
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radical-initiated P,P-metathesis reactions of diphosphanes: evidence from experimental and computational studies†
Callum
Branfoot
 a,
Tom A.
Young
b,
Duncan F.
Wass
c and
Paul G.
Pringle
*a
a,
Tom A.
Young
b,
Duncan F.
Wass
c and
Paul G.
Pringle
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: paul.pringle@bristol.ac.uk
bChemistry Research Laboratory, University of Oxford, Oxford OX1 3TA, UK
cCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff CF10 3AT, UK
First published on 28th April 2021
Abstract
By combining the diphosphanes Ar2P–PAr2, where Ar = C6H5, 4-C6H4Me, 4-C6H4OMe, 3,5-C6H3(CF3)2, it has been shown that P,P-metathesis generally occurs rapidly under ambient conditions. DFT calculations have shown that the stability of unsymmetrical diphosphanes Z2P–PZ′2 is a function of the difference between the Z and Z′ substituents in terms of size and electronegativity. Of the mechanisms that were calculated for the P,P-metathesis, the most likely was considered to be one involving Ar2P˙ radicals. The observations that photolysis increases the rate of the P,P-metatheses and TEMPO inhibits it, are consistent with a radical chain process. The P,P-metathesis reactions that involve (o-Tol)2P–P(o-Tol)2 are anomalously slow and, in the absence of photolysis, were only observed to take place in CHCl3 and CH2Cl2. The role of the chlorinated solvent is ascribed to the formation of Ar2PCl which catalyses the P,P-metathesis. The slow kinetics observed with (o-Tol)2P–P(o-Tol)2 is tentatively attributed to the o-CH3 groups quenching the (o-Tol)2P˙ radicals or inhibiting the metathesis reaction sterically.
Introduction
Dynamic and reversible chemical reactions have garnered increasing attention in medicine and biology,1 supramolecular chemistry2 and materials science,3–11 and they are key to the rapidly growing field of dynamic covalent chemistry (DCC).1,12,13 In DCC, disulfides play a leading role, by virtue of the weakness of the S–S bond and their ready availability via chemical synthesis and their presence in natural products.12,14–16 In the last decade, several other main group element–element bonds (O–O,17,18 N–N19 and Se–Se9,20,21) have been investigated for their potential in dynamic chemistry with a view to their application in the field of polymeric materials. A notable omission from this group is diphosphanes (P2R4) which contain a P–P bond whose strength (ca. 52 kcal mol−1)22 is intermediate between S–S (ca. 62 kcal mol−1)23 and Se–Se (ca. 40 kcal mol−1)22 and therefore diphosphanes should be candidates for DCC.Diphosphanes have received considerable attention as targets themselves and also as undesired byproducts in the synthesis of tertiary phosphines.24–31 Several methods to prepare diphosphanes are available, including: Wurtz-type reductive coupling of chlorophosphines (with Li, Na, K, Mg and Hg);32–34 salt metathesis between chlorophosphine and LiPR2;26,27 dehydrocoupling of HPR2;35–39 chlorosilane elimination;40 P–N/P–P bond metathesis.41 The chemistry of diphosphanes can be divided into two categories: reactions where the P–P bond remains intact and reactions where the P–P bonds are cleaved.42 Reactions involving P–P cleavage are pertinent to the chemistry described in this article and include additions of X2 to the R2P–PR2 to give monophos species R2PX (e.g. X = H, Cl) and additions of R2P–PR2 to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C or C
C or C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds (diphosphination) to give diphos species (Scheme 1). It has also been shown that diphosphanes containing P–N bonds can add to CS or CO bonds.43,44 Mechanisms have been proposed for some of the P–P cleavage reactions including radical chain processes.43,45
C bonds (diphosphination) to give diphos species (Scheme 1). It has also been shown that diphosphanes containing P–N bonds can add to CS or CO bonds.43,44 Mechanisms have been proposed for some of the P–P cleavage reactions including radical chain processes.43,45
 | ||
| Scheme 1 Some reactions involving P–P cleavage of tetraphenyldiphosphane.42 | ||
Diphosphane metathesis has not been systematically investigated, but an early 31P NMR study by Harris et al. showed that rapid metathesis occurred between Me2P–PMe2 and (F3C)2P–P(CF3)2 in CH2Cl2 to form Me2P–P(CF3)2.46,47 Gilheany et al.48 have shown that the calculated high energy barrier to P,P-metathesis via a concerted mechanism is not compatible with the observed rapid kinetics for the Me2P–P(CF3)2 system and suggested that pathways involving impurities in the diphosphanes may facilitate the P,P-metathesis process. Grubba et al.26 have succeeded in the preparation of a variety of pure, unsymmetrical diphosphanes containing P–C(alkyl) and P–N bonds. By employing low temperatures, they avoided unwanted P,P-metatheses that would lead to symmetrical diphosphanes contaminating the products. Moreover, they reported that R2PCl or R2PLi can catalyse diphosphane metathesis. We have reported that diphosphane metathesis was a side reaction in the attempted synthesis of some unsymmetrical diphosphanes.27
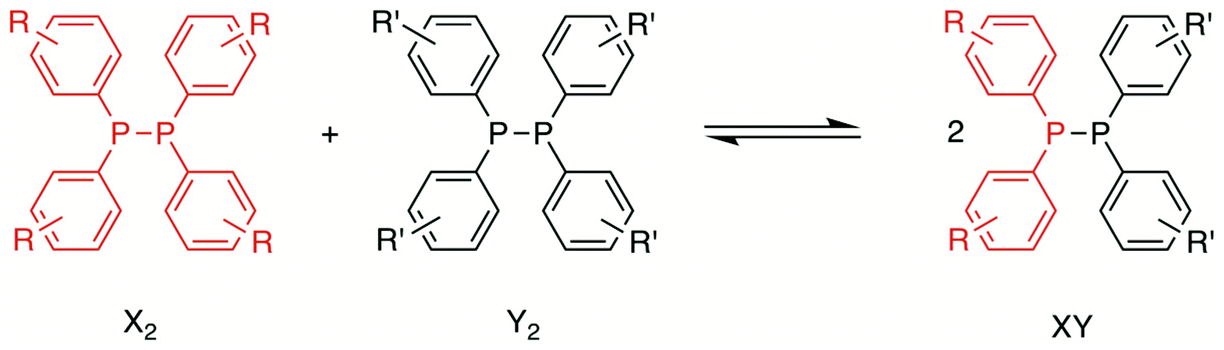
In this study, we have investigated the P,P-metathesis reactions of tetra-aryldiphosphanes (Scheme 2) using 31P{1H} NMR spectroscopy and computational methods, with a view to exploring the dynamic nature of these P–P bonds. This has provided some new insights into the diphosphane metathesis reaction.
 | ||
| Scheme 2 | ||
Results and discussion
Thermodynamics of diphosphane metathesis
 | ||
| Chart 1 | ||
When two of the diphosphanes A2–E2 were mixed in CDCl3 at ambient temperature, a reaction ensued to give an equilibrium mixture of homodiphosphanes X2 and Y2, and the heterodiphosphane XY (Scheme 2). The data for the diphosphane metathesis reactions are given in Table 1.
| Entry | X2 | Y2 | K | Timea/min | 31P NMR data for XY | ||
|---|---|---|---|---|---|---|---|
| a Time by which equilibrium had been established according to 31P NMR spectroscopy. b In THF. c In CDCl3. d In C6D6. e Estimated value (see details in ESI†). | |||||||
| 1b | A2 | B2 | 4 | 30 | −14.9 | −16.4 | J PP = 162 Hz |
| 2c | A2 | C2 | 5 | 20 | −15.9 | −18.4 | J PP = 159 Hz |
| 3d | A2 | D2 | 30e | 90 | −15.9 | −8.5 | J PP = 177 Hz |
| 4d | B2 | C2 | 4 | 15 | −16.8 | −18.8 | J PP = 156 Hz |
| 5b | B2 | D2 | 15 | 20 | −11.7 | −15.7 | J PP = 165 Hz |
| 6b | C2 | D2 | 3 | 20 | −10.5 | −15.4 | J PP = 173 Hz |
| 7c | A2 | E2 | 4 | 700 | −16.6 | −35.3 | J PP = 152 Hz |
| 8c | B2 | E2 | 4 | 1000 | −19.1 | −36.2 | J PP = 149 Hz |
| 9c | C2 | E2 | 4 | 2800 | −19.0 | −36.9 | J PP = 148 Hz |
| 10c | D2 | E2 | 30 | 4000 | −17.9 | −29.5 | J PP = 156 Hz |
The 31P{1H} NMR data for the XY species (Table 1) show that the chemical shifts for the two signals are close to the respective X2 and Y2 with 1JXY ≃160 Hz. For example, Fig. 1 is the 31P{1H} NMR spectrum obtained after mixing diphosphanes B2 and C2 (entry 4, Table 1) and shows the singlets for the reactants along with an AB pattern for the product BC; the spectra for the other diphosphane combinations are given in the ESI.†
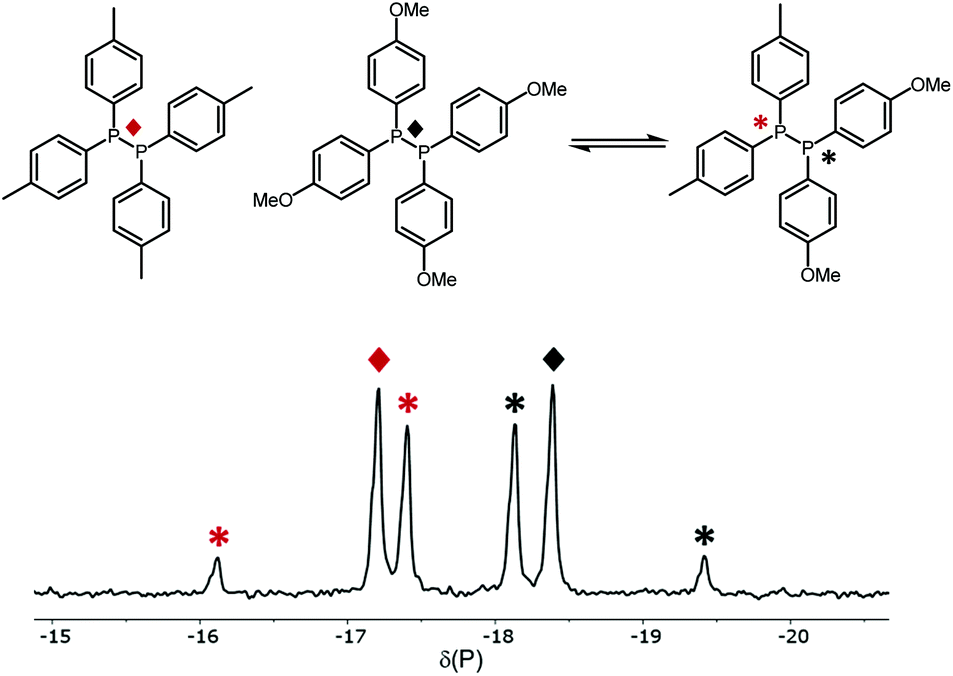 | ||
| Fig. 1 The 31P{1H} NMR spectrum of the P,P-metathesis reaction between (p-Tol)2P–P(p-Tol)2 (B2) and (p-Anisyl)2P–P(p-Anisyl)2 (C2). | ||
The pure heterodiphosphane AE was prepared in order to establish the P,P-metathesis equilibrium with A2 and E2 (Scheme 2) from the opposite direction. Grubba et al.26 have prepared a range of heterodiphosphanes (Z2P–PZ′2) by the addition of Z2PLi to Z′2PCl at low temperatures. However, our attempts to extend this route to the preparation of AE were unsuccessful, even with the reaction mixture maintained at −78 °C throughout; instead, an equilibrium mixture of A2, E2 and AE was obtained. This contrasting behaviour may be due to the lack of thermodynamic stability of heterodiphosphane AE compared with the Grubba heterodiphosphanes26 (see below). The desired AE was prepared in >96% purity (after recrystallisation from methanol) via the protection/deprotection method (Scheme 3) that we previously developed for the synthesis of heterodiphosphanes.27,49 Solutions of pure AE in CDCl3 do indeed equilibrate to a mixture of A2, E2 and AE (see below for further details).
 | ||
| Scheme 3 | ||
The diphosphane equilibrium constants for the P,P-metatheses (Scheme 2) fall in the range 3–30 (see Table 1), which corresponds to ΔG values between −0.5 and −2 kcal mol−1, demonstrating that the diphosphanes in this study are very close in energy and insensitive to the steric and electronic effects of the aryl substituents. The equilibria that do not involve E2 were established rapidly (see Table 1) despite the dilution (12.5 mM) which is a useful property for the potential application of diphosphanes in dynamic chemistry.1 The anomalously slow P,P-metatheses involving E2 are discussed later.
 | ||
| Scheme 4 | ||
| Entry | Z | Z′ | ΔG/kcal mol−1 |
|---|---|---|---|
| 1 | Me | H | −0.5 |
| 2 | Me | NH2 | −2.9 |
| 3 | Me | OH | −2.9 |
| 4 | Me | OMe | −3.7 |
| 5 | Me | F | −6.6 |
| 6 | Me | CF3 | −10.5 |
| 7 | Ph | H | 0.8 |
| 8 | Ph | Me | −0.7 |
| 9 | Ph | NH2 | 1.3 |
| 10 | Ph | NMe2 | −0.8 |
| 11 | Ph | OMe | −4.7 |
| 12 | Ph | F | −6.1 |
| 13 | Ph | CF3 | −4.4 |
| 14 | t Bu | H | −13.4 |
| 15 | t Bu | Me | −3.3 |
| 16 | t Bu | Ph | −4.0 |
| 17 | t Bu | NH2 | −6.8 |
| 18 | t Bu | NMe2 | −6.8 |
| 19 | t Bu | OMe | −11.2 |
| 20 | t Bu | F | −15.4 |
| 21 | t Bu | CF3 | −3.4 |
| 22 | o-Tol | Ph | 1.2 |
Two gross trends emerge from the data in Table 2: (1) a bias in favour of the XY species is calculated when there is a large electronegativity difference between the X and Y fragments; thus Me2P–P(CF3)2 is predicted (and experimentally observed50) to be highly favoured (Table 2, entry 6); (2) a strong bias in favour of the XY species is calculated when there is a large difference in steric bulk between the X and Y fragments; thus tBu2P–PH2 is predicted to be highly favoured (Table 2, entry 13). The largest calculated ΔG in favour of the XY species is for tBu2P–PF2 (Table 2, entry 20), at an apparent apotheosis of both trends. The observed trends in the calculated relative stabilities of the XY species in Scheme 4 can be rationalised as follows: (a) electronegative Z substituents (such as F) will lead to repulsive electrostatic interactions between the δ+ charges generated on the adjacent P atoms in diphosphane X2; (b) bulky Z′ substituents (such as tBu) will lead to steric congestion in diphosphane Y2; (c) in XY, both of the P–P bond-destabilising effects identified in (a) and (b) will be reduced and, in some cases, a destabilising, repulsive, electrostatic (δ+/δ+) interaction is replaced by a stabilising, attractive, electrostatic (δ+/δ−) interaction.
The calculated ΔG values for some of the Grubba heterodiphosphanes26 (Table 2, entries 16 and 18) suggest that these should have high thermodynamic stability with respect to the corresponding homodiphosphanes. By contrast, the heterodiphosphine AE, as reported above, is calculated to be of similar energy to the homodiphosphanes A2 and E2 (Table 2, entry 22).
Computational mechanistic studies of diphosphane metathesis
The mechanism of the diphosphane metathesis shown in Scheme 5 has been previously investigated computationally by Gilheany et al.48 The lowest energy route that they could find involved the ionic intermediate shown in Scheme 5 but the calculated barrier of 36.5 kcal mol−1 was too high to explain the rapidity of the P,P-metathesis that is observed experimentally.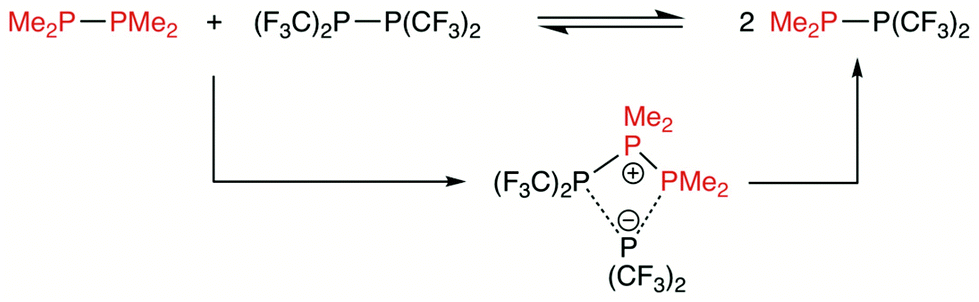 | ||
| Scheme 5 Gilheany mechanism for a P,P-heterometathesis. | ||
We have investigated the mechanism of diphosphane metathesis computationally using the degenerate exchange reactions of Me2P–PMe2 (Scheme 6). Tetramethyldiphosphane was chosen in order to limit the number of rotamers and thus the conformational noise in the calculations. It was hoped to distinguish between the three plausible pathways (i–iii) shown in Scheme 7: (i) a [2 + 2] concerted process involving a 4-membered intermediate or transition state; (ii) nucleophilic attack by one diphosphane on another to give an ionic intermediate (similar to the one suggested in Scheme 5); (iii) a [2 + 1] radical chain process.
 | ||
| Scheme 6 | ||
 | ||
| Scheme 7 | ||
The overall reaction profile calculated for the thermal rearrangement of Me2P–PMe2 (shown in Fig. 2(a) proceeds via intermediate int1 depicted in its lowest energy conformation in Fig. 2(b). The structure of int1 can be viewed in terms of the two resonance forms int1A and int1B shown in Fig. 2(c) (further details are given in the ESI†). The ionic form int1A is analogous to the intermediate suggested for the P,P-heterometathesis shown in Scheme 5 and the covalent int1B contains an unusual central P(V) moiety. The profile shown in Fig. 2 corresponds to pathway (ii) of Scheme 7, and notably, attempts to locate a synchronous, σ-bond metathesis pathway (corresponding to pathway (i) in Scheme 7) were unsuccessful; no imaginary normal modes corresponding to the 2 bond-breaking/forming vibrations are present along the relaxed, symmetric P4 ring expansion surface. The energy of int1 of 44.1 kcal mol−1 for the PMe2 species is lower for the analogous PPh2 intermediate (ΔG = 38.1 kcal mol−1, see ESI†) but this is calculated to be not lowered by solvation with CH2Cl2. Thus, the calculated very high barrier is incompatible with the rapidity of the Ar2P–PAr2 metathesis reactions that is observed experimentally.
 | ||
| Fig. 2 (a) Reaction free energy profile for the P,P-homometathesis of Me2P–PMe2. Calculations performed at the PBE0-D3BJ/def2-TZVPP/PBE0-D3BJ/def2-SVP level of theory. (b) Calculated structures of ts1 and int1. Note: ts1 and ts2 are the same; key distances given in Å. (c) Resonance forms of int1. | ||
The radical chain pathway (iii) in Scheme 7 requires homolysis of a P–P bond. Thermolysis is not viable under ambient conditions because the calculated P–P bond enthalpy is of the order of 52 kcal mol−1. However, DFT calculations for Ph2P–PPh2 (A2) suggest that excitation from S0 → S1 or S0 → S2 would result in rapid homolysis of the P–P bond, yielding phosphanyl radicals (Ar2P˙) via intersystem crossing from S1 or S2 to the first excited triplet state (T1) of A2; the T1 state is dissociative, and would lead to spontaneous radical formation. From the Ar2P˙, the P,P-metathesis barrier generating XY species is only ∼5 kcal mol−1, such that once radicals are generated an equilibrium would be expected to be established rapidly (see ESI† for details).
The DFT results suggested that Ar2P˙ radicals (generated photolytically) were viable intermediates in the P,P-metathesis reactions and so this hypothesis was pursued experimentally.
Qualitative experimental kinetic studies of diphosphane metathesis
The aryl diphosphane metathesis reactions with A2–D2 occurred readily (typically <1 h) under ambient conditions in common organic solvents (C6D6, THF, PhMe, PhCl, CDCl3 and CD2Cl2). By contrast, when the diphosphane E2, containing o-tolyl substituents, was one of the reaction partners, P,P-metathesis was initially only observed to take place in CDCl3 (typically over 6–12 h) and CD2Cl2 (typically over more than 24 h); the anomalous behaviour observed for the P,P-metatheses involving E2 will be discussed after consideration of the P,P-metatheses involving A2–D2.When cold (−78 °C) THF solutions of B2 and C2 were mixed and the P,P-metathesis reaction monitored by low-temperature 31P NMR spectroscopy, it was found that only traces of BC were detected at −80 °C after 1 h. By raising the temperature in increments of 20 °C, it was established that the P,P-metathesis at −20 °C progressed at a convenient rate to monitor the approach to P,P-metathesis equilibrium over a period of 2 h by 31P NMR spectroscopy (see Fig. 3). Although a detailed kinetic study was not attempted, the data do not appear to fit a reaction order of 1, 2 or 0.5 (see ESI†).
 | ||
| Fig. 3 (a) Plot of heterodiphosphane BC formation as a function of time over 2 h; (b) 31P{1H} NMR spectra of the reaction between B2 and C2 at −20 °C in THF, shown in 10 min increments over the course of 1 h (with inverse-gated decoupling). See Fig. 1 for the peak assignments. | ||
Evidence for radicals in the general P,P-metathesis reactions
In light of the computational results above suggesting that radicals may be involved in the mechanism of P,P-metathesis, experiments were carried out to determine if there was evidence of their presence. Two pieces of experimental evidence support the involvement of radicals in the P,P-metathesis process.(1) When A2 and C2 were mixed in the presence of the radical scavengers 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or tri(t-butyl)phenol (TTBP), the progress of the P,P-metathesis reactions was greatly inhibited. Thus, a mixture of A2 and C2 proceeded smoothly to the metathesis equilibrium over a period of 2 h in CDCl3, at −20 °C (with 35% conversion at 1 h); by contrast, when the same A2/C2 mixture was prepared in the presence of 5 equiv. of TEMPO, there was no formation of AC detected after 1 h. Moreover, when the reaction mixture containing TEMPO was allowed to warm to ambient temperature, <10% of AC was detected even after 16 h. Instead, in the presence of TEMPO, two singlet 31P resonances at +30.5 and +31.5 ppm were observed (ca. 10% of the total signal intensity), consistent with the formation of Ar2P(O)-TEMPO (Ar = Ph, p-anisyl) adducts (Scheme 8) which would be expected to be formed by the interception of the phosphanyl radicals (Ar2P˙) by TEMPO.51
 | ||
| Scheme 8 | ||
(2) When A2 and B2 were mixed in a standard NMR tube in THF at ambient temperatures, equilibration was essentially complete within ca. 15 min. However, when the same A2/B2 mixture in THF was prepared in an amberised NMR tube, to minimise the transmittance of UV and visible radiation, the equilibration to the same degree took ca. 150 min. This is consistent with diphosphane photodissociation being a significant component of the mechanism of P,P-metathesis.
The UV–vis spectra of A2, C2 and E2 are shown in Fig. 4. Each spectrum has a major absorbance between 225 and 250 nm, and a shoulder at ca. 270 nm. TD-DFT calculations for A2 were carried out using a range of functionals (CAM-B3LYP, PBE0 and M06-2X) and good agreement with experiment was found with CAM-B3LYP. These calculations suggest that the major peak in each spectrum corresponds to coincident aromatic π → π* and S0 → S2 (see Fig. 5(b)) transitions and the shoulder peak corresponds to S0 → S1 (see Fig. 5(a)). The transition electron density plots (Fig. 5) indicate that both transitions result in a significant delocalisation of electron density from the P lone pairs. The first triplet state, accessible from either S1 or S2via intersystem crossing, is dissociative, leading to spontaneous formation of phosphinyl radicals. There are small differences in the three spectra shown in Fig. 4. As expected, the p-OMe groups in C2 produce a bathochromic shift in the π → π* band and there is a long ‘tail’ that extends towards the visible region, which explains why C2 is pale yellow.
 | ||
| Fig. 4 UV–vis spectra of A2, C2 and E2 in CH2Cl2 (20 μM). | ||
 | ||
| Fig. 5 Transition electron density difference plots with the CAM-B3LYP/def2-TZVPP//PBE0-D3BJ/def2-SVP level of theory. (a) S0 → S1 and (b) S0 → S2. Red and blue correspond to areas of depleted and excess electron density, respectively. Isosurface plotted at a contour of 0.003 au. | ||
The evidence in support of radicals being present led to the radical chain mechanism proposal in Scheme 9 where homolysis of the Ar2P–PAr2 is followed by attack of the Ar2P˙ on the second diphosphane Ar′2P–PAr′2. The termination steps are then the radical couplings with P–P bond formation.
 | ||
| Scheme 9 | ||
The photodissociation requires UV radiation and it appears that there is sufficient background UVA-UVB radiation (280–400 nm) for the initiation step to proceed.
The two degenerate propagating steps, where Ar2P˙ and Ar′2P˙ react with their respective dimers Ar2P–PAr2 and Ar′2P–PAr′2, are omitted from Scheme 9. The reverse P,P-metathesis process would also be initiated by photodissociation of Ar2P–PAr′2. The P,P-metathesis reactions generally proceed with a high degree of chemoselectivity but traces of Ar2PH were often evident after several days of reaction which would arise from termination via abstraction of an H radical, presumably from the solvent.
The radical mechanism for diphosphane metathesis in Scheme 9 is reminiscent of the radical mechanism for disulfide metathesis (Scheme 10) arrived at from calculations by Ruipérez et al.52 and from experimental observations by Asua et al.53 DFT and MD calculations were applied to two mechanisms of diaryldisulfide metathesis: a [2 + 2] concerted metathesis and a [2 + 1] radical chain reaction initiated by sulfanyl radicals (ArS˙).47 The transition state in the concerted process was calculated to be too high in energy to be viable, whereas the radical process was viable provided the ArS˙ radicals could be accessed. This reasoning mirrors ours for the mechanisms proposed for diphosphane metatheses shown in Scheme 7.
 | ||
| Scheme 10 | ||
It has been reported that the diaryldisulfide metathesis between diphenyldisulfide and di(4-aminophenyl)disulfide proceeds spontaneously to equilibrium in ca. 12 h; this was accelerated by UV photolysis, and TEMPO completely inhibited the equilibration.53 These observations made on the disulfide metathesis reactions (and analogous diselenide studies)54 are very similar to our observations on the diphosphane metatheses.
Anomalous kinetics with (o-Tol)2P–P(o-Tol)2
The four P,P-metathesis reactions between tetra(o-tolyl)diphosphane (E2) and A2–D2 did not appear to proceed at all in THF, PhMe, PhCl, PhF or C6D6 under ambient conditions after 72 h but do proceed in the chloroalkanes CH2Cl2 and CHCl3, albeit slowly (see Table 1, entries 7–10). To probe this anomalous behaviour further, we have investigated the A2/E2 system (Fig. 6) in more detail including approaching the equilibrium starting from an A2/E2 mixture and starting from isolated AE. | ||
| Fig. 6 31P{1H} NMR spectra for the mixture of A2 and E2: (a) in toluene after 48 h, showing no detectable P,P-metathesis; (b) in CDCl3 after 6 h, showing equilibrium with AE has been established. | ||
The progress of the A2/E2 metathesis in CDCl3 was followed by 31P NMR spectroscopy (see ESI† for details) with a spectrum recorded every 10 min for 12 h (see Fig. 7). The approach to equilibrium was measured from integration of the 31P NMR signals to produce the plot shown in Fig. 7. Analysis of the curve revealed that it was not consistent with the integrated equations for an order of reaction of 0, ½, 1 or 2; this indicates that the kinetics are complex, which would be expected if more than one mechanistic pathway was operating (see below).
 | ||
| Fig. 7 (a) Plot of the data obtained from integration of the 31P{1H} NMR signals for the AE formed in the reaction between diphosphanes A2 and E2; (b) 31P{1H} NMR spectra in CDCl3 obtained by sampling over 12 h. See Fig. 6 for the peak assignments. | ||
Evidence for radicals in the anomalous (o-Tol)2P–P(o-Tol)2 metathesis reactions
There are several pieces of evidence supporting a role for radicals in the P,P-metathesis equilibrium shown in Fig. 6:(1) The lack of any reaction between A2 and E2 in THF after 72 h was replaced by 30% AE formation when the solution was irradiated with a UV lamp for 2 h. Similarly, no equilibration was observed in a THF solution of pure AE over a period of 72 h but proceeded smoothly over 2 h upon irradiation of the same solution with a UV lamp. This suggests that the photodissociation mechanism shown in Scheme 6 for the general P,P-metathesis (Table 1, entries 1–6) is viable for the A2/E2 metathesis but requires a more intense source of UV radiation than background UV for the initiation step.
(2) The slow A2/E2 metathesis equilibration in CDCl3 which takes place over a period of 12 h was extinguished in the presence of 4 equiv. of the radical scavenger TEMPO, as no AE was detected after 16 h.
(3) Pulse-sonication (20 kHz) at 0 °C of a CHCl3 or CH2Cl2 solution of A2 and E2 reduced the time to establish equilibrium from 12 h to <1 h. However, sonolysis had no accelerating effects on the reaction of A2 with E2 in THF or toluene. Fritze and von Delius reported the metathesis of disulfides (Scheme 10) in chloroform was accelerated by sonolysis and suggested that this proceeded via RS˙ radicals formed from the reaction of disulfides with Cl˙ generated by sonolytic chloroform degradation.16
(4) The onset of equilibration of solutions of AE in CDCl3 was preceded by an induction period of up to 6 h; moreover, the time required to establish the P,P-metathesis equilibrium from mixtures of A2/E2 in CDCl3 varied from 6 to 12 h. These observations of rate-inconsistency are typical of radical processes.55
In addition to the evidence for a radical-promoted process, it is shown below that there is evidence of chlorophosphine involvement in the A2/E2 metathesis.
Evidence for chlorophosphine intermediacy in the anomalous (o-Tol)2P–P(o-Tol)2 metathesis reactions
Under the conditions where a mixture of A2 and E2 in CDCl3 would reach equilibrium with AE within 12 h, the presence of H2O (100 equiv.) suppressed the reaction to the extent that no AE was detected even after 20 h (indicating that <1% AE had been formed). Instead, several minor signals (ca. 20% in total) were present and those at δ+ 23.5, +21.4 were tentatively assigned to Ar2P(O)H (Ar = Ph or o-Tol). These observations are consistent with the water reacting rapidly with the Ar2PCl compounds that are formed. It is notable that the addition of water to the P,P-metathesis reaction mixture of A2 with C2 in CDCl3 had no discernible effect on the rapid formation of AC which indicates that Ar2PCl is not involved in the P,P-metathesis for this typical system.When a solution of A2 alone in CHCl3 was irradiated with UV, some conversion to Ph2PCl was observed directly. The stoichiometric reaction of A2 with (o-Tol)2PCl in CHCl3 immediately produced a mixture of A2, AE, and Ar2PCl (Ar = Ph, o-Tol) (Scheme 11) along with several unidentified P-containing species. After 4 h, E2 was detected along with small amounts of secondary phosphines Ar2PH (Ar = Ph, o-Tol). These observations confirm that Ar2PCl compounds are plausible promoters of P,P-metathesis.
 | ||
| Scheme 11 P,P-Metathesis promoted by Ar2PCl. | ||
To explore whether Ar2PCl could catalyse P,P-metathesis, a sub-stoichiometric amount (5 mol%) of Ph2PCl was added to a chloroform solution of pure AE. The equilibration (Scheme 11) was then monitored by 31P{1H} spectroscopy and compared with the same process carried out in the absence of Ph2PCl. In the absence of Ph2PCl, equilibrium was established in ca. 70 h, whereas in the presence of 5 mol% of Ph2PCl, equilibrium was established in ca. 7 h (see ESI†).
The key observations on the P,P-metathesis reaction between E2 and A2 are that: (1) the solvent (CHCl3 or CH2Cl2) is critical for the reaction to proceed under ambient conditions; (2) the reaction has the characteristics of being radical-initiated; (c) Ar2PCl catalyses the P,P-metathesis.
It is known that CHCl3 is a source of Cl˙ radicals (especially under photolysis56–60 or sonolysis61–64) and therefore the process shown in Scheme 12 is proposed in which the role of Cl˙ is twofold: (a) to generate Ar2PCl which promotes the P,P-metathesis process (Scheme 11); (b) to form Ar2P˙ and initiate a radical chain P,P-metathesis process similar to Scheme 9. DFT studies (see ESI†) show that initiation by attack of Cl˙ on Ar2P–PAr2 generating Ar2P˙ (Scheme 12) is feasible: ΔG = −35.3 kcal mol−1 and ΔG‡ = 3.4 kcal mol−1 (from the Ph2P–PPh2⋯Cl˙ association complex).
 | ||
| Scheme 12 | ||
The slow rate of the P,P-metathesis reaction between the o-tolyl diphosphane E2 and A2 contrasts with the rapid rate of the P,P-metathesis reaction between the isomeric p-tolyl diphosphane B2 and A2. The explanation for the lower rates therefore lies in the proximity of the o-CH3 group to the P-reaction centre. We have proposed that photolytic Ph2P˙ radical formation initiates the P,P-metathesis reactions of A2 (see above) and, since the UV spectrum of E2 resembles that of A2 (see Fig. 4), both Ph2P˙ and (o-Tol)2P˙ radicals should be similarly accessible. We propose two tentative explanations for the role of the o-CH3 groups in E2. (1) The bulky o-tolyl substituents may be shielding E2 from radical attack by Ph2P˙ radicals or hindering radical attack by (o-Tol)2P˙ on A2. Some support for a steric effect of an ortho-alkyl group on the rate of radical-initiated P,P-metathesis comes from calculations of ΔG‡ for Ar2P˙ attack on Ar2P–PAr2 where Ar = Ph, o-C6H4Me or o-C6H4tBu which gave values of 5.5, 6.7 and 13.3 kcal mol−1 respectively (see ESI† for details). These show that, although the calculated ΔG‡ values for Ph and o-C6H4Me are similar, for o-C6H4tBu, the calculated value is significantly greater. (2) Alternatively, the o-CH3 groups may provide a means of quenching the radical chain by H˙ intramolecular abstraction from the proximal CH3 group to produce a benzylic radical by a process that is illustrated in Scheme 13. Calculations show that this would be highly unfavourable energetically (by 14.4 kcal mol−1) for the ground state (o-Tol)2P˙ radical but the initially formed P-centred diphosphane radical would have sufficient energy for H transfer to take place (see ESI† for details). The energetic C-centred radical could then abstract H˙ and the diphosphane reform thereby quenching the P,P-metathesis.
 | ||
| Scheme 13 | ||
Both of these explanations are speculative and further work would be required to establish their veracity.
Conclusions
DFT calculations have shown that the thermodynamic stability of unsymmetrical diphosphanes (Z2P–PZ′2), with respect to their symmetrical analogues (Z2P–PZ2 and Z′2P–PZ′2), should increase with increasing difference in the electronegativity and/or steric bulk of the PZ2 and PZ′2 constituents. In these terms, the observed stability of Me2P–P(CF3)2 and tBu2P–PPh2 can be understood.The unsymmetrical diphosphanes (Ar2P–PAr′2) have been shown to be in finely balanced equilibria with their symmetrical analogues Ar2P–PAr2 and Ar′2P–PAr′2 which reflects their insensitivity to the aryl substituents employed (Me, MeO, CF3). Significantly, the P,P-metathesis equilibria with the tetra-aryldiphosphanes were generally established rapidly (<10 min) at ambient temperatures, in a range of solvents, even in dilute solution; furthermore, the rate was increased by photolysis and inhibited by radical scavengers. The experimental and computational evidence points to the involvement of Ar2P˙ radicals generated photochemically.
Exceptional behaviour has been observed in the P,P-metathesis reactions involving (o-Tol)2P–P(o-Tol)2. These were only observed under UV-radiation or when the reactions were carried out in CHCl3 and CH2Cl2. The evidence supporting the involvement of Ar2PCl in the metatheses with (o-Tol)2P–P(o-Tol)2 in chlorinated solvents includes the effect of the presence of H2O: no P,P-metathesis is observed and the formation of hydrolysis products Ar2P(O)H is detected. By contrast, the P,P-metatheses involving other Ar2P–PAr2 proceed uninhibited by the presence of water and no Ar2P(O)H species are detected.
The facility with which diphosphanes undergo P,P-metathesis is similar to the S,S-metathesis with disulfides and Se,Se-metathesis with diselenides. This augurs well for the application of diphosphanes in dynamic covalent chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Engineering and Physical Sciences Research Council through the EPSRC Centre for Doctoral Training in Advanced Composites for Innovation and Science (grant number EP/L0160208/1). We thank the Department of Chemistry at Oxford and the Centre for Computational Chemistry in Bristol for access to computing facilities.References
- S. Ulrich, Acc. Chem. Res., 2019, 52, 510–519 CrossRef CAS PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- S. Nevejans, N. Ballard, M. Fernández, B. Reck, S. J. García and J. M. Asua, Polymer, 2019, 179, 121670 CrossRef CAS.
- H. Geng, Y. Wang, Q. Yu, S. Gu, Y. Zhou, W. Xu, X. Zhang and D. Ye, ACS Sustainable Chem. Eng., 2018, 6, 15463–15470 CrossRef CAS.
- M. Chen, L. Zhou, Y. Wu, X. Zhao and Y. Zhang, ACS Macro Lett., 2019, 8, 255–260 CrossRef CAS.
- J. Xia, T. Li, C. Lu and H. Xu, Macromolecules, 2018, 51, 7435–7455 CrossRef CAS.
- J. Liu, Y. Liu, Y. Wang, J. Zhu, J. Yu and Z. Hu, Mater. Today Commun., 2017, 13, 282–289 CrossRef CAS.
- C. Luo, X. Shi, Z. Lei, C. Zhu, W. Zhang and K. Yu, Polymer, 2018, 153, 43–51 CrossRef CAS.
- M. Irigoyen, A. Fernández, A. Ruiz, F. Ruipérez and J. M. Matxain, J. Org. Chem., 2019, 84, 4200–4210 CrossRef CAS PubMed.
- J.-C. Lai, L. Li, D.-P. Wang, M.-H. Zhang, S.-R. Mo, X. Wang, K.-Y. Zeng, C.-H. Li, Q. Jiang, X.-Z. You and J.-L. Zuo, Nat. Commun., 2018, 9, 2725 CrossRef PubMed.
- I. Azcune and I. Odriozola, Eur. Polym. J., 2016, 84, 147–160 CrossRef CAS.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- P. Frei, R. Hevey and B. Ernst, Chem. – Eur. J., 2019, 25, 60–73 CrossRef CAS PubMed.
- R. Martin, A. Rekondo, A. R. De Luzuriaga, P. Casuso, D. Dupin, G. Cabañero, H. J. Grande and I. Odriozola, Smart Mater. Struct., 2016, 25, 084017 CrossRef.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS PubMed.
- U. F. Fritze and M. von Delius, Chem. Commun., 2016, 52, 6363–6366 RSC.
- Y. Chen and R. P. Sijbesma, Macromolecules, 2014, 47, 3797–3805 CrossRef CAS.
- M. V. Encina, E. Lissi, M. Sarasúa, L. Gargallo and D. Radic, J. Polym. Sci., Polym. Lett. Ed., 1980, 18, 757–760 CrossRef CAS.
- K. L. Berkowski, S. L. Potisek, C. R. Hickenboth and J. S. Moore, Macromolecules, 2005, 38, 8975–8978 CrossRef CAS.
- H. Xu, W. Cao and X. Zhang, Acc. Chem. Res., 2013, 46, 1647–1658 CrossRef CAS PubMed.
- C. Liu, J. Xia, S. Ji, Z. Fan and H. Xu, Chem. Commun., 2019, 55, 2813–2816 RSC.
- N. K. Kildahl, J. Chem. Educ., 1995, 72, 423–424 CrossRef CAS.
- I. V. Koval, Russ. Chem. Rev., 1994, 63, 735–750 CrossRef.
- S. Burck, D. Gudat and M. Nieger, Angew. Chem., Int. Ed., 2004, 43, 4801–4804 CrossRef CAS PubMed.
- S. Burck, K. Götz, M. Kaupp, M. Nieger, J. Weber, J. S. auf der Günne and D. Gudat, J. Am. Chem. Soc., 2009, 131, 10763–10774 CrossRef CAS PubMed.
- N. Szynkiewicz, Ł. Ponikiewski and R. Grubba, Dalton Trans., 2018, 47, 16885–16894 RSC.
- D. L. Dodds, M. F. Haddow, A. G. Orpen, P. G. Pringle and G. Woodward, Organometallics, 2006, 25, 5937–5945 CrossRef CAS.
- G. Bettermann, H. Buhl, R. Schmutzler, D. Schomburg and U. Wermuth, Phosphorus Sulfur, 1983, 18, 77–80 CrossRef CAS.
- N. A. Giffin, A. D. Hendsbee, T. L. Roemmele, M. D. Lumsden, C. C. Pye and J. D. Masuda, Inorg. Chem., 2012, 51, 11837–11850 CrossRef CAS PubMed.
- D. Gudat, Acc. Chem. Res., 2010, 43, 1307–1316 CrossRef CAS PubMed.
- M. Blum, O. Puntigam, S. Plebst, F. Ehret, J. Bender, M. Nieger and D. Gudat, Dalton Trans., 2016, 45, 1987–1997 RSC.
- A. H. Cowley, Chem. Rev., 1965, 65, 617–634 CrossRef CAS.
- H. Nöth and H.-J. Vetter, Chem. Ber., 1963, 96, 1479–1484 CrossRef.
- K. Issleib and W. Seidel, Chem. Ber., 1959, 92, 2681–2694 CrossRef CAS.
- A. K. King, A. Buchard, M. F. Mahon and R. L. Webster, Chem. – Eur. J., 2015, 21, 15960–15963 CrossRef CAS PubMed.
- S. Molitor, J. Becker and V. H. Gessner, J. Am. Chem. Soc., 2014, 136, 15517–15520 CrossRef CAS PubMed.
- M. C. Fermin and D. W. Stephan, J. Am. Chem. Soc., 1995, 117, 12645–12646 CrossRef CAS.
- V. P. W. Böhm and M. Brookhart, Angew. Chem., Int. Ed., 2001, 40, 4694–4696 CrossRef.
- R. Waterman, Organometallics, 2007, 26, 2492–2494 CrossRef CAS.
- G. Fritz, in Advances in Inorganic Chemistry, ed. H. J. Emeleus and A. G. Sharpe, Academic Press, 1987, vol. 31, pp. 171–214 Search PubMed.
- K. O. Feldmann and J. J. Weigand, J. Am. Chem. Soc., 2012, 134, 15443–15456 CrossRef CAS PubMed.
- A. D. Gorman, J. A. Cross, R. A. Doyle, T. R. Leonard, P. G. Pringle and H. A. Sparkes, Eur. J. Inorg. Chem., 2019, 2019, 1633–1639 CrossRef CAS.
- N. A. Giffin, A. D. Hendsbee and J. D. Masuda, Dalton Trans., 2016, 45, 12636–12638 RSC.
- N. Szynkiewicz, L. Ponikiewski and R. Grubba, Chem. Commun., 2019, 55, 2928–2931 RSC.
- S. Kawaguchi and A. Ogawa, Asian J. Org. Chem., 2019, 8, 1164–1173 CrossRef CAS.
- R. K. Harris, E. M. Norval and M. Fild, Dalton Trans., 1979, 825–831 Search PubMed.
- A. A. M. Ali and R. K. Harris, Dalton Trans., 1988, 2775–2780 RSC.
- A. Molloy, G. Sánchez-Sanz and D. Gilheany, Inorganics, 2016, 4, 36 CrossRef.
- D. L. Dodds, J. Floure, M. Garland, M. F. Haddow, T. R. Leonard, C. L. McMullin, A. G. Orpen and P. G. Pringle, Dalton Trans., 2011, 40, 7137–7146 RSC.
- L. R. Avens, L. V. Cribbs and J. L. Mills, Inorg. Chem., 1989, 28, 205–211 CrossRef CAS.
- T. Heurich, Z.-W. Qu, S. Nožinović, G. Schnakenburg, H. Matsuoka, S. Grimme, O. Schiemann and R. Streubel, Chem. – Eur. J., 2016, 22, 10102–10110 CrossRef CAS PubMed.
- J. M. Matxain, J. M. Asua and F. Ruipérez, Phys. Chem. Chem. Phys., 2016, 18, 1758–1770 RSC.
- S. Nevejans, N. Ballard, J. I. Miranda, B. Reck and J. M. Asua, Phys. Chem. Chem. Phys., 2016, 18, 27577–27583 RSC.
- S. Ji, W. Cao, Y. Yu and H. Xu, Angew. Chem., Int. Ed., 2014, 53, 6781–6785 CrossRef CAS PubMed.
- X. Xu and P. D. Pacey, Phys. Chem. Chem. Phys., 2001, 3, 2836–2844 RSC.
- B. M. Harvey and P. E. Hoggard, Photochem. Photobiol., 2014, 90, 1234–1242 CrossRef CAS PubMed.
- K. Tsujikawa, H. Segawa, K. Kuwayama, T. Yamamuro, T. Kanamori, Y. T. Iwata and T. Ohmori, Forensic Toxicol., 2020, 38, 475–480 CrossRef CAS.
- K. E. Maudens, S. M. R. Wille and W. E. Lambert, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 848, 384–390 CrossRef CAS PubMed.
- C. Crabb and L. McDonald, US Pat, 3641169A, 1968 Search PubMed.
- S. Kawai, Yakugaku Zasshi, 1966, 86, 1125–1132 CrossRef CAS PubMed.
- H. M. Hung and M. R. Hoffmann, J. Phys. Chem. A, 1999, 103, 2734–2739 CrossRef CAS.
- V. Sáez, M. D. Esclapez, P. Bonete, D. J. Walton, A. Rehorek, O. Louisnard and J. González-García, Ultrason. Sonochem., 2011, 18, 104–113 CrossRef PubMed.
- B. Park, E. Cho, H. Park and J. Khim, Jpn. J. Appl. Phys., 2011, 50, 07HE10 CrossRef.
- M. Lim, Y. Son, J. Yang and J. Khim, Jpn. J. Appl. Phys., 2008, 47, 4123 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dt01013a |
| This journal is © The Royal Society of Chemistry 2021 |