration of binding energies. Photoemission spectra were taken at room temperature. The spectra obtained at high resolution were analysed by deconvolution into simple components. The U4f7/2 components were fitted to Gaussian/Lorentzian functions. In all cases, a standard Shirley baseline was employed to separate the real signal from the background. The deconvolutions have been carried out by using XPS peak41 software.Grazing incidence X-ray diffraction
The grazing incidence X-ray diffraction (GIXRD) measurements were carried out on a Rigaku SmartLab diffractometer using a 9 kW copper rotating anode X-ray source (Cu-Kα radiation λ = 0.15418 nm). It was equipped with a parabolic multilayer mirror in the primary beam, a set of axial divergence eliminating Soller slits in both the incident and diffracted beam (acceptance 5°) and a parallel beam Soller slit collimator (acceptance 0.5°). A HighPix-3000 2D hybrid pixel single photon-counting detector was located in the diffracted beam. The constant incidence angle of the primary beam ω = 3.0° was used for the measurement. Origin 2019b was used to retrace the figures. ICSD database was used to assign the different phases present in the films.Results and discussion
Before discussing the impact of the mixed H2O/H2-plasmas on uranium oxide films, it is important to understand the impact of pure plasmas. In two recent papers, it was shown that a pure H2O plasma oxidizes both UO2 and U2O5 to UO3 in 10 min at a sample temperature of 400 °C. For longer exposures of UO2 to a pure H2O plasma it was shown that the UO3 formed initially and was slowly reduced back to U2O5. It has also recently been shown that UO3 exposed to a pure H2 plasma at a sample temperature of 400 °C for 10 min was quantitatively converted to UO2.30 Exposing U2O5 to a pure H2 plasma under the same conditions also leads to the formation of UO2 while exposing UO2 to a pure H2 plasma leaves the oxide film unchanged.In this paper, we perform all plasma exposures for 10 min and at a sample temperature of 400 °C. Based on that found in the previous studies, the plasma-induced reactions are well advanced but not necessarily complete after 10 min. Nevertheless, the use of a common exposure time for all oxides and plasma compositions allows direct comparison of the results.
Properties of mixed H2O/H2 plasma
Before discussing the impact of the mixed H2O/H2 plasma on different uranium oxides, it is necessary to address the properties of the mixed plasma. Hydrogen is introduced into the plasma chamber together with water and both gases are subjected to the ECR plasma. Fig. 1 shows the evolution of three crucial gas species, O2, H2O and H2, as the plasma is switched on and the hydrogen partial pressure is gradually increased. The RGA-Mass spectrometer is placed outside the plasma generator and only detects species escaping the plasma generator. As the pressure is very low, the probability for bimolecular reactions between plasma constituents outside the plasma generator is very low. Consequently, the detected products will reflect chemistry inside the plasma generator. | ||
| Fig. 1 Pressures of molecular products in a mixed H2O/H2 plasma in a system where the H2 pressure in the plasma generator is first stepwise increased and thereafter stepwise decreased. | ||
Before the plasma is switched on and while the plasma chamber is loaded with pure H2O, only water is detected. When the plasma is switched on the water signal decreases and H2 and O2 are also detected. These two products originate from the recombination of H atoms and O atoms, respectively. Both H atoms and O atoms are produced in a water plasma (in addition to hydroxyl radicals). After 200 s, H2 is added to the plasma chamber with a relatively low partial pressure(I). The immediate response of the system is a slight decrease in the O2 pressure, a slight increase in the H2 pressure and a significant increase in the H2O pressure. After 250 s the partial pressure of H2 is increased for the second time (II). The immediate response is a further decrease in the O2 pressure, an increase in the H2 pressure and a rapid initial increase in the H2O pressure followed by an even larger drop in the pressure. This pattern is reproduced, although with a smaller initial increase and the following decrease in the H2O pressure for the following three incremental steps in the H2 pressure (III, IV, and V). The transient behaviour of H2O could partly be due to a slow response of the system when the H2 pressure is increased. After the third incremental increase in the H2 pressure, the O2 pressure is insignificant. The H2 supply is thereafter reduced in three steps (VI, VII, and VIII) and the system gradually recovers from the impact of H2 in the plasma generator. The decrease in the O2 pressure with increasing H2 pressure is partly due to the direct reaction between the O atom and H2 producing water, and the reaction between the O atom and the H atom producing hydroxyl radical. The gradual increase in the H2 pressure will also have an impact on the energy deposition in the plasma, and as the H2 pressure increases the primary production of the water plasma products will decrease. The consequence of this is that the oxidant concentration decreases both in absolute and relative terms with increasing H2 pressure. When increasing the H2 fraction in the plasma chamber of a mixed H2O/H2 plasma, the plasma changes from containing both oxidants and reductants to a purely reducing plasma.
UO2
A number of UO2 films prepared by sputter deposition were exposed to mixed H2O/H2 gas plasmas under the standard conditions (10 min exposure at 400 °C) and characterized by means of XPS. The U4f, O1s, and valence band regions of the samples before and after exposure were mutually compared. The resulting U4f spectra are shown in Fig. 2. At 0, 3, and 10% H2, the core level peaks shift towards higher binding energy as compared to the reference lines of the precursor UO2. For the pure water plasma reaction (0% H2), this was attributed previously28 to a chemical shift showing the oxidation of UO2 (U(IV)) into UO3 (U(VI)). A quite similar effect is observed for 3 and 10% H2. However, at 20% H2, the core level peak is similar to that of UO2. A more detailed view reveals that it is broadened, developing intensity at low binding energy (BE), and positioned between the peaks of UO3 and UO2. This gives a first hint of the formation of an intermediate oxidation state (U(V)).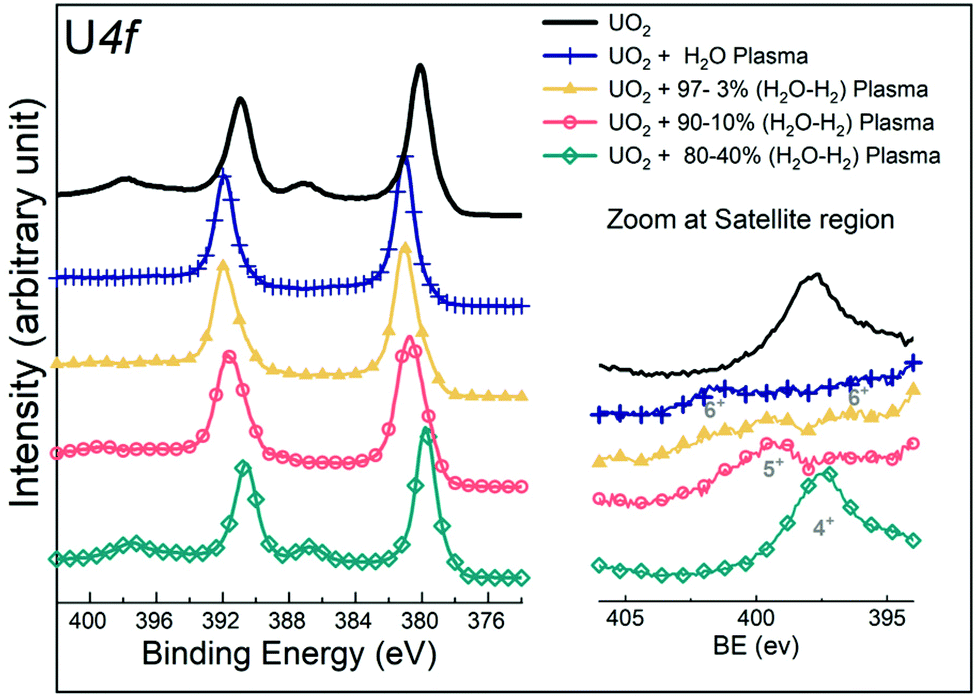 | ||
| Fig. 2 U4f core level X-ray photoemission spectra of uranium recorded for the precursor UO2 film and for the UO2 films exposed to pure water plasma and a mixed gas plasma of water and hydrogen. | ||
Besides the main lines, a convenient indicator of the oxidation state is the satellites on the high binding energy side. Although their intensities are much lower compared to the main lines, they can much better resolve individual species. In Fig. 2 (right), when the UO2 film is exposed to a pure water plasma, the typical satellite peak of U(VI) appears. With increasing H2 concentration up to 10% (hereafter referred to as 10% H2 in the H2O/H2 mixed plasma) in the mixed gas plasma, it is gradually replaced by the U(V) satellite peak. At 20% H2 in a mixed plasma, the satellite peak of U(IV) is finally formed. These data consistently show a decreasing oxidation power of the water plasma with increasing H2 content.
The valence band spectra and O1s peaks of the plasma-exposed UO2 films are plotted in Fig. 3. Regardless of the H2 concentration, the intensity of the U5f peak originally present at 1.2 eV below the Fermi level decreases after the exposure. We have previously concluded that the loss of the U5f intensity is due to the occupancy of the 5f states decreasing with uranium oxidation (nominally 2 for U(IV), 1 for U(V) and 0 for U(VI)).30 The intensity loss is strongest for the pure water plasma exposure, pointing to strong oxidation (close to U(VI)). Even when the H2 concentration in the plasma feed gas is as low as 3%, the U5f intensity is much higher compared to the peak observed for the pure water plasma exposure. At 20% H2, the intensity is almost the same as for the unexposed film. This shows that the oxidizing effect of the water plasma is suppressed by hydrogen, up to the point that practically no oxidation takes place at 20% H2. In Fig. 3c the U5f bands are normalized to the same height and shifted to the same lower binding energy side for easier comparison. The spectra can be divided into two groups. One group contains broad peaks and is observed for the untreated and 20% H2 in the H2O/H2 mixed plasma films. The other group has narrow peaks and is observed for all other plasma processed films. The different peak widths are attributed to the different 5f counts, leading to different final state multiplets: the broad shape corresponds to the 5f2 ground state of U(IV) while the narrow peak corresponds to the 5f1 ground state of U(V). This shows that for the 0, 3, and 10% H2 treated films, UO2 is oxidized into a mixture of U(V) (giving 5f1) and U(VI) (giving no peaks at all because of the 5f0 configuration). The result is a pure 5f1 signal. For 20% H2 in a mixed plasma almost no oxidation takes place and the pure 5f2 is observed, just as for the untreated film. All the plasma treated films have a featureless and broad O2p band (dominating the valence band spectra in the energy range 3–7 eV), except the one exposed to the 20% H2 in a mixed plasma, which exhibits the same two-peak shape and low intensity as UO2 (observed for the untreated films).
 | ||
| Fig. 3 X-ray photoemission spectra of the valence band (a) and O1s core level line (b) recorded for the precursor and plasma treated films. Panel (c) captures the details of the 5f states normalized and shifted in energy for comparison as described in the text. Films exposed to pure water plasma and a mixed gas plasma of water and hydrogen. | ||
The observed evolution of the U oxidation state is corroborated by the O1s spectra for different treatment conditions (Fig. 3b). The O1s signal of the original UO2 film lies at 530.1 eV. When the film is exposed to a pure H2 plasma under the conditions used in this work, the resulting XPS spectra are identical to UO2, proving that the oxide is unchanged. After exposure to the pure water plasma, the O1s signal strongly broadens and shifts towards the lower binding energy, as expected for UO3. Moreover, it acquires the shape typical of UO3.30 The broadening is attributed to the crystal structure of UO3, having inequivalent oxygen atoms (in contrast to UO2). With increasing hydrogen concentration, the broadening becomes less pronounced. The O1s peak width after exposure to 10% H2 in mixed plasma is between the O1s peak widths observed for UO3 and UO2. This is typical of U2O5, as shown previously.30 The sample exposed to 20% H2 in mixed plasma has an O1s peak of the same width as the original UO2, but only slightly shifted to the lower binding energy (by – 0.3 eV).
UO3
Thin films of UO3 were prepared by exposing UO2 films to atomic oxygen at 400 °C for 10 min. The preparation procedure has been described in detail in a previous paper.30 Each film was then exposed to a mixed H2O/H2 plasma at 400 °C for 10 min. The corresponding regions of U4f probed with XPS are presented in Fig. 4. | ||
| Fig. 4 U4f core level XPS spectra recorded for the precursor UO3 film and the films with plasma treatment. | ||
It was shown previously that UO3 is prone to reduction when exposed to pure water plasma.28 This was attributed to the atomic hydrogen, also being present in the pure water plasma. The effect is, though, very weak and longer exposure times than used in the present study are needed (30–60 min (ref. 27)) to observe partial reduction of U(VI) into U(V). For mixed H2O/H2 plasma, the reduction becomes more pronounced.
The U4f peaks shift to lower binding energy and broaden: the U(V) component grows at the expense of the U(VI) component at higher BE. After exposure to 20% H2 in a mixed plasma the U(VI) component is still the dominating peak, while after exposure to 50% H2 in a mixed plasma the U(V) peak becomes the most prominent one. After exposure to the 20% H2 in a mixed plasma, the satellite peak of U(V) appears. For exposures to 30% H2 or higher H2 concentration, the U(V) satellite completely replaces the U(VI) satellite (data not shown). For the sake of clarity, we only plotted the result for the film exposed to 50% H2 in a mixed plasma.
At still higher H2 concentrations, the surface is further reduced and the U(IV) satellite appears. However the reduction is not complete even for 90% H2, and a residual U(V) satellite remains. It is only when the UO3 film is exposed to a pure H2 plasma that it is fully reduced to U(IV).
The valence band and O1s spectra for the UO3 films exposed to mixed H2O/H2 plasmas are plotted in Fig. 5. From these spectral features it is, as expected, evident that the degree of reduction increases with increasing H2 content of the mixed plasma. The O2p band loses its broad symmetrical shape, decreases in intensity and eventually takes the two-peak shape of UO2 (Fig. 3) after exposure to a mixed plasma containing 90% H2. In addition, the U5f peak increases in intensity due to the enhanced 5f population (n5f) after the reduction of U(VI) to U(V) and then U(IV) (n5f = 0, 1 and 2, respectively). After exposure of pure UO3 to the mixed plasma the O1s signal is identical to the signal before exposure. With increasing H2 concentration, the O1s peak narrows and shifts towards higher binding energies. The exposure to 30% H2 in a mixed plasma (not shown) yields a similar O1s spectrum for the 50% H2 in a mixed plasma. This is attributed to the presence of an intermediate oxidation state (U(V)), stable over an extended range of reducing conditions. It is only when UO3 is exposed to 90% H2 in a mixed plasma that the peak narrows even more and takes the shape typical of UO2.
 | ||
| Fig. 5 a) Valence band and b) O1s core level XPS spectra for the precursor and plasma treated UO3 films. | ||
U2O5
Films of pure U2O5 (where U(V) is the only oxidation state of uranium) have been prepared by exposing thin films of UO3 to 30% H2 in a mixed plasma. The synthesized U2O5 films were subsequently exposed to a mixed H2O/H2 plasma with variable feed gas compositions at 400 °C for 10 min. The U4f spectra of these films are presented in Fig. 6. Pure water plasma has an oxidizing effect on U2O5, manifested by the shifted main line and the respective satellite, being a hallmark of U(VI).30 When H2 is introduced in the feed gas of the plasma, the oxidation of U2O5 is less pronounced. After exposure to 20% H2 in a mixed plasma, the U(VI) main line component is smaller than after exposure to the pure water plasma. In addition, the U(VI) satellite peak is less developed, and the U(V) satellite peak more intense. When U2O5 is exposed to 40% H2 in a mixed plasma, the resulting U4f is almost identical to the unexposed U2O5 film, showing only the U(V) satellite peak. After exposure to 50% H2 in a mixed plasma, the U4f peaks undergo an additional shift towards lower binding energy, showing that uranium is now reduced to U(IV). Accordingly, the satellite peak of U(IV) replaces the U(V) satellite peak (Fig. 5). However, this reduction does not advance any further for the film exposed to 80% H2 in a mixed plasma. Even though the U(IV) satellite line is solely present after exposure to 80% H2 in a mixed plasma, the full width at half maximum of the main peak is larger than that for pure UO2. Hence, there is still a noticeable U(V) component in the main line (and a weak residual U(V) satellite peak may be masked by the strong U(IV) satellite peak). This shows that the reduction is not complete. | ||
| Fig. 6 U4f core level XPS spectra recorded for the original U2O5 film and U2O5 films exposed to plasma with variable H2O–H2 composition. | ||
The effect of plasma exposure is also evident in Fig. 7 where the XPS survey spectra of the valence band and the O1s peak of each exposed sample are presented. As discussed previously,28 after the oxidation of U2O5 with the water plasma, the intensity of the U5f peak at 1.2 eV decreases without changing its width, which implies that the product after plasma exposure is a mixture of U(V) and U(VI). U(V) gives the 5f1 signal while U(VI) has no 5f emission at all. U(V) can either oxidize or reduce, depending on the redox potential of the environment. With increasing H2 concentration, the reduction in the U5f intensity is much less pronounced than for the water plasma, which implies that U2O5 is less oxidized. Upon exposure to mixed plasmas with 50% or more H2, the U5f line grows and the O2p band loses its broad shape and becomes similar to the characteristic spectrum of UO2. U2O5 is clearly reduced under such conditions.
 | ||
| Fig. 7 Valence band and O1s XPS spectra recorded for unexposed and plasma treated U2O5 films. | ||
Fig. 7 also shows that the O1s peak of U2O5 becomes broader after exposure to the water plasma and shifts to lower binding energy. As discussed above, this indicates the oxidation of U2O5 into UO3. After exposure to 20% H2 in a mixed plasma the peak is slightly narrower. After exposure to 40% H2 in a mixed plasma, the O1s line becomes again narrower and superimposes to the original peak of U2O5, showing that the film does not react at all. For plasma containing 50–80% H2, the O1s peak narrows even more and shifts to higher binding energy, characteristic of UO2.
Oxidation state distribution as a function of H2 concentration
To get more quantitative information, we deconvoluted individual U4f7/2 lines into the respective spectra of pure UO2, U2O5, and UO3. This gives fractions of U(IV), U(V), and U(VI) as a function of H2 concentration by the plasma treatment of UO2, U2O5, and UO3 precursors. The results are plotted in Fig. 8. | ||
| Fig. 8 Fractions of different uranium oxidation states (obtained by decomposition of the U4f7/2 spectra) as a function of H2 concentration in the plasma feed gas. The results from the unexposed films (marked by dashed lines) are included for comparison. | ||
The relative fractions of U(IV), U(V) and U(VI) after 10 minutes exposure to a mixed H2O/H2 plasma as a function of H2 concentration in the plasma feeding gas provides interesting information on the uranium oxide and mixed plasma system. UO2 is shown to be oxidized by the mixed plasma provided the H2 content is below 40%. In other words, 40% H2 is sufficient to suppress oxidation by increasing the reduction rate of U(V) at the surface, in combination with decreasing the rate of U(IV) oxidation (through increasing the flux of reductants and decreasing the flux of oxidants as seen in Fig. 1).
For UO3 it is obvious that reduction in the 10 min time scale occurs only in presence of H2. In general, the rate and extent of reduction appear to be increasing with increasing H2 concentration up to 30%. Between 30 and 70% H2 the major product is U(V) and the extent of reduction appears to be concentration independent. At around 90% H2, reduction proceeds all the way to U(IV) which becomes the major product.
As can be seen, the oxidation of UO2 and the reduction of UO3 proceed via the formation of U(V). Interestingly, U(V) (U2O5) is quantitatively reduced to U(IV) at an H2 concentration of 50% and higher, while it is oxidized to U(VI) at an H2 concentration from around 40% and lower. The rate and extent of the oxidation reaction within the 10 min time scale increase with decreasing H2 concentration.
The fact that U(V) is never a major product after 10 min of UO2 oxidation while the reduction of UO3 involves U(V) as the major product in a wide H2 concentration range provides interesting information about the kinetics of the processes. As described above, long-term exposure of UO2 to a pure water plasma initially leads to oxidation affording U(V) and U(VI). After reaching a state of almost pure U(VI), prolonged exposure leads to the slow formation of U(V). Interestingly, UPS data (not shown here) being more surface sensitive than XPS reveal the formation of U(V) and U(VI) during the initial oxidation of UO2, but the slow formation of U(V) from U(VI) could never be confirmed by this method. Therefore, it was concluded that the U(V) formed upon reduction of U(VI) was formed deeper in the film and thereby displayed a lower reactivity towards the water plasma constituents. The present XPS data corroborate this hypothesis since the reduction of UO3 mainly yields U(V) in a very wide H2 concentration range and proceeds to U(IV) only at around 90% H2. At the same time, U2O5 exposed to mixed plasma is quantitatively already converted to U(IV) at 50% H2, i.e., at a considerably lower reducing power.
Grazing incidence X-ray diffraction
It is naturally very interesting to compare the in situ spectroscopic information with results of ex situ structural study by X-ray diffraction. Fig. 9 displays grazing incidence X-ray diffraction (GIXRD) data for the UO2 film deposited on a gold substrate (pattern A) and for the UO2 film exposed to an oxygen plasma (pattern B) and a water plasma (pattern C). In pattern A, indexing the peaks to the structures of UO2 (ICSD#35204, space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (no. 226), a = 5.4682(21)) and gold (ICSD#259286, space group Fm m (no. 226), a = 4.0787(1)) is reported. GIXRD reveals that the UO2 film produced under the conditions used in this work is highly textured with a preferential orientation of the [111] direction perpendicular to the substrate plane. When the crystalline UO2 films (pattern A) are oxidized to UO3 by the O2 plasma, the crystallinity is lost and the obtained UO3 film is amorphous (pattern B) after 10 min, and remains amorphous also after 20 min exposure. Exposing the UO3 film to a mixed 30% H2 in a mixed plasma for 10 min (pattern D1) or performing two consecutive exposures of 20 min duration (pattern D2) shows that the relative importance of oxidation and reduction reactions changes with the exposure time. A mixed plasma exposure for 10 min leads to the formation of the fluorite structure similar to UO2. After two consecutive 20 min exposures, pattern D2 changes to a completely different structure. A fit of the pattern was done with the structure of δ-U2O5 (ICSD#77702, Pmma, no. 51, a = 31.71, b = 8.29, c = 6.73)34 based on XPS-UPS analyses showing pure U(V). However as the orthorhombic structure of δ-U2O5 is not very different from α-U3O8,35 the latter cannot be excluded by XRD analysis. The difference in the oxygen atom content and position in the lattice can be better analysed by the neutron diffraction technique. Other techniques such as micro-Raman spectroscopy36 to differentiate the uranium oxides present in the films would be also useful but maybe not sufficient to support the XRD analyses.
m (no. 226), a = 5.4682(21)) and gold (ICSD#259286, space group Fm m (no. 226), a = 4.0787(1)) is reported. GIXRD reveals that the UO2 film produced under the conditions used in this work is highly textured with a preferential orientation of the [111] direction perpendicular to the substrate plane. When the crystalline UO2 films (pattern A) are oxidized to UO3 by the O2 plasma, the crystallinity is lost and the obtained UO3 film is amorphous (pattern B) after 10 min, and remains amorphous also after 20 min exposure. Exposing the UO3 film to a mixed 30% H2 in a mixed plasma for 10 min (pattern D1) or performing two consecutive exposures of 20 min duration (pattern D2) shows that the relative importance of oxidation and reduction reactions changes with the exposure time. A mixed plasma exposure for 10 min leads to the formation of the fluorite structure similar to UO2. After two consecutive 20 min exposures, pattern D2 changes to a completely different structure. A fit of the pattern was done with the structure of δ-U2O5 (ICSD#77702, Pmma, no. 51, a = 31.71, b = 8.29, c = 6.73)34 based on XPS-UPS analyses showing pure U(V). However as the orthorhombic structure of δ-U2O5 is not very different from α-U3O8,35 the latter cannot be excluded by XRD analysis. The difference in the oxygen atom content and position in the lattice can be better analysed by the neutron diffraction technique. Other techniques such as micro-Raman spectroscopy36 to differentiate the uranium oxides present in the films would be also useful but maybe not sufficient to support the XRD analyses.
 | ||
| Fig. 9 Room-temperature grazing incidence XRD data of the UO2 film on Au substrate (A). UO3 is obtained by the oxidation of UO2 by atomic oxygen (B). Effects of mixed 30% H2 in the mixed plasma (D1, D2) on the structure of UO3 are compared with UO2 kept in water plasma for 10 min (C). Insets in the figures show corresponding XPS data. | ||
Oxidation of UO2 by the water plasma for 10 min (pattern C) shows that the UO2 concentration decreased substantially at a lower intensity peak at 28.04° in 2θ, while a new and highly textured phase is formed and characterized by a strong intensity peak at 21.48°. This peak could be assigned to cubic UO337 whose Bragg positions are reported in the figure for comparison but it could be also linked to δ-U2O5,34 α-U3O835 or β-U3O8.38 The limited number of Bragg peaks and their low intensities prevents us from unambiguous identification of the crystal structure type. As suggested by the XPS-UPS results and in agreement with the XRD analysis, sample C is not pure and contains at least 2 phases of which only the cubic structure expected of UO2 can be clearly identified.
As has been shown in the spectral analysis above and in the tentative deconvolution, U(V) is formed as a major product upon the 10 min exposure of UO3 to a mixed H2O/H2 plasma in a fairly wide H2 concentration range. When starting with U(V) (U2O5) it is evident that fairly high H2 concentrations are needed to reduce U(V) to U(IV) in 10 min. In the plasma induced oxidation of UO2, U(V) is never observed as a major product after 10 min and it seems that once U(V) is formed in the oxidation of UO2, it is oxidized further to U(VI). One possible explanation may be that compounds containing U(V) and U(VI) have similar crystal structures: UO3, U3O8 (a mixed oxide with 66% U(V) and 33% U(VI)) and U2O5 (pure U(V) oxide) all exist in a layered structure (see the additional material), while U(IV) mostly exists in a cubic (fluorite) structure.31–33 Because of the similarity in the crystal structure, reduction of UO3 to U3O8 then to U2O5 does not require any major structure reorganization. However, when oxidizing UO2 to U2O5 a major structural transformation is required. This is also the case when reducing U(V) to U(IV). This would partly explain why the reduction of UO3 with mixed H2 in a mixed plasma appears to have U(V) as the major product after 10 min exposure within a wide range of H2 concentrations and why fairly high H2 concentrations are needed to reduce U2O5 to U(IV) within 10 min. The kinetic limitations would also account for the fact that U(V) is never observed as the major product in plasma induced oxidation of UO2. Given the fairly short exposure times used in this work, we can only rationalize the structural reorganization in terms of a kinetic barrier.
Conclusions
In this work, we have studied the impact of mixed H2O/H2 plasma of variable composition on thin uranium oxide films under UHV conditions. The starting compositions of the films used were UO2, U2O5, and UO3. The analysis of the films was based on XPS and grazing incidence XRD. The results clearly show that the reducing power of the mixed plasma applied for 10 min increases with increasing H2 concentration. At the same time the oxidizing power inherent to the water plasma is diminished both in absolute and relative terms when increasing the H2 concentration. The changes in the plasma redox properties are mainly attributed to recombination reactions within the plasma generator. In all cases, the oxidation proceeds as a consecutive reaction: U(IV) ↔ U(V) ↔ U(VI). The direct transformation U(IV) ↔ U(VI) does not occur. This is also supported by the deconvolution result showing that the maximum concentration of [U(VI)] is reached when [U(IV)] is at its minimum.The XPS results show that U(V) is formed as a major product upon exposure of UO3 to a mixed H2O/H2 plasma in a fairly wide H2 concentration range. When starting with U(V) (U2O5) appreciable H2 concentrations are needed to reduce U(V) to U(IV) in 10 minutes. In the plasma induced oxidation of UO2, U(V) is never observed as a major product after 10 min and it would seem that once U(V) is formed during the oxidation of UO2 it is rapidly oxidized further to U(VI). Formation of U(V) is easier, obtained by the reduction of UO3 by a mixed H2O/H2 plasma. It could not be prepared by the oxidation of UO2 with the water plasma. Once U(V) is obtained from the UO3 reduction, it is relatively stable as it shows lower reactivity than UO2 for oxidation and UO3 for reduction. The grazing incidence X-ray diffraction analysis shows that there is a considerable impact of the plasma and heating conditions on the crystallographic structure of the films in line with the change of the oxidation state. This structural difference may be the main kinetic barrier for plasma induced transfer between U(IV) and U(V) in both directions. Further characterisation by micro-Raman spectroscopy and transmission electron microscopy would be helpful to identify different types of uranium oxides present on the substrate.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Swedish Nuclear and Fuel Waste Management Company (SKB) is gratefully acknowledged for financial support. This work has been partially supported by the ENEN+project that has received funding from the Euratom research and training Work Programme 2016–2017–1#755576). E. T.-Ch. acknowledges the support of “Nano-materials Centre for Advanced Applications,” Project No. CZ.02.1.01/0.0/0.0/15_003/0000485, financed by ERDF. We want to thank Prof. L. Havela for the fruitful discussion about the characterization of the films and F. Huber for the technical support.Notes and references
- J. Nowotny and L. C. Dufour, Mater. Sci. Monogr., 1988, 47, 275 Search PubMed.
- D. Rai, A. R. Felmy and J. L. Ryan, Inorg. Chem., 1990, 29, 260–264 CrossRef CAS.
- S. Svensk Kaernbraenslefoersoerjning Ab, Final storage of spent nuclear fuel - KBS-3, Swedish Nuclear Fuel Supply Co/Div KBS, Sweden, 1983 Search PubMed.
- J. Bruno and R. C. Ewing, Elements, 2006, 2, 343–349 CrossRef CAS.
- H. Kleykamp, J. Nucl. Mater., 1985, 131, 221–246 CrossRef CAS.
- J. W. T. Spinks and R. J. Woods, An Introduction to Radiation Chemistry, 1990 Search PubMed.
- D. W. Shoesmith, J. Nucl. Mater., 2000, 282, 1–31 CrossRef CAS.
- D. W. Shoesmith and S. Sunder, J. Nucl. Mater., 1992, 190, 20–35 CrossRef CAS.
- F. King, L. Ahonen, C. Taxen, U. Vuorinen and L. Werme, Copper corrosion under expected conditions in a deep geologic repository, Report 1404-0344, Sweden, 2001 Search PubMed.
- N. R. Smart, D. J. Blackwood and L. Werme, The anaerobic corrosion of carbon steel and cast iron in artificial groundwaters, Report 1404-0344, Sweden, 2001 Search PubMed.
- E. Ekeroth, M. Granfors, D. Schild and K. Spahiu, J. Nucl. Mater., 2020, 531, 151981 CrossRef CAS.
- T. Eriksen and M. Jonsson, The effect of hydrogen on dissolution of spent fuel in 001 mol/dm-3 NaHCO3 solution, Report 1404-0344, Sweden, 2007 Search PubMed.
- P. Carbol, The effect of dissolved hydrogen on the dissolution of 233U doped UO2(s) high burn-up spent fuel and MOX fuel, Report 1404-0344, Sweden, 2005 Search PubMed.
- S. Röllin, K. Spahiu and U. B. Eklund, J. Nucl. Mater., 2001, 297, 231–243 CrossRef.
- S. Nilsson and M. Jonsson, J. Nucl. Mater., 2008, 372, 160–163 CrossRef CAS.
- M. E. Broczkowski, J. J. Noël and D. W. Shoesmith, J. Nucl. Mater., 2005, 346, 16–23 CrossRef CAS.
- S. Kastriot, L. Werme and U. B. Eklund, Radiochim. Acta, 2000, 88, 507–512 CrossRef.
- N. L. Hansson, P. L. Tam, C. Ekberg and K. Spahiu, J. Nucl. Mater., 2021, 543, 152604 CrossRef CAS.
- M. Trummer, S. Nilsson and M. Jonsson, J. Nucl. Mater., 2008, 378, 55–59 CrossRef CAS.
- K. Spahiu, U.-B. Eklund, D. Cui and M. Lundström, MRS Proc., 2002, 713, JJ14.15 Search PubMed.
- L. Bauhn, N. Hansson, C. Ekberg, P. Fors, R. Delville and K. Spahiu, J. Nucl. Mater., 2018, 505, 54–61 CrossRef CAS.
- A. Puranen, A. Barreiro, L. Z. Evins and K. Spahiu, MRS Adv., 2017, 2, 681–686 CrossRef CAS.
- M. Jonsson, F. Nielsen, O. Roth, E. Ekeroth, S. Nilsson and M. M. Hossain, Environ. Sci. Technol., 2007, 41, 7087–7093 CrossRef CAS PubMed.
- M. Trummer, O. Roth and M. Jonsson, J. Nucl. Mater., 2009, 383, 226–230 CrossRef CAS.
- S. Nilsson and M. Jonsson, J. Nucl. Mater., 2008, 374, 290–292 CrossRef CAS.
- P. Fors, P. Carbol, S. Van Winckel and K. Spahiu, J. Nucl. Mater., 2009, 394, 1–8 CrossRef CAS.
- G. El Jamal, T. Gouder, R. Eloirdi and M. Jonsson, Dalton Trans., 2021, 50, 4796–4804 RSC.
- G. El Jamal, T. Gouder, R. Eloirdi and M. Jonsson, Dalton Trans., 2021, 50, 729–738 RSC.
- A. Fridman, Plasma Chemistry, Cambridge University Press, 2008 Search PubMed.
- T. Gouder, R. Eloirdi and R. Caciuffo, Sci. Rep., 2018, 8, 8306 CrossRef CAS PubMed.
- C. A. Colmenares, Prog. Solid State Chem., 1975, 9, 139–239 CrossRef CAS.
- G. C. Allen and N. R. Holmes, J. Nucl. Mater., 1995, 223, 231–237 CrossRef CAS.
- M. Molinari, N. A. Brincat, G. C. Allen and S. C. Parker, Inorg. Chem., 2017, 56(8), 4468–4473 CrossRef PubMed.
- N. C. Baenziger, Iowa State Coll. J. Sci., 1952, 27, 126–128 Search PubMed ISCJAF loop.
- P. Taylor, D. D. Wood and A. M. Duclos, J. Nucl., Mater., 1992, 189, 116–123 CrossRef CAS.
- M. L. Palacios and S. H. Taylor, Appl. Spectrosc., 2000, 54, 1372–1378 CrossRef CAS.
- E. Wait and J. Inorg, Nucl. Chem., 1955, 1, 309–312 CrossRef CAS.
- B. O. Loopstra, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26, 656–657 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dt01020d |
| This journal is © The Royal Society of Chemistry 2021 |